

Abstract
We recently proposed a role for the 2-pore-domain K+ (K2P) channel TREK-1 in the regulation of cytokine release from alveolar epithelial cells (AECs) by demonstrating decreased IL-6 secretion from TREK-1 deficient cells, but the effects of altered TREK-1 expression on other inflammatory mediators remain poorly understood. We now examined the role of TREK-1 in TNF-α-induced MCP-1 release from human A549 cells. We hypothesized that TREK-1 regulates TNF-α-induced MCP-1 secretion via c-Jun N-terminal kinases (JNK)- and protein kinase-C (PKC)-dependent pathways. In contrast to IL-6 secretion, we found that TREK-1 deficiency resulted in increased MCP-1 production and secretion, although baseline MCP-1 gene expression was unchanged in TREK-1 deficient cells. In contrast to TREK-1 deficient AECs, overexpression of MCP-1 had no effect on MCP-1 secretion. Phosphorylation of JNK1/2/3 was increased in TREK-1 deficient cells upon TNF-α stimulation, but pharmacological inhibition of JNK1/2/3 decreased MCP-1 release from both control and TREK-1 deficient cells. Similarly, pharmacological inhibition of PKC decreased MCP-1 secretion from control and TREK-1 deficient cells, suggesting that alterations in JNK and PKC signaling pathways were unlikely the cause for the increased MCP-1 secretion from TREK-1 deficient cells. Furthermore, MCP-1 secretion from control and TREK-1 deficient cells was independent of extracellular Ca2+ but sensitive to inhibition of intracellular Ca2+ reuptake mechanisms. In summary, we report for the first time that TREK-1 deficiency in human AECs resulted in increased MCP-1 production and secretion, and this effect appeared unrelated to alterations in JNK-, PKC- or Ca2+-mediated signaling pathways in TREK-1 deficient cells.
Keywords: TREK-1, MCP-1, TNF-α, chemokine, epithelium, acute lung injury
Introduction
In addition to immune cells, alveolar epithelial cells (AECs) play a central role in the regulation of lung inflammation induced by infectious as well as non-infectious agents including viruses, bacteria, allergens, inhaled toxins, mechanical stretch, and hyperoxia. The chemokine (C-C motif) ligand-2 (CCL2), also referred to as Monocyte Chemotactic Protein-1 (MCP-1), plays an important role in these processes by promoting the recruitment of monocytes, and possibly neutrophils, to the lung [1]. An increase in MCP-1 concentrations in the lung tissue and in broncho-alveolar lavage (BAL) fluid has been documented in allergen- [2], virus- [3], exotoxin- [4,5], and endotoxin [6]-induced lung inflammation, and in cystic fibrosis [7]. Interestingly, dependent on the type of stimulus, MCP-1 may exert pro- [6] or anti-inflammatory [8] properties in the lung. MCP-1 is secreted by both immune cells and AECs [9,10], but the molecular pathways regulating MCP-1 secretion from AECs remain poorly understood. The pathological findings observed in patients with acute lung injury (ALI) and Acute Respiratory Distress Syndrome (ARDS) include high levels of TNF-α in the BAL fluid [11] and correlate with patient mortality rates [12]. We recently provided evidence that the 2-pore domain (K2P) potassium TREK-1 may play a regulatory role in TNF-α-induced mediator secretion from AECs [13,14]. We found that TREK-1 deficiency resulted in a decrease in IL-6 and an increase in MCP-1 release. The decrease in IL-6 release from TREK-1 deficient AECs was associated with alterations in protein kinase-C signaling, but the molecular mechanisms underlying the TNF-α-induced increase in MCP-1 release from TREK-1 deficient AECs remains unknown. In this study we show for the first time that TREK-1 deficiency in human AECs resulted in an increase in MCP-1 production and secretion. This increase in MCP-1 secretion from TREK-1 deficient cells appeared unrelated to alterations in JNK- and PKC-mediated signaling pathways. Furthermore, the increased release of MCP-1 from AECs was independent of the extracellular calcium concentration, but sensitive to changes in the intracellular calcium concentration.
Materials and methods
Cell culture
Human A549 alveolar epithelial cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were cultured in DMEM (Gibco, Carlsbad, CA) supplemented with 10% FBS (Gibco), 1% Penicillin/Streptomycin (Gibco), 20 mM HEPES (Sigma Aldrich, St. Louis, MO) and 2 mM L-Glutamine (Gibco). Once cells were 80-90% confluent they were treated with TNF-α (5 ng/ml; R&D Systems) at 37 °C. Cell viability was determined to be > 90% under all conditions by Trypan blue staining.
Development of a stable TREK-1 shRNA and a TREK-1 over-expressing A549 cell line
A stable TREK-1-deficient A549 cell line using a commercially available pRFP-C-RS vector (Origene, TREK-1 specific probe #FI348008; control scrambled peptide #TR30015) was developed as previously described [14].
A stable TREK-1 over-expressing A549 cell line was created using an Origene TrueORF Gold cDNA Clones and Precision Shuttle Vector system (cat #RC210180) by following to the manufacturer’s instructions. Details of the pCMV6-Entry vector containing a DDK-tag for detection are available on the Origene website (www.origene.com/cdna/trueorf/destinationvector.mspx). Briefly, 3 x 105 cells were grown in 6 well plates prior to transfection until cells reached 60-70% confluence in DMEM medium supplemented with 10% FBS, 20 mM HEPES and 2 mM L-Glutamine. Cells were transfected with the DNA probe provided by the manufacturer using the Turbofectin 8.0 transfection system and incubated for 24 hours at 37 °C. To select for positively transfected cells, cells were cultured in T75 flasks in DMEM medium (10% FBS, 1% Penicillin/Streptomycin, 20 mM HEPES and 2 mM L-Glutamine) supplemented with 0.5 mg/mL G418. As a control, non-transfected A549 cells were cultured in parallel under the same conditions. TREK-1 over-expression was confirmed by Western Blot using the anti-DDK antibody provided by the manufacturer, and by real time PCR using a TaqMan Gene Expression assay (Roche). Primer sets for human TREK-1 were purchased from IDT, IA [14].
MCP-1 ELISA measurements
Initially, 1 x 105 A549 cells were seeded in 12 well culture plates and grown to 80-90% confluence. Cells were then incubated in complete culture medium in the presence or absence of TNF-α (5 ng/ml) for 2, 6 or 24 hours at 37 °C. In experiments using the PKC inhibitor calphostin C (0.2 μM in the presence of a 8 W light source; Sigma), the JNK inhibitor SP600125 (10 μM, Sigma), the translation inhibitor cycloheximide (0.2 μg/ml, Sigma) and the Ca2+ reuptake inhibitor thapsigargin (0.5 μM; Sigma), cells were incubated with each inhibitor for 30 min prior to stimulation with TNF-α. When MCP-1 measurements were performed in the absence of extracellular Ca2+, cells were incubated in DMEM without Ca2+ (Gibco, cat #21068-028) supplemented with 10% FBS (Gibco), 1% Penicillin/Streptomycin (Gibco), 20 mM HEPES (Sigma) and 2 mM L-Glutamine (Gibco) during TNF-α stimulation. Cell viability was assessed after 2, 6 and 24 hours using Tryp an blue staining and was consistently > 90%. Furthermore, total intracellular protein concentrations were measured in each experiment using the Bradford assay and remained consistent under all experimental conditions, suggesting that non-specific leakage of intracellular proteins did not occur. Supernatants were collected at 2, 6 and 24 hours, and MCP-1 concentrations were determined using BD Bioscience OptEIA species-specific MCP-1 ELISA kits.
Gene expression by real-time PCR
Total RNA was isolated from 2 x 106 A549 cells using a High Pure RNA Isolation Kit (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s instructions. Single-stranded DNA was synthesized from 1μg total RNA and Reverse Transcription PCR was performed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA) according to the manufacturer’s instructions. Real-Time PCR was performed using a TaqMan Gene Expression assay (Roche). Primer sets for human MCP-1 were purchased from IDT, IA (Forward: agtctctgccgcccttct, Reverse: gtgac-tggggcattgattg). In preliminary experiments we confirmed that HGPRT levels were unchanged between control and TREK-1 deficient A549 cells and, therefore, MCP-1 mRNA levels of control and TREK-1 deficient cells were normalized to HGPRT expression. Data were expressed as fold change in MCP-1 mRNA expression of TREK-1 deficient cells compared to control cells. A fold change greater than 2-fold was considered as significant. All experiments were repeated a minimum of 3 times and each sample was run in triplicate.
Western blot analysis
Antibodies for total JNK and phospho JNK were purchased from Cell Signaling and used in 1: 1000 dilutions as recommended by the manufacturer. GAPDH (1: 2000, Cell Signaling) was used as an internal loading control. Western blots to confirm TREK-1 overexpression in transfected A549 cells were performed using the DDK-tag antibody (1: 1000) provided by the Origene TrueORF Gold cDNA Clones and Precision Shuttle Vector system (cat # RC210180). Initially, 8 x 104 cells were seeded into 12-well tissue culture plates (MidWest Science, St. Louis, MO). Once cells reached 80-90% confluence, they were lysed on ice in RIPA buffer (50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, 0.1% SDS) with a protease inhibitor cocktail (Roche, Burlington, NC). Lysates were centrifuged at 4 °C and 17,000 g for 15 min, and total protein concentrations were measured using the Bradford assay (BioRad, Hercules, CA). A total of 45-60 μg protein of each sample was separated by sodium dodecyl sulfate (SDS)- polyacrylamide gel electrophoresis (PAGE) on 4-12% NuPage Bis-Tris gradient gels (Invitrogen) and transferred onto nitrocellulose membranes at 35 mV for 2 hours. All membranes were blocked in 5% non-fat dry milk in Tris-buffered saline (Bio-Rad) containing 0.1% Tween-20 for 1 h at 37 °C. The membranes were then incubated overnight with the indicated primary antibodies at 4 °C. The next day, membranes were incubated for 1 hour with the following secondary antibodies: for total JNK and phospho JNK immunoblots we used an anti-goat HRP-conjugated IgG antibody (1: 5000, Santa Cruz), and for GAPDH we used an anti-rabbit HRP-conjugated IgG (1: 5000, Cell Signaling). Bands were visualized by enhanced chemiluminescence with ECL SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Rockford IL). Band densitometry measurements to determine relative quantities of protein were performed using ImageJ 1.42 software for Windows.
Statistical analysis
All values were expressed as means ± SEM. Student’s t-test was used to compare means of two different groups. Real time PCR data were analyzed using the ΔΔCt method, and a change in gene expression greater than 2-fold was considered significant. Additionally, in cytokine and phosphorylation studies, ANOVA analysis was used to compare means of different groups. All statistical analyses were performed using SigmaStat 3.5 software and a p-value of p ≤ 0.05 was considered significant.
Results
TREK-1 deficient AECs secrete increased amounts of MCP-1
Efficiency of TREK-1 knockdown was previously documented in A549 cells by Western blot, real time PCR and confocal microscopy [14]. TREK-1 overexpression was assessed by detection of a DDK-tag by Western blot (Figure 1A) and by real-time PCR (Figure 1B; 10.7-fold increase in TREK-1 mRNA expression compared to control cells; a change in mRNA expression greater than 2-fold was considered significant).
Figure 1.
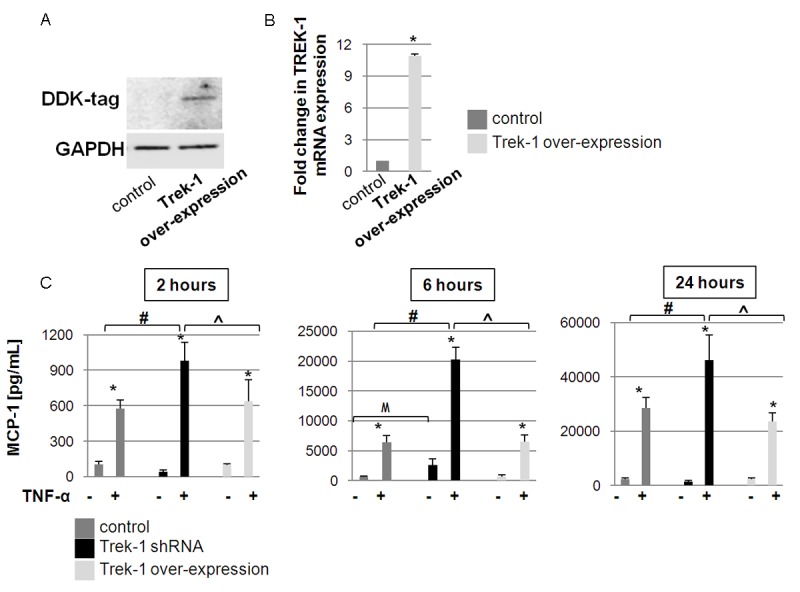
MCP-1 secretion from TREK-1 deficient and TREK-1 over-expressing cells: A shows a positive DDK-tag signal confirming Trek-1 overexpression in A549 cells by Western blot. GAPDH was used as a loading control. B shows a 10.7-fold increase in TREK-1 mRNA expression in TREK-1 overexpressing A549 cells compared to control cells by real time PCR (*a fold change greater than 2-fold from control cells was considered significant). C shows a time course of MCP-1 secretion from control, TREK-1 deficient, and TREK-1 overexpressing A549 cells following TNF-α treatment. *compared to untreated control cells; #comparison between TNF-α-treated control and TREK-1 shRNA transfected cells; ^comparison between TNF-α-treated TREK-1 shRNA transfected cells and TREK-1 overexpressing cells; Mcomparison between unstimulated control and unstimulated TREK-1 shRNA transfected cells; n = 4-12, p ≤ 0.05.
We previously reported that TREK-1 deficient mouse alveolar epithelial cells (AECs) showed an increase in MCP-1 secretion after 24 hours of TNF-α stimulation [13]. We now examined the time course of baseline and TNF-α-induced MCP-1 release from human A549 cells following 2, 6 and 24 hours of TNF-α stimulation (Figure 1C). To determine the role of TREK-1 in MCP-1 secretion, we compared the amounts of MCP-1 released from control, TREK-1 deficient and TREK-1 overexpressing A549 cells. A TNF-α dose of 5 ng/ml was chosen as a stimulus based on the results obtained in our previous studies [13,14]. Stimulation of control, TREK-1 deficient, and TREK-1 overexpressing A549 cells with TNF-α resulted in an increase in MCP-1 secretion in a time-dependent manner. Importantly, MCP-1 secretion from TREK-1 deficient cells was more pronounced at all 3 time points compared to both control and TREK-1 overexpressing cells. Furthermore, we observed no differences in MCP-1 release between control and TREK-1 overexpressing cells at baseline or in TNF-α treated cells. Therefore, for the remainder of this study, we focused on the differences in MCP-1 secretion between control and TREK-1 deficient cells.
MCP-1 release from TREK-1 deficient AECs is regulated at translational level
To study whether MCP-1 was predominantly pre-formed and stored, or newly synthesized, we compared intracellular and extracellular MCP-1 concentrations between untreated and TNF-α stimulated (6 hours) A549 cells (Figure 2A). Even without stimulation, TREK-1 deficient cells secreted slightly more MCP-1 protein over 6 hours in culture than control cells. After TNF-α stimulation, an increased MCP-1 concentration was detected in both the pellets and the supernatants of TREK-1 deficient cells when compared to controls.
Figure 2.
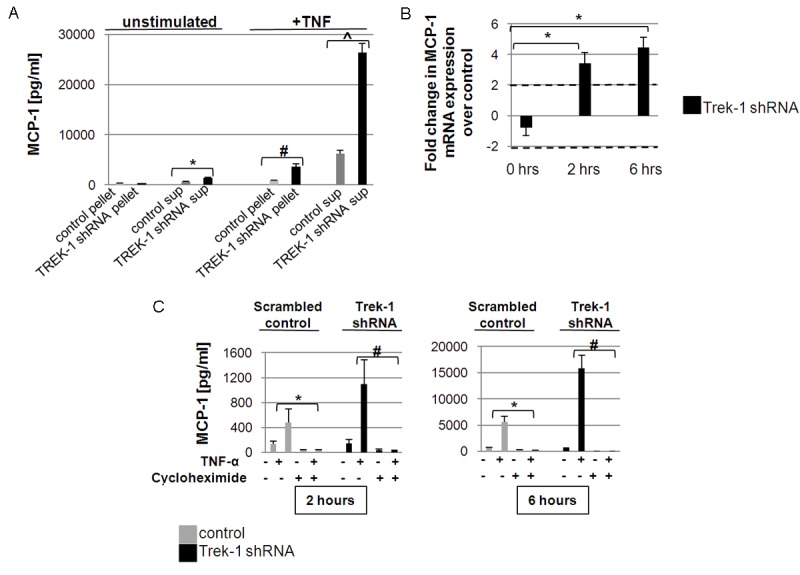
TREK-1 production is regulated at transcriptional and translational level: A shows MCP-1 concentrations in cell pellets and culture supernatants (sup) in untreated cells and after 6 hours TNF-α treatment. *comparison of MCP-1 concentrations in the supernatants of unstimulated control and TREK-1 deficient A549 cells; #comparison of MCP-1 concentrations in the pellets of TNF-α stimulated control and TREK-1 deficient A549 cells; ^comparison of MCP-1 concentrations in the supernatants of TNF-α stimulated control and TREK-1 deficient A4549 cells; n = 4, p ≤ 0.05). B shows no significant change in baseline MCP-1 mRNA expression between unstimulated control and TREK-1 deficient A549 cells by real time PCR. However, stimulation of A549 cells with TNF-α (5ng/mL) resulted in an increase in MCP-1 mRNA expression in TREK-1 deficient cells after 2 and 6 hours when compared to control cells at each time point (n = 3, *a greater than 2-fold change was considered significant). C shows inhibition of MCP-1 secretion by cycloheximide after 2 and 6 hours of TNF-α stimulation. *comparison of cycloheximide-exposed and unexposed control cells; #comparison of cycloheximide-exposed and unexposed TREK-1 deficient cells; n = 3, p ≤ 0.05.
To determine whether the increase in MCP-1 secretion from TREK-1 deficient cells was due to increased MCP-1 gene transcription, we compared MCP-1 mRNA levels between control and TREK-1 deficient cells at baseline and after TNF-α stimulation (Figure 2B) using real time PCR. We detected no difference in baseline MCP-1 gene expression between untreated control and TREK-1 deficient cells (-0.7 ± 0.5-fold change in mRNA expression compared to untreated controls; a change in gene expression greater than 2-fold was considered significant). However, after 2 and 6 hours of TNF-α stimulation, we found a 3.4 and 4.5-fold increase in MCP-1 gene expression, respectively, in TREK-1 deficient cells when compared to equally treated controls.
In a next set of experiments we evaluated whether the increase in MCP-1 secretion from TREK-1 deficient cells was due to alterations in MCP-1 protein translation. We used cycloheximide, an inhibitor of protein translation, to study the contribution of protein translation to MCP-1 secretion from control and TREK-1 deficient A549 cells after 2 and 6 hours of TNF-α stimulation (Figure 2C). Inhibition of protein translation with cycloheximide decreased MCP-1 secretion by > 95% in both control and TREK-1 deficient A549 cells after 2 and 6 hour of TNF-α stimulation.
Collectively, these data suggest that the majority of secreted MCP-1 was newly synthesized after TNF-α stimulation rather than stored within the cells, and these results point towards TREK-1 as an important regulator of MCP-1 protein synthesis and secretion from AECs.
C-Jun N-terminal kinases (JNK) signaling was altered in TREK-1 deficient cells
TNF-α-induced activation of JNK kinases (JNK1, JNK2, JNK3) is an important step in epithelial cytokine secretion and in the development of ALI/ARDS [15-17]. To investigate whether alterations in JNK signaling in TREK-1 deficient cells contributed to the increase in MCP-1 secretion from these cells, we used Western blot experiments to study the time course of JNK1/2/3 phosphorylation in control and TREK-1 deficient A549 cells (Figure 3A). Densitometry analysis summarizing normalized phospho JNK1/2/3 to total JNK1/2/3 ratios of 4 separate experiments is shown in Figure 3B. TNF-α treatment resulted in increased JNK1/2/3 phosphorylation in both control and TREK-1 deficient cells after 10 min, 2 hours, and 6 hours when compared to untreated control cells (Figure 3B). However, TREK-1 deficient cells showed stronger JNK1/2/3 phosphorylation than equally treated control cells after 10 min and 2 hours of TNF-α stimulation.
Figure 3.
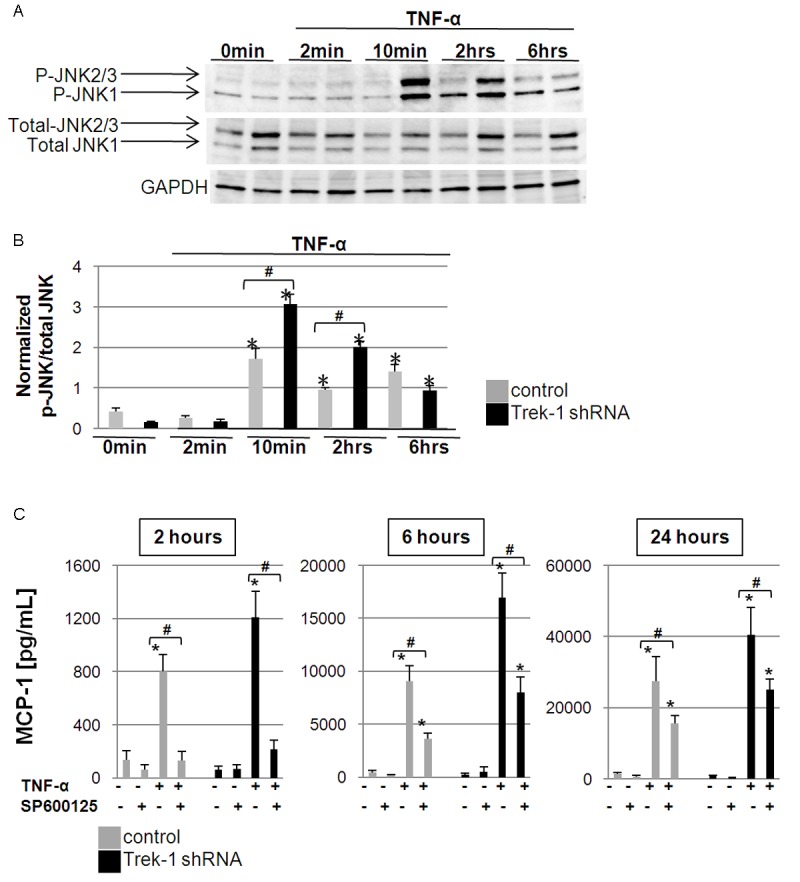
MCP-1 secretion is JNK-dependent: A shows a 6-hour time course of total and phosphorylated JNK1/2/3 expression upon TNF-α stimulation. GAPDH was used as a loading control. B summarizes densitometry analysis of phosphorylated to total JNK (p-JNK/total JNK) ratios of 4 separate experiments. *compared to unstimulated control cells; #comparison of control and TREK-1 deficient cells within each time point; p ≤ 0.05. C shows inhibition of MCP-1 secretion by the broad spectrum JNK inhibitor SP600125 in control and TREK-1 deficient A549 cells. *compared to untreated control cells at each time point; #comparison of SP600125-exposed and -unexposed TNF-α stimulated cells; n = 3, p ≤ 0.05.
To determine whether these alterations in JNK phosphorylation in TREK-1 deficient cells contributed to the increased MCP-1 release from these cells, we studied the effect of the broad spectrum JNK1/2/3 inhibitor SP600125 [18,19] on MCP-1 release after 2, 6 and 24 hours of TNF-α stimulation(Figure 3C). Inhibition of JNK1/2/3 decreased TNF-α-induced MCP-1 secretion from both control and TREK-1 deficient cells after 2, 6 and 24 hours of TNF-α stimulation. Interestingly, although the percentage of inhibition of MCP-1 release was similar between control and TREK-1 deficient cells at each time point, the inhibitory effect of SP60012 on MCP-1 secretion was more pronounced after 2 hours of TNF-α stimulation than after 6 and 24 hours (Figure 3C). SP600125 alone had no effect on baseline MCP-1 release from untreated control and TREK-1 deficient cells at any of the time points studied.
Collectively, these data suggest that TNF-α-induced JNK1/2/3 phosphorylation was increased in TREK-1 deficient cells, and that MCP-1 release from both control and TREK-1 deficient cells was JNK-dependent. However, since inhibition of JNK1/2/3 decreased MCP-1 release from control and TREK-1 deficient cells to a similar degree, alterations in JNK signaling were unlikely the cause for the increased MCP-1 release from TREK-1 deficient cells.
Secretion of MCP-1 from A549 cells was protein kinase C (PKC)-dependent
In addition to JNK signaling, PKC is another key regulatory kinase mediating TNF-α-induced inflammatory effects [20-22], and we previously reported a role for PKCθ in regulation of IL-6 secretion form TREK-1 deficient AECs [14]. To determine whether alterations in PKC signaling in TREK-1 deficient AECs could explain the increase in MCP-1 secretion from these cells, we studied the effects of the PKC inhibitor calphostin C on MCP-1 release after 2, 6 and 24 hours of TNF-α stimulation (Figure 4). We found that calphostin C inhibited TNF-α-induced MCP-1 secretion from both control and TREK-1 deficient cells in a time-dependent fashion. Within each time point, the degree of inhibition of MCP-1 release was similar between control and TREK-1 deficient cells, and was most pronounced after 24 hours of TNF-α stimulation. Interestingly, calphostin C alone also inhibited baseline MCP-1 release from both unstimulated control and TREK-1 deficient cells after 6 and 24 hours, but not after 2 hours. Collectively, these data suggest that MCP-1 release from A549 cells was PKC-dependent. However, since calphostin C had a similar inhibitory effect on control and TREK-1 deficient cells, alterations in PKC signaling were unlikely related to the increase in MCP-1 secretion from TREK-1 deficient cells.
Figure 4.
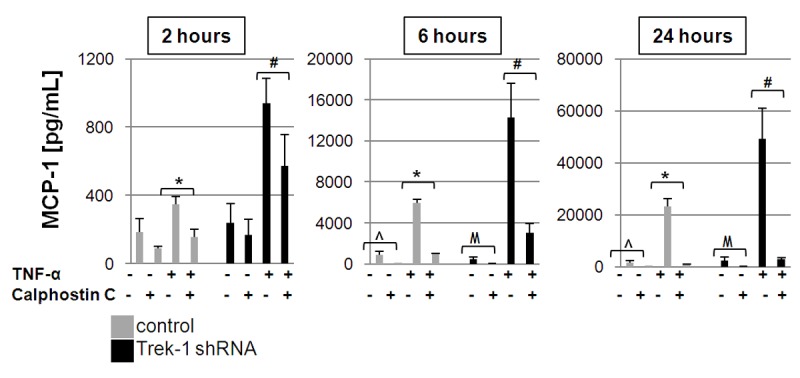
MCP-1 secretion is PKC-dependent: Figure 4 shows inhibition of MCP-1 secretion by the PKC inhibitor calphostin C in control and TREK-1 deficient A549 cells. *comparison of calphostin C-exposed and unexposed TNF-α stimulated control cells within each time point; #comparison of calphostin C-exposed and unexposed TNF-α stimulated TREK-1 deficient cells within each time point; ^comparison of calphostin C-exposed control cells to untreated control cells; Mcomparison of calphostin C-exposed TREK-1 deficient cells to untreated TREK-1 deficient cells; n = 3, p ≤ 0.05.
MCP-1 secretion from A549 cells was dependent on the intracellular, but not the extracellular Ca2+ concentration
To determine whether the increase in MCP-1 secretion from TREK-1 deficient cells was related to alterations in the extra- or intracellular Ca2+ concentration, we measured TNF-α-induced MCP-1 release from control and TREK-1 deficient A549 cells in the presence and absence of extracellular Ca2+ (Figure 5A), and after inhibition of intracellular Ca2+ reuptake mechanisms by thapsigargin (Figure 5B).
Figure 5.
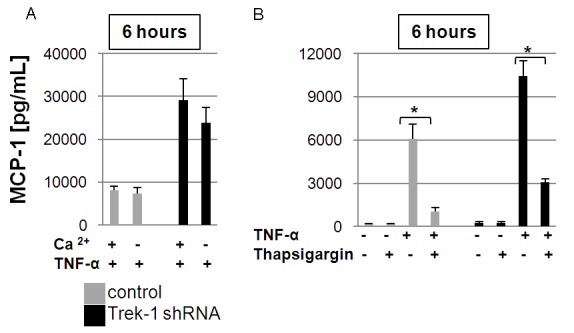
MCP-1 secretion is sensitive to changes in the intracellular but not extracellular Ca2+ concentration: Lowering of the extracellular Ca2+ concentration did not affect MCP-1 secretion from control or TREK-1 deficient A549 cells after 6 hours of TNF-α stimulation (A). Inhibition of intracellular Ca2+ reuptake mechanisms by thapsigargin decreased MCP-1 secretion to a similar degree in control and TREK-1 deficient cells after 6 hours of TNF-α stimulation (B). *comparison of thapsigargin-exposed and unexposed TNF-α stimulated control cells; #comparison of thapsigargin-exposed and unexposed TNF-α stimulated TREK-1 deficient cells; n = 3, p ≤ 0.05.
First, to study the effect of extracellular Ca2+ on MCP-1 release, we measured MCP-1 secretion from cells cultured in a physiologic Ca2+ concentration (20 mg/dl) and from cells exposed to a low extracellular Ca2+ concentration (Ca2+-free DMEM with 10% FBS, final Ca2+ concentration 1 mg/dl). Lowering of the extracellular Ca2+ concentration did not affect TNF-α-induced MCP-1 secretion from either control or TREK-1 deficient cells (Figure 5A). However, inhibition of Ca2+ reuptake mechanisms by thapsigargin decreased TNF-α-induced MCP-1 secretion from both control and TREK-1 deficient A549 cells to a similar degree (Figure 5B). These data suggest that TNF-α-induced MCP-1 secretion occurred irrespective of the extracellular Ca2+ concentration, but was dependent on intracellular Ca2+ signaling mechanisms. However, since the degree of inhibition by thapsigargin was similar in control and TREK-1 deficient cells, alterations in intracellular Ca2+ signaling were unlikely responsible for the increase in MCP-1 secretion from TREK-1 deficient cells.
Disussion
Despite our best clinical and research efforts, the mortality rates of patients with acute lung injury (ALI) and Acute Respiratory Distress Syndrome (ARDS) remain high [23,24]. The two main treatment regimens for ALI/ARDS, mechanical ventilation and oxygen supplementation, are life-saving interventions but are also known to further propagate lung injury [25,26], a finding supported by data recently published by our group [16,27]. From both clinical [11,28] and animal [29] studies we learned that an increase in inflammatory mediators, in particular TNF-α, MCP-1 and IL-6, contributes to the pathological changes observed in ALI/ARDS. However, the specific mechanisms leading to these findings remain unclear. The role of TNF-α as a pro-inflammatory cytokine in ALI/ARDS is widely accepted, and TNF-α levels in the BAL fluid correlate with patient mortality rates [28,30]. In contrast, the contribution of MCP-1 to lung injury is much more controversial. Its main role in inflammation appears to consist in mononuclear cell and neutrophil recruitment [1,31], but MCP-1 has also been involved in regulation of mucin secretion from airway epithelial cells [32] and in lung repair mechanisms [33]. In humans, lipopolysaccharide (LPS)- and lipotheicoic acid (LTA)-induced MCP-1 secretion promoted pulmonary inflammation [6], and MCP-1 levels in the BAL fluid of patients with ARDS correlated with mortality rates [34]. Furthermore, elevated levels of MCP-1 have been associated with the development of idiopathic pulmonary fibrosis [4]. On the other hand, MCP-1 protected against lethal endotoxemia in mice [35], and in mouse and pig models of influenza pneumonitis, neutralization of MCP-1 enhanced lung damage and impeded alveolar repair mechanisms [36]. MCP-1 may also play a critical role in the resolution of lung inflammation since it enhanced apoptotic cell removal by macrophages in vitro and in vivo [37], promoted fibroblast survival [38], and facilitated clearance of pneumococcal bacteria from the lung [39].
In previous studies we identified the 2-pore domain potassium (K2P) channel TREK-1 as a new potential therapeutic target against ALI/ARDS. Using the Luminex system, we found that mouse AECs responded to a deficiency in TREK-1 expression with a decrease in IL-6 secretion and an increase in MCP-1 release [13], and in a follow-up study we investigated potential mechanisms related to the decrease in IL-6 secretion from TREK-1 deficient AECs [14]. However, the role of TREK-1 in the regulation of MCP-1 secretion from human AECs remained unexplored. In this study, we show for the first time that TREK-1 deficiency in human A549 cells resulted in an increase in TNF-α-induced MCP-1 production and secretion. We developed stable TREK-1 deficient and TREK-1 overexpressing A549 cell lines to study potential differences in TNF-α-mediated signaling mechanisms (JNK-, PKC- and Ca2+-mediated pathways) between control and TREK-1 deficient cells that could be responsible for the increase in MCP-1 secretion from these cells.
As shown in Figure 1C, stimulation of control, TREK-1 deficient and TREK-1 overexpressing cells with TNF-α resulted in an increase in MCP-1 secretion in a time-dependent fashion. However, the increase in MCP-1 release from TREK-1 deficient cells was more pronounced than from control and TREK-1 overexpressing cells. Interestingly, after 6 hours of culture, TREK-1 deficient cells secreted increased amounts of MCP-1 even in the absence of TNF-α, suggesting that this effect may not be exclusive to this particular stimulus (Figure 2A). Nevertheless, stimulation of cells with TNF-α significantly increased both production and secretion of MCP-1 as seen in the pellet to supernatant ratios depicted in Figure 2A. The differences in baseline MCP-1 release between control and TREK-1 deficient cells were unlikely related to alterations in baseline gene transcription, since baseline MCP-1 mRNA levels were unchanged between untreated controls and TREK-1 deficient cells (Figure 2B). This is particularly interesting since in our previous study a decrease in IL-6 secretion from TREK-1 deficient cells was accompanied by a decrease in baseline IL-6 mRNA expression [14]. However, after TNF-α stimulation we detected higher levels of MCP-1 mRNA in TREK-1 deficient cells than in equally treated controls (Figure 2B). The fact that TNF-α stimulation can induce MCP-1 gene transcription in A549 cells has previously been described [40]. However, whether the activation of specific transcription factors is altered in TREK-1 deficient cells thus resulting in increased MCP-1 mRNA and protein levels in these cells, needs to be addressed in future studies. Boekhoudt et al. reported that in cultured fibroblasts TNF-α regulated MCP-1 gene expression via NFκB- and SP-1-dependent pathways [41], and similar regulatory mechanisms were described in A549 cells [42]. In our study, MCP-1 release seemed mostly regulated at the translational level, since inhibition of translation by cycloheximide virtually abolished TNF-α-induced MCP-1 secretion from control and TREK-1 deficient cells.
TNF-α is known to exert its proinflammatory effects via several signaling pathways, including JNK [43] and protein kinase C (PKC) [44]. Importantly, MCP-1 secretion from AECs has been reported to be JNK-dependent in different types of lung inflammation, including ALI [45,46] and asthma [47]. Our data show increased phosphorylation of JNK1/2/3 isoforms in TREK-1 deficient cells after TNF-α stimulation, and a requirement of JNK activation for MCP-1 secretion (Figure 3A-C). While it is intriguing to speculate that increased phosphorylation of JNK led to the observed increase in MCP-1 secretion form TREK-1 deficient cells, this appears unlikely since pharmacological inhibition of JNK reduced MCP-1 secretion from control and TREK-1 deficient cells to similar degrees (Figure 3C).
In addition to JNK, activation of PKC is one of the cornerstones of TNF-α signaling in the lung [36,48]. We previously proposed that alterations in PKC signaling in TREK-1 deficient cells may lead to a decrease in IL-6 secretion form these cells [14]. In this study, we investigated whether similar alterations in PKC signaling could be responsible for the increase in MCP-1 secretion from TREK-1 deficient cells. Importantly, in a rat model of ALI TNF-α-induced MCP-1 secretion was reported to be PKC-dependent [49]. Similarly, MCP-1 release from lung fibroblasts was also PKC- dependent [50], and inhibition of MCP-1 attenuated the proinflammatory effects of MCP-1 on macrophages [50] and on vascular smooth muscle cells [50]. Similar to JNK, our data show that PKC activation was required for MCP-1 secretion, but inhibition of PKC with calphostin C deceased MCP-1 from control and TREK-1 deficient cells to similar degrees. These results suggest that alterations in PKC signaling in TREK-1 deficient cells were unlikely the cause for the increase in MCP-1 secretion form these cells. Of note, currently 3 families of PKC isoforms, including 12 different isoenzymes, have been described in the literature [51]. Further studies will be necessary to determine which specific isoforms are predominantly involved in MCP-1 secretion from AECs. Importantly, one way of classifying PKC isoforms is to divide them into Ca2+-dependent (α, β, γ) and Ca2+-independent (δ, ε, η, θ) subtypes [52], and we previously reported that IL-6 secretion form TREK-1 deficient AECs occurred independently of the intra- and extracellular Ca2+ concentration [14]. Similar to IL-6 release, secretion of MCP-1 was not affected by lowering the extracellular Ca2+ concentration (Figure 5A). In contrast to the results obtained with IL-6 in our previous study [14], inhibition of intracellular Ca2+ reuptake mechanisms by thapsigargin decreased TNF-α-induced MCP-1 secretion (Figure 5B). However, since thapsigargin was equally effective in inhibiting MCP-1 release from control and TREK-1 deficient cells, it is unlikely that alterations in intracellular Ca2+ signaling mechanisms in TREK-1 deficient cells were responsible for the increase in MCP-1 secretion from these cells. Collectively, the difference in sensitivity to the extra- and intracellular Ca2+ concentration between IL-6 and MCP-1 secretion is quite surprising since generally cytokine release is thought to be a Ca2+-dependent process [53]. It is important to keep in mind, however, that most studies were performed in secretory or inflammatory cells [54,55], and little is currently known about the mechanisms regulating cytokine secretion from AECs. In mast cells [56] and eosinophils [57], secretion of MCP-1 was described as Ca2+-dependent. In the ocular system, inhibition of Ca2+ influx by nilvadipine but not diltiazem, two structurally different L-type Ca2+ channel antagonists, inhibited MCP-1 release [58]. Therefore, the Ca2+ requirement for MCP-1 secretion may be cell type- and tissue-specific. Of note, it is important to remember that even in a “low Ca2+” solution the addition of 10% FBS to the culture medium will result in a final Ca2+ concentration of about 1 mg/dl of Ca2+ ions (assuming a normal serum Ca2+ concentration of 10 mg/dl), whereas our standard culture medium contained 20 mg/dl of Ca2+ (Gibco).
In conclusion, this is the first report demonstrating a role for TREK-1 in MCP-1 secretion from human AECs. We are proposing a potential role for this K2P channel in the development of alveolar inflammation by regulating MCP-1 secretion. In addition to limitations imposed by using a cultured cell line (A549), we recognize that the increase in MCP-1 release from TREK-1 deficient cells may not be TNF-α specific, and based on our previous data we now know that TREK-1 deficiency differentially regulates mediator secretion from AECs. It is, therefore, intriguing to speculate that TREK-1 could be a key regulator in the development of alveolar inflammation during ALI/ARDS. Nevertheless, the specific mechanisms by which TREK-1 affects inflammatory mediator production and secretion, including MCP-1 and IL-6, remain to be determined. In the light of this current and our previous study [14], a model where TREK-1 regulates mediator secretion simply by altering the resting membrane potential and thereby changing the driving force for Ca2+ to enter a cell, appears quite unlikely. One must consider the possibility of TREK-1 acting more as a “regulatory molecule” rather than a simple potassium-permeable pore in the cell membrane, potentially similar to what is now widely accepted for the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) [59-61].
Acknowledgements
This study was supported by a grant from the Le Bonheur Children’s Foundation Research Institute (CFRI) and the National Institutes of Health Grants: HL094366 (CMW) and 5K12HD047349-09 (AS).
Disclosure of conflict of interest
None.
References
- 1.Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect Immun. 2011;79:2567–77. doi: 10.1128/IAI.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H, Xin Y, Tang Y, Shao G. Zinc suppressed the airway inflammation in asthmatic rats: effects of zinc on generation of eotaxin, MCP-1, IL-8, IL-4, and IFN-gamma. Biol Trace Elem Res. 2012;150:314–21. doi: 10.1007/s12011-012-9493-7. [DOI] [PubMed] [Google Scholar]
- 3.Narasaraju T, Ng HH, Phoon MC, Chow VT. MCP-1 antibody treatment enhances damage and impedes repair of the alveolar epithelium in influenza pneumonitis. Am J Respir Cell Mol Biol. 2010;42:732–43. doi: 10.1165/rcmb.2008-0423OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mercer PF, Johns RH, Scotton CJ, Krupiczojc MA, Konigshoff M, Howell DC, McAnulty RJ, Das A, Thorley AJ, Tetley TD, Eickelberg O, Chambers RC. Pulmonary epithelium is a prominent source of proteinase-activated receptor-1-inducible CCL2 in pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:414–25. doi: 10.1164/rccm.200712-1827OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Persoz C, Achard S, Momas I, Seta N. Inflammatory response modulation of airway epithelial cells exposed to formaldehyde. Toxicol Lett. 2012;211:159–63. doi: 10.1016/j.toxlet.2012.03.799. [DOI] [PubMed] [Google Scholar]
- 6.van Zoelen MA, Verstege MI, Draing C, de Beer R, van't Veer C, Florquin S, Bresser P, van der Zee JS, te Velde AA, von Aulock S, van der Poll T. Endogenous MCP-1 promotes lung inflammation induced by LPS and LTA. Mol immunol. 2011;48:1468–76. doi: 10.1016/j.molimm.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Kieninger E, Vareille M, Kopf BS, Blank F, Alves MP, Gisler FM, Johnston SL, Edwards MR, Regamey N. Lack of an exaggerated inflammatory response on virus infection in cystic fibrosis. Eur Respir J. 2012;39:297–304. doi: 10.1183/09031936.00054511. [DOI] [PubMed] [Google Scholar]
- 8.Zisman DA, Kunkel SL, Strieter RM, Tsai WC, Bucknell K, Wilkowski J, Standiford TJ. MCP-1 protects mice in lethal endotoxemia. J Clin Invest. 1997;99:2832–6. doi: 10.1172/JCI119475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paine R 3rd, Rolfe MW, Standiford TJ, Burdick MD, Rollins BJ, Strieter RM. MCP-1 expression by rat type II alveolar epithelial cells in primary culture. J Immunol. 1993;150:4561–70. [PubMed] [Google Scholar]
- 10.Hasan Z, Rahman M, Palani K, Syk I, Jeppsson B, Thorlacius H. Geranylgeranyl transferase regulates CXC chemokine formation in alveolar macrophages and neutrophil recruitment in septic lung injury. Am J Physiol Lung Cell Mol Physiol. 2013;304:L221–9. doi: 10.1152/ajplung.00199.2012. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Lopez A, Garcia-Prieto E, Batalla-Solis E, Amado-Rodriguez L, Avello N, Blanch L, Albaiceta GM. Lung strain and biological response in mechanically ventilated patients. Intensive Care Med. 2012;38:240–7. doi: 10.1007/s00134-011-2403-1. [DOI] [PubMed] [Google Scholar]
- 12.Lee YL, Chen W, Chen LY, Chen CH, Lin YC, Liang SJ, Shih CM. Systemic and bronchoalveolar cytokines as predictors of in-hospital mortality in severe community-acquired pneumonia. J Crit Care. 2010;25:176, e7–13. doi: 10.1016/j.jcrc.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Schwingshackl A, Teng B, Ghosh M, West AN, Makena P, Gorantla V, Sinclair SE, Waters CM. Regulation and function of the two-pore-domain (K2P) potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2012;302:L93–L102. doi: 10.1152/ajplung.00078.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwingshackl A, Teng B, Ghosh M, Lim KG, Tigyi G, Narayanan D, Jaggar JH, Waters CM. Regulation of Interleukin-6 Secretion by the Two-Pore-Domain Potassium (K2P) Channel Trek-1 in Alveolar Epithelial Cells. Am J Physiol Lung Cell Mol Physiol. 2013;304:L276–86. doi: 10.1152/ajplung.00299.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Yang Y, Yang H, Capo-Aponte JE, Tachado SD, Wolosin JM, Reinach PS. NF-kappaB feedback control of JNK1 activation modulates TRPV1-induced increases in IL-6 and IL-8 release by human corneal epithelial cells. Molecular vision. 2011;17:3137–46. [PMC free article] [PubMed] [Google Scholar]
- 16.Makena PS, Gorantla VK, Ghosh MC, Bezawada L, Balazs L, Luellen CL, Parthasarathi K, Waters CM, Sinclair SE. Lung injury caused by high tidal volume mechanical ventilation and hyperoxia is dependent on oxidant-mediated c-Jun NH2-terminal kinase (JNK) activation. J Appl Physiol. 2011;111:1467–76. doi: 10.1152/japplphysiol.00539.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hah YS, Kang HG, Cho HY, Shin SH, Kim UK, Park BW, Lee SI, Rho GJ, Kim JR, Byun JH. JNK signaling plays an important role in the effects of TNF-alpha and IL-1beta on in vitro osteoblastic differentiation of cultured human periosteal-derived cells. Mol Bio Rep. 2013;40:4869–81. doi: 10.1007/s11033-013-2586-3. [DOI] [PubMed] [Google Scholar]
- 18.Park SR, Kim PH, Lee KS, Lee SH, Seo GY, Yoo YC, Lee J, Casali P. APRIL stimulates NF-kappaB-mediated HoxC4 induction for AID expression in mouse B cells. Cytokine. 2013;61:608–13. doi: 10.1016/j.cyto.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pauloin A, Chanat E. Prolactin and epidermal growth factor stimulate adipophilin synthesis in HC11 mouse mammary epithelial cells via the PI3-kinase/Akt/mTOR pathway. Biochim Biophys Acta. 2012;1823:987–96. doi: 10.1016/j.bbamcr.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 20.Gordon R, Anantharam V, Kanthasamy AG, Kanthasamy A. Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J Neuroinflammation. 2012;9:82. doi: 10.1186/1742-2094-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JK, Lee SM, Suk K, Lee WH. A novel pathway responsible for lipopolysaccharide-induced translational regulation of TNF-alpha and IL-6 expression involves protein kinase C and fascin. J Immunol. 2011;187:6327–34. doi: 10.4049/jimmunol.1100612. [DOI] [PubMed] [Google Scholar]
- 22.Deng B, Xie S, Wang J, Xia Z, Nie R. Inhibition of protein kinase C beta(2) prevents tumor necrosis factor-alpha-induced apoptosis and oxidative stress in endothelial cells: the role of NADPH oxidase subunits. J Vasc Res. 2012;49:144–59. doi: 10.1159/000332337. [DOI] [PubMed] [Google Scholar]
- 23.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 24.Zimmerman JJ, Akhtar SR, Caldwell E, Rubenfeld GD. Incidence and outcomes of pediatric acute lung injury. Pediatrics. 2009;124:87–95. doi: 10.1542/peds.2007-2462. [DOI] [PubMed] [Google Scholar]
- 25.Fan E, Villar J, Slutsky AS. Novel approaches to minimize ventilator-induced lung injury. BMC medicine. 2013;11:85. doi: 10.1186/1741-7015-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMullen SM, Meade M, Rose L, Burns K, Mehta S, Doyle R, Henzler D. Partial ventilatory support modalities in acute lung injury and acute respiratory distress syndrome-a systematic review. PLoS One. 2012;7:e40190. doi: 10.1371/journal.pone.0040190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makena PS, Luellen CL, Balazs L, Ghosh MC, Parthasarathi K, Waters CM, Sinclair SE. Pre-Exposure to Hyperoxia Causes Increased Lung Injury and Epithelial Apoptosis in Mice Ventilated with High Tidal Volumes. Am J Physiol Lung Cell Mol Physiol. 2010;299:L711–9. doi: 10.1152/ajplung.00072.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107:1062–73. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 29.Nagato AC, Bezerra FS, Lanzetti M, Lopes AA, Silva MA, Porto LC, Valença SS. Time course of inflammation, oxidative stress and tissue damage induced by hyperoxia in mouse lungs. Int J Exp pathol. 2012;93:269–78. doi: 10.1111/j.1365-2613.2012.00823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raymondos K, Martin MU, Schmudlach T, Baus S, Weilbach C, Welte T, Krettek C, Frink M, Hildebrand F. Early alveolar and systemic mediator release in patients at different risks for ARDS after multiple trauma. Injury. 2012;43:189–95. doi: 10.1016/j.injury.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 31.Young SK, Arndt PG. c-Jun NH2-terminal kinase regulates lipopolysaccharide-induced pulmonary mononuclear cell recruitment via CCL2. Exp Lung Res. 2009;35:682–700. doi: 10.3109/01902140902853168. [DOI] [PubMed] [Google Scholar]
- 32.Monzon ME, Forteza RM, Casalino-Matsuda SM. MCP-1/CCR2B-dependent loop upregulates MUC5AC and MUC5B in human airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2011;300:L204–15. doi: 10.1152/ajplung.00292.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298:L715–31. doi: 10.1152/ajplung.00361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin WC, Lin CF, Chen CL, Chen CW, Lin YS. Prediction of outcome in patients with acute respiratory distress syndrome by bronchoalveolar lavage inflammatory mediators. Exp Biol Med (Maywood) 2010;235:57–65. doi: 10.1258/ebm.2009.009256. [DOI] [PubMed] [Google Scholar]
- 35.Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90:559–605. doi: 10.1152/physrev.00029.2009. [DOI] [PubMed] [Google Scholar]
- 36.Oenema TA, Kolahian S, Nanninga JE, Rieks D, Hiemstra PS, Zuyderduyn S, Halayko AJ, Meurs H, Gosens R. Pro-inflammatory mechanisms of muscarinic receptor stimulation in airway smooth muscle. Respir Res. 2010;11:130. doi: 10.1186/1465-9921-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka T, Terada M, Ariyoshi K, Morimoto K. Monocyte chemoattractant protein-1/CC chemokine ligand 2 enhances apoptotic cell removal by macrophages through Rac1 activation. Biochem Biophys Res Commun. 2010;399:677–82. doi: 10.1016/j.bbrc.2010.07.141. [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Das AM, Seideman J, Griswold D, Afuh CN, Kobayashi T, Abe S, Fang Q, Hashimoto M, Kim H, Wang X, Shen L, Kawasaki S, Rennard SI. The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am J Respir Cell Mol Biol. 2007;37:121–8. doi: 10.1165/rcmb.2005-0253OC. [DOI] [PubMed] [Google Scholar]
- 39.Winter C, Taut K, Srivastava M, Langer F, Mack M, Briles DE, Paton JC, Maus R, Welte T, Gunn MD, Maus UA. Lung-specific overexpression of CC chemokine ligand (CCL) 2 enhances the host defense to Streptococcus pneumoniae infection in mice: role of the CCL2-CCR2 axis. J Immunol. 2007;178:5828–38. doi: 10.4049/jimmunol.178.9.5828. [DOI] [PubMed] [Google Scholar]
- 40.Pechkovsky DV, Goldmann T, Ludwig C, Prasse A, Vollmer E, Muller-Quernheim J, Zissel G. CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir Res. 2005;6:75. doi: 10.1186/1465-9921-6-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boekhoudt GH, Guo Z, Beresford GW, Boss JM. Communication between NF-kappa B and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J Immunol. 2003;170:4139–47. doi: 10.4049/jimmunol.170.8.4139. [DOI] [PubMed] [Google Scholar]
- 42.Carpenter LR, Moy JN, Roebuck KA. Respiratory syncytial virus and TNF alpha induction of chemokine gene expression involves differential activation of Rel A and NF-kappa B1. BMC Infect Dis. 2002;2:5. doi: 10.1186/1471-2334-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maniatis NA, Sfika A, Nikitopoulou I, Vassiliou AG, Magkou C, Armaganidis A, Roussos C, Kollias G, Orfanos SE, Kotanidou A. Acid-induced acute lung injury in mice is associated with P44/42 and c-Jun N-terminal kinase activation and requires the function of tumor necrosis factor alpha receptor I. Shock. 2012;38:381–6. doi: 10.1097/SHK.0b013e3182690ea2. [DOI] [PubMed] [Google Scholar]
- 44.Siflinger-Birnboim A, Johnson A. Protein kinase C modulates pulmonary endothelial permeability: a paradigm for acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;284:L435–51. doi: 10.1152/ajplung.00106.2002. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Gigliotti F, Bhagwat SP, Maggirwar SB, Wright TW. Pneumocystis stimulates MCP-1 production by alveolar epithelial cells through a JNK-dependent mechanism. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1495–505. doi: 10.1152/ajplung.00452.2006. [DOI] [PubMed] [Google Scholar]
- 46.Wei L, Matsumoto H, Yamaguchi H. Propofol attenuates lipopolysaccharide-induced monocyte chemoattractant protein-1 production through p38 MAPK and SAPK/JNK in alveolar epithelial cells. J Anesth. 2013;27:366–73. doi: 10.1007/s00540-012-1539-7. [DOI] [PubMed] [Google Scholar]
- 47.Wuyts WA, Vanaudenaerde BM, Dupont LJ, Demedts MG, Verleden GM. Involvement of p38 MAPK, JNK, p42/p44 ERK and NF-kappaB in IL-1beta-induced chemokine release in human airway smooth muscle cells. Respir Med. 2003;97:811–7. doi: 10.1016/s0954-6111(03)00036-2. [DOI] [PubMed] [Google Scholar]
- 48.Boehringer N, Hagens G, Songeon F, Isler P, Nicod LP. Differential regulation of tumor necrosing factor-alpha (TNF-alpha) and interleukin-10 (IL-10) secretion by protein kinase and phosphatase inhibitors in human alveolar macrophages. Eur Cytokine Netw. 1999;10:211–8. [PubMed] [Google Scholar]
- 49.Zhao X, Shi C, Wang X, Andersson R. Protein kinase C modulates the pulmonary inflammatory response in acute pancreatitis. Respir Physiol Neurobiol. 2006;152:16–26. doi: 10.1016/j.resp.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Yuan C, Xu M, Chen G, Fu Y, Deng X. [Protein kinase C mediates thrombin-induced monocyte chemoattractant protein-1 release from human lung fibroblasts] . Nan Fang Yi Ke Da Xue Xue Bao. 2012;32:1250–4. [PubMed] [Google Scholar]
- 51.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol. 2000;279:L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 52.Culp DJ, Zhang Z, Evans RL. Role of calcium and PKC in salivary mucous cell exocrine secretion. J Dent Res. 2011;90:1469–76. doi: 10.1177/0022034511422817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wirtz HR, Dobbs LG. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science. 1990;250:1266–9. doi: 10.1126/science.2173861. [DOI] [PubMed] [Google Scholar]
- 54.Logan MR, Odemuyiwa SO, Moqbel R. Understanding exocytosis in immune and inflammatory cells: the molecular basis of mediator secretion. J Allergy Clin Immunol. 2003;111:923–32. quiz 33. [PubMed] [Google Scholar]
- 55.Ma HT, Beaven MA. Regulators of Ca(2+) signaling in mast cells: potential targets for treatment of mast cell-related diseases? Adv Exp Med Biol. 2011;716:62–90. doi: 10.1007/978-1-4419-9533-9_5. [DOI] [PubMed] [Google Scholar]
- 56.Zaidi AK, Thangam ER, Ali H. Distinct roles of Ca2+ mobilization and G protein usage on regulation of Toll-like receptor function in human and murine mast cells. Immunology. 2006;119:412–20. doi: 10.1111/j.1365-2567.2006.02450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabroe I, Hartnell A, Jopling LA, Bel S, Ponath PD, Pease JE, Collins PD, Williams TJ. Differential regulation of eosinophil chemokine signaling via CCR3 and non-CCR3 pathways. J Immunol. 1999;162:2946–55. [PubMed] [Google Scholar]
- 58.Ishida S, Koto T, Nagai N, Oike Y. Calcium channel blocker nilvadipine, but not diltiazem, inhibits ocular inflammation in endotoxin-induced uveitis. Jpn J Ophthalmol. 2010;54:594–601. doi: 10.1007/s10384-010-0862-5. [DOI] [PubMed] [Google Scholar]
- 59.Van de Weert-van Leeuwen PB, van Meegen MA, Speirs JJ, Pals DJ, Rooijakkers SH, Van der Ent CK, Terheggen-Lagro SW, Arets HG, Beekman JM. Optimal Complement-Mediated Phagocytosis of Pseudomonas aeruginosa by Monocytes is CFTR-Dependent. Am J Respir Cell Mol Biol. 2013 doi: 10.1165/rcmb.2012-0502OC. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 60.Bodas M, Min T, Vij N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. Am J Physiol Lung Cell Mol Physiol. 2011;300:L811–20. doi: 10.1152/ajplung.00408.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castellani S, Guerra L, Favia M, Di Gioia S, Casavola V, Conese M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: role of ezrin and the RhoA/ROCK pathway. Lab Invest. 2012;92:1527–40. doi: 10.1038/labinvest.2012.123. [DOI] [PubMed] [Google Scholar]