

Abstract
Background
Variable function and expression of drug transporters have been proposed as mechanisms contributing to variable response to drug therapy. Block of the HERG channel, encoding IKr, can lead to serious arrhythmias, and a key drug-blocking site in HERG has been identified on the intracellular face of the pore. We begin to advance the hypothesis that active drug uptake enhances IKr block.
Methods and Results
Reverse transcriptase–polymerase chain reaction identified expression in the human atrium and ventricle of 14 of 31 candidate drug uptake and efflux transporters, including OCTN1 (SLC22A4), a known uptake transporter of the HERG channel blocker quinidine. In situ hybridization and immunostaining localized OCTN1 expression to cardiomyocytes. The IC50 for quinidine block of IKr in CHO cells transfected with HERG alone was significantly higher than cells transfected with HERG + OCTN1 (0.66 ± 0.15 μM versus 0.14 ± 0.06 μM [52% absolute increase in drug block; 95% confidence interval, 0.4–0.64 μM]), and this effect was further potentiated by a common genetic variant of OCTN1, L503F. In the absence of OCTN1, quinidine block could be 91% ± 5% washed out, but with the transporter, washout was incomplete (57% ± 6%). OCTN1 coexpression also facilitated HERG block by flecainide and ibutilide, but not erythromycin.
Conclusions
Coexpression of the organic cation transporter, OCTN1, expressed in human cardiac myocytes, intensifies quinidine-induced HERG block. These findings establish a critical hypothesis that variable drug transporter activity may be a potential risk factor for torsade de pointes.
Keywords: antiarrhythmia agents, arrhythmia, pharmacology, pharmacokinetics, ion channels
INTRODUCTION
Drug concentration achieved at the molecular effector site is a key determinant of the magnitude of any drug effect.1–3 Accordingly, variable drug metabolism is one well-recognized determinant of variable drug action. More recently, delivery of drug to and removal from intracellular molecular effector sites by xenobiotic transporters has become increasingly well recognized as a modulator of intracellular drug concentration.4–7 Voltage-gated ion channels (including calcium, potassium, and sodium channels) represent a class of molecular targets in which sites of drug block have been identified at the intra-cellular face of the pore region.8–13 Ion channel blockers display considerable interpatient variability in clinical response ranging from suppression of arrhythmia to life-threatening proarrhythmia.14–17 One of the best studied examples of pro-arrhythmia is exaggerated prolongation of the QT interval and the development of torsade de pointes (TdP) through antagonism of the HERG (or KCNH2) potassium channel by anti-arrhythmic medications (eg, quinidine, dofetilide, sotalol, ibutilide) and medications with “noncardiovascular” indications (eg, erythromycin, thioridazine). Indeed, drug-induced QT interval prolongation has been one of most common causes of prescription drug withdrawal or relabeling over the last decade.18 Although numerous risk factors for drug-induced QT interval prolongation have been identified, including gender, hypokalemia, hypomagnesemia, bradycardia, and ion channel mutations, the risk of TdP remains variable among patients with the same risk profile and equivalent QT intervals.19–24
A key site of drug block for the HERG channel has been localized to the intracellular face of the protein.10,25,26 Increasing evidence from other organ systems, including the testis, placenta, and vascular endothelium of the blood–brain barrier, indicate that drug access to intracellular sites of drug action is controlled by xenobiotic transporters that mediate drug uptake and efflux.27–32 Efflux transporters that export drug against a concentration gradient from cells to the extra-cellular space are encoded primarily by genes belonging to the ATP-binding cassette (ABC) superfamily, which includes MDR1 (formally known as ABCB1, encoding P-glycoprotein) and the multidrug resistance proteins (MRPs). Conversely, uptake transporters usually deliver medication or endogenous substrates from the bloodstream to the extracellular space or from the extracellular space to the inside of cells and include the organic anion polypeptide (eg, OATP, SLCO), organic anion (eg, OAT, SLC22A), and organic cation (OCT, SLC22A) families.2 Recently, Grube et al showed that OCTN2 localized to the vascular endothelium in the heart, which suggests that delivery of drug to the extracellular surface of the myocyte is an active process.33
In this study, we first examined whether mRNA for drug efflux and uptake transporters could be detected in the human atrium and ventricle. We then focused on the human organic cation/ergothioneine uptake transporter, OCTN1 (SLC22A4), a known transporter of the TdP-associated antiarrhythmic quinidine.34 Second, we determined the localization of OCTN1 in the heart by in situ hybridization and immunohistochemistry. We then tested the hypothesis that coexpression of OCTN1 would potentiate block of the HERG potassium channel. Finally, we compared HERG block in cells expressing wild-type OCTN1 with those expressing a known common functional variant of OCTN1, L503F.35
METHODS
Analysis of Transporter Expression in the Human Heart
Complementary single-strand DNA was generated from commercially available RNA samples (Transcriptor First Strand cDNA synthesis kit; Roche Laboratories, Nutley, NJ) isolated from healthy human kidney, liver, left ventricular, and left atrial tissue samples (Biochain, Hayward, CA). We probed for cardiac expression of mRNAs encoding drug efflux and uptake transporters using polymerase chain reaction (Taq polymerase; Roche Laboratories) relative to either transporter mRNA expression from the liver or kidney as a positive control. The final 50-μL polymerase chain reaction contained 10 mM Tris-HCl, 1.5 mM MgCl, 50 mM KCl with 200 μM dATP, dCTP, dGTP, and dTTP. Template cDNA was added as 150 ng of RNA equivalent per 50 μL reaction, and the final primer concentration was 10 mM. Amplicons were sized on a 2% agarose gel and those showing cardiac expression were excised from the gel, purified, subcloned into a pGEMTEasy vector (Promega, Madison, WI), and sequenced to verify that the anticipated product had been amplified. Primer sequences can be found in Table 1.
TABLE 1.
Primer Sequences Used for Reverse Transcriptase–polymerase Chain Reaction Screening for Drug Efflux and Uptake Transporters
| Transporter | Forward primer | Reverse primer | Control tissue |
|---|---|---|---|
| Efflux | |||
| MDR1 (ABCB1) | TCAGAATGGCAGAGTCAAGGAGC | TAGCAAGGCAGTCAGTTACAGTCCA | Liver |
| MRP2 (ABCC2) | AAGATTATAGAGTGCGGCAGCCCT | CCTGGGTAGTAGGTTCATGGGTGTT | Liver |
| MRP3 (ABCC3) | TCAGCACCCCTGCAGATCATCC | CACACTCTGGGGGTCAAGTTCCTC | Liver |
| MRP4 (ABCC4) | TCAAGTCCGTTCCGAAGGCA | GCATTTAACTGGTGGCCTGCA | Liver |
| MRP5 (ABCC5) | CCTGGAACCTCCGCTCAGAGAA | CTGGGTGCTGGTGTTTGGAAGTAG | Liver |
| MRP7 (ABCC7) | TCCAGCAGACCA TCTGCAAACG | GAGGTAGAGCCCCAGAGGAGAATGT | Liver |
| BCRP (ABCG2) | TGCATCTTGGCTGTCATGGC | TGTGCAACAGTGTGATGGCAAG | Liver |
| Uptake | |||
| OATP-B (SLC21A9) | AATAATGACCTGCTCCGAAACCG | ACTCAGAGGAGGTACTGCTGTGGCT | Liver |
| OCTN1 (SLC22A4) | TTCTTATCCATTGGTCTGGTCATGC | TGGGAACAACGAATTTCTCCACA | Kidney |
| OCTN2 (SLC22A5) | ATGCGGGACTACGACGAGGTGA | TGAAGGAGCCCAACAGCACACC | Liver |
| OAT3 (SLC22A8) | CCGCAACCTTCCCTGGTCGT | CCAGGTTCCACTCGGTCACAATG | Kidney |
| OCT3 (SLC22A3) | TTCCGCTTCCTGCAAGGTGTA | CCCCCTATAATTCCCAGGCG | Kidney |
| PGT (SLCO2A1) | CGCTCGGTCTTCGGCAACAT | AGGTGTACTGGTAGGGCTCGGAGAG | Kidney |
| PepT2 (SLC15A2) | AGCCAACAAAATGTCCATTGCG | GAAAGGTGGAGGAGATCAGCTCTGA | Kidney |
Localization of OCTN1
In situ hybridization and immunohistochemistry were performed on human atrial samples obtained from consenting patients undergoing mitral valve surgery. Healthy human kidney tissue was obtained at autopsy from the Vanderbilt Pathology Service. The tissues were snap-frozen with liquid nitrogen within 5 minutes of surgical removal, embedded in paraffin, and 5-μm sections were placed on charged slides.
For in situ hybridization, OCTN1 cDNA was subcloned into the pGEMT Easy vector (Promega). Template for the antisense probe was formed using a restriction digest upstream of the T7 promoter on the vector. Template for the sense probe was generated using a restriction site downstream of the SP6 promoter on the vector. Digoxigenin-labeled RNA probes (approximately 600 bp) for the OCTN1 gene were generated by in vitro transcription using the Message Machine system (Ambion, Austin, TX). Tissue sections were subsequently deparaffinized, fixed in 4% paraformaldehyde, and treated with proteinase K (5 μg/mL) before an acetic anhydride/ ethanolamine wash. Labeled probe (sense or antisense, 1 μg/mL of hybridization solution) was hybridized overnight at 50°C before washing, ribonuclease treatment, and incubation with an antidigoxigenin alkaline phosphatase antibody (1:500 dilution). Substrate color development (nitro blue tetrazolium chloride and 5-bromo-4-chloro-3-indolyl phosphate) was monitored by light microscopy using a Zeiss Axiophot (Carl Zeiss: MicroImaging, Thornwood, NY) inverted microscope equipped with a Photometrics CoolSnap (Tucson, AZ) digital camera and stopped after 24 hours.
For immunohistochemistry, samples were deparaffinized, rehydrated, and placed in a decloaker with a citrate buffer at pH 6.0. The tissue was rinsed with water and subsequently incubated with 0.03% hydrogen peroxide for 20 minutes followed by a 20-minute protein block (Dako, Carpinteria, CA). The rabbit antihuman anti-OCTN1 antibody targeted against the carboxyl terminus of OCTN1 (Santa Cruz Biotechnology, Santa Cruz, CA; proprietary amino acid sequence) was diluted 1:40 in Dako’s antibody diluent and incubated on the sample overnight at 4°C. It was then rinsed with water and Dako’s Rabbit Envision was placed on tissue followed by DAB and a hematoxylin counterstain. Samples were visualized on a LEICA DM IRBE (Leica, Solms, Germany).
Using a Nikon digital camera, DXM 1220C (Tokyo, Japan) with NIS-Elements BR 2.30, optical settings were maintained between samples.
Electrophysiology Studies
For these studies, Chinese Hamster Ovary (CHO) cells were cotransfected (Fugene 6; Roche Laboratories) with separate plasmids, one encoding HERG and a second the green fluorescent protein (GFP) or a bicistronic plasmid encoding eGFP and either wild-type OCTN1 or the L503F OCTN1 variant. OCTN1 cDNA was subcloned into the HindIII site of the expression vector pEGFP-C1 (Clontech Laboratories Inc., Mountain View, CA), and the L503F variant was introduced by site-directed mutagenesis of this clone. Whole cell voltage clamp to assess IKr was then performed in cells demonstrating green fluorescence. The ratio of wild-type or variant OCTN1 and HERG plasmids relative to Fugene 6 was 2 μg:2 μg:12 μL. Whole cell voltage clamp recordings were performed 48 to 72 hours after transfection.
Whole Cell Voltage Clamp Protocol
In all experiments, the intracellular pipette filling solution contained (in mM): KCl, 110; HEPES, 10; K4 BAPTA, 5; K2 ATP, 5; and MgCl2, adjusted to pH 7.2 with KOH yielding a final intracellular K+ concentration of 145 mM. The bath (Normal Tyrode’s) solution contained (in mM): NaCl, 145; KCl, 4; CaCl2, 1.8; MgCl2, 1; HEPES, 10; and glucose, 10 adjusted to pH 7.35 with NaOH. Experiments took place at room temperature (22°C). Samples were obtained at 1 to 10 kHz after antialias filtering at half or less of the sampling frequency using an Axopatch 200B Amplifier (Molecular Devices, Sunnyvale, CA). The holding potential was maintained at −80 mV. The standard protocol to obtain current-voltage relations and activation curves consisted of 1-second steps that were imposed in 10-mV increments between the holding potential and +60 mV followed by 1 second at −40 mV to measure deactivating tail currents. The cycle time for protocols was 15 seconds or slower. Under these conditions, IKr is stable for >60 minutes in the absence of a drug intervention. After collection of baseline data, quinidine was added to the perfusate while pulsing continued. IKr was monitored, and when a new steady state was reached (generally in 5 minutes), the maximum IKr amplitude at −40mV in the presence of quinidine was determined. Quinidine concentration was increased in stepwise increments from 0.01 μM to 5 μM in the same cell. The IC50 and 95% confidence intervals for each group were calculated using the Hill equation. The effect of three other IKr blockers, erythromycin (38 μM), ibutilide (20 nM), and flecainide (4 μM), was also assessed in wild-type OCTN1-transfected and non-OCTN1-transfected cells. The effect of temperature on OCTN1-mediated transport of quinidine 0.05 μM was assessed in OCTN1− and OCTN1+ cells at 37°C using a HCC100 chamber heating element (Dagan Corporation, Minneapolis, MN).
The time course of washout after stable block by quinidine 0.5 μM, corresponding to the IC50, was established in the following fashion. After removal of drug from the extracellular solution (Valve Bank 8.2 Perfusion System; Automated Scientific, Berkeley, CA), a rapid pulse protocol was initiated. This consisted of a holding potential at −80 mV, a 500-μs activation pulse to +40 mV, followed by a 500-μs hyperpolarization pulse to −40 mV to observe deactivating tail currents at a cycle time of 3 seconds.36
RESULTS
Transporter Expression and Localization
Thirty-one uptake and efflux transporters were screened for expression in human atrium and ventricle using reverse transcriptase–polymerase chain reaction (Fig. 1). Liver or kidney expression (positive controls) was detected in 31 of 31 and heart expression in 14 of 31; no transcript was expressed in a chamber-specific fashion. Efflux transporters showing expression in human heart included MDR1 (P-glycoprotein), MRP2, MRP3, MRP5, MRP7, and the Breast Cancer Resistance Protein (BCRP). Uptake transporters expressed in the heart included OCTN1, OCTN2, OCT3, OAT3, the prostaglandin transporter PGT, and the peptide transporter PepT2.
FIGURE 1.

Expression of drug efflux and uptake transporters. Reverse transcriptase–polymerase chain reaction using RNA isolated from healthy human atrial (A) and ventricular (V) samples was compared with samples isolated from healthy human liver or kidney as a positive control (C; see Table 1 for positive control). Water control (W) appears in the far left lane.
Localization of OCTN1
Figure 2A shows staining in myocytes and vascular endothelium with an antisense probe directed against OCTN1, whereas Figure 2B shows absent staining with the sense control. Immunostaining (Fig. 3) with an anti-OCTN1 antibody revealed expression in kidney (as expected) as well as in myocytes. Thus, OCTN1 is positioned to modulate access of substrate drugs to the intracellular space of the cardiomyocyte.
FIGURE 2.
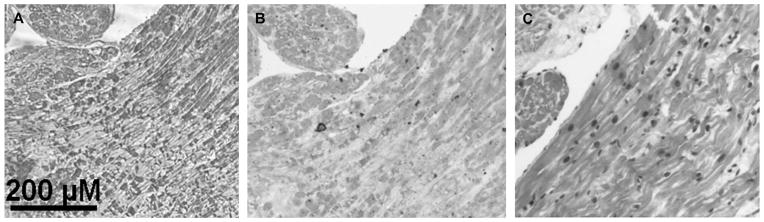
In situ hybridization of OCTN1. Digoxigenin-labeled RNA probe demonstrates localization of OCTN1 mRNA in vascular endothelial cells and atrial cardiomyocytes (A). An atrial tissue sample treated with a sense control is shown (B), whereas (C) staining is shown with hematoxylin and eosin.
FIGURE 3.
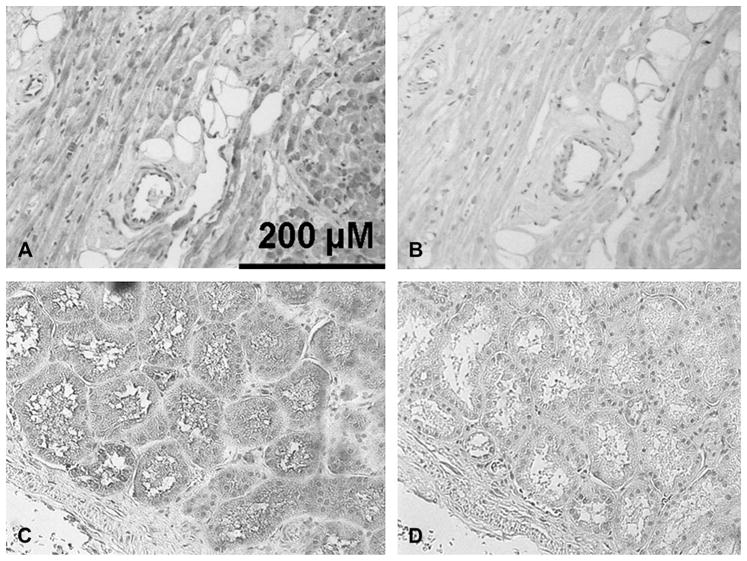
Anti-OCTN1 immunos-taining in human atrium. (A) With antibody. Note the absence of chromagen staining on the blood vessel. (B) An atrial tissue sample treated with the secondary antibody alone as a negative control. (C) Healthy human kidney with antibody. (D) Negative control with healthy human kidney. Background hematoxylin stain was used for all four experiments.
Coexpression of OCTN1 Potentiates IKr Drug Block
Before drug, IKr amplitude and gating were comparable in cells transfected with HERG + GFP (designated OCTN1−) and with HERG + wild-type or variant OCTN1 (Table 2).
TABLE 2.
Comparison of Baseline Electrophysiology in CHO Cells Expressing HERG Alone, Wild-type OCTN1, and L503F OCTN1*
| OCTN1− | WT OCTN1+ | L503F OCTN1+ | |
|---|---|---|---|
| Peak tail current | 10.2 ± 1.8 | 10.9 ± 1.9 | 9.1 ± 1.7 |
| Time constant of activation | 257 ± 21 | 255 ± 41 | 280 ± 25 |
| Time constant of deactivation | 204 ± 10 | 222 ± 21 | 221 ± 27 |
| V1/2 (mV) | −1.01 ± 0.29 | 1.38 ± 3.08 | −1.56 ± 3.30 |
n = 7 each; all P values > 0.75.
OCTN1−, cells transfected with HERG alone; OCTN1+, cells transfected with HERG + OCTN1; L503F OCTN1+, cells transfected with HERG + the L503F OCTN1 variant. Peak tail current: pA/pF at −40 mV after a depolarizing pulse to 60 mV. Time constant of activation (msec): monoexponential fit to activating current during a pulse to 40 mV. Time constant of deactivation: (msec): monoexponential fit to deactivating current at −40 mVafter a depolarizing pulse to 40 mV. V1/2 defined as the voltage at which IKris half maximal.
Examples of the effect of quinidine washin on IKr block in OCTN1− and OCTN1+ (wild-type or variant) are shown in Figures 4A–C. Exposure to the OCTN1 substrate/IKr blocker quinidine produced greater current suppression in the cells expressing wild-type OCTN1 versus the OCTN1− cells (Fig. 4D). The IC50 was 0.14 ± 0.006 μM in OCTN1+ cells. In OCTN1− cells, the IC50 was much higher (0.66 ± 0.15 μM; 52% increase in drug block; 95% confidence interval, 0.4–0.64 μM). When cells expressing the OCTN1 L503F variant were exposed to 0.01 μM quinidine, tail current was reduced 56.2% ± 1.1%, a threefold increase in block over cells expressing wild-type OCTN1 (P < 0.05) (Figure 4D). The slopes were similar—HERG alone: 0.57 ± 0.08; OCTN1 WT: 0.52 ±0.08; and L503F OCTN1: 0.42 ±0.09 (P = 0.449). We also conducted experiments at 37°C, which replicated the findings at room temperature. At 37°C, OCTN1– cells displayed a 30% ± 1% reduction in IKr, whereas OCTN1+ cells displayed a 68% ± 14% reduction on exposure to quinidine (0.5 μM). This corresponds to a difference of 38%; at 22°C, the difference was 22%.
FIGURE 4.

Functional effects of coexpressing OCTN1 with HERG. (A) Response of IKr in an OCTN1– cell exposed to serially increasing quinidine concentrations. (B) Response of IKr in an OCTN1+ cell exposed to serially increasing quinidine concentrations. (C) Response of IKr in an L503F OCTN1+ cell exposed to serially increasing quinidine concentrations. (D) Dose–response curves.
To further demonstrate the point that the same extra-cellular concentration of drug generates divergent effects at the intracellular site of action, we assessed IV relationships in OCTN1− and WT OCTN1+ cells exposed to a submaximal blocking concentration of quinidine (0.5 μM). As shown in Figure 5A, not only was current markedly reduced with coexpression of the transporter, but the voltage-dependence of steady-state activation was markedly shifted (V1/2: 3.33 ± 5.6 mV versus −10.4 ± 7.4 mV for OCTN1− versus OCTN1 WT), a difference that was not present predrug (Table 2).
FIGURE 5.

Further evidence in support of a role for OCTN1 in mediating IKr block. (A) The data shown here demonstrate that coex-pression of the transporter shifts the voltage-dependence of steady-state activation in the presence of quin-idine 0.5 μM (left). The amplitude at which IKr becomes half maximal (right) in the presence of quinidine 0.5 μM (V1/2: 3.33 ± 5.6 mV versus −10.4 ± 7.4 mV). (B) Washout of drug block: This experiment shows that coexpression of OCTN1 resulted in incomplete recovery of tail current after washout of quinidine 0.5 μM (n = 5 in both groups).
Further data attesting to the role of the transporter in mediating drug effects were obtained in washout experiments. We reasoned that in the presence of the transporter, drug leaving the cell might be taken up more rapidly back into the cytosol, so we compared washout of IKr block after withdrawal of quinidine from the medium in cells with and without coexpression of OCTN1. Figure 5B shows that cells expressing wild-type OCTN1 treated with quinidine failed to completely recover from drug block during washout compared with OCTN1− cells (57 ± 6 versus 91% ± 5%; P < 0.002).
To test whether the effect of coexpression of the transporter might exert some nonspecific effect on structurally unrelated drugs sharing the same intracellular binding side or if the effect was unique to methanesulfanilamide HERG blockers, we determined the effect of OCTN1 coexpression on the extent of IKr inhibition by the known blockers erythromycin, ibutilide, and flecainide. Coexpression of OCTN1 enhanced block by approximately 25% in the presence of either ibutilide (20 nM) or flecainide (4 μM), similar to the OCTN1 effect observed with 0.5 μM quinidine. However, coexpression of OCTN1 did not alter block by 38 μM erythromycin compared with OCTN1− control (44% ± 7% versus 41% ± 7%; P = 0.76).
DISCUSSION
We report that the drug uptake transporter OCTN1 is expressed in cardiomyocytes. When wild-type transporter was coexpressed with HERG, IKr block by quinidine, ibutilide, and flecainide was enhanced and washout impaired. The common OCTN1 variant L503F, with an allele frequency of 42% in populations of European descent, and lower allele frequencies in African American and Mexican American populations,35 increased the extent of quinidine block. Thus, these studies establish a new hypothesis for a mechanism contributing to variability in IKr block and thus potentially altered susceptibility to drug-related TdP: variable drug access to an intracellular binding site on the HERG channel.
Drug Uptake in the Heart
In mice in which mdr1a, encoding the efflux transporter P-glycoprotein, was knocked out, we have previously reported marked cardiac accumulation of quinidine relative to wild-type mice (1.4 ±0.2 versus 5.3 ±1.5 μg/g tissue).37 More recently, Grube et al have assessed the expression and function of OCTN2, another cationic uptake transporter, in the heart. Using in situ hybridization, they showed that OCTN2 was expressed in the vascular endothelium and not myocytes. Furthermore, expression of OCTN2 was reduced in ventricular tissue isolated from patients with either ischemic or dilated cardiomyopathy relative to tissue isolated from nonfailing myocardium.33 Taken together, the data suggest that drug delivery to membrane-bound and intracellular molecular targets in cardiac cells (myocytes or those in vascular tissue) is a regulated process. Thus, a new series of candidate genes have been identified as potential modulators of variable cardiovascular drug actions.
Precedent in Other Therapeutic Areas
Work in drug-resistant epilepsy provides further support for the idea that access to intracellular drug target sites on ion channels is modulated by drug uptake and efflux transport proteins. The drug efflux transporters MDR1, MRP3, and MRP5 have been reported to be upregulated in drug resistant seizure disorder, and this has been implicated as a mechanism for drug resistance.38 Tishler et al showed that brain slices from patients with medically intractable epilepsy demonstrated higher mRNA expression of MDR1. In that study, steady-state brain concentrations of the sodium channel blocker phenytoin were 75% lower in patients with higher mRNA levels of MDR1.39 In a rat model of intractable epilepsy, Clinickers et al studied orally administered oxcarbazepine (a sodium channel blocker) administered alone and in the presence of both verapamil and probenecid, inhibitors of P-glycoprotein, and MRP transporters, respectively. When oxcarbazepine was administered alone, it failed to produce an antiepileptic effect but was able to prevent epileptic activity when coadministered with either verapamil or probenecid.40
Intracellular Drug-Binding Sites for Ion Channels
Many drugs block HERG, which accounts for its importance in preclinical and postmarketing drug safety programs. The mechanism for the susceptibility of this channel to drug block by a wide array of structurally divergent compounds has been explored in a series of studies from the Sanguinetti laboratory.10,25,26,41 Alanine scanning mutagenesis implicated two aromatic residues on the intracellular aspect of S6 as high-affinity binding sites for many drugs and identified other residues also accessible from the interior of the cell as potential modulators of drug block. Interestingly, other studies have similarly identified intracellular sites important for drug block in other K+ channels, sodium channels, and calcium channels.42,43 Furthermore, the S6 sequence in the HERG channel includes two prolines (absent in other ion channels); these are thought to kink the S6 helix to allow an unusually wide “mouth” to the intracellular face of the pore region and thus allow unusually large drugs to access the binding/ blocking sites.
Work in the drug transport field has generally used radiolabeled drugs to probe drug-transporter interactions. The experiments we present here indicate that we can exploit the very promiscuity of the HERG channel to assess the extent to which a HERG blocker is transported by a given drug efflux or uptake molecule. Another important implication of the concept we advance is that traditional plasma concentration monitoring approach to guide therapy can be confounded by variable drug access to an intracellular site of action.
Limitations
A key question raised by this work is how variability in a given transport molecule determines net drug effect in a given patient. We did examine the potential that L503F might modulate risk of quinidine-related TdP in a collection of 158 samples (cases and controls) from the Vanderbilt Acquired Long QT Syndrome Registry14 and no effect was observed in this small and underpowered experiment. It is likely that variable function or expression of other transport molecules may also modulate drug access to intracellular sites of action. In this study, we did not assess the expression level of transporters in the heart. However, our results are in general agreement with a study by Nishimura et al, which reported relative expression levels of uptake and efflux transporters in the heart using real-time polymerase chain reaction.44 Furthermore, the lack of an effect of OCTN1 on the extent of IKr block by erythromycin indicates that this is not a nonspecific effect of coexpressing OCTN1 with HERG. Finally, CHO cells express an endogenous protein with 85% sequence homology to human OCTN1; several laboratories demonstrated that the endogenous protein does not transport carnitine and have used this cell line for heterologous expression of OCTN1 (and other related OCTs) for drug transport studies.45–49 To support the notion that the endogenous transporter is not involved in xenobiotic transport, our laboratory previously demonstrated that cimetidine 5 mM (an inhibitor of rat and human OCTN1) inhibits uptake of ion channel antagonists in CHO cells transfected with human OCTN1, but did not alter ion channel block in the absence of human OCTN1.45,50
Summary
We report that coexpression of the organic cation uptake transporter, OCTN1, which is expressed in cardiac myocytes, with the HERG potassium channel enhances the degree of drug block conferred by a prototypical torsades-inducing drug, quinidine. These findings implicate variable drug access to intracellular sites of action as a mechanism contributing to variable drug effects, including drug toxicity.
Acknowledgments
This work is supported by a Training Grant in Clinical Pharmacology (5T32 GM07569) through the National Institute of General Medical Sciences (NIGMS), the Pharmacogenomics of Arrhythmia Therapy Center Grant (U01HL65962) from the National Heart Lung and Blood Institute, and the Pharmacogenetics of Membrane Transporters Grant (U01GM61390) from the National Institute of General Medical Sciences.
We thank Sam Wells, PhD, of the Vanderbilt Imaging Core; Pam Wirth and Melissa Dowling of the Vanderbilt Immunohistochemistry Core; and Nancy J. Brown, MD, Katherine T. Murray, MD, Kenneth B. E. Gagnon, PhD, and Bjorn C. Knollmann, MD, PhD.
Footnotes
The authors report no conflict of interest.
References
- 1.Jazwinska-Tarnawska EO, Echowska-Juzwenko K, Niewinski P, et al. The influence of CYP2D6 polymorphism on the antiarrhythmic efficacy of propafenone in patients with paroxysmal atrial fibrillation during 3 months propafenone prophylactic treatment. Int J Clin Pharmacol Ther. 2001;39:288–292. doi: 10.5414/cpp39288. [DOI] [PubMed] [Google Scholar]
- 2.Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson GR. Drug Metabolism and variability among patients in drug response. N Engl J Med. 2005;352:2211–2221. doi: 10.1056/NEJMra032424. [DOI] [PubMed] [Google Scholar]
- 4.Haas DW, Smeaton LM, Shafer RW, et al. Pharmacogenetics of long-term responses to antiretroviral regimens containing efavirenz and/or nelfina-vir: an adult AIDS clinical trials group study. J Infect Dis. 2005;192:1931–1942. doi: 10.1086/497610. [DOI] [PubMed] [Google Scholar]
- 5.Woodahl EL, Yang Z, Bui T, et al. Multidrug resistance gene G1199A polymorphism alters efflux transport activity of P-glycoprotein. J Pharmacol Exp Ther. 2004;310:1199–1207. doi: 10.1124/jpet.104.065383. [DOI] [PubMed] [Google Scholar]
- 6.Schinkel AH, Wagenaar E, Mol CAAM, et al. P-glycoprotein in the blood–brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadeque AJM, Wandel C, He H, et al. Increased drug delivery to the brain by P-glycoprotein inhibition. Clin Pharmacol Ther. 2000;68:231–237. doi: 10.1067/mcp.2000.109156. [DOI] [PubMed] [Google Scholar]
- 8.Roden DM, Balser JR, George AL, Jr, et al. Cardiac ion channels. Ann Rev Physiol. 2002;64:431–475. doi: 10.1146/annurev.physiol.64.083101.145105. [DOI] [PubMed] [Google Scholar]
- 9.Zhou M, Morais-Cabral JH, Mann S, et al. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 10.Mitcheson JS, Chen J, Lin M, et al. A structural basis for drug-induced long QT syndrome. PNAS. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeola SW, Rich TC, Uebele VN, et al. Molecular analysis of a binding site for quinidine in a human cardiac delayed rectifier K+ channel: role of S6 in antiarrhythmic drug binding. Circ Res. 1996;78:1105–1114. doi: 10.1161/01.res.78.6.1105. [DOI] [PubMed] [Google Scholar]
- 12.MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 1990;250:276–279. doi: 10.1126/science.2218530. [DOI] [PubMed] [Google Scholar]
- 13.Schuster A, Lacinova L, Klugbauer N, et al. The IVS6 segment of the L-type calcium channel is critical for the action dihydropyridines and phenylalkylamines. EMBO J. 1996;15:2365–2370. [PMC free article] [PubMed] [Google Scholar]
- 14.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 15.Donger C, Denjoy I, Berthet M, et al. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation. 1997;96:2778–2781. doi: 10.1161/01.cir.96.9.2778. [DOI] [PubMed] [Google Scholar]
- 16.Priori SG, Napolitano C, Schwartz PJ, et al. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 17.Napolitano C, Schwartz PJ, Brown AM, et al. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol. 2000;11:691–696. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 18.Lasser KE, Allen PD, Woolhandler SJ, et al. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287:2215–2220. doi: 10.1001/jama.287.17.2215. [DOI] [PubMed] [Google Scholar]
- 19.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 20.Haverkamp W, Breithardt G, Camm AJ, et al. The potential for QT prolongation and proarrhythmia by non-antiarrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Eur Heart J. 2000;21:1216–1231. doi: 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]
- 21.Makkar RR, Fromm BS, Steinman RT, et al. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 22.Houltz B, Darpo B, Edvardsson N, et al. Electrocardiographic and clinical predictors of torsades de pointes induced by almokalant infusion in patients with chronic atrial fibrillation or flutter: a prospective study. PACE. 1998;21:1044–1057. doi: 10.1111/j.1540-8159.1998.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 23.Gbadebo TD, Trimble RW, Khoo MSC, et al. Calmodulin inhibitor W-7 unmasks a novel electrocardiographic parameter that predicts initiation of torsade de pointes. Circulation. 2002;105:770–774. doi: 10.1161/hc0602.103724. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 25.Mitcheson JS, Chen J, Sanguinetti MC. Trapping of a methanesulfona-nilide by closure of the HERG potassium channel activation gate. J Gen Physiol. 2000;115:229–240. doi: 10.1085/jgp.115.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry M, Stansfeld PJ, Leaney J, et al. Drug binding interactions in the inner cavity of hERG channels: molecular insights from structure–activity relationships of clofilium and ibutilide analogs. Mol Pharmacol. 2006;69:509–519. doi: 10.1124/mol.105.016741. [DOI] [PubMed] [Google Scholar]
- 27.Grégoire D, Christine F, Patrick H, et al. Efavirenz does not interact with the ABCB1 transporter at the blood–brain barrier. Pharm Res. 2006;V23:1525–1532. doi: 10.1007/s11095-006-0279-5. [DOI] [PubMed] [Google Scholar]
- 28.Tohyama K, Kusuhara H, Sugiyama Y. Involvement of multispecific organic anion transporter, Oatp14 (Slc21a14), in the transport of thyroxine across the blood–brain barrier. Endocrinology. 2004;145:4384–4391. doi: 10.1210/en.2004-0058. [DOI] [PubMed] [Google Scholar]
- 29.Hebert MF, Dowling ALS, Gierwatowski C, et al. Association between ABCB1 (multidrug resistance transporter) geneotype and post-liver transplantation renal dysfunction in patients receiving calcineurin inhibitors. Pharmacogenetics. 2003;13:661–674. doi: 10.1097/00008571-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Nakai D, Nakagomi R, Furuta Y, et al. Human liver-specific organic anion transporter, LST-1, mediates uptake of pravastatin by human hepatocytes. J Pharmacol Exp Ther. 2001;297:861–867. [PubMed] [Google Scholar]
- 31.Dalton TP, He L, Wang B, et al. Identification of mouse SLC39A8 as the transporter responsible for cadmium-induced toxicity in the testis. PNAS. 2005;102:3401–3406. doi: 10.1073/pnas.0406085102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sata R, Ohtani H, Tsujimoto M, et al. Functional analysis of organic cation transporter 3 expressed in human placenta. J Pharmacol Exp Ther. 2005;315:888–895. doi: 10.1124/jpet.105.086827. [DOI] [PubMed] [Google Scholar]
- 33.Grube M, Meyer zu Schwabedissen HEU, Prager D, et al. Uptake of cardiovascular drugs into the human heart: expression, regulation, and function of the carnitine transporter OCTN2 (SLC22A5) Circulation. 2006;113:1114–1122. doi: 10.1161/CIRCULATIONAHA.105.586107. [DOI] [PubMed] [Google Scholar]
- 34.Yabuuchi H, Tamai I, Nezu JI, et al. Novel membrane transporter OCTN1 mediates multispecific, bidirectional, and pH-dependent transport of organic cations. J Pharmacol Exp Ther. 1999;289:768–773. [PubMed] [Google Scholar]
- 35.Urban TJ, Giacomini KM, Risch N. Haplotype structure and ethnic-specific allele frequencies at the OCTN locus: implications for the genetics of Crohn’s disease. Inflamm Bowel Dis. 2005;11:78–79. doi: 10.1097/00054725-200501000-00014. [DOI] [PubMed] [Google Scholar]
- 36.Yang T, Snyders D, Roden DM. Drug block of IKr: model systems and relevance to human arrhythmias. J Cardiovasc Pharmacol. 2001;38:737–744. doi: 10.1097/00005344-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Fromm MF, Kim RB, Stein CM, et al. Inhibition of P-Glycoprotein mediated drug transport: a unifying mechanism to explain the interaction between digoxin and quinidine. Circulation. 1999;99:552–557. doi: 10.1161/01.cir.99.4.552. [DOI] [PubMed] [Google Scholar]
- 38.Dombrowski SM, Desai SY, Marroni M, et al. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia. 2001;42:1501–1506. doi: 10.1046/j.1528-1157.2001.12301.x. [DOI] [PubMed] [Google Scholar]
- 39.Tishler D, Weingberg K, Hinton D, et al. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia. 1995;36:1–6. doi: 10.1111/j.1528-1157.1995.tb01657.x. [DOI] [PubMed] [Google Scholar]
- 40.Clinckers R, Smolders I, Meurs A, et al. Quantitative in vivo microdialysis study on the influence of multidrug transporters on the blood–brain barrier passage of oxcarbazepine: concomitant use of hippocampal monoamines as pharmacodynamic markers for the anticonvulsant activity. J Pharmacol Exp Ther. 2005;314:725–731. doi: 10.1124/jpet.105.085514. [DOI] [PubMed] [Google Scholar]
- 41.Kamiya K, Niwa R, Mitcheson JS, et al. Molecular determinants of hERG channel block. Mol Pharmacol. 2006;69:1709–1716. doi: 10.1124/mol.105.020990. [DOI] [PubMed] [Google Scholar]
- 42.Du LP, Li MY, Tsai KC, et al. Characterization of binding site of closed-state KCNQ1 potassium channel by homology modeling, molecular docking, and pharmacophore identification. Biochem Biophys Res Commun. 2005;332:677–687. doi: 10.1016/j.bbrc.2005.04.165. [DOI] [PubMed] [Google Scholar]
- 43.Drolet B, Simard C, Mizoue L, et al. Human cardiac potassium channel DNA polymorphism modulates access to drug-binding site and causes drug resistance. J Clin Invest. 2005;115:2209–2213. doi: 10.1172/JCI23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nisimura M, Naito S. Tissue specific mRNA expression profiles of human ATP binding cassette and solute carrier superfamilies. Drug Metab Pharmacokinet. 2005;20:452–477. doi: 10.2133/dmpk.20.452. [DOI] [PubMed] [Google Scholar]
- 45.Wu X, George RL, Huang W, et al. Structural and functional characteristics and tissue distribution pattern of rat OCTN1, an organic cation transporter, cloned from placenta. Biochim Biophys Acta Biomembr. 2000;1466:315–327. doi: 10.1016/s0005-2736(00)00189-9. [DOI] [PubMed] [Google Scholar]
- 46.di San Filippo CA, Wang Y, Longo N. Functional domains in the carnitine transporter OCTN2, defective in primary carnitine deficiency. J Biol Chem. 2003;278:47776–47784. doi: 10.1074/jbc.M307911200. [DOI] [PubMed] [Google Scholar]
- 47.Suhre WM, Ekins S, Chang C, et al. Molecular determinants of substrate/ inhibitor binding to the human and rabbit renal organic cation transporters hOCT2 and rbOCT2. Mol Pharmacol. 2005;67:1067–1077. doi: 10.1124/mol.104.004713. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, Groves CE, Bahn A, et al. Relative contribution of OAT and OCT transporters to organic electrolyte transport in rabbit proximal tubule. Am J Physiol Renal Physiol. 2004;287:F999–F1010. doi: 10.1152/ajprenal.00156.2004. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Shirahatti NV, Mahadevan D, et al. A conserved glutamate residue in transmembrane helix 10 influences substrate specificity of rabbit OCT2 (SLC22A2) J Biol Chem. 2005;280:34813–34822. doi: 10.1074/jbc.M506342200. [DOI] [PubMed] [Google Scholar]
- 50.Yang T, Yang P, Liu K, et al. Facilitation of drug block of the human cardiac K+ channel Kv1.5 by a human heart organic cation uptake transporter (OCTN1) Biophysics. 2006;90:2574. [Google Scholar]