

Abstract
This report describes an algorithm developed to predict the pathogenicity of copy number variants (CNVs) in large sample cohorts. CNVs (genomic deletions and duplications) are found in healthy individuals and in individuals with genetic diagnoses, and differentiation of these two classes of CNVs can be challenging and usually requires extensive manual curation. We have developed PECONPI, an algorithm to assess the pathogenicity of CNVs based on gene content and CNV frequency. This software was applied to a large cohort of patients with genetically heterogeneous non-syndromic hearing loss to score and rank each CNV based on its relative pathogenicity. Of 636 individuals tested, we identified the likely underlying etiology of the hearing loss in 14 (2%) of the patients (1 with a homozygous deletion, 7 with a deletion of a known hearing loss gene and a point mutation on the trans allele and 6 with a deletion larger than 1 Mb). We also identified two probands with smaller deletions encompassing genes that may be functionally related to their hearing loss. The ability of PECONPI to determine the pathogenicity of CNVs was tested on a second genetically heterogenous cohort with congenital heart defects (CHDs). It successfully identified a likely etiology in 6 of 355 individuals (2%). We believe this tool is useful for researchers with large genetically heterogeneous cohorts to help identify known pathogenic causes and novel disease genes.
Keywords: Copy number variation, genetic heterogeneity
INTRODUCTION
The recent advent of high resolution, genome-wide copy number analysis using chromosomal microarray platforms to detect small genomic deletions and duplications has revolutionized both the diagnosis of genetic disorders and the discovery of disease genes [Vissers et al., 2004; Beaudet and Belmont, 2008]. Copy number variations (CNVs) can be detected by both comparative genomic hybridization (CGH) and single nucleotide polymorphism (SNP) array platforms, which share the ability to scan across the genome at high resolution. Multiple studies have shown that there is considerable copy number variation among normal human subjects, comprising a significant percentage of the genome at the structural level [Iafrate et al., 2004; Sebat et al., 2004; Tuzun et al., 2005; Conrad et al., 2006; McCarroll et al., 2006; Redon et al., 2006; Sebat, 2007]. Therefore, it has become clear that CNVs can occur in genes but still be clinically benign, while the deletion or duplication of other genes may directly cause disease. CNV-calling algorithms have been developed to automate the process of detecting CNV intervals from array hybridization [Komura et al., 2006; Colella et al., 2007; Marioni et al., 2007; Wang et al., 2007], however; these detection algorithms do not discern the benign from the pathogenic CNVs.
CNV analysis in large cohorts of individuals with specific phenotypes can be used to uncover deletions or duplications of specific genes that contribute to the phenotype. This is particularly useful for phenotypes that are genetically heterogeneous, since CNV detection provides an unbiased analysis across the entire genome. For Mendelian disorders, the deletion of a causative gene within a pathogenic CNV may be a rare event. However, the identification of rare deletion events can lead to the discovery of candidate genes to be screened for point mutations, a more common form of gene disruption. It is laborious to peruse through CNV data for each patient of a large cohort. Moreover, the discrimination of pathogenic CNVs from benign CNVs is not always clear. The large number of CNVs identified per individual (24 CNVs on average on a half-million marker SNP array) [Redon et al., 2006] and cohort sizes spanning many hundreds or thousands of patients turns the process of unmasking pathogenic CNVs into a high-throughput task. We have developed Perl Copy Numbers of Potential Interest (PECONPI), an algorithm to ranks CNVs to help researchers prioritize their search for the pathogenic CNVs. PECONPI utilizes the properties of each CNV to rank them based on their potential to be damaging. We developed a logistic regression based algorithm to measure the performance of PECONPI’s heuristic algorithm.
Both algorithms were benchmarked on a cohort of 100 patients with known pathogenic CNVs as described in Supplementary Information (in supporting information online). For this study, we chose to focus on only the copy number losses (deletions) and not duplications, as gene deletions are more commonly the cause of genetic disease and their mechanisms are better understood than duplications [Emanuel and Shaikh 2001]. We applied PECONPI to the CNVs of a 636 proband cohort with nonsyndromic sensorineural hearing loss (NSSNHL), a common, genetically heterogeneous disorder. We also used PECONPI to prioritize the CNVs from a cohort of 355 probands with congenital heart disease (CHD), another genetically hetereogenous disorder (see Supplementary Information). The ability to delineate pathogenic CNVs is important for gene discovery in heterogeneous disorders. The contribution of a deleterious CNV containing a dominant disease gene (resulting in haploinsufficiency) to a disorder is direct and clear, however a heterozygous deletion of a gene can also unmask a recessive mutation on the trans allele and lead to the discovery of a new genetic locus for a disorder.
In this study, we demonstrate the utility of PECONPI to identify pathogenic gene deletions in NSSNHL. We determined a likely underlying molecular diagnosis in 14 cases of NSSNHL as well as some candidate genes that require further validation. Our work with these common, genetically heterogeneous disorders may have relevance for investigators using genome-wide CNV analysis to study common, genetically complex human traits such as autism and schizophrenia [Sebat et al., 2007; Szatmari et al., 2007; Marshall et al., 2008; Walsh et al., 2008; Weiss et al., 2008].
SUBJECTS AND METHODS
Sample Selection
NSSNHL Cohort
The bilateral sensorineural hearing loss cohort is comprised of 636 children with NSSNHL presenting at The Genetics for Hearing Loss Clinic at CHOP for a genetic evaluation of their hearing loss or through the Laboratory of Molecular Medicine (LMM) at the Harvard Medical School – Partners Healthcare Center for Genetics and Genomics (HPCGG). All probands and families studied were either voluntarily enrolled in an IRB-approved protocol of informed consent (CHOP, LMM) or analyzed through an anonymous discarded tissue IRB-approved protocol (LMM). All probands studied had a confirmed nonsyndromic bilateral sensorineural hearing loss as based upon clinical history, exam, review of records and audiometric testing. Other pre-screening and selection factors are described in the Supplementary Information (in supporting information online). All individuals with confirmed homozygous or compound heterozygous causative mutations were excluded from the analysis, with the exception of patients with GJB6 deletions, who were retained in the cohort as positive controls.
CHD Cohort
This cohort consists of 355 probands with apparent isolated congenital heart defects (CHDs) ascertained through the Department of Cardiothoracic Surgery at CHOP and voluntarily enrolled in an IRB-approved protocol of informed consent. All probands had standard cytogenetic analysis performed through G-banding and a subset had targeted fluorescent in situ hybridization (FISH) or CGH studies performed when requested clinically. This cohort was selected due to the genetic heterogeneity of CHDs, with some being caused by large CNVs (e.g. 22q11.21 deletion) and most being of unknown etiology, echoing the issues faced in the hearing loss cohort. The analysis of this cohort is detailed in the Supplementary Information (in supporting information online).
Controls Cohort
The CNVs of 2,026 normal probands run on the Illumina 550 BeadChip [Shaikh et al., 2009], were used as control data. During quality control, 16 controls were filtered out, because SD(log2R)>= 0.35. We withheld a randomly selected cohort of 100 probands from this control cohort for cross validation (described in Supporting Information online). The remaining 1,910 probands were used in our control cohort.
SNP Genotyping
All the NSSNHL probands were analyzed using HumanHap550-v3 BeadChip at the Center for Applied Genomics at CHOP, using recommended quality control protocols as previously described [Steemers and Gunderson 2007]. The CHD cohort consisted of probands run on the 550-v3 as well as the 610-Quad BeadChip. The healthy controls from the Center for Applied Genomics were genotyped on the 550 BeadChip. To analyze the data across cohorts using different SNP arrays, the probes unique to either the 550 or the 610 platforms were removed. The raw data were normalized to reduce the variation between BeadChip arrays and then the clustering algorithm was run to evaluate the positions and to genotype the locus of each SNP probe. All samples had GenCall genotyping call rates ≥ 0.98 with a standard deviation of log2R ratios ≤ 0.35.
CNV Calling
PennCNV was used to call the CNVs for the NSSNHL cohort, CHD cohort, and the controls cohort [Wang et al., 2007]. All calls are reported as the minimal CNV size, which is defined by the interval between the first and last abnormal SNP in the call. For both NSSNHL and CHD cohorts, the SNP array data and CNV calls of the probands were de-identified so that the investigators identifying and scoring potential pathogenic associations were unaware of any clinical information (i.e. previous molecular test results, patient or family history or severity of hearing loss) pertaining to the proband being tested.
PECONPI Software
PECONPI is a software program that takes any set of CNV calls and ranks the CNVs based upon their likelihood to be pathogenic using a multidimensional algorithm. PECONPI’s scoring algorithm is based upon the largely intuitive analysis of genetic variants for their potential to be pathogenic. For example, calls that have no gene involvement or that overlap with regions of known benign CNV (as demonstrated in controls and/or literature references) tend to be less suspicious for pathogenicity, whereas CNVs that are large and/or that encompass multiple genes have greater potential to be pathogenic [Lee et al., 2007]. The PECONPI score is the final value reported for each CNV call. PECONPI reports the CNV information, a breakdown of subscores, and the final PECONPI score in an Excel spreadsheet. Each row represents an input CNV call, and the rows are ranked in order of predicted pathogenicity. Screenshots of the PECONPI graphical user interface (GUI) can be found in Supplementary eFig. 1–3 (in supporting information online). The default parameters were used for the analysis of all the cohorts. From these parameters, a score of 25 points or higher corresponds roughly to a deletion CNV involving exonic DNA and little control or literature CNV overlap.
Input Files
Because PECONPI uses data from many different databases, it requires the user to supply the path to flat files containing information about the sample CNVs, control CNVs, known genes, and SNP markers. Users can include additional information such as CNVs reported in the literature (see Supplementary Information (in supporting information online)). To eliminate systematic errors of CNV-calling, the control CNVs are called with the same calling algorithm as the sample CNVs. The controls cohort were run on the Illumina 550 BeadChip SNP array while the samples were run on a higher density Illumina 610-Quad BeadChip SNP array, we chose to only use markers common to both platforms for our CNV-calling and subsequent analysis.
Scoring Algorithm
PECONPI analyzes each sample CNV and assigns it a set of subscores reflecting the CNV's coverage in control patients and literature, its gene content, and its coverage in the sample cohort. The sum of these subscores determines the final PECONPI score for the given CNV call. The PECONPI software flowchart is illustrated in Supplementary eFig. 4 (in supporting information online). The PECONPI subscores and parameters are detailed in Supplementary eTable I (in supporting information online), and we used these default values in our analysis. PECONPI scores and outputs deletions and duplications separately. Many of the subscores are based upon a coverage metric, which is defined as the percentage of the sample CNV (or any other interval) that overlaps with one or more different intervals as measured by physical genomic distance. The weight and the maximum contribution of each subscore can be adjusted. A more detailed description of additional data available in the PECONPI analysis is described in the supplementary information. The performance of PECONPI was compared to a statistically modeled pathogenic CNV prediction algorithm we developed and both algorithms performed similarly.
Validation of OTOF deletion
In order to clone the breakpoints of a homozygous OTOF gene deletion identified on the array and by PECONPI, we designed primers using the SNPs boundaries as guides. We designed a forward primer using Primer3 [Rozen and Skaletsky 2000] in exon 15 (5’- ACTCGGACAAGGTCAACGAC-3’) and paired it with a previously published reverse primer of exon 48 (5’-GAAAGAGTCCAAGCCACTGAAA-3’) [Migliosi et al., 2002]. DNA from the proband’s original and repeated sample, both parental samples and a control were diluted to a concentration of 20ng/µl and PCR amplified with 1.5mM MgCl2 using these primers under the following conditions: 94°C for 5 min then 36 cycles of 94°C for 30 sec, 62.3°C for 45 sec, 72°C for 30 sec followed by 72°C for 4 min and a 4°C hold. The products were visualized on an agarose gel. PCR products were purified (Exosap) and direct sequenced with both the forward and reverse primers. All sequencing products were analyzed with BLAT alignment tool [Kent 2002]. Additionally, parental samples were run on the array to verify carrier status.
Validation of USH2A deletion
The 170kb deletion that included the USH2A gene was validated using FISH (fluorescence in situ hybridization). Metaphase spreads were prepared from peripheral blood lymphocytes and probed with fluorescently labeled BACs and fosmids for FISH. One BAC (RP11–195B13) and two fosmids (G248P80743F1, G248P8306F6) were identified using the UCSC browser (hg18) to map within the deleted region. All clones were obtained through CHORI BACPAC Resources (Oakland, CA). BAC and fosmid DNA was isolated (PureLink HiPure Plasmid Filter Purification Kits, Invitrogen, Carlsbad, CA) and labeled by nick translation (Nick Translation Reagent Kit, Vysis, Inc.) in the presence of Spectrum Orange dUTP (Vysis, Inc.). Commercial FISH probes (Vysis, Inc.) for 1p telomere (TelVysion1p) were used. FISH probes were hybridized to metaphase preparations overnight; slides were then washed and counterstained with DAPI using standard protocols. USH2A sequencing was performed as previously described [van Wijk et al., 2004].
RESULTS
NSSNHL Cohort Analysis
Our cohort of 636 probands with NSSNHL had 6,449 deletion CNVs after the exclusion criteria was applied as described in Materials and Methods. Three samples with known heterozygous GJB6 deletions served as positive controls. GJB6 is a gap junction protein that is abundant in the inner ear and necessary for sensory transduction [Wangemann 2006]. We input the sample set of deletion CNVs along with the control set of 29,026 deletion CNVs, and ran the PECONPI analysis. The top ranking CNV calls in the NSSNHL cohort based on the PECONPI score are listed in Table I. The distribution of PECONPI scores for all the deletion CNVs of the NSSNHL cohort is illustrated in Fig. 1. There were 84 calls from 92 probands whose PECONPI score was ≥ 29. From the pathogenicity analysis, this score cutoff corresponds to a 1% false positive rate and 90% true positive rate. Within this group, there were deletions of 9 known hearing loss genes, including GJB6, OTOF and USH2A.
Table I.
PECONPI scores from the NSSNHL cohort
This selected PECONPI output focuses on the 35 top-ranking deletion CNVs from 636 NSSNHL probands. The highly ranked large deletions (> 1MB in size) most likely represents undetected syndromic forms of HL including microdeletion syndromes. Rank 3 highlights a homozygous deletion of OTOF. Ranks 23 and 32 include CNTNAP2 deletions. Rank 24 highlights a heterozygous USH2A deletion.
| Rank | Sample ID | Sample Chr | Sample State | Sample Size | Sample #ofSNPs |
Bands | Literature Ovlp% |
Controls# | Total Control Coverage |
Corrected Control Coverage |
Perfect Matches |
Cohort Redundancy |
Genes# | Unique Genes# |
Unique Genes ()=intronic |
Conserved Element # |
Conservation Value |
PECONPI score |
Hyperlink |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HLS407P | Chr6 | hom | 25,868 | 4 | 6q14.1 | 2 | 2 | *MYO6; *SENP6 | 3 | 1000 | 50.7 | link | ||||||
| 2 | HLS372P | Chr9 | het | 4,606,233 | 1,114 | 9q31.1 - q31.2 | 4% | 60 | 8% | 13 | 25 | 24 | *ABCA1; ALDOB; BAAT; C9orf125; CYLC2; FCMD; FSD1L; GRIN3A; MRPL50; *NIPSNAP3A; NIPSNAP3B; OR13C2; OR13C3; OR13C4; OR13C8; OR13C9; OR13D1; OR13F1; PPP3R2; PRG-3; RNF20; SLC44A1; *SMC2; *ZNF189 |
46.6 | link | ||||
| 3 | HLS034P | Chr2 | hom | 21,823 | 5 | 2p23.3 | 1 | 1 | OTOF | 4 | 500 | 45.7 | link | ||||||
| 4 | HLB428 | Chr22 | het | 1,872,299 | 498 | 22q11.21 - q11.23 | 6% | 83 | 19% | 12 | 17 | 14 | BCR; GNAZ; *MAPK1; PPIL2; PPM1F; PRAME; RAB36; RTDR1; SDF2L1; SUHW1; *SUHW2; UBE2L3; VPREB1; *YPEL1 |
45.0 | link | ||||
| 5 | HLS385P | Chr2 | het | 1,045,881 | 70 | 2q11.2 | 44% | 5 | 17 | 17 | ANKRD23; ANKRD36; ANKRD39; ARID5A; ASCC3L1; *CNNM3; CNNM4; FAHD2B; *FLJ10081; *KIAA1641; KIAA1754L; LINCR; LMAN2L; LOC51252; LOC90342; NCAPH; SEMA4C |
44.7 | link | ||||||
| 6 | HLB393 | ChrX | hom | 141,109 | 17 | Xq28 | 3 | 3 | GABRA3; MAGEA10; MAGEA5 | 43.1 | link | ||||||||
| 7 | HLB399 | Chr17 | het | 2,016,861 | 217 | 17q23.2 | 1% | 1 | 12 | 12 | APPBP2; *BCAS3; BRIP1; C17orf64; C17orf82; INTS2; NACAL; PPM1D; TBX2; TBX4; *THRAP1; *USP32 |
42.6 | link | ||||||
| 8 | HLS285 | ChrX | hom | 104,766 | 12 | Xp11.22 | 1 | 1 | PHF8 | 42.2 | link | ||||||||
| 9 | HLS155P | Chr13 | het | 294,448 | 118 | 13q12.11 | 42% | 4 | 2 | 2 | CRYL1; *GJB6 | 1 | 500 | 42.2 | link | ||||
| 9 | HLS170P | Chr13 | het | 294,448 | 118 | 13q12.11 | 42% | 4 | 2 | 2 | CRYL1; *GJB6 | 1 | 500 | 42.2 | link | ||||
| 11 | HLB393 | Chr13 | het | 283,158 | 117 | 13q12.11 | 43% | 4 | 2 | 2 | CRYL1; *GJB6 | 1 | 500 | 42.1 | link | ||||
| 12 | HLB105 | Chr3 | het | 2,694,289 | 397 | 3q13.13 | 6% | 2 | 0% | 8 | 8 | DPPA2; DPPA4; DZIP3; GUCA1C; MORC1; PVRL3; RETNLB; TRAT1 | 42.0 | link | |||||
| 13 | HLB90 | Chr3 | hom | 37,535 | 3 | 3p21.2 | 1 | 2 | 2 | RAD54L2; TEX264 | 41.3 | link | |||||||
| 14 | HLB447 | Chr11 | hom | 27,454 | 4 | 11q13.1 | 2 | 2% | 1 | 1 | 1 | PACS1 | 41.2 | link | |||||
| 15 | HLS123P | Chr2 | hom | 14,084 | 3 | 2p13.1 | 4 | 4 | DOK1; HTRA2; LOC130951; LOXL3 | 40.4 | link | ||||||||
| 16 | HLB122 | ChrX | hom | 2,902 | 4 | Xq28 | 1 | 1 | *TMLHE | 40.4 | link | ||||||||
| 17 | HLS123P | Chr7 | hom | 12,203 | 3 | 7q21.12 | 1 | 1 | *ABCB1 | 40.4 | link | ||||||||
| 18 | HLS403P | Chr3 | hom | 11,533 | 3 | 3q21.1 | 1 | 1 | CCDC14 | 40.4 | link | ||||||||
| 19 | HLS219P | Chr10 | hom | 5,536 | 3 | 10q22.2 | 1 | 1 | SYNPO2L | 40.4 | link | ||||||||
| 20 | HLB421 | ChrX | het | 1,626,888 | 183 | Xp22.31 | 25% | 6 | 17% | 6 | 6 | HDHD1A; PNPLA4; STS; VCX; VCX2; VCX3A | 40.2 | link | |||||
| 21 | HLS489P | Chr4 | het | 548,229 | 177 | 4q35.1 - q35.2 | 2% | 2 | 6 | 6 | CYP4V2; DKFZP564J102; F11; KLKB1; SORBS2; TLR3 | 38.7 | link | ||||||
| 22 | HLB273 | Chr17 | het | 548,740 | 73 | 17q21.31 | 29% | 1 | 5 | 5 | CRHR1; IMP5; KIAA1267; *MAPT; STH | 38.1 | link | ||||||
| 23 | HLS253P | Chr7 | het | 514,993 | 102 | 7q35 | 1 | 1 | 1 | *CNTNAP2 | 37.8 | link | |||||||
| 24 | HLS112P | Chr1 | het | 170,107 | 54 | 1q41 | 1 | 1 | USH2A | 53 | 500 | 37.1 | link | ||||||
| 25 | HLS152P | Chr2 | het | 504,909 | 124 | 2q23.3 | 3% | 1 | 1 | RPRM | 37.0 | link | |||||||
| 26 | HLB167 | Chr3 | het | 472,934 | 83 | 3q25.1 | 6 | 6 | C3orf44; EIF2A; *SELT; *SERP1; SIAH2; TSC22D2 | 36.7 | link | ||||||||
| 27 | HLB451 | Chr14 | het | 432,880 | 86 | 14q21.3 | 21% | 2 | 21% | 1 | 1 | 1 | C14orf155 | 35.9 | link | ||||
| 28 | HLB394 | Chr8 | het | 297,668 | 182 | 8p22 | 69% | 15 | 1% | 2 | 1 | 1 | *TUSC3 | 35.8 | link | ||||
| 29 | HLS154P | Chr22 | het | 507,491 | 143 | 22q11.22 | 71 | 8% | 8% | 10 | 3 | 3 | PRAME; SUHW1; *SUHW2 | 35.8 | link | ||||
| 30 | HLS139P | Chr4 | het | 453,855 | 60 | 4q22.1 | 1 | 1 | MGC48628 | 35.5 | link | ||||||||
| 31 | HLS404P | Chr16 | het | 521,944 | 46 | 16p11.2 | 9% | 27 | 27 | ALDOA; ASPHD1; BOLA2B; C16orf53; C16orf54; CCDC95; CDIPT; *CORO1A; DOC2A; FAM57B; GDPD3; GIYD2; HIRIP3; KCTD13; LOC124446; MAPK3; MAZ; MVP; PPP4C; PRRT2; QPRT; SEZ6L2; SPN; *SULT1A3; TAOK2; TBX6; YPEL3 |
34.8 | link | |||||||
| 32 | HLS087P | Chr7 | het | 281,596 | 63 | 7q35 | 1 | 1 | 1 | *CNTNAP2 | 34.7 | link | |||||||
| 33 | HLB099 | Chr2 | hom | 182,150 | 6 | 2q11.2 | 100% | 4 | 3 | 3 | ANKRD36; FAHD2B; *KIAA1641 | 34.7 | link | ||||||
| 34 | HLS098P | Chr1 | het | 238,644 | 85 | 1q43 | 25% | 1 | 1 | *FMN2 | 34.4 | link | |||||||
| 35 | HLB71 | Chr16 | het | 234,330 | 70 | 16p13.3 | 21% | 2 | 2 | *ADCY9; TFAP4 | 34.3 | link |
Figure 1. Distribution of PECONPI scores.
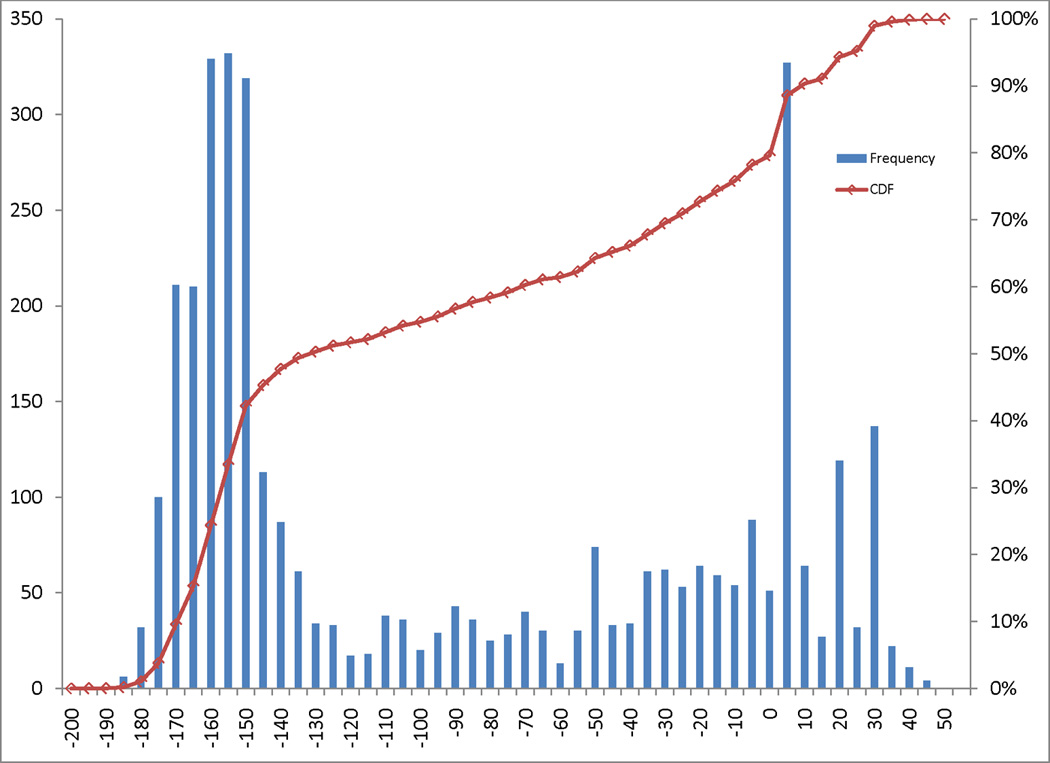
The distribution of PECONPI scores are illustrated in a histogram. Each bar represents a five point score spread, with the height indicating the frequency of that score. The red square boxes correspond to the percentile rank of each score, with 100% representing the top score. Scores with 25 or greater points corresponded to the top four percent of all calls.
Validation of Known GJB6 Deletions
PECONPI ranked all three known GJB6 containing CNVs in the top 0.3 percentile of deletion CNVs. A deletion CNV containing GJB6 has been seen in 1% of North Americans [Putcha et al., 2007]. This deletion was identified in one control cohort sample. All the deletions also span part of CRYL1, a lambda crystalline gene whose product is thought to play a role in the uronate cycle. Two probands with the GJB6 deletion CNV also have a previously identified mutation in GJB2 on the trans allele and one proband is a confirmed carrier of the deletion CNV with no other associated mutation on the trans GJB6 or GJB2 allele.
Identification of Known Hearing Loss Genes and Loci
PECONPI was able to rank a number of deletion CNVs encompassing genes that are known to cause hearing loss including, Otoferlin (OTOF), a gene involved in an autosomal recessive form of hearing loss (OMIM #601071) (non-syndromic recessive auditory neuropathy or NSRAN) [Yasunaga et al., 1999; Varga et al., 2003]. Usherin (USH2A), Cadherin 23 (CDH23) and G-Protein-Coupled Receptor 98 (GPR98), which all cause recessive forms of Usher Syndrome (2A, 1D and 2C respectively). Myosin VI (MYO6), Lipoxygenase Homology Domain-containing 1 (LOXHD1), Otoancorin (OTOA), and Stereocilin (STRC) cause forms of autosomal recessive non-syndromic BSNHL.
The top scoring deletion CNV (50.7) was a novel homozygous deletion of the MYO6 locus that involves 2 distal exons and the 3’ UTR of SENP6 through the 5’ UTR and first intron of the gene. MYO6 can cause both autosomal recessive and autosomal dominant SNHL. This homozygous deletion is also present in an affected sibling. Although the coding region of MYO6 was not involved in this deletion, in mice it has previously been shown that disruption of the regulatory/promoter region of MYO6 causes autosomal recessive deafness[Avraham et al., 1995].
The third ranked call highlighted a homozygous deletion of 5 SNPs (22kb) in OTOF, a gene involved in an autosomal recessive form of hearing loss termed NSRAN (OMIM #601071) [Yasunaga et al., 1999; Varga et al., 2003]. Mutations in OTOF have been described, all of which have caused a bilateral sensorineural hearing loss (BLSNHL) with auditory neuropathy [Houseman et al., 2001; Migliosi et al., 2002; Mirghomizadeh et al., 2002]. These deletions were confirmed to be the cause of the proband’s deafness. Standard molecular testing for hearing loss genes (GJB2, GJB6, the A1555G mitochondrial mutation, SLC26A4) was negative. Since other standard evaluations (ophthalmologic exam, renal and thyroid function testing, CT/MRI studies) were normal, no syndromic etiology was suspected. The patient’s parents are second cousins of Indian descent with no family history of hearing loss.
A homozygous deletion phenotype of OTOF has not yet been described. Sequencing and BLAT alignment of the breakpoint revealed a deletion involving exons 16–48 extending eight bases into exon 48 (NM_194248.1:g.73735_100486del) (Fig. 2) [Yasunaga et al., 2000] SNP array analysis of parental samples demonstrated that both were heterozygous carriers of the deletion. While deletions in this gene have not been previously reported, the same form of hearing loss is seen in association with all other homozygous or compound heterozygous mutations of OTOF. PECONPI accurately ranked this homozygous deletion of OTOF third out of 6,449 deletion CNVs.
Figure 2. Intragenic HomozygousOTOF Deletion.

Top: View adapted from UCSC browser illustrating the 30kb intragenic homozygous deletion affecting exons 17–48. The deletion affects both the long and short isoform of OTOF. Bottom: Gel showing presence of PCR product for multiple exons in OTOF. The child with HL fails multiple reactions, with parents and controls successful in all exons.
Three different types of Usher Syndrome were unmasked in three high-ranking heterozygous deletion CNVs scoring above 30 that involved USH2A, CDH23 and GPR98. Usher Syndrome is a heterogeneous diagnosis both in the underlying genetic mutations and the phenotypic severity that often results in hearing loss and retinitis pigmentosa, a form of progressive blindness. In one proband, a large heterozygous multi-exon deletion of USH2A was predicted to be pathogenic by PECONPI. Point mutations in USH2A cause Usher Syndrome type IIA (OMIM #276901), a recessive disorder with clinical symptoms of congenital hearing loss and retinitis pigmentosa. However, deletions have not been reported and would not be identified using standard sequencing protocols. The patient's audiometric test showed a moderately severe BLSNHL, but he was otherwise healthy. Standard molecular testing for hearing loss genes was negative and all other standard evaluations (ophthalmologic exam, temporal bone CT, renal and thyroid function testing) were normal. Subsequent to the PECONPI ranking of the heterozygous deletion in USH2A, sequencing of the gene revealed a heterozygous nonsense mutation in exon 63, c.13130C>A (p.S4377X) in the proband and the father. FISH analysis confirmed the heterozygous deletion in both the proband and his mother (ish del(1)(q41q41)(W12–739L2,W12–859K12, RP11–195B13 × 1)) (Fig. 3).
Figure 3. USH2A deletion.

Top: Heterozygous deletion of USH2A. View adapted from UCSC browser illustrating 170kb intragenic deletion in proband, affecting exons 22–32/33. Two small control CNVs are present within the deleted interval. The deletion affects both isoforms of USH2A. Locations of Illumina 550k BeadChip SNP markers are shown below gene. Bottom: FISH Validation of USH2A deletion. FISH images showing single copy deletion of USH2A in proband and mother (top right, bottom) and normal copy number in father (top left)
Several known autosomal recessive hearing loss genes lie in complex genomic regions where there is significant control and literature deletion CNV overlap and thus did not score as highly using PECONPI. These included three heterozygous multi-exon OTOA deletion CNVs of 19 SNPs (164kb) on 16p12.2, one homozygous and two heterozygous STRC loci deletion CNVs of 3 SNPs (25kb) on 15q15.3. OTOA (OMIM*607038) encodes an inner ear protein and mutations in OTOA were found to co-segregate with DFNB22 in a consanguineous Palestinian family with moderate to severe prelingual sensorineural recessive hearing loss [Zwaenepoel et al., 2002]. Large homozygous deletions of this region have previously been described in DFNB22 in a consanguineous Palestinian family where 1% of the control population carried the heterozygous deletion [Shahin et al., 2010]. Subsequent Sanger sequencing of the three probands with a heterozygous OTOA CNV unmasked a novel missense mutation, c.1249 C>T (p.L417F), on the trans allele of OTOA in one proband and an affected sibling also carrying the deletion that was not seen in 188 control chromosomes analyzed. The two remaining probands with an OTOA heterozygous deletion did not have subsequent findings and are suspected to be carriers. Deletions of STRC (OMIM*606440) have also previously been reported to co-segregate with DFNB16 [Verpy et al., 2001; Zhang et al., 2007; Knijnenburg et al., 2009]. The three CNVs of 15q15.3 deleting STRC were confirmed by Sanger sequencing to involve the coding region of STRC and causative of all three proband’s hearing loss [Francey et al., 2012]. A subsequent cohort was further analyzed for mutations in STRC by CNV analysis and Sanger sequencing. A total of 13 patients had a large STRC locus deletion as a contributing factor to their hearing loss [Francey et al., 2012].
A heterozygous deletion of 5 SNPs on 18q21.1 that encompasses at least 3 exons of the LOXHD1 gene (OMIM *613072) was also identified. LOXHD1 is expressed in the mechanosensory hair cells in the inner ear and mutations in LOXHD1 were recently identified as the cause of DFNB77, a progressive form of autosomal recessive NSSNHL [Grillet et al., 2009; Edvardson et al., 2011]. Subsequent Sanger sequencing of the 40 coding exons identified a heterozygous point mutation in exon 30, c. 4714 C>T (p.R1572X). The truncating mutation is the same mutation identified previously in nine patients with this form of hearing loss from two unrelated Ashkenazi Jewish families [Edvardson et al., 2011]. The proband identified in this study was also of Ashkenazi Jewish decent.
Syndromic and Other Large Deletions
Among the top ranking PECONPI scores there were 6 heterozygous deletion CNVs >1Mb. These include a 4.6 Mb deletion of 9q31.1-q31.2 that spans 26 genes and 1 open reading frame, a 1.8 Mb deletion of 22q11.21-q11.23 located distal to the DiGeorge Syndrome/velocardiofacial syndrome locus (DGS [MIM 188400]/VCFS [MIM 192430]) that has previously been described [Ben-Shachar et al., 2008], a 1 Mb deletion of 2q11.2 spanning ARID5A, a 2 Mb deletion within 17q23.2, containing 10 genes and 2 open reading frames, which has recently been described in a patient with a larger microdeletion of 17q22-q23.2 presenting with multiple anomalies including sensorineural hearing loss [Nimmakayalu et al., 2011], a 2.6 Mb deletion of 3q13.13, and a 1.6 Mb deletion of Xp22.31.
Other High-Scoring Deletions
In addition to these established and novel HL deletions, PECONPI identified many candidate genes contained within CNVs that scored well above the 1% false positive rate cutoff (see Table I). Further studies will be carried out on the remaining candidate CNV to evaluate their potential pathogenicity.
CHD cohort
The top PECONPI scoring deletions can be found in Supplementary Table II. PECONPI successfully identified and prioritized the pathogenic CNVs in 2 probands with known pathogenic 22q11.21 deletions. It identified 4 additional probands with 22q11.21 deletions that were not previously diagnosed. These 6 chromosomal 22q11.21 deletions were top ranking calls (all equally scored with rank 3). The top two ranking deletions were large, previously clinically unidentified 8p23.1 deletions known to cause CHD due to GATA4 haploinsufficency [Pehlivan et al., 1999]. The detailed analysis of this cohort can be found in Supplementary Information.
DISCUSSION
There is one published pathogenic CNV prediction tool designed for a cohort of patients with intellectual disability that utilizes a Naïve-Bayes classifier. This approach was based more on the gene expression, genomic architecture and conservation of the region[Hehir-Kwa et al., 2010]. In this paper, we detail a more gene centric approach and use information that is already known from other CNV databases to predict the pathogenicity of the CNV. The use of CNV databases for normal controls has played an important role in the clinical call of pathogenic CNVs and was not incorporated in the Naïve-Bayes method previously described. It allows for the discrimination of dosage sensitive areas. For instance if the gene was in an often deleted region, it should be less dosage sensitive than one in a region that has never been observed to be in a deletion CNV of normal individuals.
In this paper, we describe the development of algorithms that predict the pathogenicity of CNVs. Separating benign from pathogenic deletions or duplications is complex, and the tools we present allow high throughput analysis with discovery of deletions or duplications that underlie the disease phenotype. Utilization of CNVs to screen for disease-causing genes in cohorts with a given phenotype has been previously carried out, however, in general the separation of benign from pathogenic CNVs relies on the presence of parental samples to look for familial benign variants [Greenway et al., 2009].
PECONPI presents a tool for evaluating the potential pathogenicity of a variant in the absence of parental samples. The algorithm makes several assumptions. Each CNV is independent of the patient. It follows that the pathogenicity of each CNV is also assumed to be independent of the patient’s other CNV calls. This may not always be an accurate assumption, as is the case for functional loss of the genes involved in parallel pathways that may result in a phenotype unobserved with a functional loss of the genes individually. Furthermore, this method will rank rare CNVs as more pathogenic than common CNVs. This assumption may not always hold as rare CNVs may be benign and common CNVs may unmask a recessive mutation.
We have demonstrated that PECONPI is potentially a powerful tool for gene discovery in genetically heterogeneous disorders. It can provide the user with a ranked list of pathogenic CNVs, which helps to streamline the identification of candidate disease genes. It is not biased towards previously linked genes and has been demonstrated to have the ability to unearth pre-existing and novel disease genes.
In the NSSNHL cohort, PECONPI successfully identified known pathogenic CNVs containing a gene previously implicated in hearing loss (GJB6) and ranked these CNVs very high. It also identified candidate pathogenic CNVs that contain genes associated with hearing loss (OTOF and USH2A). Heterozygous deletions such as that in USH2A are easily missed with conventional molecular sequencing. This highlights the importance of genome-wide scans for CNVs and the downstream analysis of ranking and discriminating pathogenic CNVs. This study demonstrates PECONPI’s ability to distill calls of relevance from any cohort with a common phenotype of presumed genetic etiology or contribution.
The success and specificity of finding all known hearing loss genes within the top percent underscores the promise of other candidate genes in this group of high-scoring CNVs. In this way, PECONPI will be a helpful tool in identifying candidate genes for disorders of unknown etiology. Among these candidate genes in the NSSNHL cohort CNVs is CNTNAP2, a potassium channel clustering protein, which has been suspected as a hearing loss gene [Mustapha et al., 1998; Poliak et al., 1999; Nakabayashi and Scherer 2001; Poliak et al., 2003] and CALN1, a signaling pathway protein found in the DFNB39 locus. PECONPI helps identify genes of interest for screening and further analysis. However, a CNV-focused study for gene discovery can only identify disorders dependent on total gene or intragenic deletions. As CNVs were not detected in GJB2, the most common NSSNHL gene, this important hearing loss gene would not have been identified in this study. The use of CHD as a second disease cohort that also manifests a high degree of genetic heterogeneity underscores the ability of PECONPI to identify and rank pathogenic CNVs.
In this study, we used PECONPI to find rare pathogenic CNVs to discover genes that might be more commonly affected by point mutations. PECONPI can be adapted to respond to these and other scenarios, as the scoring parameters can all be adjusted based upon the population and disorders being studied. Gains in copy number can also be pathogenic and should be studied further. PECONPI is a useful tool to discriminate pathogenic from benign CNVs from a cohort with similar phenotype. Given the large amount of CNV data generated from increasingly higher resolution array studies, PECONPI will be a useful tool to facilitate the identification of pathogenic calls and candidate genes. PECONPI can provide researchers with an accurate and rapidly produced list of candidate genes and loci for clinical disorders that do not lend themselves to standard linkage or association gene discovery approaches.
Supplementary Material
ACKNOWLEDGMENTS
We thank the many hundreds of families and children who have graciously agreed to participate in this study. This work was supported in part by NIH/NIDCD grants R01DC005247 (IDK), R33DC008630 (IDK), U01HG006546 (IDK, NBS), Ring Chromosome 20 Foundation Grant (NBS), T32HG000046 (EAT), T32DC005363 (MAB) and T32GM008638 (LKC).
Footnotes
SUPPLEMENTAL ONLINE MATERIAL
The supplemental data includes two PDF files, one containing elaborated methods, validation, and software specifications and another including additional figures and tables.
WEB RESOURCES
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ UCSC Genome Browser, http://genome.ucsc.edu/
REFERENCES
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat Genet. 1995;11:369–375. doi: 10.1038/ng1295-369. [DOI] [PubMed] [Google Scholar]
- Beaud AL, Belmont JW, et al. Array-based DNA diagnostics: let the revolution begin. Annu Rev Med. 2008;59:113–129. doi: 10.1146/annurev.med.59.012907.101800. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, Patel MS, Hummel M, Amato S, Tartaglia N, Berg J, Sutton VR, Lalani SR, Chinault AC, Cheung SW, Lupski JR, Patel A. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet. 2008;82:214–221. doi: 10.1016/j.ajhg.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Jalas C, Shaag A, Zenvirt S, Landau C, Lerer I, Elpeleg O. A deleterious mutation in the LOXHD1 gene causes autosomal recessive hearing loss in Ashkenazi Jews. Am J Med Genet A. 2011;155A:1170–1172. doi: 10.1002/ajmg.a.33972. [DOI] [PubMed] [Google Scholar]
- Emanuel BS, Shaikh TH. Segmental duplications: an 'expanding' role in genomic instability and disease. Nat Rev Genet. 2001;2:791–800. doi: 10.1038/35093500. [DOI] [PubMed] [Google Scholar]
- Francey LJ, Conlin LK, Kadesch HE, Clark D, Berrodin D, Sun Y, Glessner J, Hakonarson H, Jalas C, Landau C, Spinner NB, Kenna M, Sagi M, Rehm HL, Krantz ID. Genome-wide SNP genotyping identifies the Stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am J Med Genet A. 2012;158A:298–308. doi: 10.1002/ajmg.a.34391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla-Dieck Mde L, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, Webster JA, Kahrizi K, Najmabadi H, Kimberling WJ, Stephan D, Bahlo M, Wiltshire T, Tarantino LM, Kuhn P, Smith RJ, Muller U. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehir-Kwa JY, Wieskamp N, Webber C, Pfundt R, Brunner HG, Gilissen C, de Vries BB, Ponting CP, Veltman JA. Accurate distinction of pathogenic from benign CNVs in mental retardation. PLoS Comput Biol. 2010;6:e1000752. doi: 10.1371/journal.pcbi.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman MJ, Jackson AP, Al-Gazali LI, Badin RA, Roberts E, Mueller RF. A novel mutation in a family with non-syndromic sensorineural hearing loss that disrupts the newly characterised OTOF long isoforms. J Med Genet. 2001;38:E25. doi: 10.1136/jmg.38.8.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg J, Oberstein SA, Frei K, Lucas T, Gijsbers AC, Ruivenkamp CA, Tanke HJ, Szuhai K. A homozygous deletion of a normal variation locus in a patient with hearing loss from non-consanguineous parents. J Med Genet. 2009;46:412–417. doi: 10.1136/jmg.2008.063685. [DOI] [PubMed] [Google Scholar]
- Komura D, Shen F, Ishikawa S, Fitch KR, Chen W, Zhang J, Liu G, Ihara S, Nakamura H, Hurles ME, Lee C, Scherer SW, Jones KW, Shapero MH, Huang J, Aburatani H. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res. 2006;16:1575–1584. doi: 10.1101/gr.5629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Iafrate AJ, Brothman AR. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat Genet. 2007;39:S48–S54. doi: 10.1038/ng2092. [DOI] [PubMed] [Google Scholar]
- Marioni JC, Thorne NP, Valsesia A, Fitzgerald T, Redon R, Fiegler H, Andrews TD, Stranger BE, Lynch AG, Dermitzakis ET, Carter NP, Tavare S, Hurles ME. Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007;8:R228. doi: 10.1186/gb-2007-8-10-r228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Migliosi V, Modamio-Hoybjor S, Moreno-Pelayo MA, Rodriguez-Ballesteros M, Villamar M, Telleria D, Menendez I, Moreno F, Del Castillo I. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J Med Genet. 2002;39:502–506. doi: 10.1136/jmg.39.7.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirghomizadeh F, Pfister M, Apaydin F, Petit C, Kupka S, Pusch CM, Zenner HP, Blin N. Substitutions in the conserved C2C domain of otoferlin cause DFNB9, a form of nonsyndromic autosomal recessive deafness. Neurobiol Dis. 2002;10:157–164. doi: 10.1006/nbdi.2002.0488. [DOI] [PubMed] [Google Scholar]
- Mustapha M, Chardenoux S, Nieder A, Salem N, Weissenbach J, el-Zir E, Loiselet J, Petit C. A sensorineural progressive autosomal recessive form of isolated deafness, DFNB13, maps to chromosome 7q34-q36. Eur J Hum Genet. 1998;6:245–250. doi: 10.1038/sj.ejhg.5200177. [DOI] [PubMed] [Google Scholar]
- Nakabayashi K, Scherer SW. The human contactin-associated protein-like 2 gene (CNTNAP2) spans over 2 Mb of DNA at chromosome 7q35. Genomics. 2001;73:108–112. doi: 10.1006/geno.2001.6517. [DOI] [PubMed] [Google Scholar]
- Nimmakayalu M, Major H, Sheffield V, Solomon DH, Smith RJ, Patil SR, Shchelochkov OA. Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am J Med Genet A. 2011;155A:418–423. doi: 10.1002/ajmg.a.33827. [DOI] [PubMed] [Google Scholar]
- Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS, Hing AV. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet. 1999;83:201–206. [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Bejjani BA, Bleoo S, Booker JK, Carey JC, Carson N, Das S, Dempsey MA, Gastier-Foster JM, Greinwald JH, Jr, Hoffmann ML, Jeng LJ, Kenna MA, Khababa I, Lilley M, Mao R, Muralidharan K, Otani IM, Rehm HL, Schaefer F, Seltzer WK, Spector EB, Springer MA, Weck KE, Wenstrup RJ, Withrow S, Wu BL, Zariwala MA, Schrijver I. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet Med. 2007;9:413–426. doi: 10.1097/gim.0b013e3180a03276. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Sebat J. Major changes in our DNA lead to major changes in our thinking. Nat Genet. 2007;39:S3–S5. doi: 10.1038/ng2095. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Shahin H, Walsh T, Rayyan AA, Lee MK, Higgins J, Dickel D, Lewis K, Thompson J, Baker C, Nord AS, Stray S, Gurwitz D, Avraham KB, King MC, Kanaan M. Five novel loci for inherited hearing loss mapped by SNP-based homozygosity profiles in Palestinian families. Eur J Hum Genet. 2010;18:407–413. doi: 10.1038/ejhg.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O'Hara R, Casalunovo T, Conlin LK, D'Arcy M, Frackelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield JP, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB, Zackai EH, Chiavacci RM, Grundmeier R, Rappaport EF, Grant SF, White PS, Hakonarson H. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–49. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, Goedken R, Segre A, Pericak-Vance MA, Cuccaro ML, Gilbert JR, Wright HH, Abramson RK, Betancur C, Bourgeron T, Gillberg C, Leboyer M, Buxbaum JD, Davis KL, Hollander E, Silverman JM, Hallmayer J, Lotspeich L, Sutcliffe JS, Haines JL, Folstein SE, Piven J, Wassink TH, Sheffield V, Geschwind DH, Bucan M, Brown WT, Cantor RM, Constantino JN, Gilliam TC, Herbert M, Lajonchere C, Ledbetter DH, Lese-Martin C, Miller J, Nelson S, Samango-Sprouse CA, Spence S, State M, Tanzi RE, Coon H, Dawson G, Devlin B, Estes A, Flodman P, Klei L, McMahon WM, Minshew N, Munson J, Korvatska E, Rodier PM, Schellenberg GD, Smith M, Spence MA, Stodgell C, Tepper PG, Wijsman EM, Yu CE, Roge B, Mantoulan C, Wittemeyer K, Poustka A, Felder B, Klauck SM, Schuster C, Poustka F, Bolte S, Feineis-Matthews S, Herbrecht E, Schmotzer G, Tsiantis J, Papanikolaou K, Maestrini E, Bacchelli E, Blasi F, Carone S, Toma C, Van Engeland H, de Jonge M, Kemner C, Koop F, Langemeijer M, Hijmans C, Staal WG, Baird G, Bolton PF, Rutter ML, Weisblatt E, Green J, Aldred C, Wilkinson JA, Pickles A, Le Couteur A, Berney T, McConachie H, Bailey AJ, Francis K, Honeyman G, Hutchinson A, Parr JR, Wallace S, Monaco AP, Barnby G, Kobayashi K, Lamb JA, Sousa I, Sykes N, Cook EH, Guter SJ, Leventhal BL, Salt J, Lord C, Corsello C, Hus V, Weeks DE, Volkmar F, Tauber M, Fombonne E, Shih A, Meyer KJ. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, Hoefsloot LH, Cremers FP, Cremers CW, Kremer H. Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet. 2004;74:738–744. doi: 10.1086/383096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verpy E, Masmoudi S, Zwaenepoel I, Leibovici M, Hutchin TP, Del Castillo I, Nouaille S, Blanchard S, Laine S, Popot JL, Moreno F, Mueller RF, Petit C. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat Genet. 2001;29:345–349. doi: 10.1038/ng726. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, Wilcox ER, Petit C. OTOF encodes multiple long and short isoforms: genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet. 2000;67:591–600. doi: 10.1086/303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Malekpour M, Al-Madani N, Kahrizi K, Zanganeh M, Lohr NJ, Mohseni M, Mojahedi F, Daneshi A, Najmabadi H, Smith RJ. Sensorineural deafness and male infertility: a contiguous gene deletion syndrome. J Med Genet. 2007;44:233–240. doi: 10.1136/jmg.2006.045765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaenepoel I, Mustapha M, Leibovici M, Verpy E, Goodyear R, Liu XZ, Nouaille S, Nance WE, Kanaan M, Avraham KB, Tekaia F, Loiselet J, Lathrop M, Richardson G, Petit C. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc Natl Acad Sci U S A. 2002;99:6240–6245. doi: 10.1073/pnas.082515999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.