

Abstract
Purpose: The EGFR tyrosine kinase inhibitors (TKIs) demonstrate efficacy in NSCLC patients whose tumors harbor activating EGFR mutations. However, patients who initially respond to EGFR TKI treatment invariably develop resistance to the drugs. Known mechanisms account for approximately 70% of native and acquired EGFR TKI resistance. In the current study we investigated a novel mechanism of NSCLC resistance to erlotinib. Experimental Design: The mechanisms of acquired erlotinib resistance were evaluated by microarray analysis in thirteen NSCLC cell lines and in vivo in mice. Correlations between plasma neutrophil gelatinase associated lipocalin (NGAL) levels, erlotinib response and the EGFR mutational status were assessed in advanced stage NSCLC patients treated with erlotinib. Results: In 5 of 13 NSCLC cell lines NGAL was significantly upregulated. NGAL knockdown in erlotinib-resistant cells increased erlotinib sensitivity in vitro and in vivo. NGAL overexpression in erlotinib-sensitive cells augmented apoptosis resistance. This was mediated by NGAL-dependent modulation of the pro-apoptotic protein Bim levels. Evaluation of the plasma NGAL levels in NSCLC patients that received erlotinib revealed that patients with lower baseline NGAL demonstrated a better erlotinib response. Compared to patients with wild type EGFR, patients with activating EGFR mutations had lower plasma NGAL at baseline and weeks 4 and 8. Conclusions: Our studies uncover a novel mechanism of NGAL-mediated modulation of Bim levels in NSCLC that might contribute to TKI resistance in lung cancer patients. These findings provide the rationale for the further investigations of the utility of NGAL as a potential therapeutic target or diagnostic biomarker.
Keywords: Lung cancer, effectors of apoptosis, survival factors, EGFR, erlotinib resistance
Introduction
The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), such as gefitinib and erlotinib, are therapeutic agents for management of non-small cell lung cancer (NSCLC) [1-5]. Activating mutations in the TK domain of the EGFR gene serve as major determinants of response to EGFR TKI therapy [6-9], however, clinical benefit of EGFR TKIs is also reported in patients without EGFR mutations [10]. Unfortunately, patients who initially respond to EGFR TKI treatment, invariably develop secondary resistance to these agents. Specifically, a somatic T790M mutation in exon 20 of EGFR accounts for approximately 50% of acquired erlotinib resistance in patients with activating EGFR mutations [11,12]. Other mechanisms of acquired TKI resistance include amplification of the MET proto-oncogene [13], overexpression of HER2 [14] or CXCR4 [15], increased HGF production [16], activation of IGFR1 [17], amplification of MAPK1 [18], loss of PTEN [19] and development of an EMT phenotype [20,21] that altogether are found in approximately 20% of lung cancer patients. Finally, in as many as 30% of EGFR TKI resistant lung cancers the mechanisms of resistance remain unknown [21]. Thus, a more complete understanding of mechanisms of native and acquired TKI resistance would allow for improved outcomes for NSCLC patients.
Establishment of drug-resistant lung cancer cell lines and comparative investigations with their parental cells is a useful approach to elucidate the mechanisms of acquired drug resistance [22]. We developed NSCLC cell lines with acquired erlotinib resistance by culturing the cells in the presence of increasing concentrations of erlotinib. We analyzed these cells by microarray gene expression profiling and found that LCN2 (lipocalin-2) gene that encodes the protein neutrophil gelatinase-associated lipocalin (NGAL), was highly upregulated in NSCLC cells with acquired erlotinib resistance. This gene was selected for investigation because of the known capacity of NGAL to bind gelatinase/matrix metalloproteinase-9 (MMP-9) [23] and mediate apoptosis resistance [24]. NGAL, originally identified in human neutrophils as a 25-kDa protein associated with MMP-9, belongs to the family of lipocalin proteins. This family shares a common tertiary structure that confers the ability to bind and transport a wide variety of lipophilic substances, such as retinoids, fatty acids, cholesterol and prostaglandins [25]. NGAL expression is detected in normal lung tissues [26,27] and has been found to be altered in several malignancies, where its elevation is associated with increased invasiveness and metastasis, as well as poor prognosis [28-32]. In this study, we investigated the role of NGAL in native and acquired resistance to erlotinib in NSCLC.
Materials and methods
Cell lines and cell culture
The cell lines were obtained from American Type Culture Collection (Rockville, MD). Cells were cultured at 37°C in an atmosphere of 5% CO2 in RPMI-1640 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products, Woodland, CA), 100 units/mL penicillin/streptomycin and 2 mM glutamine (Invitrogen, Carlsbad, CA). The cells were genotyped regularly (every three months) utilizing Promega Cell ID System (Promega, Madison, WI), and the number of post-genotyping passages was limited to eight. All cell lines were tested and found negative for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit; Lonza, Walkersville, MD).
Generation of NSCLC cells with acquired resistance to erlotinib
To study the mechanisms of acquired erlotinib resistance, thirteen NSCLC cell lines were cultured in the culture medium described above supplemented with increasing concentrations of erlotinib to develop acquired resistance. The starting concentration of erlotinib was 1.5 μM and as soon as the cells demonstrated no growth disadvantage in erlotinib-containing medium, the concentration of the drug was increased by 1 μM to a final concentration of 20 μM. At this point the cells were tested for erlotinib IC50 (H441 - 18.3 μM, up from 3.3 μM for the parental cells and H358 - 10.8 μM, up from 2.8 μM) and used in subsequent experiments.
Microarray expression profiling
5 x 105 parental and erlotinib-resistant H441 cells were plated in 192 cm2 flasks in duplicates and incubated for 72 h. Cells were washed twice with PBS and RNA was isolated using RNeasy Mini kit (Qiagen, Valencia, CA) with the on-column DNase digestion according to Qiagen protocol. RNA quantity and quality were assessed utilizing Agilent 2100 Bioanalyzer prior to microarray experiments. Microarray analysis was performed using Affymetrix U133 Plus 2 chips. Microarray data was analyzed using dChip and R software packages.
Analysis of erlotinib-resistant cells for secondary somatic EGFR mutations
Cells with acquired erlotinib resistance were tested for the presence of secondary somatic T790M mutation in exon 20 of EGFR. Exon 20 of EGFR was amplified by PCR utilizing 10 ng of genomic DNA isolated from cells with acquired erlotinib resistance. The PCR fragment was isolated and sequenced. The sequence analysis demonstrated the absence of T790M mutation in these cells. PCR primer sequences: forward 5’-ATCGCATTCATGCGTCTTCA-3’, reverse 5’-ATCCCCATGGCAAACTCTTG-3’.
Antibodies and reagents
Erlotinib was provided by OSI Pharmaceuticals (Farmingdale, NY). Antibodies against Bim, phospho-Bim (Ser69), PARP, p44/42 MAPK, phospho-p44/42 MAPK, Akt, phospho-Akt and apoptotic proteins (Bcl-2, Bcl-xL, Mcl-1, Puma, Bad, Bax) were purchased from Cell Signaling (Danvers, MA). NGAL antibody utilized for Western blotting was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, catalog number sc-57517, dilution 1:250). GAPDH antibody was from Advanced ImmunoChemical (Long Beach, CA). U0126 was purchased from Calbiochem (Gibbstown, NJ). PD98059, MG132 and emetine were purchased from Sigma (Saint Louis, MO). Recombinant human NGAL and the NGAL antibody utilized for immunohistochemical staining were purchased from R & D Systems (Minneapolis, MN).
Plasmids and stable cell lines
Human full-length cDNA clone of NGAL (in pCMV-XL4 vector) and NGAL shRNA constructs (in pRS vector, catalog # TR311777) were purchased from OriGene Technologies (Rockville, MD). NGAL cDNA was subsequently cloned into the pLXSN retroviral vector (Clontech Laboratories, Mountain View, CA). To generate NGAL overexpressing or NGAL shRNA stable cell lines, pLXSN-NGAL or pRS-shRNA and packaging virus were co-transfected into HEK 293T cells to generate viruses that were subsequently utilized to infect NSCLC cells. The cells were selected with G418 (for pLXSN) or puromycin (for pRS), and the expression of NGAL was assessed by Western blot. One of the four NGAL shRNA constructs utilized, construct # T1347102, significantly suppressed NGAL expression. The shRNA sequence of this construct is: 5’-GAGAACCAAGGAGCUGACUUCGGAACUAA-3’.
Western blotting
Cells were washed with PBS and lysed with RIPA buffer 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholic acid, 1 mM EDTA, 1 mM PMSF, 1X Complete protease inhibitor cocktail (Roche, Penzberg, Germany). Twenty μg of protein were loaded per lane. Cell lysates were separated by SDS-PAGE and transferred to the Immobilon-P Transfer Membrane (Millipore, Billerica, MA). Membranes were blocked with 5% milk and then incubated with primary antibodies diluted in blocking solution. Horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) and enhanced chemiluminescence (ECL) reagent (Amersham Biosciences, Piscataway, NJ) were used for protein detection.
NGAL ELISA
A human lipocalin-2/NGAL ELISA kit was purchased from R & D Systems (catalog # DLCN20). The assays were performed according to the manufacturer’s protocol. Typically, 50 μL of cell culture medium or diluted plasma was used for each assay. NGAL protein concentration was compared between cell lines using two-sample t-tests.
Flow cytometry
Cells were plated in T25 flasks, cultured overnight and then treated with erlotinib for the indicated times. Following treatment, both the floating cells and the adherent cells were collected, stained with Annexin V and PI using FITC Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s protocol and analyzed using FACScan instrument (BD Biosciences).
Real-time PCR
Cells were cultured under standard conditions, and total RNA was isolated utilizing the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA (1 μg) was then reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). The resulting cDNA samples were used in real-time PCR analysis of Bim (forward primer: 5’-agtgggtatttctcttttgacacag-3’, reverse primer: 5’-tcaatgccttctccataccagacg-3’) and β-actin (forward primer: 5’-gatgagattggcatggcttt-3’, reverse primer: 5’-caccttcaccgttccagttt-3’) utilizing IQ SYBRGreen Supermix (Bio-Rad). Expression of Bim was normalized to β-actin expression for each sample, and then fold changes were determined by comparing vector control and NGAL cells. Each assay was performed in triplicate, and the results of one representative experiment are shown.
Transfection of siRNA
Cells were plated in 12-well plates and cultured overnight. Cells were then transfected with Bim siRNA (catalog # 6461, Cell Signaling Technology) or negative control siRNA (Silencer Negative Control # 1 siRNA, Ambion Austin, TX) at 3.2 μg/well utilizing TransMessenger Transfection Reagent (Qiagen). Cells were harvested 48 hours later and analyzed by Western blot.
Murine studies
Mice were maintained in the West Los Angeles Veterans Affairs Animal Research vivarium. The animal studies institutional review board (IRB) approved all studies. In brief, 8 x 106 tumor cells (Vector control and NGAL overexpressing) were injected subcutaneously into the right suprascapular area of 8- to 10-week-old SCID mice. Ten days post-tumor inoculation, mice were treated with erlotinib (0.5 mg/kg or 1 mg/kg) or normal saline diluent daily by gavage for the duration of the experiment. Tumor volumes were monitored by measuring two bisecting diameters of each tumor with calipers. Tumor volumes were calculated using the formula: V = 0.4ab 2, with a as the larger diameter and b as the smaller diameter. We utilized a mixed effects model to examine the effects of cell type and erlotinib dose on tumor growth over time. This model included fixed effects of cell type, dose, time and the interactions of the main effects.
Immunohistochemistry
Immunohistochemical staining for NGAL was performed utilizing formalin-fixed, paraffin-embedded tissues from the UCLA Specialized Programs of Research Excellence (SPORE) in Lung Cancer tissue bank and pathology department archives. Tissue sections (4 μM thick) were cut, deparaffinized in xylene, rehydrated in alcohol, and washed twice with de-ionized H2O (dH2O). The tissues were then steamed in EDTA buffer (10 mM, pH 8.0) for 30 minutes to unmask antigens. Following cooling to room temperature and rinsing with dH2O, specimens were treated for 30 minutes with 3% H2O2 in PBS containing 1% NaN3. They were then washed with PBS containing 0.05% Tween 20 and blocked with 10% normal horse serum (NHS) (Vector Laboratories, catalog # S-2000) for 30 minutes. After blocking, the tissues were incubated with anti-NGAL antibody (catalog # AF1757, R & D Systems) diluted 1:100 in 3% NHS overnight at 4°C. They were then washed with PBS and incubated with biotinylated horse anti-goat IgG antibody (catalog # BA-9500, Vector Laboratories) diluted 1:200 in 3% NHS for 40 minutes at room temperature. After the incubation, specimens were washed with PBS and incubated with horseradish peroxidase avidin D (Vector Laboratories, catalog # A-2400)diluted 1:1000 in PBS for 30 minutes at room temperature. The tissues were then washed with PBS and incubated with DAB peroxidase substrate (Vector Laboratories, catalog # SK-4100) for 10 minutes. Next, the reaction was stopped by washing with dH2O and counterstained with dilute hematoxylin for 6 seconds, followed by dehydration with alcohol and xylene and mounting with coverslips.
Human plasma samples
Human plasma samples were collected from 107 patients in a Phase II trial designed to evaluate the efficacy of the combination of erlotinib plus celecoxib compared to erlotinib alone in patients with stage IIIB or IV NSCLC [33]. Samples were collected at City of Hope (COH) in accordance with the COH IRB requirements, and all patients provided written informed consent. Plasma samples were collected in green top Vacutainer tubes, centrifuged at 3,000 rpm for 15 minutes within one hour of collection, and supernatants were collected, aliquoted and stored at -80°C. Samples collected at baseline and during 4- and 8-week visits were utilized to measure NGAL levels by ELISA.
Statistical analysis
The Wilcoxon rank sum test was used to compare subject’s NGAL levels, as well as changes in NGAL levels between response groups. For the samples from the Phase II study, we constructed receiver operating characteristic (ROC) curves for baseline and 4-week visit NGAL values versus 8-week treatment response categories PR versus SD+PD. The area under the ROC curve (ROC AUC) was computed via numerical integration. Statistical analyses were performed using R version 2.13.1.
Results
NGAL is elevated in NSCLC cell lines with acquired erlotinib resistance
Analysis of gene expression of parental and erlotinib-resistant NSCLC cells by microarray revealed that among the genes potentially relevant in TKI resistance, the LCN2 (lipocalin-2) gene was upregulated 2-200-fold in 5 out of 13 cell lines (Figure 1A). To confirm this result, we performed Western blotting and found that the levels of the LCN2 protein product, NGAL, were also significantly increased in NSCLC cells with acquired resistance to erlotinib (Figure 1B). Because NGAL is as a secreted protein, we measured its levels in cell culture supernatants by ELISA and found that they were also elevated in erlotinib-resistant cell supernatants (Figure 1C).
Figure 1.
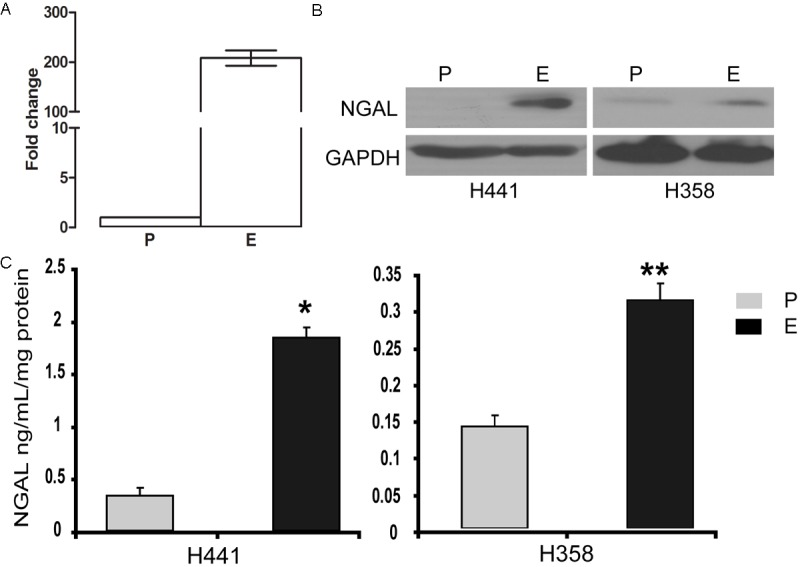
NGAL is increased in erlotinib-resistant cell lines. A: LCN2 gene expression analysis by microarray in H441 cells: P-parental cells, E-cells with acquired erlotinib resistance. B: NGAL protein levels are elevated in H358 and H441 cells with acquired resistance to erlotinib (E) compared to the respective parental cells (P) by Western blot. C: The same cells were cultured for 48 hours and secreted NGAL protein levels were assessed by ELISA. NGAL protein levels were significantly higher in erlotinib-resistant cells than in the parental cells (*p = 0.03, **p = 0.02). Error bars indicate standard deviation (SD). NGAL concentration was normalized by total protein concentration.
NGAL overexpressing NSCLC cell lines demonstrate heightened resistance to erlotinib-induced apoptosis
To study the role of NGAL in erlotinib resistance in both EGFR mutant and wild type cell lines, NGAL overexpressing H3255 (L858R activating EGFR mutation) and H441 (wild type EGFR) NSCLC cell lines were generated. NGAL overexpression was confirmed by Western blot and ELISA (Figure 2A and 2B respectively). Interestingly, H3255 cells bearing an activating EGFR mutation, consistently expressed lower levels of NGAL than the wild-type EGFR H441 cells (Figure 2C). To determine if NGAL mediates erlotinib resistance, cells were treated with erlotinib, and PARP cleavage was evaluated as an indicator of apoptosis. Erlotinib treatment resulted in prominent PARP cleavage in parental and vector control cells, but in contrast the apoptotic response was decreased or abolished in NGAL overexpressing cells (Figure 2D), suggesting that increased NGAL confers resistance to erlotinib-induced apoptosis in NSCLC cells. To further confirm this result, we also assessed apoptosis in erlotinib-treated cells by Annexin V and PI staining and flow cytometry. Compared to vehicle control-treated cells, the apoptosis levels in response to erlotinib treatment were significantly lower in NGAL overexpressing cells (Figure 2E). These data suggest that an NGAL-dependent erlotinib resistance mechanism may be operative in NSCLC cells with either wild type or mutated EGFR.
Figure 2.
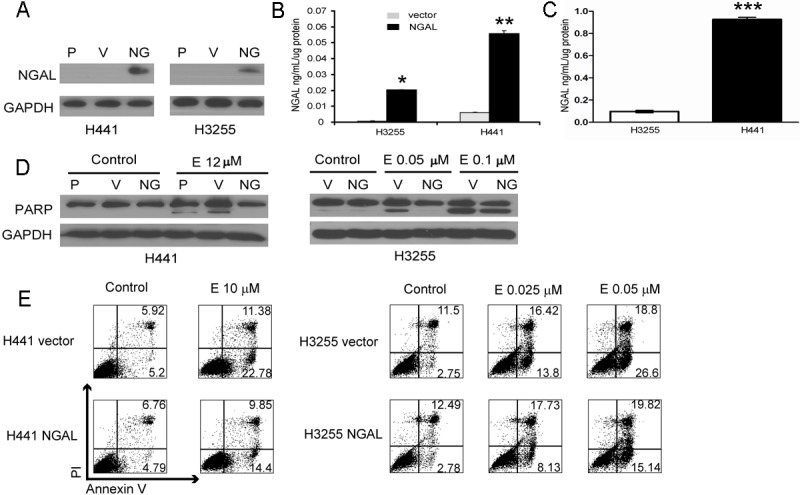
Ectopic expression of NGAL leads to increased apoptosis resistance in NSCLC cells. A: NGAL levels are elevated in in cell lysates from H441 and H3255 cells with ectopic NGAL expression (NG) compared to parental (P) and vector control (V) cells. B: Supernatants from the same cells were tested for NGAL levels by ELISA and demonstrated elevated secreted NGAL levels; *p ≤ 10-8, **p ≤ 10-5. C: Supernatants from H3255 cells that bear activating EGFR mutation and are highly sensitive to erlotinib, have significantly lower NGAL levels compared to H441 cells that have the wild-type EGFR (***p < 0.0001). D: Parental (P), vector control (V) and NGAL overexpressing (NG) cells were treated with the vehicle control (Control) or erlotinib (E) at indicated concentrations for 24 (H3255) or 48 hours (H441) and analyzed for cleaved PARP as an indicator of apoptosis by Western blot. Reduced PARP cleavage in response to erlotinib treatment was observed in NGAL-overexpressing cells. E: The same cells treated as described above were then stained with PI and Annexin V and assessed for apoptosis by flow cytometry. Apoptosis resistance was significantly heightened in NGAL-overexpressing cells.
NGAL mediates apoptosis resistance by decreasing Bim protein levels
Bcl-2 interacting mediator of cell death (Bim) is a pro-apoptotic protein that contains the BH3 domain and belongs to Bcl-2 family of proteins [34]. Bim activates apoptosis by binding to and antagonizing the activity of anti-apoptotic members of the Bcl-2 family and is known to have three major isoforms generated by alternative splicing and designated EL (extra-long), L (long) and S (short); ectopic expression of Bim isoforms leads to cell death via a caspase-dependent mechanism [34]. Recent studies identified Bim as one of the most important effectors of apoptosis induced by TKIs, including erlotinib [35-38], and BIM silencing can reverse gefitinib-induced apoptosis [39]. To determine whether Bim is involved in NGAL-dependent resistance to erlotinib, we first assessed Bim protein levels in H3255 and H441 NGAL overexpressing cells by Western blotting. In both NGAL overexpressing cell lines the total Bim protein levels were decreased compared to the vector control cells (Figure 3A), indicating that NGAL overexpression potently reduces Bim protein levels. To further clarify the role of Bim in NGAL-mediated erlotinib resistance, Bim expression was knocked down with siRNA in H3255 and H441 cells, and apoptosis sensitivity to erlotinib was evaluated by measuring PARP cleavage. We found that Bim knockdown resulted in significant reduction of erlotinib-induced apoptosis (Figure 3B), suggesting that Bim is an essential mediator of erlotinib-induced apoptosis and that its downregulation by NGAL might confer NSCLC cells resistant to erlotinib. Furthermore, to assess the potential role of Bcl2 family proteins in NGAL-mediated erlotinib-induced apoptosis resistance, we evaluated the expression of anti-apoptotic proteins Puma, Bad and Bax as well as Bim binding partners Bcl-2, Bcl-xL and Mcl-1, in NGAL overexpressing cells, and found that none of these proteins were modulated by NGAL over-expression (Figure 3C).
Figure 3.
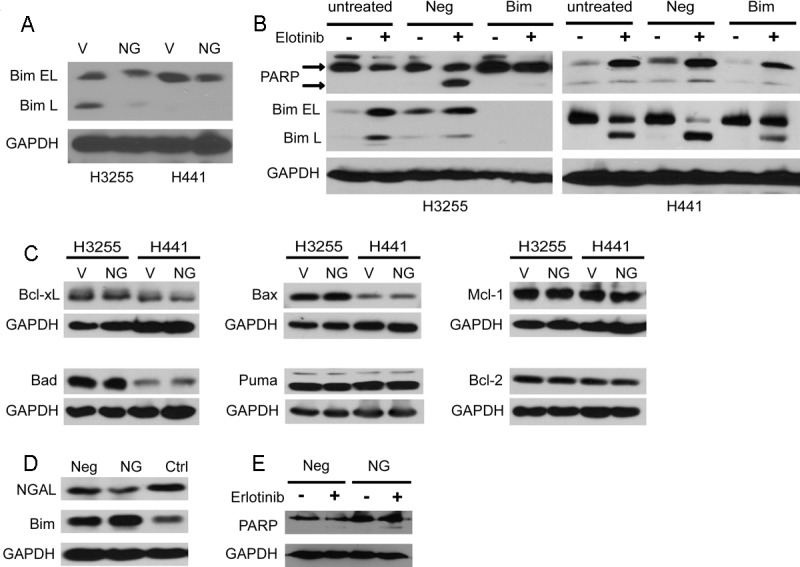
NGAL-mediated decrease of Bim protein is responsible for apoptosis resistance. A: Bim expression is downregulated in NGAL overexpressing H3255 and H441 cells (NG) compared to vector control cells (V). Bim EL: Bim extra-long isoform; Bim L: Bim long isoform. B: H3255 and H441 cells were either untreated or transfected with negative control siRNA (Neg) or Bim siRNA (Bim). After 48 hours, cells were treated with erlotinib (8 μM for H441 and 0.05 μM for H3255) or vehicle control (DMSO) for 24 hours. Cell lysates were analyzed for Bim and PARP cleavage by Western blot. C: Expression of apoptosis effector proteins Bcl-xL, Bax, Mcl-1, Bad, Puma and Bcl-2 NGAL is not altered in NGAL overexpressing (NG) H3255 and H441 cells compared to vector control (V). D: NGAL knockdown by shRNA leads to an increase in Bim levels in H358 erlotinib-resistant cells. Neg - negative control shRNA; NG-NGAL shRNA; Ctrl - non-transduced cells. E: NGAL knockdown increases the apoptosis sensitivity of cells with acquired resistance to erlotinib. H358 erlotinib-resistant cells were transduced with NGAL shRNA (NG) or negative control shRNA (Neg) and treated with erlotinib (10 μM) for 72 hours. Cell lysates were analyzed for cleaved PARP protein by Western blot.
Down-regulation of NGAL expression decreases resistance to erlotinib-induced apoptosis
To further confirm the role of NGAL in conferring apoptosis resistance to cells with acquired resistance to erlotinib, we knocked down NGAL expression in erlotinib-resistant H358 cells by shRNA. Decreased NGAL levels were observed in the cell lysates of NGAL shRNA cells compared to negative control shRNA-transduced and non-transduced erlotinib-resistant H358 cells. Similarly, Bim protein levels were increased in NGAL shRNA cells (Figure 3D). When these cells were treated with erlotinib, apoptosis resistance was decreased in NGAL shRNA cells compared to negative control shRNA cells as measured by PARP cleavage (Figure 3E).
NGAL-dependent decrease of Bim protein in NSCLC cells is mediated by the ERK pathway
The MAPK/Erk pathway is one of the major signaling cascades activated following EGFR stimulation. In addition, this pathway has been reported to regulate Bim protein levels, predominantly by regulating Bim protein stability and apoptotic activity [40-42]. These studies have demonstrated that phosphorylation of Bim serine 69 (Ser69) by Erk targets the protein for proteasomal degradation. To determine whether the MAPK/Erk pathway is involved in NGAL-mediated reduction of Bim protein levels, the phosphorylation status of Bim Ser69 was determined in NGAL overexpressing and vector control H3255 and H441 cells. Whether cultured under serum-containing or serum-free conditions, Erk phosphorylation was increased in NGAL overexpressing cells compared to vector control cells (Figure 4A). Erlotinib treatment led to a notable decrease in Erk phosphorylation and an increase in Bim protein levels. The increase of Bim occurred at earlier time points and to a greater extent in control cells compared to NGAL overexpressing cells (Figure 4A). To further confirm that the MAPK/Erk pathway is involved in NGAL-dependent Bim downregulation, NGAL overexpressing H3255 and H441 cells were treated with the MEK inhibitors U0126 or PD98056. Inhibition of the MAPK/Erk pathway increased the total Bim protein and concomitantly reduced the phosphorylated Bim levels (Figure 4B and 4C). Taken together, these results suggest that NGAL promotes downregulation of Bim by enhancing Erk activation. To confirm that Bim was degraded via the ubiquitin-proteasome pathway, H3255 and H441 NGAL overexpressing and vector control cells were treated with the proteasome inhibitor, MG132, together with the protein synthesis inhibitor, emetine. Emetine treatment alone caused a reduction in Bim protein levels. However, with the addition of MG132, Bim protein levels were increased, and the extent of the increase was greater in NGAL overexpressing cells compared to vector control cells (Figure 4D), suggesting that NGAL-dependent reduction of Bim protein levels was mediated by Bim phosphorylation by Erk and subsequent degradation via the ubiquitin-proteasome pathway. Furthermore, we investigated the effect of exogenous NGAL on Bim levels and Erk activation in NSCLC. Our results indicate that addition of recombinant NGAL to the culture medium induces Erk phosphorylation and concomitantly decreases Bim protein levels in H3255 and H441 cells (Figure 4E and 4F), consistent with our results in NGAL overexpressing cell lines.
Figure 4.
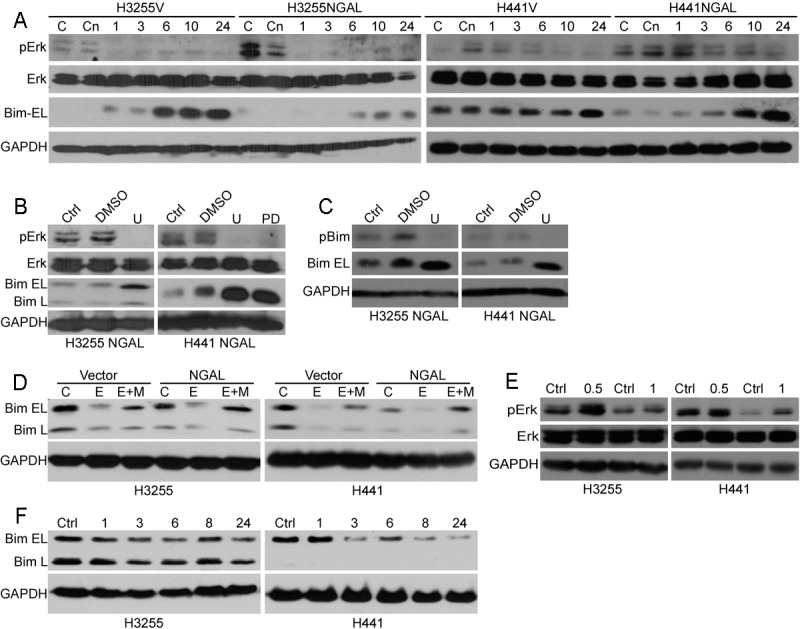
NGAL confers erlotinib resistance by enhancing ERK pathway activity and decreasing Bim protein levels. A: H3255 and H441 cells ectopically expressing NGAL (NGAL) and vector controls (V) were treated with erlotinib (0.1 μM for H3255 and 10 μM for H441) or vehicle control (DMSO) for 1-24 hours, as indicated. Cell lysates were analyzed for Erk pathway activation and Bim protein levels by Western blot. C - untreated cells in medium supplemented with 10% FBS. Cn - untreated cells in serum-free medium. B: NGAL overexpressing H3255 and H441 cells were treated with the MEK inhibitors U0126 (U, 10 μM) or PD98059 (PD, 40 μM) or the diluent (DMSO) for 24 hours. Cell lysates were analyzed for Erk pathway activation and Bim protein levels by Western blot. Ctrl-untreated cells. C: The same cells were treated with MEK inhibitor U0126 (U, 10 μM) or diluent (DMSO) for 4 hours, and the cell lysates were analyzed for phospho-Bim (pBim) and Bim extra-long isoform (Bim EL) levels by Western blot. D: NGAL overexpressing (NGAL) and vector control (Vector) H3255 and H441 cells were treated with emetine (E, 10 μM) or emetine plus MG132 (E+M, 100 μM) for 5 hours in serum-free medium. Cell lysates were analyzed for Bim isoform levels by Western blot. C - diluent (DMSO) treated control cells. E: H3255 and H441 cells were serum-depleted for 2 hours and treated with recombinant NGAL (105 ng/mL) in serum-free medium for 0.5 and 1 hour. Cell lysates were analyzed for Erk activation by Western blot. F: The cells were treated with recombinant NGAL (105 ng/mL) for 1-24 hours as indicated, and cell lysates were analyzed for Bim isoform levels by Western blot. Ctrl - IgG control.
NGAL confers resistance to erlotinib in vivo
To determine the effect of NGAL on erlotinib resistance in vivo, H3255 vector control or NGAL overexpressing cells were injected subcutaneously into SCID mice. Ten days post tumor inoculation the mice were treated with erlotinib or diluent. Tumors derived from vector control and NGAL overexpressing cells had similar growth curves when mice were treated with diluent. Administration of erlotinib led to a significant reduction in tumor volume in vector control cells-derived tumors, whereas no reduction in tumor volume was observed in tumors derived from NGAL overexpressing cells (Figure 5A). After mice were euthanized, the expression levels of NGAL, Bim, phospho-Erk and phospho-Akt were measured in tumors. We found that consistent with our in vitro results, Bim was decreased and Erk phosphorylation was increased in tumors derived from NGAL overexpressing cells (Figure 5B). Additionally, in contrast to our in vitro results (data not shown), Akt phosphorylation was increased, indicating that Akt pathway activation may be an additional mediator of erlotinib resistance in vivo (Figure 5B).
Figure 5.
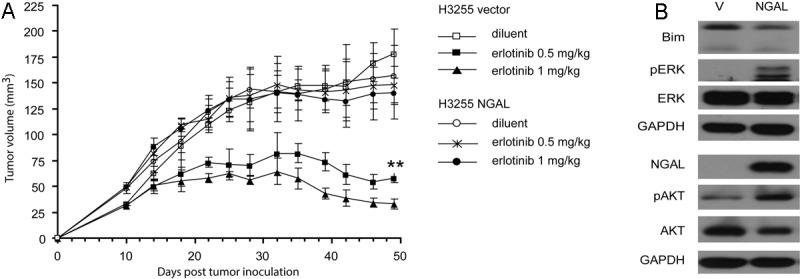
NGAL confers resistance to erlotinib-induced tumor growth inhibition in SCID mice. A: H3255 Vector control and NGAL overexpressing cells were injected subcutaneously into SCID mice (5 mice per group). Ten days post-tumor inoculation, mice were treated with erlotinib at 0.5 mg/kg or 1 mg/kg daily, and tumor volumes were measured. Tumors derived from NGAL overexpressing cells were significantly more resistant to erlotinib treatment than vector control-derived tumors (**p < 0.001). Error bars represent the standard deviation. B: After mice were sacrificed, the diluent-treated tumors were homogenized, and tumor lysates were analyzed for Erk and Akt pathway activation and NGAL and Bim protein by Western blot.
NGAL is expressed in human NSCLC in situ
To confirm that NGAL is expressed in human lung cancer, we performed immunohistochemical analysis of NGAL expression in lung cancer tissues. We found that NGAL is expressed in both adenocarcinomas (Figure 6A-D) and squamous cell carcinomas (Figure 6E and 6F). NGAL staining was also detected in the regions surrounding lung cancer cells in mucinous invasive adenocarcinoma. This pattern is consistent with our studies of NGAL overexpressing cell lines in which we found increased NGAL levels, both intracellularly and secreted into the culture medium.
Figure 6.
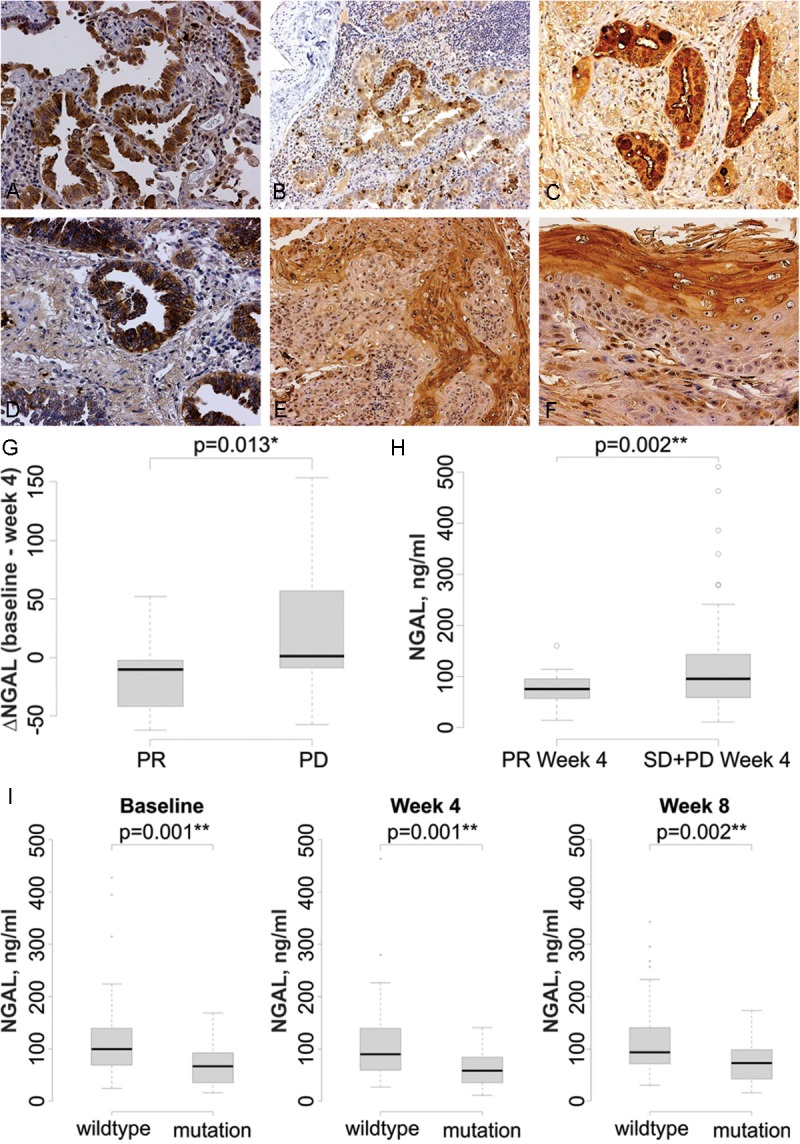
NGAL is detected in lung cancer specimens and is elevated in patients who do not respond to erlotinib. NGAL expression is detected by immunohistochemistry in the in situ component of adenocarcinoma (A) and the invasive component of adenocarcinoma (B). C: NGAL is present in the cytoplasm of mucin-producing cells, as well as in the surrounding regions, in a mucinous invasive adenocarcinoma. D: Strong cytoplasmic NGAL staining in non-mucinous invasive adenocarcinoma. E: Invasive squamous cell carcinoma. F: Greater expression in the keratinizing portion of an invasive squamous cell carcinoma. The magnification is 200X in A, C, D and F and 100X in B and E. G. Change in serum NGAL levels after erlotinib treatment. Following 4 weeks of treatment NGAL levels were significantly decreased in the PR group and increased in the PD group. H: In patients that had a PR, the 4 week plasma NGAL level is significantly lower than that in SD+PD patients (p = 0.002). I: Plasma NGAL levels are lower in patients with mutant EGFR at baseline (**p = 0.001), week 4 (**p = 0.001) and week 8 (***p = 0.002) compared to patients with wild type EGFR.
Lung cancer patients who fail to respond to erlotinib demonstrate elevated NGAL plasma levels
To further investigate the ability of NGAL to function as a marker for resistance to TKI therapy, we evaluated plasma samples collected from a randomized phase II trial that evaluated treatment with erlotinib in advanced stage NSCLC [33]. We measured NGAL levels by ELISA in plasma collected prior to therapy and following 4 and 8 weeks of erlotinib treatment in 107 patients (Table 1). After 4 weeks of treatment the partial response (PR) group demonstrated a significant reduction of NGAL compared to the pre-treatment levels, whereas in the progressive disease (PD) group NGAL was significantly elevated (Figure 6G). Furthermore, the absolute NGAL levels after 4 weeks were significantly elevated in the plasma of patients who did not have a PR (Figure 6H). These data suggest that NGAL may have a role in regulating the response to erlotinib in NSCLC patients.
Table 1.
Patient characteristics
| Total (%) | |
|---|---|
| Gender | |
| Female | 57 (53.3) |
| Male | 50 (46.7) |
| Age at randomization | |
| Mean ± SD | 63.3 ± 10.1 |
| Median | 65 |
| Min-Max | 30-81 |
| Race | |
| White | 72 (67.3) |
| Asian | 26 (24.3) |
| Black | 5 (4.7) |
| American Indian | 1 (0.9) |
| UK/Not reported | 3 (2.8) |
| Ethnicity | |
| Hispanic | 7 (6.5) |
| Non-Hispanic | 100 (93.5) |
| Smoking status | |
| Never | 40 (37.4) |
| Currently | 6 (5.6) |
| Previously | 61 (57.0) |
| ECOG | |
| 0 | 52 (48.6) |
| 1 | 55 (51.4) |
| Stage | |
| IIIb | 10 (9.3) |
| IV | 97 (90.7) |
| Previous Treatment | |
| Surgery | 89 (84.0) |
| Chemo | 94 (87.8) |
| Radiation | 45 (42.1) |
| Clinic | |
| 01 | 97 (90.6) |
| 05 | 10 (9.4) |
| Histology | |
| Adenocarcinoma | 64 |
| NSCLC, NOS | 31 |
| Squamous | 11 |
| Unknown | 1 |
| EGFR mutation status | |
| Mutation positive | 26 |
| Wild type | 58 |
| Unknown | 23 |
NGAL plasma level correlates with EGFR mutation status
It has been reported that NGAL expression is down-regulated by EGFR activation in pancreatic and ovarian cancer [43,44]. We found that plasma collected from NSCLC patients with activating EGFR mutations from the Phase II trial (n = 26) described above had lower NGAL levels at baseline and weeks 4 and 8 compared to patients with wild type EGFR (Figure 6I).
Discussion
Our studies of gene expression in NSCLC cells with acquired resistance to erlotinib revealed a strong upregulation of LCN2 gene encoding the protein NGAL in erlotinib-resistant cells, which was also confirmed by Western blotting. These cells did not bear secondary EGFR mutations that might confer acquired resistance to TKIs including erlotinib [6,11]. Similarly, our analysis of the microarray data did not reveal upregulation of c-Met, HER2 or CXCR4 that has been associated with the acquired resistance to TKIs [13-15]. Our subsequent experiments utilizing NSCLC cell lines with ectopic expression or shRNA knockdown of NGAL, indicated that elevated NGAL expression leads to increased resistance to erlotinib induced apoptosis, while downregulation of NGAL augments erlotinib sensitivity in NSCLC. Ectopic expression of NGAL in H3255 cells that bear an activating L858R EGFR mutation and are sensitive to EGFR TKIs led to a significant reduction of their sensitivity to erlotinib.
We investigated the mechanism of this effect and found that it was mediated by NGAL-dependent modulation of the pro-apoptotic protein Bim. The levels of Bim were decreased in NGAL overexpressing cells and increased when NGAL expression was suppressed. Similarly, treatment of the cells with exogenous NGAL led to a decline of Bim levels and concomitantly induced Erk phosphorylation. Expression of several other important apoptosis effector proteins was not altered in response to NGAL. Bim phosphorylation by Erk has been reported to target it for ubiquitination and subsequent proteasomal degradation [42]. In accord with these studies, we observed increased Erk1/2 phosphorylation with resultant decreased total Bim protein both in NGAL overexpressing cells and in the cells treated with exogenous NGAL. Treatment with MEK inhibitors led to a decrease in pErk and pBim levels and an increase in total Bim protein. In addition, treatment of the cells with proteasome inhibitors increased total Bim protein levels, and this effect was more pronounced in NGAL overexpressing cells. Taken together, these data strongly support the observation that NGAL decreases Bim levels by promoting Erk phosphorylation and protein turnover. Previous reports also implicated the PI3K/Akt pathway in Bim regulation at the level of transcription via phosphorylation-dependent inactivation of the transcription factor FOXO3a, which is required for BIM gene expression [45,46]. However, we did not observe similar Akt activation by NGAL in vitro.
Consistent with the studies describing Bim as a major effector of TKI-induced apoptosis [35-38], our experiments demonstrated a significant attenuation of erlotinib-induced apoptosis in NSCLC cells by Bim knockdown. In accord with our in vitro data, we found the NGAL-mediated decrease of Bim protein levels and increase of Erk phosphorylation in lysates of NGAL overexpressing xenograft tumors, suggesting that similar mechanisms accounted for erlotinib resistance in vivo. Interestingly, in contrast to our in vitro results, Akt phosphorylation was increased in vivo in tumors derived from NGAL overexpressing cells, which could be due to contributions from the tumor microenvironment.
Together, the in vitro and in vivo studies suggest that the NGAL-mediated suppression of Bim protein may contribute to failure to respond to erlotinib treatment. This notion is supported by previous reports indicating that secondary EGFR mutations [35] or deletion polymorphism of the BIM gene [47] attenuated Bim expression, which correlated with inferior response to TKIs. Given the fact that aberrant Erk and Akt signaling have been identified as major determinants of EGFR TKI sensitivity [48], and we did not find Bim RNA upregulation in our microarray studies, our results suggest that the predominant mechanism of Bim regulation in NSCLC is via NGAL/Erk-dependent modulation of Bim protein turnover. Thus, our studies document an inverse relationship between Bim and NGAL levels in NSCLC and define the mechanisms of this relationship. Because the cause of EGFR TKI acquired resistance is unknown in approximately one third of cases, we posit that NGAL upregulation may represent one of these as yet undefined mechanisms of resistance.
Previous reports indicate that mucin-producing in situ adenocarcinoma of the lung show the strongest NGAL staining by immunohistochemistry, with other adenocarcinomas showing moderate to strong staining. Squamous cell and large cell carcinomas were either negative or weakly positive [27]. NGAL expression can also be detected in other cells or tissues including neutrophils and lymph nodes [27]. To further investigate the pattern of NGAL expression in NSCLC we performed immunohistochemical staining of NSCLC specimens for NGAL and found that it was significantly upregulated in adenocarcinomas and, to a lesser extent, in squamous cell carcinomas, as well as in directly adjacent non-neoplastic tissues. Importantly, the highest levels of NGAL were detected in aggressive invasive carcinomas.
By evaluation of the NGAL levels in plasma of lung cancer patients that were treated with erlotinib we found correlation of these levels with the response to the drug and other clinical parameters. Utilizing the specimens from a Phase II trial, we found that patients that had lower NGAL levels at baseline, demonstrated a better overall response to erlotinib treatment. Furthermore, the patients whose NGAL levels increased at weeks 4 and 8 of erlotinib therapy, were found to have a poorer tumor response than those whose NGAL levels decreased or did not change. These findings were corroborated by the analysis of NGAL levels in additional 22 patients with advanced NSCLC who were treated with erlotinib [49] (data not shown). We also evaluated NGAL levels in patient samples collected in the ECOG 3503 trial designed to identify downstream markers of EGFR linked signaling pathways that are predictive of response to erlotinib [50]. Analysis of this latter sample set did not show any correlation between NGAL levels and erlotinib response, possibly because only 3 patients out of 61 had a complete or partial response. Our findings indicate that NGAL levels in patients correlate with the EGFR mutational status. In addition, if NGAL in the circulation is produced by the tumor cells, the reduction of NGAL levels following treatment could reflect a reduction in tumor burden. Similarly, plasma might not adequately reflect the fluctuations of intratumoral NGAL levels and therefore might not constitute the best means to assess this parameter. Also, although our initial results are significant, the changes of plasma NGAL levels were modest as well as the number of patients analyzed were insufficient to firmly establish the clinical significance of NGAL testing. Thus, additional studies analyzing larger cohorts of patients are needed to determine the clinical significance of NGAL as an early biomarker of response to erlotinib in NSCLC.
We next assessed if the EGFR mutational status of the tumors correlated with the patient’s NGAL levels. We found that patients whose tumors bore activating EGFR mutations, had lower plasma NGAL levels at baseline as well as weeks 4 and 8 compared to patients with wild type EGFR. Concomitantly, the former patients demonstrated a better clinical response to erlotinib treatment, however, we were unable to separately assess the inputs of reduced NGAL levels and the activating mutations in enhanced sensitivity to erlotinib.
In summary, we investigated NGAL-mediated erlotinib resistance in NSCLC. We found that both NGAL overexpression and treatment with exogenous NGAL activated Erk phosphorylation and led to a reduction of Bim protein levels in NSCLC cells, resulting in diminished sensitivity to erlotinib-induced apoptosis. These findings were further confirmed in a murine xenograft tumor model, in which we observed enhanced erlotinib resistance in tumors with ectopic expression of NGAL. Thus, our results highlight a potential NGAL/Bim-dependent mechanism of TKI resistance that is operative in a subset of patients with advanced NSCLC. These findings suggest that interference with NGAL-mediated Bim downregulation may be beneficial in overcoming TKI resistance. The possibilities of such interference will require further investigation.
Acknowledgements
This study was supported by the following grants: NIH/National Center for Advancing Translational Science UCLA CTSI Grant Number UL1TR000124, UCLA SPORE in Lung Cancer P50 CA90388, Tobacco Related Disease Program # 15RT-0152, UCLA Training Tumor Immunology Training Grant NIH/NCI T32-CA009120-36 and American Thoracic Society/LUNGevity Foundation Research Grant LC-06-003. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility that is supported by NIH awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA. We thank Ju-Whei Lee (Dana-Farber Cancer Institute) for assistance with the ECOG 3503 trial data analysis.
References
- 1.Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290:2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 2.Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] . J. Clin. Oncol. 2003;21:2237–2246. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 4.Sandler A. Clinical experience with the HER1/EGFR tyrosine kinase inhibitor erlotinib. Oncology (Williston Park) 2003;17:17–22. [PubMed] [Google Scholar]
- 5.Perez-Soler R. The role of erlotinib (Tarceva, OSI 774) in the treatment of non-small cell lung cancer. Clin Cancer Res. 2004;10:4238s–4240s. doi: 10.1158/1078-0432.CCR-040017. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 8.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 10.Yoshioka H, Hotta K, Kiura K, Takigawa N, Hayashi H, Harita S, Kuyama S, Segawa Y, Kamei H, Umemura S, Bessho A, Tabata M, Tanimoto M Okayama Lung Cancer Study Group. A phase II trial of erlotinib monotherapy in pretreated patients with advanced non-small cell lung cancer who do not possess active EGFR mutations: Okayama Lung Cancer Study Group trial 0705. J Thorac Oncol. 2010;5:99–104. doi: 10.1097/JTO.0b013e3181c20063. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 12.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 14.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, Riely GJ, Kris MG, Miller VA, Ladanyi M, Politi K, Pao W. HER2 Amplification: A Potential Mechanism of Acquired Resistance to EGFR Inhibition in EGFR-Mutant Lung Cancers That Lack the Second-Site EGFRT790M Mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB, Ko YG, Lee JS, Lee SJ, Lee JC, Park MJ. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene. 2013;32:209–221. doi: 10.1038/onc.2012.37. [DOI] [PubMed] [Google Scholar]
- 16.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, Uehara H, Mitsudomi T, Yatabe Y, Nakamura T, Sone S. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 17.Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, Christensen JG, Wong KK, Gray NS, Janne PA. Resistance to Irreversible EGF Receptor Tyrosine Kinase Inhibitors through a Multistep Mechanism Involving the IGF1R Pathway. Cancer Res. 2013;73:834–843. doi: 10.1158/0008-5472.CAN-12-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, Tadi M, Rogers A, Repellin C, Capelletti M, Maertens O, Goetz EM, Letai A, Garraway LA, Lazzara MJ, Rosen N, Gray NS, Wong KK, Janne PA. Reactivation of ERK Signaling Causes Resistance to EGFR Kinase Inhibitors. Cancer Discov. 2012;2:934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto C, Basaki Y, Kawahara A, Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K, Kuwano M, Ono M. Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib-resistant lung cancer cells harboring epidermal growth factor receptor-activating mutations. Cancer Res. 2010;70:8715–8725. doi: 10.1158/0008-5472.CAN-10-0043. [DOI] [PubMed] [Google Scholar]
- 20.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, Nilsson MB, Gudikote J, Tran H, Cardnell RJ, Bearss DJ, Warner SL, Foulks JM, Kanner SB, Gandhi V, Krett N, Rosen ST, Kim ES, Herbst RS, Blumenschein GR, Lee JJ, Lippman SM, Ang KK, Mills GB, Hong WK, Weinstein JN, Wistuba II, Coombes KR, Minna JD, Heymach JV. An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002003. 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koizumi F, Shimoyama T, Taguchi F, Saijo N, Nishio K. Establishment of a human non-small cell lung cancer cell line resistant to gefitinib. Int J Cancer. 2005;116:36–44. doi: 10.1002/ijc.20985. [DOI] [PubMed] [Google Scholar]
- 23.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 24.Tong Z, Wu X, Ovcharenko D, Zhu J, Chen CS, Kehrer JP. Neutrophil gelatinase-associated lipocalin as a survival factor. Biochem J. 2005;391:441–448. doi: 10.1042/BJ20051020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flower DR. The lipocalin protein family: a role in cell regulation. FEBS Lett. 1994;354:7–11. doi: 10.1016/0014-5793(94)01078-1. [DOI] [PubMed] [Google Scholar]
- 26.Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics. 1997;45:17–23. doi: 10.1006/geno.1997.4896. [DOI] [PubMed] [Google Scholar]
- 27.Friedl A, Stoesz SP, Buckley P, Gould MN. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. Histochem J. 1999;31:433–441. doi: 10.1023/a:1003708808934. [DOI] [PubMed] [Google Scholar]
- 28.Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treat. 2008;108:389–397. doi: 10.1007/s10549-007-9619-3. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y, Yokoi K, Li H, Gao J, Hu L, Liu B, Chen K, Hamilton SR, Fan D, Sun B, Zhang W. NGAL expression is elevated in both colorectal adenoma-carcinoma sequence and cancer progression and enhances tumorigenesis in xenograft mouse models. Clin Cancer Res. 2011;17:4331–4340. doi: 10.1158/1078-0432.CCR-11-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du ZP, Lv Z, Wu BL, Wu ZY, Shen JH, Wu JY, Xu XE, Huang Q, Shen J, Chen HB, Li EM, Xu LY. Neutrophil gelatinase-associated lipocalin and its receptor: independent prognostic factors of oesophageal squamous cell carcinoma. J Clin Pathol. 2011;64:69–74. doi: 10.1136/jcp.2010.083907. [DOI] [PubMed] [Google Scholar]
- 31.Liu MF, Jin T, Shen JH, Shen ZY, Zheng ZC, Zhang ZL, Xu LY, Li EM, Xu HX. NGAL and NGALR are frequently overexpressed in human gliomas and are associated with clinical prognosis. J Neurooncol. 2011;104:119–127. doi: 10.1007/s11060-010-0486-0. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Fan Y, Mei Z. NGAL and NGALR overexpression in human hepatocellular carcinoma toward a molecular prognostic classification. Cancer Epidemiol. 2012;36:e294–299. doi: 10.1016/j.canep.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 33.Reckamp KKM, Cristea MC, Dowell J, Gardner B, Milne G, Fouladi Rad S, Figlin R, Elashoff R, Dubinett SM. Randomized, placebo-controlled, phase II trial of EGFR and COX-2 inhibition in advanced non-small cell lung cancer. J. Clin. Oncol. 2010;28:65s. [Google Scholar]
- 34.O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, Tenen DG, Kobayashi S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007;4:1669–1679. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4:1681–1689. doi: 10.1371/journal.pmed.0040316. discussion 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng J, Shimamura T, Perera S, Carlson NE, Cai D, Shapiro GI, Wong KK, Letai A. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67:11867–11875. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 38.Gong Y, Somwar R, Politi K, Balak M, Chmielecki J, Jiang X, Pao W. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Zhou S, Li X, Wang D, Wang Y, Zhou C, Schmid-Bindert G. Gefitinib-Resistance Is Related to BIM Expression in Non-Small Cell Lung Cancer Cell Lines. Cancer Biother Radiopharm. 2013;28:115–123. doi: 10.1089/cbr.2012.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phospho- rylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 41.Hubner A, Barrett T, Flavell RA, Davis RJ. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol Cell. 2008;30:415–425. doi: 10.1016/j.molcel.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–18816. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 43.Lim R, Ahmed N, Borregaard N, Riley C, Wafai R, Thompson EW, Quinn MA, Rice GE. Neutrophil gelatinase-associated lipocalin (NGAL) an early-screening biomarker for ovarian cancer: NGAL is associated with epidermal growth factor-induced epithelio-mesenchymal transition. Int J Cancer. 2007;120:2426–2434. doi: 10.1002/ijc.22352. [DOI] [PubMed] [Google Scholar]
- 44.Tong Z, Chakraborty S, Sung B, Koolwal P, Kaur S, Aggarwal BB, Mani SA, Bresalier RS, Batra SK, Guha S. Epidermal growth factor down-regulates the expression of neutrophil gelatinase-associated lipocalin (NGAL) through E-cadherin in pancreatic cancer cells. Cancer. 2011;117:2408–2418. doi: 10.1002/cncr.25803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moller C, Alfredsson J, Engstrom M, Wootz H, Xiang Z, Lennartsson J, Jonsson JI, Nilsson G. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood. 2005;106:1330–1336. doi: 10.1182/blood-2004-12-4792. [DOI] [PubMed] [Google Scholar]
- 46.Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278:49795–49805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 47.Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, Soh S, Lee WH, Huang JW, Allen JC Jr, Woo XY, Nagarajan N, Kumar V, Thalamuthu A, Poh WT, Ang AL, Mya HT, How GF, Yang LY, Koh LP, Chowbay B, Chang CT, Nadarajan VS, Chng WJ, Than H, Lim LC, Goh YT, Zhang S, Poh D, Tan P, Seet JE, Ang MK, Chau NM, Ng QS, Tan DS, Soda M, Isobe K, Nothen MM, Wong TY, Shahab A, Ruan X, Cacheux-Rataboul V, Sung WK, Tan EH, Yatabe Y, Mano H, Soo RA, Chin TM, Lim WT, Ruan Y, Ong ST. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18:521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 48.Amann J, Kalyankrishna S, Massion PP, Ohm JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart Salmon J, Kim YH, Pollack JR, Yanagisawa K, Gazdar A, Minna JD, Kurie JM, Carbone DP. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–235. [PubMed] [Google Scholar]
- 49.Reckamp KL, Krysan K, Morrow JD, Milne GL, Newman RA, Tucker C, Elashoff RM, Dubinett SM, Figlin RA. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clin Cancer Res. 2006;12:3381–3388. doi: 10.1158/1078-0432.CCR-06-0112. [DOI] [PubMed] [Google Scholar]
- 50.Kolesar J, Brahmer J, Lee J, Guaglianone P, Patel J, Keppen M, Hidalgo M, Carbone D, Schiller J. Final results of ECOG 3503: A pilot study to determine if downstream markers of EGFR linked signaling pathways predict response to erlotinib (OSI-774) in the first-line treatment of patients with advanced non-small cell lung cancer (NSCLC) J. Clin. Oncol. 2007;25:7588. [Google Scholar]