

Abstract
Trastuzumab emtansine (T-DM1) is an antibody–drug conjugate (ADC) composed of multiple molecules of the antimicrotubule agent DM1 linked to trastuzumab, a humanized anti–human epidermal growth factor receptor 2 (HER2) monoclonal antibody. Pharmacokinetics data from phase I (n = 52) and phase II (n = 111) studies in HER2-positive metastatic breast cancer patients show a shorter terminal half-life for T-DM1 than for total trastuzumab (TTmAb). In this work, we translated prior preclinical modeling in monkeys to develop a semi-mechanistic population pharmacokinetics model to characterize T-DM1 and TTmAb concentration profiles. A series of transit compartments with the same disposition parameters was used to describe the deconjugation process from higher to lower drug-to-antibody ratios (DARs). The structure could explain the shorter terminal half-life of T-DM1 relative to TTmab. The final model integrates prior knowledge of T-DM1 DARs from preclinical studies and could provide a platform for understanding and characterizing the pharmacokinetics of other ADC systems.
Breast cancer is the most common cancer among women worldwide, affecting 10–12% of women per year.1 In the United States alone, there are ∼230,480 new diagnoses and 39,520 deaths annually.2 Approximately 25% of women who develop breast cancer have human epidermal growth factor receptor 2–positive (HER2-positive) tumors overexpressing HER2 receptors.2 Prior to the availability of HER2-directed therapy, patients with HER2-positive breast cancer had a 5.7-fold greater likelihood of recurrence and an 11.1-fold greater likelihood of disease-related death as compared with patients with normal levels of HER2.3
Trastuzumab is a humanized anti-HER2 monoclonal antibody (mAb). It is a well-established agent indicated for the treatment of patients with metastatic breast cancer having tumors that overexpress HER2 protein.4–6 The response rate to trastuzumab is significant, particularly when it is administered in combination with chemotherapy. However, a substantial proportion of patients do not respond to the drug, and some patients develop resistance within the first year of treatment or experience a relapse after an initial clinical response.5,7 Therefore, alternative treatments are required to prolong the survival of patients with HER2-positive metastatic breast cancer.
Antibody–drug conjugates (ADCs) are antibodies bearing covalently-bound cytotoxic agents. They are designed to target antigen-specific cells to enhance efficacy and reduce the systemic toxicity associated with using the cytotoxic agent alone. For example, maytansine, a highly potent antimitotic agent, was explored as a therapeutic agent, but its development was discontinued because of its severe dose-limiting toxicity (e.g., gastrointestinal, hepatic, and neurotoxicity).8 On the other hand, maytansine-based cytotoxic drugs are widely used as part of many ADCs. These include IMGN901,9 IMGN388, SAR3914, BT-062, and BIIB015.10 Trastuzumab emtansine (T-DM1) is an ADC in which DM1, a potent antimicrotubule agent derived from maytansine, is covalently linked to trastuzumab by an MCC (4-N-maleimidomethyl cyclohexane-1-carboxylate) linker (a thioether nonreducible linker; 4-N-maleimidomethyl cyclohexane-1-carboxylate).11,12 It is hypothesized that, once T-DM1 binds to HER2, the complex undergoes receptor-mediated internalization, resulting in intracellular release of DM1-containing catabolites and subsequent cellular apoptosis.11,13
The characterization of drug disposition is critical for evaluating the determinants of efficacy and toxicity. To our knowledge, clinical pharmacokinetics models describing the disposition of ADC and total antibody have yet to be reported. The purpose of this study is to develop a semi-mechanistic population pharmacokinetics model of T-DM1 in patients with HER2-positive metastatic breast cancer. Pharmacokinetics data for T-DM1 and total trastuzumab antibody (TTmAb) were obtained from a phase I dose-escalation study and a phase II study. The trastuzumab assay measures total antibody concentrations, and the T-DM1 assay recognizes trastuzumab with at least one covalently attached DM1 molecule. The clearance of ADC is apparently faster than that of TTmAb (Figure 1). Preclinical pharmacokinetics studies in monkeys suggest that this inconsistency is the result of a DM1 deconjugation process, transforming parts of the ADCs into unconjugated trastuzumab.14 Our final structural model provides a platform for understanding this inconsistency in ADC and TTmAb disposition, on the basis of mechanisms underlying their behavior. It also helps in identifying the magnitude of interindividual variability and the influence of patient characteristics.
Figure 1.

Time course of T-DM1 (gray) and TTmAb (black) for cycle 1 of the 3.6 mg/kg once every 3 weeks treatment regimen.15 Symbols represent observed mean concentrations and error bars represent SD. The assay for TTmAb measures both conjugated and unconjugated antibody, whereas the T-DM1 assay recognizes conjugated antibody with at least one covalently attached DM1 molecule. The apparent difference in elimination half-life is hypothesized to result from T-DM1 deconjugation, with the ultimate formation of unconjugated trastuzumab. MCC, 4-N-maleimidomethyl cyclohexane-1-carboxylate; SD, standard deviation; T-DM1, trastuzumab emtansine; TTmAb, total trastuzumab antibody.
RESULTS
A total of 53 patients completed the phase I open-label dose-escalation study; the distribution of patients within each dose level and regimen is listed in Table 1. A majority of the patients received 2.4 mg/kg once weekly or 3.6 mg/kg once every 3 weeks. In the phase II study, all 111 patients received 3.6 mg/kg of the drug once every three weeks. Patient demographics and baseline characteristics are shown in Table 2; as is seen from the data presented, most of the patients presented with three or more metastatic sites.
Table 1.
Phase I and phase II study dosage regimens
| Dose (mg/kg) | Number of patients |
|---|---|
| Phase I study | |
| Weekly dosing | |
| 1.2 | 3 |
| 1.6 | 3 |
| 2 | 3 |
| 2.4 | 16 |
| 2.9 | 3 |
| Every-3-weeks dosing | |
| 0.3 | 3 |
| 0.6 | 1 |
| 1.2 | 1 |
| 2.4 | 1 |
| 3.6 | 15 |
| 4.8 | 3 |
| Phase II study | |
| Every-3-weeks dosing | |
| 3.6 | 111 |
Table 2.
Patient demographics and baseline characteristics
| Median (range) | n (%) | |
|---|---|---|
| Age, years | 54 (28–82) | — |
| Weight (kg) | 69.5 (37–137) | — |
| CLCR (ml/min) | 93.7 (35.1–190) | — |
| Total bilirubin (mg/dl) | 0.44 (0.1–1.7) | — |
| Tumor burdena (cm) | 8.15 (1.0–53.1) | — |
| Distinct sites of metastasis | ||
| 1 | 15 (9.1) | |
| 2 | 38 (23.2) | |
| 3+ | 111 (67.7) |
CLCR, creatinine clearance.
Sum of longest diameters for all measurable tumors according to RECIST.31
Pharmacokinetics model
An earlier noncompartmental analysis of antibody–drug conjugate and total antibody concentration–time profiles after the first dose conducted for phase I patients in the once-every-3-weeks treatment regimen indicated a concentration-dependent clearance (CLT-DM1 = 0.0618 ± 0.0135 vs. 0.0202 ± 0.0011 and CLTTmAb = 0.0480 ± 0.0066 vs. 0.00761 ± 0.00377 l/h for the 0.3 and 4.8 mg/kg dose levels, respectively).15 Similar findings were obtained for the once-weekly treatment regimen (data not shown). Noncompartmental analysis and model-fitting criteria for different structural pharmacokinetic models suggested that a nonlinear elimination process was required to characterize phase I data. One- and two-compartment models with linear elimination failed to capture the extended disposition phase of the ADC and total antibody concentrations. For some antibodies that exhibit nonlinear behavior, models of target-mediated drug disposition (TMDD) have been used to describe the time course of antibody concentrations.16 However, parameters of a TMDD model were not identifiable, possibly because of relatively incomplete sampling during the terminal disposition phase. Both rapid binding and Michaelis–Menten (MM) approximations of the full TMDD model are capable of describing saturable elimination processes. The MM model tends to perform better for pharmacokinetic profiles when true elimination phases are not fully characterized, and therefore we included an MM elimination pathway in the model.17 A parallel linear first-order elimination pathway was also evaluated; however, this failed to improve model-fitting criteria and the Akaike Information Criterion increased by 374 points. On the basis of the chemistry of the ADC and a preclinical pharmacokinetics model developed for monkeys,14 seven T-DM1 compartments were originally used to define the distribution of the various species of the ADC. The final number of compartments was optimized by removing one ADC transit compartment at a time and comparing model diagnostic plots, the algorithm objective function, and parameter estimates. A comparison of Akaike Information Criterion values for models containing from 1 to 7 transit compartments is shown in Supplementary Table S1 online.
The model-building process was based on phase I data, and the final pharmacokinetics model is shown in Figure 2. The fraction of the dose entering the system as unconjugated antibody (fr0) was estimated during model fitting. A first-order intercompartmental transfer rate constant (ktd) was used to define the dissociation of DM1 from drug–antibody conjugates. Initial values of fr0 and ktd were obtained from a single-dose monkey study wherein the disposition and residence time of each molecular species were evaluated.14 Given that most of the patients had received previous treatment with trastuzumab, an initial trastuzumab concentration was measurable in some patients, and these values were fixed as initial conditions for the unconjugated trastuzumab compartment (M0 in Figure 2). A simultaneous linear and nonlinear elimination process was also evaluated to address the slight bias in population-fitted TTmAb at higher concentrations (Supplementary Figure S1d online); however, this permutation of the model failed to produce any significant improvement in the modeling criteria.
Figure 2.
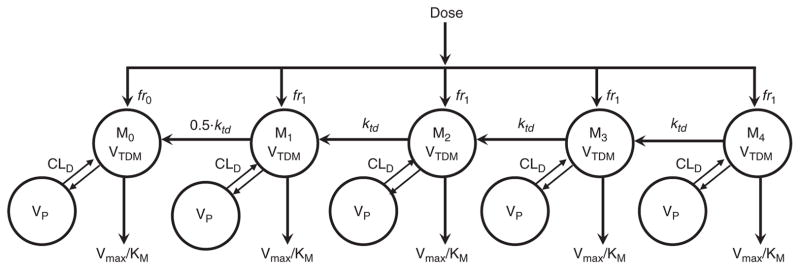
Semi-mechanistic pharmacokinetics model of T-DM1 and TTmAb disposition. CLD, intercompartmental distribution clearance; fr, fraction; KM, Michaelis–Menten affinity constant; ktd, interspecies first-order transfer rate constant; Mi, transit compartment concentration of T-DM1 where i represents a bin or category of drug:antibody ratios; T-DM1, trastuzumab emtansine; TTmAb, total trastuzumab antibody; VC, central compartment volume of distribution; Vmax, Michaelis–Menten capacity constant; VP, peripheral compartment volume of distribution; VTDM, volume of distribution of T-DM1.
Final parameter estimates were obtained by simultaneously fitting the model to both phase I and phase II data. Parameters were precisely estimated, with relative standard error values of <12% for all fixed-effect parameters and <25% for interindividual variability terms (Table 3). As expected for macromolecules, the volume of distribution of T-DM1 (VTDM) was estimated to be relatively low, and its value (3.21 l) is similar to the average adult plasma volume (3.0 l) and to a previously reported estimate for trastuzumab (2.95 l).18 The initial percentage of dose entering the system as unconjugated antibody was estimated to be 4.5%. Of note, the estimate for the first-order rate constant of DM1 dissociation from T-DM1 (ktd = 0.350/day or 0.015/h) is similar to the value estimated in a T-DM1 pharmacokinetics study in monkeys (0.013/h).14 The MM affinity constant (KM) was estimated (with good precision) to be 11.1 mg/l, well within the observed concentration range (0.04–150 mg/l), albeit with large interindividual variability (129%). The MM capacity term (VMAX) was modeled as the product of KM and CLint (VMAX = KM·CLint) and was estimated to be 8.33 mg/day. Model diagnostic plots are shown in the Supplementary Figures S1–S3 online, and observed versus individual- and population-fitted drug concentration plots were reasonable for both T-DM1 and TTmAb, with the exception of a slight bias for the population-fitted TTmAb concentrations (Supplementary Figure S1 online). Individual and population standardized weighted residuals as a function of time for both T-DM1 and TTmAb seem to be equally distributed about zero, and no trends were observed (Supplementary Figure S2 online). Potential covariates, including body weight, creatinine clearance, total bilirubin, and tumor burden (Table 2), were evaluated for each model parameter; however, no physiologically or mathematically relevant covariates were identified. A visual predictive check was conducted for internal model validation. The 5th, 50th, and 95th percentiles of model-simulated concentrations are in good agreement with observed clinical data (Figure 3); however, there is a slight tendency to overpredict the 95th percentile and under-predict the 5th percentile at later time points for TTmAb, whereas the median values of the data are well predicted for the whole treatment period. Individual pharmacokinetic profiles were captured well, and representative individual fitted curves are shown in Supplementary Figure S3 online.
Table 3.
Final parameter estimates for T-DM1 in metastatic breast cancer patients
| Parameter (unit) | Definition | Estimate | CV (%) | IIV (%) | % CV on IIV |
|---|---|---|---|---|---|
| fr0 | Fractional dose input into TTmab compartment | 0.0450 | 3.71 | 63.6 | 23.5 |
| VTDM (l) | Volume of T-DM1 and TTmab compartments | 3.21 | 1.54 | 17.5 | 13.4 |
| ktd (d−1) | First-order transfer rate constant from higher to lower T-DM1 species | 0.350 | 1.84 | 15.5 | 20.5 |
| KM (mg/l) | Michaelis–Menten affinity constant | 11.1 | 11.3 | 129 | 14.2 |
| CLint (l/d) | Intrinsic clearance | 0.751 | 6.56 | 75.8 | 14.3 |
| CLD (l/d) | Distributional clearance | 0.576 | 9.61 | 63.1 | 24.4 |
| VP (l) | Peripheral compartment volume of distribution | 1.70 | 5.81 | 62.3 | 15.1 |
| VMAX (mg/d) | Michaelis–Menten capacity constant | 8.33 | — | — | — |
CV, coefficient of variance; IIV, interindividual variability; T-DM1, trastuzumab emtansine; TTmab, total trastuzumab antibody.
Figure 3.

Visual predictive check of the final model for T-DM1 and TTmAb. Shaded areas are 90% confidence intervals around each simulated percentile (5, 50, and 95%). Dashed and solid lines are simulated and observed medians, respectively. Ninety percent confidence intervals around each percentile were generated in two steps. In step 1, the 5th, 50th, and 95th percentiles were calculated for each simulated trial (n = 100). In step 2, the 5th, 50th, and 95th percentiles were calculated for each percentile from step 1 for all trials. Panels show (a) T-DM1 multiple doses, (b) T-DM1 single dose, (c) TTmAb multiple doses, and (d) TTmAb single dose. T-DM1, trastuzumab emtansine; TTmAb, total trastuzumab antibody.
DISCUSSION
T-DM1 is an ADC bearing up to eight molecules of DM1 covalently bound to a single antibody molecule.14,19 The pharmacokinetics of T-DM1 and total trastuzumab antibody (TTmAb) are complex and, to our knowledge, such profiles have yet to be characterized in humans using a semi-mechanistic model. T-DM1 species with a high drug-to-antibody ratio (DAR) are converted to conjugates with a lower DAR because of deconjugation. As a consequence, the apparent systemic clearance is greater for ADCs with a higher DAR. The deconjugation of DM1 from ADCs with high DAR results in an accumulation of ADCs with lower DAR, thereby increasing their apparent residence times. Simulations of individual compartments (Mi) representing bins or categories of different DAR levels are shown in Figure 4 for up to 500 h after i.v. bolus administration, and compartments representing greater DARs (M3 and M4) clearly exhibit lower net exposures as compared with bins representing lower DARs M1 and M2; Figure 4a). In Figure 4, T-DM1 concentration is the sum of all T-DM1 species, and TTmAb represents the sum of T-DM1 and unconjugated trastuzumab concentrations (Figure 4b). The concentration of unconjugated trastuzumab (M0) gradually increases over time, and therefore concentrations of total T-DM1 decrease faster than those of the total antibody. These profiles, including the relatively slow formation of unconjugated antibody (M0), are in good qualitative agreement with preclinical pharmacokinetics studies in monkeys, wherein the plasma concentration of each T-DM1 species was measured over time after an i.v. bolus dose of the ADC.16
Figure 4.
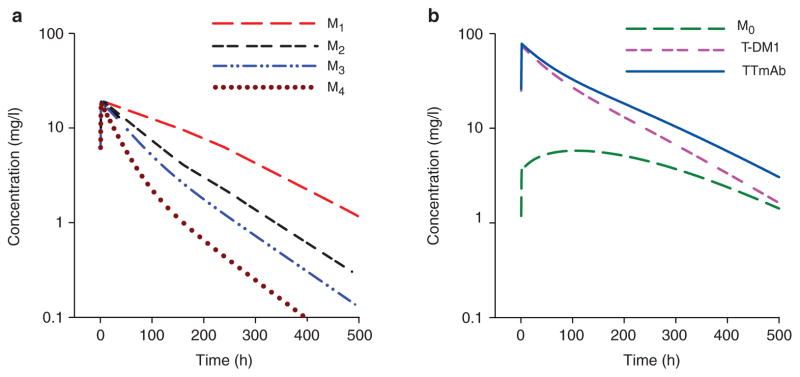
Simulations of individual transit compartments (Mi) for the 3.6 mg/kg treatment regimen, using the proposed semi-mechanistic model (Figure 2) and population mean parameter estimates (Table 3). (a) Individual transit compartments: M1 (long dash), M2 (dash-dash-space), M3 (dash-dot-dot), and M4 (dotted). (b) T-DM1 (dash-dash-space) is the sum of all T-DM1 species concentrations (Mi≥1), TTmAb (solid line) is the sum of conjugated and unconjugated monoclonal antibody concentrations, and M0 (long dash), represents concentrations of unconjugated antibody. T-DM1, trastuzumab emtansine; TTmAb, total trastuzumab antibody.
The shorter half-life of T-DM1 relative to TTmAb suggested unique distribution and/or elimination processes; however, merely assigning different disposition parameters would be rather empirical in nature. To better characterize ADC and total antibody concentrations simultaneously, transit compartments were used to emulate the distribution of molecular species of the ADC. Transit compartments are often used to describe temporal delays or distributions in the pharmacokinetics and pharmacodynamics of many drugs.20,21 A transit compartment model is an extension of the tanks-in-series model wherein N identical compartments with similar residence times are assumed.22,23 The response of a bolus or an impulse of the tanks-in-series model follows an Erlang distribution:
where MN is the Nth compartment and τ is the transit time.24 Bischoff and colleagues provided an early example of applying transit compartments to pharmacokinetic lag times, by constructing an enterohepatic cycling delay in a physiology-based model for methotrexate.25 Savic and colleagues successfully applied a transit compartment approach to characterize delays in drug absorption after oral bolus administration.26 In their model, the ultimate absorption compartment was defined as the final transit compartment, and an explicit function was used to approximate the tanks-in-series model:
where F is bioavailability, ktr is the inverse transit time, n is the optimal number of transit compartments, and ka is the first-order absorption rate constant. For cellular or molecular distributions, the model output is governed by the sum of all transit compartments. Simeoni and colleagues used a transit compartment approach to characterize the delay between drug exposure and response (inhibition of tumor growth), and tumor volume was defined by the sum of all transit compartments.27 Harker and colleagues also modeled peripheral platelet counts using the transit compartment approach, wherein the total peripheral platelets were defined as the sum of all transit compartments.28
Our final model (Figure 2) incorporates transit compartments, in a manner similar to those of the tumor and platelet pharmacodynamic models, to characterize the pharmacokinetics of T-DM1 and TTmAb concentrations. The final number of compartments was optimized through trial and error to attain a parsimonious model. Although an explicit approximation is possible for simple kinetic delays,26 ordinary differential equations were retained to emulate the distribution of ADCs. This model structure allowed for the implementation of a single set of elimination parameters for both T-DM1 and TTmAb, despite the fact that the half-life of T-DM1 is shorter in duration. The binding of DM1 to trastuzumab does not alter the binding affinity of T-DM1 to HER2 receptors.15 This also suggests that a single apparent volume of distribution could be specified that would be applicable for both T-DM1 and unconjugated trastuzumab.
In summary, T-DM1 is a conjugate of a cytotoxic agent (DM1) covalently linked to a humanized-antibody that selectively targets HER2 receptors on breast cancer cells. Preclinical pharmacokinetic modeling efforts in monkeys were used as an initial approach to developing a clinical semi-mechanistic disposition model that would highlight the translational potential of coupling meaningful animal studies with modeling and simulation. The proposed model successfully described the time courses of T-DM1 and TTmAb concentrations simultaneously and captured the extended terminal half-life of total antibody relative to T-DM1 as a consequence of the deconjugation process of DM1-related products from the ADC. Although no physiologically relevant covariates were identified, the proposed model should provide a platform for further studies seeking to evaluate the safety and efficacy of T-DM1 in a quantitative manner. For example, the model structure and post hoc individual parameter values may be used to access the unobserved interindividual variability in the release of DM1, the metrics of which could be compared to clinical outcomes and the frequency and severity of adverse events. The final model could also provide a platform for evaluating the pharmacokinetic properties of other ADC systems.
METHODS
Patient population and study design
Patients with locally advanced and/or metastatic breast cancer, previously treated with a trastuzumab-containing regimen for 60 days without any evident improvement, were included in a phase I open-label multicenter study.15 Escalating dose levels were evaluated for two treatment schedules: once weekly and once every 3 weeks. Doses ranged from 1.2 to 2.9 mg/kg (1.2, 1.6, 2.0, 2.4, and 2.9 mg/kg) in the once-weekly study arm (n = 28) and from 0.3 to 4.8 mg/kg (0.3, 0.6, 1.2, 2.4, 3.6, and 4.8 mg/kg) in the once-every-3-weeks regimen (n = 24). In a phase II single-arm, open-label study,29 3.6 mg/kg of T-DM1 was given once every 3 weeks to 111 patients with HER2-positive metastatic breast cancer that had progressed despite trastuzumab therapy. T-DM1 was administered by i.v. infusion over a period of 90 min for the first dose and over a period of 30 min for subsequent doses. Concentrations of T-DM1 and total trastuzumab (conjugated and unconjugated T-DM1) were measured throughout the treatment course, with a frequent sampling schedule in cycles 1 and 4, and at pre- and post-infusion time points in all other cycles (see Krop et al.15 for details). For the phase II study, blood samples were collected every week during cycles 1 and 4; at other periods of the study, peak and trough concentration samples were collected for both T-DM1 and total antibody. Because most of the study patients had previously been treated with trastuzumab, baseline trastuzumab concentrations were also measured. The studies were approved by the respective site institutional review boards, and each study is in conformity with the assurances filed with and approved by the Department of Health and Human Services. All patients provided written informed consent before enrollment.
Assay
A validated enzyme-linked immunosorbent assay was used to measure T-DM1 concentrations in serum samples from patients, using an anti-DM1 mAb as the coat capture reagent and biotinylated recombinant HER2 extracellular domain (ECD) and horseradish peroxidase–conjugated streptavidin for detection. The T-DM1 assay was designed to measure T-DM1 containing one or more covalently bound DM1 molecules but not to measure unconjugated trastuzumab. A validated enzyme-linked immunosorbent assay was used to measure the total concentration of trastuzumab (conjugated and unconjugated T-DM1) in serum samples from patients, using recombinant HER2 ECD as capture reagent and peroxidase-conjugated F(ab)2 goat anti-human immunoglobulin G Fc for detection. The assay was designed to measure T-DM1 with one or more covalently bound DM1 molecules and unconjugated trastuzumab capable of binding to HER2 ECD. Serum samples were quantified against an assay standard curve prepared from T-DM1, with an average DAR of ∼3.5. The minimum quantifiable concentration in human serum for T-DM1 and TTmAb was 0.04 mg/l.
Pharmacokinetics model
ADCs are designed such that more than one drug molecule binds to an antibody to form an ADC conjugate. The DAR for T-DM1 can range from 0 to 8, with the average DAR being 3.5.14,19 After T-DM1 administration, DM1 molecules are assumed to dissociate at a relatively slow rate from the T-DM1 conjugate, resulting in a reduction in the DAR over time. Molecular species with a greater DAR exhibit a shorter residence time as compared with species with a lower DAR. On the basis of this mechanism, Leipold et al.14 proposed a pharmacokinetics model to simultaneously describe the time courses of plasma concentrations of individual T-DM1 DAR species after i.v. administration in monkeys. Their model included distinct compartments to represent each T-DM1 species, with nonspecific tissue distribution from each compartment along with a single linear clearance term. Conversion among T-DM1 species (i.e., DM1 dissociation) was modeled using a first-order transfer rate constant. Fractional input into each compartment was assumed to be equal regardless of the conjugation status.
Our proposed pharmacokinetics model (Figure 2) is based on the semi-mechanistic model developed for T-DM1 kinetics in monkeys. Because only total T-DM1 concentration was measured, the number of individual T-DM1 compartments was optimized by removing one compartment at a time and comparing model-fitting criteria. In addition, the elimination of free antibody and of each T-DM1 species was described using a standard Michaelis–Menten function (CL = Vmax/Km + Conc) to capture the apparent dose-dependent clearance. A single first-order rate constant (ktd) was used to define the dissociation of DM1; however, based on the results from preclinical modeling, the dissociation of the last DM1 molecule forming unconjugated trastuzumab was fixed to 50% of ktd (0.5ktd).16,22 T-DM1 kinetics in monkeys also suggested rapid equilibration after i.v. administration, and the fractional input into each compartment was modeled as fr1 = 1 − fr0/4, where fr0 is the fraction of the dose entering as unconjugated trastuzumab. Alternative distributions of initial conditions were also evaluated and were shown not to influence model performance or results. Each T-DM1 compartment (Mi) undergoes nonspecific tissue distribution, with the distribution clearance, CLD, assumed to be identical for all T-DM1 species and for unconjugated trastuzumab. The volume of distribution was also assumed to be identical for all compartments. The final model (Figure 2) is defined by the following general system of ordinary differential equations:
where K0 represents a zero-order infusion rate constant, j = 0 (M0) or 1 (i ≥ 1), α = 0 (M0), 0.5 (M1), or 1 (i ≥ 2), and β = 0.5 (M0), 1 (3 ≥ i ≥ 1), or 0 (M4).
Data analysis
All measurements of total T-DM1 and total antibody concentrations were modeled simultaneously using a nonlinear mixed-effects modeling approach with the Monte Carlo parametric expectation maximization algorithm implemented in SADAPT v1.56 with SADAPT-TRAN interface30 (R. Bauer; available from Biomedical Simulations Resource, USC). Inter- and intra-individual variabilities were characterized, along with a full variance–covariance matrix. Relationships between patient characteristics (Table 2) and model parameters were evaluated using standard forward selection and backward elimination, guided by the magnitude of changes in the algorithm objective function. The physiologic rationale for selecting the potential covariates listed in Table 2 was based on variable association with allometry (body weight, age), renal or hepatic function (creatinine clearance, bilirubin), or target-mediated disposition (tumor burden, number of metastatic sites). Model construction and qualification were guided by goodness-of-fit plots, the precision of parameter estimates, and an internal visual predictive check. Residual variability was modeled using the additive plus proportional variance model, Var = σ1+ σ2 × Ypred, where σ1, and σ2 are the estimated variance model parameters, and Ypred is the model-predicted concentration at any point in time. Internal model qualification was achieved by visual predictive check simulating 100 trials of 126 patients (Monte Carlo simulation in NONMEM VI) based on individual patient dosing and sampling records for the 3.6 mg/kg treatment regimen.
Supplementary Material
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The ADC T-DM1 can bear up to 8 molecules of DM1 covalently bound to a single antibody molecule of trastuzumab. Relative to total antibody, T-DM1 exhibits nonlinear pharmacokinetics and a shorter terminal half-life in patients with metastatic breast cancer.
WHAT QUESTION DID THIS STUDY ADDRESS?
Our study sought to determine why T-DM1 exhibits a shorter half-life than that observed for total trastuzumab.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
Our final population-based model contains a series of transit compartments to describe the deconjugation process from higher to lower DARs, which confers a greater apparent systemic clearance for ADCs with higher DARs.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS
Our structural model explains the shorter terminal half-life of T-DM1 relative to total antibody on the basis of a single set of parameters and may provide a platform for understanding and characterizing the disposition, and possibly the toxicity, of other ADC systems.
Acknowledgments
This research was funded, in part, by a research contract from F. Hoffmann–La Roche Ltd and National Institutes of Health grant GM57980 (to D.E.M).
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/cpt
AUTHOR CONTRIBUTIONS V.L.C., F.S.S., J.T., S.R.G., M.G., N.F., and D.E.M. wrote the manuscript; V.L.C., F.S.S., J.M.H., and N.F. analyzed the data.
CONFLICT OF INTEREST F.S.S. and N.F. are employees of F. Hoffmann–La Roche, and J.T. and S.R.G. are employees of Genentech (a member of the Roche Group), the manufacturer of T-DM1. J.M.H. has served as an unpaid consultant to Genentech and is currently employed by Pfizer. M.G. is an employee of Bristol-Myers Squibb. D.E.M. has served as a paid consultant to Genentech Inc. V.L.C. declared no conflict of interest.
References
- 1.Benson JR, Jatoi I, Keisch M, Esteva FJ, Makris A, Jordan VC. Early breast cancer. Lancet. 2009;373:1463–1479. doi: 10.1016/S0140-6736(09)60316-0. [DOI] [PubMed] [Google Scholar]
- 2.Altekruse SF, et al. SEER Cancer Statistics Review 1975–2007. 2010 < http://seer.cancer.gov/csr/1975_2007>.
- 3.Press MF, et al. HER-2/neu gene amplification characterized by fluorescence in situ hybridization: poor prognosis in node-negative breast carcinomas. J Clin Oncol. 1997;15:2894–2904. doi: 10.1200/JCO.1997.15.8.2894. [DOI] [PubMed] [Google Scholar]
- 4.Genentech. Drug datasheet: Herceptin (trastuzumab) 2008. [Google Scholar]
- 5.Slamon DJ, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 6.Smith I, et al. HERA study team. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369:29–36. doi: 10.1016/S0140-6736(07)60028-2. [DOI] [PubMed] [Google Scholar]
- 7.Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res. 2006;8:215. doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blum RH, et al. A therapeutic trial of maytansine. Cancer Clin Trials. 1978;1:113–117. [PubMed] [Google Scholar]
- 9.Ishitsuka K, Jimi S, Goldmacher VS, Ab O, Tamura K. Targeting CD56 by the maytansinoid immunoconjugate IMGN901 (huN901-DM1): a potential therapeutic modality implication against natural killer/T cell malignancy. Br J Haematol. 2008;141:129–131. doi: 10.1111/j.1365-2141.2008.07000.x. [DOI] [PubMed] [Google Scholar]
- 10.Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 11.Lewis Phillips GD, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68:9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 12.Remillard S, Rebhun LI, Howie GA, Kupchan SM. Antimitotic activity of the potent tumor inhibitor maytansine. Science. 1975;189:1002–1005. doi: 10.1126/science.1241159. [DOI] [PubMed] [Google Scholar]
- 13.Erickson HK, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66:4426–4433. doi: 10.1158/0008-5472.CAN-05-4489. [DOI] [PubMed] [Google Scholar]
- 14.Leipold D, Bender B, Xu K, Theil FP, Tibbitts J. Understanding the de-conjugation of Trastuzumab-MCC-DM1 through application of a multicompartmental model of individual drug:antibody species in cynomolgus monkey. Am Assoc Cancer Res Annual Meeting. 2009;100:2914. [Google Scholar]
- 15.Krop IE, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28:2698–2704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 16.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 17.Yan X, Mager DE, Krzyzanski W. Selection between Michaelis-Menten and target-mediated drug disposition pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2010;37:25–47. doi: 10.1007/s10928-009-9142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P. Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol. 2005;56:361–369. doi: 10.1007/s00280-005-1026-z. [DOI] [PubMed] [Google Scholar]
- 19.Wakankar AA, et al. Physicochemical stability of the antibody-drug conjugate Trastuzumab-DM1: changes due to modification and conjugation processes. Bioconjug Chem. 2010;21:1588–1595. doi: 10.1021/bc900434c. [DOI] [PubMed] [Google Scholar]
- 20.Mager DE, Jusko WJ. Pharmacodynamic modeling of time-dependent transduction systems. Clin Pharmacol Ther. 2001;70:210–216. doi: 10.1067/mcp.2001.118244. [DOI] [PubMed] [Google Scholar]
- 21.Yates JW. Mathematical properties and parameter estimation for transit compartment pharmacodynamic models. Eur J Pharm Sci. 2008;34:104–109. doi: 10.1016/j.ejps.2008.02.122. [DOI] [PubMed] [Google Scholar]
- 22.Sun YN, Jusko WJ. Transit compartments versus gamma distribution function to model signal transduction processes in pharmacodynamics. J Pharm Sci. 1998;87:732–737. doi: 10.1021/js970414z. [DOI] [PubMed] [Google Scholar]
- 23.Heinrich R, Neel BG, Rapoport TA. Mathematical models of protein kinase signal transduction. Mol Cell. 2002;9:957–970. doi: 10.1016/s1097-2765(02)00528-2. [DOI] [PubMed] [Google Scholar]
- 24.Ette EI, Williams PJ. Pharmacometrics: The Science of Quantitative Pharmacology. Wiley; Hoboken, NJ: 2007. [Google Scholar]
- 25.Bischoff KB, Dedrick RL, Zaharko DS, Longstreth JA. Methotrexate pharmacokinetics. J Pharm Sci. 1971;60:1128–1133. doi: 10.1002/jps.2600600803. [DOI] [PubMed] [Google Scholar]
- 26.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 27.Simeoni M, et al. Predictive pharmacokinetic-pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 2004;64:1094–1101. doi: 10.1158/0008-5472.can-03-2524. [DOI] [PubMed] [Google Scholar]
- 28.Harker LA, et al. Effects of megakaryocyte growth and development factor on platelet production, platelet life span, and platelet function in healthy human volunteers. Blood. 2000;95:2514–2522. [PubMed] [Google Scholar]
- 29.Burris HA, III, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 30.Bulitta JB, Bingölbali A, Shin BS, Landersdorfer CB. Development of a new pre- and post-processing tool (SADAPT-TRAN) for nonlinear mixed-effects modeling in S-ADAPT. AAPS J. 2011;13:201–211. doi: 10.1208/s12248-011-9257-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Therasse P, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and treatment of cancer, National cancer Institute of the United States, National cancer Institute of canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.