

Abstract
Hearing impairment affects 1 in 650 newborns, making it the most common congenital sensory impairment. Autosomal recessive nonsyndromic sensorineural hearing impairment (ARNSHI) comprises 80% of familial hearing impairment cases. Mutations in GJB2 account for a significant number of ARNSHI (and up to 50% of documented recessive (e.g. more than 1 affected sibling) hearing impairment in some populations). Mutations in the GJB2 gene are amongst the most common causes of hearing impairment in populations of various ethnic backgrounds. Two mutations of this gene, 35delG and 167delT, account for the majority of reported mutations in Caucasian populations, especially those of Mediterranean and Ashkenazi Jewish background. The 235delC mutation is most prevalent in East Asian populations. Some mutations are of less well characterized significance. The V37I missense mutation, common in Asian populations, was initially described as a polymorphism and later as a potentially pathogenic mutation. We report here on 15 unrelated individuals with ARNSHI and homozygosity for the V37I GJB2 missense mutation. Nine individuals are of Chinese ancestry, two are of unspecified Asian descent, one is of Japanese descent, one individual is of Vietnamese ancestry, one of Philippine background and one of Italian and Cuban/Caucasian background. Homozygosity for the V37I GJB2 mutation may be a more common pathogenic missense mutation in Asian populations, resulting in mild to moderate sensorineural hearing impairment. We report a presumed haplotype block specific to East Asian individuals with the V37I mutation encompassing the GJB2 gene that may account for the high prevalence in East Asian populations.
Keywords: GJB2, V37I, China, haplotype, hearing impairment
INTRODUCTION
Hearing impairment is the most common congenital sensory impairment, affecting 1 in 650 newborns [Mehl and Thomson, 2002]. Approximately 50% of cases are attributable to genetic causes with 70% occurring without other abnormalities and thus termed nonsyndromic hearing impairment. Autosomal recessive nonsyndromic sensorineural hearing impairment (ARNSHI) comprises 80% of familial hearing impairment cases. Mutations in the GJB2 gene are the largest genetic etiologic contributor to ARNSHI. GJB2 is believed to play a critical role in the recycling of potassium ions at their entry into hair cells during sensory transduction from the endolymph to the stria vascularis where other potassium channels pump potassium back into the endolymph [Kikuchi et al., 1995]. Certain mutations have been described frequently among various populations, including 35delG amongst individuals of Mediterranean and European decent, 167delT amongst individuals of Ashkenazi Jewish decent and 235delC amongst individuals of Asian ancestry [Wattanasirichaigoon et al., 2004]. The V37I missense mutation was first described as a polymorphism [Kelley et al., 1998] when it was found as a heterozygous variant in a sample from a control group. Then it was described as a potential pathogenic missense mutation [Bason et al., 2002; Wilcox et al., 2000]. Bruzzone et al. [2003] confirmed that the V37I mutation can result in impairment of channel activity. Snoeckx et al. [2005] reported 1,531 individuals with autosomal recessive hearing impairment, and confirmed that the V37I mutation accounted for 2.45% of the alleles in this studied population, and for 37% of Asians in the population. Even after these reports, the V37I mutation has been described both as a potentially hearing impairment-causing mutation in some studies [Dahl et al., 2006] as well as a change of unknown significance or as a non-pathogenic polymorphism [Wattanasirichaigoon et al., 2004]. While it remains possible (but unlikely given mounting evidence reported in the literature) that homozygosity for the V37I mutation may not be fully penetrant, the rare reports of individuals homozygous for this mutation without observed hearing impairment may be due to the manner in which hearing impairment is being ascertained as V37I related hearing impairment can be very mild.
We report here on 15 unrelated probands with ARNSHI who are all homozygous for the V37I GJB2 mutation, further supporting the pathogenicity of this mutation. The clinical characteristics of these probands are also reported (Table I). We have also performed haplotype analysis of this locus across individuals of Caucasian/European and Asian ancestry.
TABLE I.
Clinical and Audiologic Features of the Reported Homozygous Probands
| Proband No. | Age | Gender | Clinical Features | Audiology Findings | Temporal Bone CT/MRI | Renal Function Studies | Thyroid Function Studies | Family History | Ethnicity | Genotype |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 11y8m | F | Development: normal PE: one hypopigmented macule on back |
bilateral mild-moderate sensorineural HI diagnosed at 8 years of age |
Slight prominence to vestibular aqueduct bilaterally |
Normal | Normal | Unknown (adopted) | Chinese | V37I/V37I |
| 2*= | 3y5m | M | Development: Delay in speech and in playing skills PE: unilateral cleft lip and palate history Chromosome, SNP array, subtelomere analysis and FISH for 22q deletion normal |
bilateral mild-moderate sensorineural HI diagnosed at 3 years of age |
Not performed | Normal | Normal | Unknown (adopted) | Chinese | V37I/V37I |
| 3*= | 13y4m | F | Development: normal PE: right bundle branch block, prolonged QTC |
Bilateral mild sensorineural HI diagnosed at 13 years of age |
Normal | Normal | Normal | No Family History of HI | Vietnamese | V37I/V37I |
| 4*= | 4y11m | F | Development: Hyperactive and behind in social skills PE: within normal limits |
Bilateral mild sensorineural HI diagnosed at 3 years and 6 months of age |
Normal | Normal | Normal | Unknown (adopted) | Chinese | V37I/V37I |
| 5* | 10m | M | Development: normal PE: within normal limits |
Bilateral mild sensorineural HI diagnosed in newborn |
Normal | Normal | Normal | Sister with mild HI | Italian/Cuban Caucasian | V37I/V37I |
| 6 | 11y3m | F | Development: normal PE: within normal limits |
Bilateral mild-moderate left ear and mild right ear |
Normal | Normal | Normal | Unknown (adopted) | Chinese | V37I/V37I |
| 7*= | 6y3m | F | Development: normal PE: within normal limits |
Bilateral mild diagnosed at 5 years of age |
Normal | Normal | Normal | Unknown (adopted) | Chinese | V37I/V37I |
| 8 | 1y10m | M | Development: normal PE: within normal limits |
Bilateral mild high frequency diagnosed in newborn |
Not performed | Normal | Normal | Mother with same high frequency HI (also V37I/V37I) |
Chinese | V37I/V37I |
| 9 | 6y3m | M | Development: delayed PE: Microcephaly |
Diagnosed at 2 years of age with moderate HI progressed to severe by 6 years of age |
Normal | Not performed | Not performed | Mother with bilateral HI with onset in 20s |
Chinese/Cambodian | V37I/V37I |
| 10 | 4y6m | F | Development: normal PE: within normal limits |
Bilateral moderate HI in high frequencies diagnosed at 4 years of age |
Not performed | Not performed | Not performed | Mother with bilateral HI diagnosed in 20s with tinnitus and vestibular problems |
Philippine | V37I/V37I |
| 11 | 2y3m | F | Development: normal PE: within normal limitsMedical history: ASD, moderate myopia |
Bilateral mild to moderate HI diagnosed at 2 years of age |
Normal | Not performed | Not performed | Unknown (adopted) | Chinese | V37I/V37I |
| 12*= | 13yr7m | F | Development: normal PE: normal |
Bilateral moderate HI diagnosed at 12 years of age |
Normal | Normal | Normal | No family history of HI | Chinese | V37I/V37I |
| 13*= | 3yr | F | Development: normal | Mild bilateral hearing impairment that was considered the "lower limits of normal" |
Unknown | Unknown | Unknown | No family history of HI | Asian, unspecified | V37I/V37I |
| 14*= | 9yr | M | Development: normal | Moderately severe bilateral HI | Unknown | Unknown | Unknown | No family history of HI | Asian, unspecified | V37I/V37I |
| 15* | 9yr11m | F | Development: normal PE: normal |
Bilateral mild to moderately severe HI |
Not performed | Mostly normal, slightly elevated CO2 of 28 |
Normal | No family history of HI | Japanese | V37I/V37I |
HI: hearing impairment
PE: physical exam
QTC: heart rate-corrected QT interval
Patients haplotyped in this study
Patients homozygous for the ancestral SNP haplotype
MATERIALS AND METHODS
Probands
All probands (Table I) were enrolled in these studies under an IRB-approved protocol of informed consent at The Children’s Hospital of Philadelphia. Thirteen of the 15 V37I homozygous and eight of the 11 V37I heterozygote probands reported here (Table I), were identified from among a total cohort of 289 probands referred with bilateral sensorineural hearing impairment to The Genetics of Hearing Impairment Clinic at The Children’s Hospital of Philadelphia who were screened for GJB2 mutations (Table I). Of these probands, 271 were evaluated by Illumina SNP genotyping array. Twenty-three individuals in this cohort were of Asian ancestry as evaluated by ethnicity screening using Ancestry-Informative Marker (AIM) SNPs from the genotype data generated by the Illumina® 550K SNP array or from documented family history if genotyping data was unavailable. Those probands with heterozygous or homozygous V37I mutations were included in this study. Hearing impairment severity was categorized by the average thresholds between ears and classified in the range of: mild (20–39dB), moderate (40–69dB), severe (70–89dB) or profound (90dB). Routine dysmorphology exam, ophthalmologic exam, temporal bone CT or MRI, EKG, thyroid and renal function studies were performed on these probands. In addition to the group identified at CHOP, genetic samples from 301 children with nonsyndromic sensorineural hearing impairment from Boston Children’s Hospital were analyzed on SNP array. All samples were screened for common genetic causes of hearing impairment as described below. Of this group, fourteen were of Asian ancestry (4.7%) as determined by AIM SNPs. Of the 14 Asian probands from the Boston cohort two were found to be homozygous for V37I mutations and three were found to be heterozygous for the mutation. None of the non-Asian probands from the Boston cohort were found to be homozygous for V37I mutations. This gave a combined cohort (between CHOP and Boston Children’s Hospital) of 15 V37I homozygous and 11 V37I heterozygous probands.
Mutational analysis of GJB2, GJB6, the A1555G mitochondrial mutation and the seven common mutations in the SLC26A4 gene were screened for, as described elsewhere [Yaeger et al., 2006] Haplotype analysis across the GJB2 locus on chromosome 13q11-12 was performed on 375 individuals. The haplotype was determined by analyzing the arrayed SNPs of a 1MB segment (inclusive of 219 SNPs) that encompassed the GJB2 locus. In addition to analyzing the haplotypes of the nine probands with homozygous V37I mutations (8 Asians, 1 Caucasian), we also analyzed the haplotypes of the eight probands with a single V37I mutation (6 Asians, 1 half-Asian, and 1 Caucasian); all other Asians without V37I mutations (19); 159 Caucasian probands from our cohort for whom SNP genotyping was available; 90 randomly selected Caucasian children without V37I mutations, and 90 randomly selected Caucasian and Asian HapMap normal control samples.
Statistical analysis of the proposed haplotype in Asian V37I patients and Caucasian and Asian controls was performed using chi square testing for 2 × 2 tables with SigmaStat v3.5 (http://www.aspiresoftwareintl.com/html/sigmastat.html). As more than two alleles were represented at several SNPs in the cohort, all alleles except for the allele corresponding to the proposed haplotype were added together. If less than 80% of the expected frequencies exceeded five or if any of the expected frequencies did not exceed 1 then Yates’s correction for continuity was used. All p values were considered significant at < 0.05.
To test if haplotypes could be found in association with other GJB2 variants, chi square analysis was also performed using the same parameters as above on a cohort of 35delG patients seen in clinic and on Caucasian controls. Of these 35 patients, 15 are homozygous for the 35delG mutation, seven are heterozygous with no other known GJB2 mutation and 13 are compound heterozygotes for another GJB2 mutation.
RESULTS
The clinical features, audiological findings, family histories and ethnicities of the fifteen homozygous patients are summarized in Table I. While all probands were felt to be non-syndromic, Proband 2 had a repaired unilateral cleft lip and palate, Proband 3 was found to have a right bundle branch block, as well as a prolonged QTC secondary to a depolarization abnormality and Proband 1 demonstrated a slight prominence to her vestibular aqueduct bilaterally.
Out of our cohort of 590 probands, 26 individuals (4.4%) were found to have at least one V37I mutation. In six of these individuals the single V37I allele was the only mutation identified. In five of these probands there was a compound heterozygous V37I allele with another known GJB2 mutation. These individuals had varying severities of hearing impairment ranging from mild to moderate. Fifteen probands in the combined cohort were found to be homozygous for the V37I mutation. Of these 15 V37I probands, 14 were of Asian ancestry. Of the 41 V37I alleles identified in our hearing impairment cohort 36 (88%) were present in individuals of Asian ancestry. Of all probands from our cohort of Asian ancestry (n = 40) the 14 homozygous V37I probands with ARNSHI made up 35%.
Haplotype analysis across the GJB2 locus on chromosome 13q11-12 revealed a homozygous ancestral SNP haplotype consisting of G/A/A/A/C/G/G/G/A/G/C/A/A in nearly all the Asian V37I probands (Fig. 1). Initially we looked at all genotyped SNPs that were within 1MB of the single SNP that lies within GJB2, rs3751385. Seven out of eight Asian V37I homozygotes had completely homozygous genotypes from rs9578257 until rs945373, corresponding to a 66.2kb segment. The haplotypes were identical in seven of the eight Asian V37I homozygotes, 15 out of 16 alleles, and was not present in the single Caucasian V37I homozygote. The homozygous haplotype found in the Asian V37I homozygotes was presumed to be a V37I-specific haplotype. The six Asian heterozygotes and the single half-Asian heterozygote all had genotypes that were compatible for carrying the proposed V37I haplotype block. The Caucasian heterozygote did not carry the presumed haplotype block.
FIG. 1.
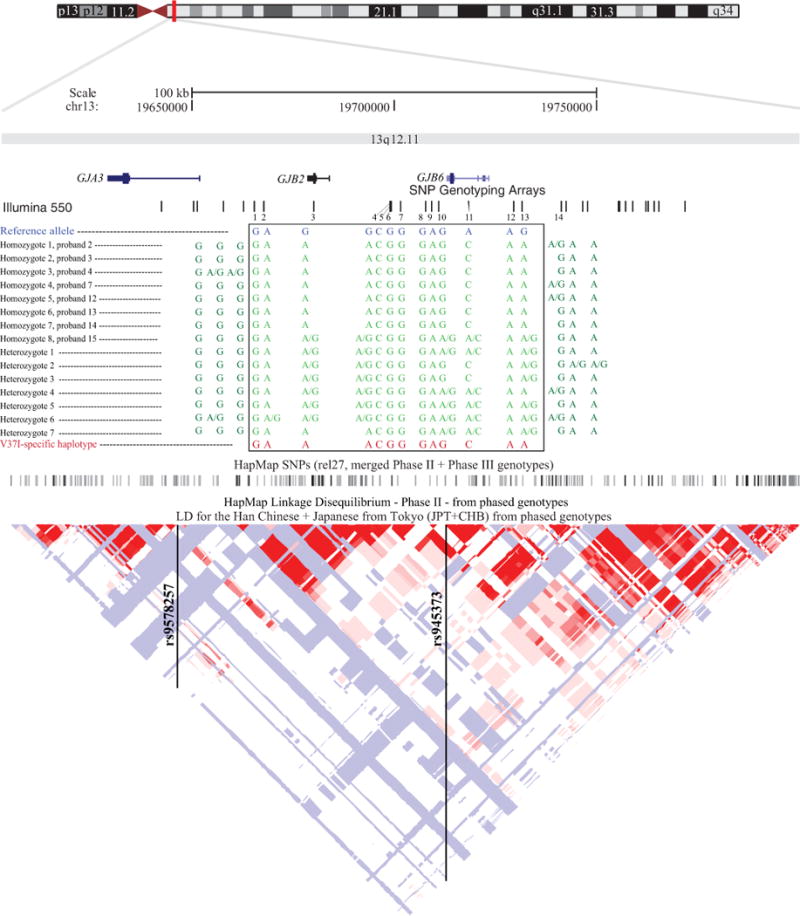
200 MB region at GJB2-GJB6 locus showing high linkage disequilibrium. Positions of the 13 SNPs comprising the proposed haplotype are shown in relation to the V37I mutation. Reference allele and the allele represented in Asian V37I probands at each SNP are shown. SNP1 = rs9578257 (15,253 bp downstream from V37I mutation), SNP2 = rs2313475 (12,834 bp downstream), SNP3 = rs3751385 (654 bp downstream ), SNP4 = rs870729 (18,269 bp upstream), SNP5 = rs11620460 (18,549 bp upstream), SNP6 = rs1889784 (18,622 bp), SNP7 = rs6490527 (20,819 bp), SNP8 = rs7981756 (27,132 bp), SNP9 = rs7330206 (28,574 bp), SNP10 = rs2065796 (30,259 bp), SNP11 = rs945369 (37,663 bp), SNP12 = rs7328044 (48,953 bp), SNP13 = rs945373 (50,949 bp). SNP14 (rs9509119; 60,433 bp upstream) is included to determine the age of the mutation.
(elements for this figure were adapted from the UCSC Genome Browser: http://genome.ucsc.edu/)
We examined all Asians without an identified V37I mutation. Of these nineteen individuals, only three (15.8%) had genotypes that were compatible with having the V37I haplotype block (due to the lack of an ability to phase these alleles, and in the absence of homozygosity across the region, we are only able to state that these genotypes are “compatible” with the V37I haplotype block). The remaining sixteen had genotypes that ruled out the possibility of having the proposed V37I haplotype. Among the entire genotyped cohort without an identified V37I mutation, 41 out of 358 individuals (11.5%) had genotypes that were compatible with having the proposed V37I haplotype. No homozygotes for this haplotype block were found among those without a V37I mutation.
Statistical analysis found nine of the 13 SNPs in the proposed haplotype to be significantly different between Asian patients and Asian controls (Table II). Between Asian patients and Caucasian controls 10 of the 13 SNPs were found to be significantly different. The one heterozygous and the one homozygous V37I Caucasian patients were excluded from this analysis.
TABLE II.
Results of SNP analysis for V37I probands and controls
| SNP | Allele | V37I Asian patients | Asian controls | Caucasian controls | P value (patients v. Asian controls) | P value (patients v. Caucasian controls) |
|---|---|---|---|---|---|---|
| rs9578257 | G OTHER |
30 0 |
201 9 |
369 105 |
0.521 | 0.008 |
| rs2313475 | A OTHER |
29 1 |
210 4 |
363 135 |
0.875 | 0.007 |
| rs3751385 | A OTHER |
23 7 |
10 204 |
86 412 |
< 0.001 | < 0.001 |
| rs870729 | A OTHER |
23 7 |
93 121 |
123 375 |
0.001 | < 0.001 |
| rs11620460 | C OTHER |
30 0 |
215 1 |
472 24 |
0.247 | 0.434 |
| rs1889784 | G OTHER |
30 0 |
212 2 |
426 64 |
0.583 | 0.068 |
| rs6490527 | G OTHER |
30 0 |
38 178 |
287 211 |
< 0.001 | < 0.001 |
| rs7981756 | G OTHER |
30 0 |
38 185 |
258 240 |
< 0.001 | < 0.001 |
| rs7330206 | A OTHER |
30 0 |
38 67 |
438 60 |
< 0.001 | 0.085 |
| rs2065796 | G OTHER |
25 5 |
38 181 |
165 321 |
< 0.001 | < 0.001 |
| rs945369 | C OTHER |
24 6 |
38 129 |
173 323 |
< 0.001 | < 0.001 |
| rs7328044 | A OTHER |
30 0 |
38 181 |
271 227 |
< 0.001 | < 0.001 |
| rs945373 | A OTHER |
23 7 |
38 195 |
100 398 |
< 0.001 | < 0.001 |
| rs9509119 | G OTHER |
25 5 |
137 81 |
334 162 |
0.045 | 0.104 |
The allelic age of V37I was estimated by using the recombination frequency of a SNP rs9509119 found 60 kb away and just outside of the shared haplotype. The method of using a nearby marker to calculate allelic age has been previously described by Van Laer et al. [2001]. The recombination fraction, θ, is the fraction of probands that did not have a recombination event between rs9509119 and the V37I mutation (θ = 0.0006). Since (1 − θ) is the fraction of chromosomes that exhibit no recombination, then (1- θ)N is the fraction of chromosomes that have not exhibited recombination over N generations. Assuming that the V37I mutation occurred on the haplotype containing the common G allele of rs9509119, 25 of the 30 chromosomes we analyzed showed no evidence of recombination. Therefore, the V37I mutation in our Asian cohort is approximately 300 generations old.
This result is comparable to the estimation of allelic age based on the carrier frequency of V37I in the Han Chinese and Japanese populations. The allelic age can be calculated based by N = −2p/(1 − p) * ln(p) * Ne, where t is the time in generations, p is the allele frequency and Ne is the effective population size [Slatkin and Rannala, 2000]. The effective population sizes are estimated in the Han Chinese (Ne = 2,620) and Japanese (Ne = 2,517) populations based on HapMap haplotype studies [Tenesa et al., 2007]. The carrier frequency of V37I is reported to be 3–6% in the Han Chinese population and 1–3% in the Japanese population [Abe et al., 2000; Dai et al., 2009; Kudo et al., 2000; Li et al., 2012]. The allelic age of V37I estimated from these studies is between 200 and 1,000 generations old.
Chi square analysis done on 35delG patients and Caucasian controls for the same area shows significant differences for 10 out of 13 SNPs (Table III). The most common SNPs for this region in our 35delG cohort were G/G/A/A/C/G/G/G/A/A/A/A/G. All 35delG patients had genotypes that were compatible with this SNP block but only 8 patients (all of whom were homozygous for the 35delG mutation) had this block of SNPs homozygously. Of the 35 35delG patients screened, a total of 15 35delG homozygotes were found, seven of whom did not have this block of SNPs in a homozygous state. As determined by Kokotas et al. [2008], the 35delG mutation was estimated to be about 700 generations old.
TABLE III.
Results of SNP analysis for 35delG probands and controls
| SNP | Allele | 35delG patients | Caucasian controls | P value |
|---|---|---|---|---|
| rs9578257 | G OTHER |
70 0 |
369 105 |
< 0.001 |
| rs2313475 | G OTHER |
46 0 |
135 363 |
< 0.001 |
| rs3751385 | A OTHER |
50 0 |
86 412 |
< 0.001 |
| rs870729 | A OTHER |
53 0 |
123 375 |
< 0.001 |
| rs11620460 | C OTHER |
70 0 |
472 24 |
0.118 |
| rs1889784 | G OTHER |
64 6 |
426 64 |
0.385 |
| rs6490527 | G OTHER |
65 5 |
287 211 |
< 0.001 |
| rs7981756 | G OTHER |
60 8 |
258 240 |
< 0.001 |
| rs7330206 | A OTHER |
65 5 |
438 60 |
0.314 |
| rs2065796 | A OTHER |
55 15 |
153 333 |
< 0.001 |
| rs945369 | A OTHER |
63 7 |
323 173 |
< 0.001 |
| rs7328044 | A OTHER |
66 4 |
271 227 |
< 0.001 |
| rs945373 | G OTHER |
60 10 |
218 280 |
< 0.001 |
DISCUSSION
In the present study, we have reported on 15 unrelated children with ARNSHI who are homozygous for V37I GJB2 mutation. Published reports suggest that biallelic V37I mutations are likely pathogenic in Asian children with ARNSHI [Bason et al., 2002; Dahl et al., 2006; Siemering et al., 2006]. Of our 15 probands, nine are of Chinese ancestry, two are of Asian descent, one is of Vietnamese ancestry, one is of Philippine background, one is of Japanese background, and one is of Italian, Cuban and Caucasian background. GJB2 gene mutation spectrums are quite different between East Asian populations such as Japanese and Chinese [Oguchi et al., 2005; Ohtsuka et al., 2003] and most European or North American populations [Huculak et al., 2006]. Amongst Asians, 235delC is the most common GJB2 mutation, in contrast to the 35delG mutation which is the most prevalent mutation in individuals of European or North American ancestry. The second most common GJB2 mutation reported in the Japanese is the V37I mutation [Cryns et al., 2004], at 16.5%.[Tsukada et al., 2010]
The significance of the V37I allele has been much debated in the literature. Snoeckx et al. [Snoeckx et al., 2005] reported 1,531 individuals from sixteen different countries with autosomal recessive mild to profound nonsyndromic hearing impairment. The majority of participants were white (90%). Asians represented 1.8% of the studied cohort. The V37I allele accounted for 2.45% of the alleles in the total study population but 37% of the alleles in Asians, which is close to the 5.2% found in our total genotyped cohort and the 35% found in our Asian cohort. Another study analyzed the GJB2 gene in 51 deaf students from Malaysia [Ruszymah et al., 2005]. Bidirectional sequencing demonstrated that 25% of childhood deafness in this population was caused by mutations in the GJB2 gene and 62% of these children demonstrated at least one V37I missense mutation. V37I missense mutations were not found in the normal control population. Huculak et al. [2006] reported GJB2 testing in 40 individuals of Chinese ancestry with bilateral sensorineural hearing impairment tested for GJB2 mutations and compared these results with 100 Chinese controls, 100 Caucasian controls and 40 Caucasian individuals with bilateral sensorineural hearing impairment. Of the 40 Chinese patients, 13 were found to be V37I homozygotes, six V37I heterozygotes, and two compound heterozygotes. One Chinese control individual was found to be a V37I homozygote and 21 were heterozygous for the V37I mutation. The frequency of the V37I allele was 43.75% in the Chinese patient chromosomes and 11.5% in the Chinese control chromosomes. Among the Caucasian controls none of the 40 individuals with BLSNHL were found to carry a V37I mutation nor any of the 100 unaffected controls. These authors concluded that the V37I allele is a common pathogenic allele in the Chinese population. This conclusion was supported by V37I functional studies in COS-7 cells [Oguchi et al., 2005] and in Xenopus oocytes [Bruzzone et al., 2003] that have shown that the V37I variant is associated with abnormal connexin 26 function. In our study, 13 of 15 homozygous V37I probands displayed a pattern of mild to moderate NSHL, one proband displayed moderately severe HL, and one proband had moderate HL that progressed to severe. These results corroborate previous studies [Cryns et al., 2004; Huculak et al., 2006; Oguchi et al., 2005; Snoeckx et al., 2005] demonstrating that the V37I missense mutation is pathogenic and results in a less severe hearing impairment phenotype than frameshift mutations such as the 35delG or 235delC mutations.
The debate in the literature on the pathogenic significance of the V37I mutation stems partially from the difficulty in comparing data from different studies. Variability of the patient cohorts and the severities of hearing impairment being assessed have made meaningful comparisons across studies challenging. In the initial reports of GJB2 mutations and their relevance to hearing impairment the patient populations typically were of non-Asian ethnic backgrounds and with severe to profound hearing impairment [Gronskov et al., 2004]. Additional reports of cohorts with some Asian ethnic participants were difficult to interpret as they did not include individuals with mild to moderate hearing impairment [Propst et al., 2006]. The V37I missense mutation was likely under-ascertained in these studies because they were biased towards identification of more severe alleles (e.g. the 35delG mutation) and were not representative of all connexin 26-related hearing impairment. While we cannot conclusively exclude the possibility that the V37I allele may be in linkage disequilibrium with an unidentified pathogenic allele, pervious studies and the findings reported here add further support to the argument that the V37I variant is a pathogenic mutation and typically results in a milder hearing impairment phenotype, often causing a mild-moderate sensorineural hearing impairment when present homozygously.
Haplotype analysis across the GJB2 locus on chromosome 13q11-12 supports an ethnic-specific haplotype which is seen in the SNP-array genotyping of all Asian probands homozygous for V37I and found to be compatible with all heterozygous Asian V37I probands. No controls, Asian or Caucasian, were found to have this haplotype homozygously. This proposed haplotype found in heterozygous and homozygous V37I Asian probands was largely found to be significantly different from the genotypes of Caucasian and Asian controls.
The genotypes found in the 35delG probands were also found to be significantly different from those of controls. Although only eight patients were found to have this possible haplotype homozygously, all patients, both homozygous and heterozygous, had genotypes which were compatible with having the haplotype. Several haplotypes have been found to be associated with specific Caucasian populations [Morell et al., 1998] although it has been proposed that there is a common haplotype from a common founder [Kokotas H et al., 2008; Van Laer et al., 2001]. Given the high prevalence of the 35delG mutation in European populations and the diverse backgrounds of our Caucasian cohort of 35delG probands it is interesting that eight of the 15 35delG homozygous patients (53.3%) are homozygous for this block of SNPs. It is possible that a long-range haplotype could be tagging the 35delG mutation.
The utility of SNP arrays in diagnosing copy number variants that are pathogenic and causative of disease phenotypes has been well documented [Kamath et al., 2009; Yau and Holmes, 2008]. Using SNP array platforms to identify disease-associated haplotypes, as in this case for the V37I mutation and ARNSHI, is feasible and adds another utilization to the platform in mutational screening. Haplotype analysis has proven a useful tool in uncovering rare causal variants that have been masked by common tag SNPs, with SNP-array genotyping data alone being successfully used to screen for the presence of pathogenic mutations at the GJB2-GJB6 locus in a known hearing impairment cohort [Wang et al., 2007]. This V37I-specific haplotype and others like it yet to be discovered could be used diagnostically in ARNSHI cases.
Acknowledgments
We would like to acknowledge the families who participated in this study. This work was supported by The Children’s Hospital of Philadelphia Institutional Develop Funds and by NIH/NIDCD Grant R33DC008630 (IDK).
References
- Abe S, Usami S, Shinkawa H, Kelley P, Kimberling W. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37:3. doi: 10.1136/jmg.37.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bason L, Dudley T, Lewis K, Shah U, Potsic W, Ferraris A, Fortina P, Rappaport E, Krantz ID. Homozygosity for the V37I Connexin 26 mutation in three unrelated children with sensorineural hearing loss. Clin Genet. 2002;61(6):459–464. doi: 10.1034/j.1399-0004.2002.610611.x. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D’Andrea P, White TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS lett. 2003;533(1–3):79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- Cryns K, Orzan E, Murgia A, Huygen PL, Moreno F, del Castillo I, Chamberlin GP, Azaiez H, Prasad S, Cucci RA, Leonardi E, Snoeckx RL, Govaerts PJ, Van de Heyning PH, Van de Heyning CM, Smith RJ, Van Camp G. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J Med Genet. 2004;41(3):147–154. doi: 10.1136/jmg.2003.013896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl HH, Tobin SE, Poulakis Z, Rickards FW, Xu X, Gillam L, Williams J, Saunders K, Cone-Wesson B, Wake M. The contribution of GJB2 mutations to slight or mild hearing loss in Australian elementary school children. J Med Genet. 2006;43(11):850–855. doi: 10.1136/jmg.2006.042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Yu F, Han B, Liu X, Wang G, Li Q, Yuan Y, Huang D, Kang D, Zhang X, Yuan H, Yao K, Hao J, He J, He Y, Wang Y, Ye Q, Yu Y, Lin H, Liu L, Deng W, Zhu X, You Y, Cui J, Hou N, Xu X, Zhang J, Tang L, Song R, Lin Y, Sun S, Zhang R, Wu H, Ma Y, Zhu S, Wu BL, Han D, Wong LJ. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009;7:26. doi: 10.1186/1479-5876-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronskov K, Larsen LA, Rendtorff ND, Parving A, Norgaard-Pedersen B, Brondum-Nielsen K. GJB2 and GJB6 mutations in 165 Danish patients showing non-syndromic hearing impairment. Genet Test. 2004;8(2):181–184. doi: 10.1089/gte.2004.8.181. [DOI] [PubMed] [Google Scholar]
- Huculak C, Bruyere H, Nelson TN, Kozak FK, Langlois S. V37I connexin 26 allele in patients with sensorineural hearing loss: evidence of its pathogenicity. Am J Med Genet A. 2006;140(22):2394–2400. doi: 10.1002/ajmg.a.31486. [DOI] [PubMed] [Google Scholar]
- Kamath B, Thiel B, Gai X, Conlin L, Munoz P, Glessner J, Clark D, Warthen D, Shaikh T, Mihei E, Piccoli D, Grant S, Hakonarson H, Krantz I, Spinner N. SNP array mapping of chromosome 20p deletions: genotypes, phenotypes, and copy number variation. Hum Mutat. 2009;30:371–378. doi: 10.1002/humu.20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley PM, Harris DJ, Comer BC, Askew JW, Fowler T, Smith SD, Kimberling WJ. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am J Med Genet. 1998;62(4):792–799. doi: 10.1086/301807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat Embryol. 1995;191(2):101–118. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]
- Kokotas H, Van Laer L, Grigoriadou M, Iliadou V, Economides J, Pomoni S, Pampanos A, Eleftheriades N, Ferekidou E, Korres S, Giannoulia-Karantana A, VanCamp G, MB P. Strong linkage disequilibrium for the frequent GJB2 35delG mutation in the Greek population. Am J Med Genet Part A. 2008;146A:2879–2884. doi: 10.1002/ajmg.a.32546. [DOI] [PubMed] [Google Scholar]
- Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe K, Kawase T, Narisawa K, Takasaka T. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet. 2000;90:5. doi: 10.1002/(sici)1096-8628(20000117)90:2<141::aid-ajmg10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Li L, Lu J, Tao Z, Huang Q, Chai Y, Li X, Huang Z, Li Y, Xiang M, Yang J, Yao G, Wang Y, Yang T, Wu H. The p.V37I Exclusive Genotype Of GJB2: A Genetic Risk-Indicator of Postnatal Permanent Childhood Hearing Impairment. PLoS One. 2012;7(5):e36621. doi: 10.1371/journal.pone.0036621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. The Colorado newborn hearing screening project, 1992–1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics. 2002;109(1):E7. doi: 10.1542/peds.109.1.e7. [DOI] [PubMed] [Google Scholar]
- Morell R, Kim H, Hood L, Goforth L, Frederici K, Fisher R, Van Camp G, Berlin C, Oddoux C, Ostrer H, Keats B, Friedman T. Mutations in the connxin26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. New Engl J Med. 1998;339:1500–1505. doi: 10.1056/NEJM199811193392103. [DOI] [PubMed] [Google Scholar]
- Oguchi T, Ohtsuka A, Hashimoto S, Oshima A, Abe S, Kobayashi Y, Nagai K, Matsunaga T, Iwasaki S, Nakagawa T, Usami S. Clinical features of patients with GJB2 (connexin 26) mutations: severity of hearing loss is correlated with genotypes and protein expression patterns. J Hum Genet. 2005;50(2):76–83. doi: 10.1007/s10038-004-0223-7. [DOI] [PubMed] [Google Scholar]
- Ohtsuka A, Yuge I, Kimura S, Namba A, Abe S, Van Laer L, Van Camp G, Usami S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet. 2003;112(4):329–333. doi: 10.1007/s00439-002-0889-x. [DOI] [PubMed] [Google Scholar]
- Propst EJ, Stockley TL, Gordon KA, Harrison RV, Papsin BC. Ethnicity and mutations in GJB2 (connexin 26) and GJB6 (connexin 30) in a multi-cultural Canadian paediatric Cochlear Implant Program. Int J Pediatr Otorhi. 2006;70(3):435–44. doi: 10.1016/j.ijporl.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Ruszymah BH, Wahida IF, Zakinah Y, Zahari Z, Norazlinda MD, Saim L, Aminuddin BS. Congenital deafness: high prevalence of a V37I mutation in the GJB2 gene among deaf school children in Alor Setar. Med J Malaysia. 2005;60(3):269–274. [PubMed] [Google Scholar]
- Siemering K, Manji SS, Hutchison WM, Du Sart D, Phelan D, Dahl HH. Detection of mutations in genes associated with hearing loss using a microarray-based approach. J Mol Diagn. 2006;8(4):483–489. doi: 10.2353/jmoldx.2006.050147. quiz 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M, Rannala B. Estimating allele age. Annu Rev Genomics Hum Genet. 2000;1:25. doi: 10.1146/annurev.genom.1.1.225. [DOI] [PubMed] [Google Scholar]
- Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, Pollak A, Ploski R, Murgia A, Orzan E, Castorina P, Ambrosetti U, Nowakowska-Szyrwinska E, Bal J, Wiszniewski W, Janecke AR, Nekahm-Heis D, Seeman P, Bendova O, Kenna MA, Frangulov A, Rehm HL, Tekin M, Incesulu A, Dahl HH, du Sart D, Jenkins L, Lucas D, Bitner-Glindzicz M, Avraham KB, Brownstein Z, del Castillo I, Moreno F, Blin N, Pfister M, Sziklai I, Toth T, Kelley PM, Cohn ES, Van Maldergem L, Hilbert P, Roux AF, Mondain M, Hoefsloot LH, Cremers CW, Lopponen T, Lopponen H, Parving A, Gronskov K, Schrijver I, Roberson J, Gualandi F, Martini A, Lina-Granade G, Pallares-Ruiz N, Correia C, Fialho G, Cryns K, Hilgert N, Van de Heyning P, Nishimura CJ, Smith RJ, Van Camp G. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77(6):945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenesa A, Navarro P, Hayes B, Duffy D, Clarke G, Goddard M, Visscher P. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007;17:7. doi: 10.1101/gr.6023607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada K, Nishio S, Usami S, Consortium TDGS. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin Genet. 2010;78:464–470. doi: 10.1111/j.1399-0004.2010.01407.x. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Coucke P, Mueller R, Caethoven G, Flothmann K, Prasad S, Chamberlin G, Houseman M, Taylor G, Heyning CVd, Fransen E, Rowland J, Cucci R, Smith R, Camp GV. A common founder for the 35delG GJB2 gene mutation in connxin26 hearing impairment. J Med Genet. 2001;38:515–518. doi: 10.1136/jmg.38.8.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanasirichaigoon D, Limwongse C, Jariengprasert C, Yenchitsomanus PT, Tocharoenthanaphol C, Thongnoppakhun W, Thawil C, Charoenpipop D, Pho-iam T, Thongpradit S, Duggal P. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals. Clin Genet. 2004;66(5):452–460. doi: 10.1111/j.1399-0004.2004.00325.x. [DOI] [PubMed] [Google Scholar]
- Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, Collins V, Wilcox LJ, McKinlay Gardner RJ, Kamarinos M, Cone-Wesson B, Williamson R, Dahl HH. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106(4):399–405. doi: 10.1007/s004390000273. [DOI] [PubMed] [Google Scholar]
- Yaeger D, McCallum J, Lewis K, Soslow L, Shah U, Potsic W, Stolle C, Krantz I. Outcomes of clinical examination and genetic testing of 500 individuals with hearing loss evaluated through a genetics of hearing loss clinic. Am Journal Med Genet A. 2006;140A:827–836. doi: 10.1002/ajmg.a.31179. [DOI] [PubMed] [Google Scholar]
- Yau C, Holmes C. CNV discovery using SNP genotyping arrays. Cytogenet Genome Res. 2008;123:307–312. doi: 10.1159/000184722. [DOI] [PubMed] [Google Scholar]