

Abstract
The enteric nervous system (ENS) forms from neural crest-derived precursors that colonize the bowel before differentiating into a network of neurons and glia that control intestinal function. Retinoids are essential for normal ENS development, but the role of retinoic acid (RA) metabolism in development remains incompletely understood. Because RA is produced locally in tissues where it acts by stimulating RAR and RXR receptors, RA signaling during development is absolutely dependent on the rate of RA synthesis and degradation. RA is produced by three different enzymes called retinaldehyde dehydrogenases (RALDH1, RALDH2 and RALDH3) that are all expressed in developing bowel. To determine the relative importance of these enzymes for ENS development, we analyzed whole mount preparations of adult (8–12 week old) myenteric and submucosal plexus stained with NADPH diaphorase (neurons and neurites), anti-TuJ1 (neurons and neurites), anti-HuC/HuD (neurons), and anti-S1003 (glia) in an allelic series of mice with mutations in Raldh1, Raldh2, and Raldh3. We found that Raldh1−/−, Raldh2+/−, Raldh3+/− (R1KOR2HetR3Het) mutant mice had a reduced colon myenteric neuron density, reduced colon myenteric neuron to glia ratio, reduced colon submucosal neuron density, and increased colon myenteric fibers per neuron when compared to wild type (WT; Raldh1WT, Raldh2WT, Raldh3WT) mice. These defects are unlikely to be due to defective ENS precursor migration since R1KOR2HetR3KO mice had increased enteric neuron progenitor migration into the distal colon compared to WT during development. RALDH mutant mice also have reduced contractility in the colon compared to WT mice. These data suggest that RALDH1, RALDH2 and RALDH3 each contribute to ENS development and function.
Keywords: Retinoid metabolism, enteric nervous system, intestinal motility, retinaldehyde dehydrogenase
Introduction
The enteric nervous system (ENS) controls most aspects of intestinal function (Wood et al., 1999). To perform this task, the ENS contains an integrated network of neurons and glia within the bowel wall estimated to contain at least 20 different types of neurons (Furness, 2006). ENS abnormalities can cause intestinal motility disorders leading to abdominal pain, constipation, diarrhea, vomiting, malnutrition and death (Faure, 2013; Obermayr et al., 2012). Elucidation of the molecular mechanisms controlling ENS development may therefore lead to new ways to prevent or treat intestinal motility disorders.
The active vitamin A metabolite retinoic acid (RA) is known to be essential for normal ENS development. Studies in vitro demonstrated that blocking RAR signaling or depriving cells of RA reduced ENS precursor proliferation, reduced neuronal differentiation, and increased neurite length (Sato and Heuckeroth, 2008). Furthermore, retinoid deficiency in vivo from E7.5 to E14.5 impaired ENS precursor colonization of the bowel leading to distal bowel aganglionosis resembling human Hirschsprung disease (Fu et al., 2010). RA affects development by binding to RAR and RXR receptors that in turn regulate transcription. Because RA is produced locally in tissues from retinol, RA signaling depends on the rate of RA synthesis and degradation (Duester, 2008). RA is synthesized by three retinaldehyde dehydrogenase enzymes expressed in the developing bowel (RALDH1, RALDH2 and RALDH3) (Sato and Heuckeroth, 2008). Previous studies demonstrated that Raldh2 homozygous null embryos die by E10.5 due to severe early heart defects, unless given supplemental RA from E7.5-E9.5 that allows survival until E14.5 (Kumar et al., 2012; Niederreither et al., 2003, 2001). However, even with early RA supplementation that allows longer survival, Raldh2 null embryos fail to form an enteric nervous system (Niederreither et al., 2002), demonstrating that Raldh2 function after E9.5 is absolutely essential for ENS development. Mice homozygous for null Raldh1 mutations are fertile and survive without any apparent developmental anomalies (Fan et al., 2003). Mice homozygous for null Raldh3 mutations die just after birth of respiratory distress secondary to choanal atresia (Dupé et al., 2003), but the role of RALDH1 and RALDH3 in ENS development has not been investigated. In this study, our goal was to determine the relative importance for ENS development of each of the three RALDH enzymes expressed in developing bowel. We found that compound Raldh mutant mice had reduced colon myenteric and submucosal neuron density, reduced colon glial cell density, increased colon myenteric neuronal fiber density per neuron, and we demonstrated defects in the peristaltic response of Raldh mutant mice to mucosal stimulation. From these data, we conclude that RALDH1, RALDH2 and RALDH3 each influence ENS development and function.
Materials and Methods
Animals
All animal experiments were in compliance with institutional animal protocols approved by the Washington University Animal Studies Committee. Raldh1−/− (Fan et al., 2003), Raldh2+/− (Mic et al., 2002), and Raldh3+/− mice (Molotkov et al., 2006) on a mixed 129SvJ, Black Swiss, and C57Bl/6 background were bred to produce Raldh1WT, Raldh2WT, Raldh3WT and Raldh1−/−, Raldh2+/−, Raldh3+/− animals, as well as several intermediate genotypes. Mice were genotyped using the PCR primers in Table 1.
Table 1.
PCR primers used for genotyping.
| Primer | Sequence |
|---|---|
| RALDH1 WT F1 | TAAAGACCTGGATAAGGCCATCACTGTGTC |
| RALDH1 WT R1 | CCGCGAGGCACCAACACATTCTCTAACGTG |
| RALDH1 mutant (Neo3′) | CGCCAAGCTCTTCAGCAATATCACGGGTAG |
| RALDH1 mutant (Neo5′) | TGCTCCTGCCGAGAAAGTATCCATCATGGC |
| RALDH2 WT F1 | GAAGCAGACAAGGTGTGTATTGCTTAGAAG |
| RALDH2 WT R1 | GCTTGCACTGCCTTGTCTATATCCACCTGT |
| RALDH2 Mutant F1 | GCCTGACCTATTGCATCTCCCG |
| RALDH2 Mutant R1 | GCCATGTAGTGTATTGACCGATTCC |
| RALDH3 F2 | CAGCCGTCACACAGTTATAGAGGTCTCTGG |
| RALDH3 WT R2 | TCGAGCGCAGCAGCCAGCTTCAGTGCTTTG |
| RALDH3 Mutant R2 | GTTGCAAAACACGTAGGTGCATGCAGGAGA |
Quantitative ENS analysis
Mice were euthanized by carbon dioxide asphyxiation followed by cervical dislocation. Myenteric and submucosal plexus samples were obtained as described previously (Viader et al., 2011). In the adult mice, sequential 2 cm long samples of myenteric plexus taken from the duodenum (starting at the junction with the pylorus) and distal colon were double-labeled with either biotinylated-anti-HuC/HuD (mouse; polyclonal; 1:250; Invitrogen A21272) and anti-S1003 (rabbit; polyclonal; 1:500; Dako), or NADPH diaphorase (NADPH-d) (Neuhuber et al., 1994) and anti-TuJ1 (rabbit; polyclonal; 1:10,000; Covance). Submucosal plexus samples (2 cm long) from the proximal small intestine (SI) and distal colon were labeled with acetylcholinesterase (AChE) as described previously (Enomoto et al., 1998), a method that stains all mouse submucosal neurons that are labeled by TuJ1 (Wang et al., 2010). In E13.5 mice, whole gut samples were labeled with anti-TuJ1 (1:10,000; Covance). Briefly, bowel was incubated in TBST (100 mM Tris, 150 mM NaCl, 0.5% Triton X-100) for 15 minutes at 37°C, blocked in 5% normal donkey serum (NDS)/TBST for 30 minutes at 37°C and then incubated in primary antibody in TBST/5% NDS overnight at 4°C. Samples to be stained with biotinylated-anti-HuC/HuD were treated with 3% H2O2 (in PBS) (20 minutes, 25°C), incubated in PBST (1x PBS, 1% Triton X-100, 30 minutes, 25°C), and then blocked using the Vector SA-Biotin Blocking Kit, per manufacturer’s protocol. Samples were subsequently blocked in 10% NDS/PBST (45 minutes, 25°C) before incubating in biotinylated-anti-HuC/HuD antibody (in 10% NDS/PBST) overnight at 4°C.
Quantification of neuron or glial cell density was done by counting all cells present within the borders of a 0.5 × 0.5 mm grid (20x objective). Neuronal fiber density was analyzed in the circular and longitudinal muscle layers that contain the myenteric plexus by counting “fibers” or “bundles” of fibers that cross perpendicular axes of the same counting grid as described previously (Wang et al., 2010). Supplemental Figure 1 shows how “fibers” and “fiber bundles” were counted. An attempt was made to stretch segments evenly for all samples. Counting was done without knowledge of the genotype. Twenty randomly selected fields were counted for each region and staining method. Data are presented per square millimeter. Neuron-to-glia ratio was determined by dividing the number of neurons by the number of glia within a single field. Quantification of extent of colon colonization by ENS precursors in E13.5 mice was accomplished by stitching 4x images using Fiji/ImageJ (Preibisch et al., 2009) and measuring colon length from the tip of the cecum to the anus and the length of the segment colonized by ENS precursors from the tip of the cecum to the most distal TuJ1+ neuronal cell body or neurite.
Light Microscopy and quantification
Samples were counted on an Olympus Bx60 or Zeiss Axioskop microscope. Images were obtained with an Olympus Bx60 or Axioplan2 microscope equipped with an Axiocam digital camera using AxioVision imaging software (Zeiss, Germany). Fiji/ImageJ (1.47d) was used to uniformly adjust contrast and brightness so that digital images appear as when observed directly through the microscope.
Functional motility studies
Colonic segments were removed and cleared of luminal contents by gentle flushing with Krebs bicarbonate buffer. Colonic segments of about 5 cm length were opened along the mesenteric attachment to make a flat sheet preparation and pinned mucosal side up in a 3-compartment organ chamber as described in detail previously (Grider, 2003; Wang et al., 2010). One mL of a Krebs-bicarbonate medium was added to each compartment (mM: 118 NaCl, 4.8 KCl, 1.2 KH2PO4, 2.5 CaCl2, 1.2 MgSO4, 25 NaH2CO3, 11 glucose). The mucosa of the central compartment was stimulated by stroking with a fine brush (2–8 strokes, 1 stroke per second). Ascending contraction of circular muscle was measured in the orad peripheral compartment and descending relaxation was measured in the caudad peripheral compartment using force-displacement transducers attached to the muscle layers. Results are expressed as grams force above or below baseline tone.
Statistical analysis
All values are expressed as mean ± SEM. Averages for each animal were used for statistical analyses. P values were determined using Sigma Plot 11.0 (Systat Software, San Jose, CA). For single comparisons, Student’s t-test or Mann-Whitney rank sum test (for data that is not normally distributed) were used. For multiple comparisons, one-way ANOVA with Holm-Sidak correction or Kruskal-Wallis one-way ANOVA on ranks (for data that is not normally distributed) with Dunn’s method of pairwise multiple comparison correction were used. Linear regression analysis was done using Sigma Plot 11.0. In all applicable figures, an asterisk (*) indicates statistical significance (p value < 0.05).
Results
Raldh mutants have reduced colon neuron and glial cell density
To determine how Raldh mutations affect ENS structure, we used mice carrying null mutations for Raldh1, Raldh2, and Raldh3 in an allelic series. Because Raldh2 null mice die by mid-gestation and Raldh3 null mice die immediately after birth, we could not evaluate ENS structure in adult Raldh2−/− or Raldh3−/− mice. The most severe adult genotype evaluated is therefore R1KOR2HetR3Het (Raldh1−/− Raldh2+/− Raldh3+/−). Previous in vitro studies suggested that retinoic acid deficiency would reduce ENS precursor proliferation and enteric neuron differentiation, without reducing glial differentiation (Sato and Heuckeroth, 2008). We therefore hypothesized that Raldh mutant mice would have reduced neuronal density and a reduced neuron-to-glia ratio throughout the bowel. To test this hypothesis, neuronal and glial cell density in the small bowel and colon were measured using HuC/HuD and S1003 double-labeled whole mount immunohistochemistry. Analysis of R1KOR2HetR3Het mice showed that small intestine myenteric neuron density is similar to WT (WT: 328.8±33.0 cells/mm2, n=5 vs. R1KOR2HetR3Het: 422.6±64.9 cells/mm2, n=5, p=0.233) whereas colon myenteric neuron density is reduced by 45% (WT: 421.8±26.0 cells/mm2, n=6 vs. R1KOR2HetR3Het: 230.7±17.5 cells/mm2, n=6, p<0.001) (Fig. 1a, b). Small intestine myenteric glial cell density was also equivalent to WT (WT: 458.9±27.1 cells/mm2, n=5 vs. R1KOR2HetR3Het: 558.7±51.8 cells/mm2, n=5, p=0.126) whereas colon glial cell density is reduced by 27% (WT: 658.9±51.5 cells/mm2, n=6 vs. R1KOR2HetR3Het: 480.7±37.3 cells/mm2, n=6, p=0.019) (Fig. 1a, c). The myenteric neuron-to-glial cell ratio is unchanged in the small intestine (WT: 0.70±0.03 neurons/glia, n=5 vs. R1KOR2HetR3Het: 0.74±0.05 neurons/glia, n=5, p=0.563), but is reduced by 27% in the colon (WT: 0.63±0.04 neurons/glia, n=6 vs. R1KOR2HetR3Het: 0.46±0.03 neurons/glia, n=6, p=0.004) (Fig. 1a, d). Neuronal cell density in the submucosal plexus was evaluated after AChE staining and was similar in WT and mutant small intestine (WT: 133.9±5.7 cells/mm2, n=6 vs. R1KOR2HetR3Het: 137.6±5.9 cells/mm2, n=10, p=0.683), but is reduced by 57% in the colon (WT: 46.9±4.4 cells/mm2, n=6 vs. R1KOR2HetR3Het: 20.1±1.4 cells/mm2, n=10, p<0.001) (Fig. 1e, f). Thus, R1KOR2HetR3Het mice have colon hypoganglionosis, but normal neuron and glia numbers in the small bowel.
Figure 1. R1KOR2HetR3Het adult mice have reduced colon enteric neuron and glial cell density.

a) Representative immunohistochemistry of whole mount preparations from WT and R1KOR2HetR3Het mice showing neurons (HuC/HuD+, red) and glia (S1003+, green) in the small intestine (SI) and colon (CO) myenteric plexus. Scale bar: 200 μm. b) Quantification of myenteric neuron density (*, p<0.001 versus WT). c) Quantification of myenteric plexus glial cell density (*, p<0.05 versus WT). d) Quantification of myenteric neuron-to-glia ratio (HuC/HuD+ cells/S1003 + cells) (*, p<0.01 versus WT). e) Representative images from WT and R1KOR2HetR3Het mice showing acetylcholinesterase (AChE+) submucosal neurons in the small intestine and colon. Scale bar: 100 μm. f) Quantification of submucosal neuron density (*, p<0.001 versus WT). Number of animals analyzed is indicated on each bar.
To determine which RALDH proteins account for the reduced density of myenteric and submucosal neurons in the colon of R1KOR2HetR3Het mice, we evaluated neuron density in mice with R1WTR2HetR3WT, R1KOR2HetR3WT, and R1KOR2HetR3Het genotypes. These genotypes were selected because they permit evaluation of Raldh1 KO or Raldh3 Het phenotypes in a sensitized Raldh2 mutant background. We limited our analyses to the colon, since we found normal small intestine neuron density in the most severe genotype that survives to adulthood (R1KOR2HetR3Het). When all genotypes are compared, we found that the colon myenteric neuron density depends primarily on Raldh2 genotype. R1WTR2HetR3WT, R1KOR2HetR3WT, and R1KOR2HetR3Het mice all have similar colon myenteric neuron density (R1WTR2HetR3WT: 211.2±35.3 cells/mm2, n=5, p<0.001 vs. WT; R1KOR2HetR3WT: 251.1±18.8 cells/mm2, n=6, p<0.001 vs. WT; R1KOR2HetR3Het: 230.7±17.5 cells/mm2, n=6, p<0.001 vs. WT) which are different from WT (Fig. 2a). In contrast, colon submucosal neuron density is affected by mutations in Raldh1, Raldh2 and Raldh3 (WT: 46.9±4.4 cells/mm2, n=6; R1WTR2HetR3WT: 41.8±4.0 cells/mm2, n=12; R1KOR2HetR3WT: 28.1±1.6 cells/mm2, n=11; R1KOR2HetR3Het: 20.1±1.4 cells/mm2, n=10) (Fig. 2b, c). Linear regression analysis of the density of submucosal neurons as a function of the number of alleles yields a statistically significant correlation (average colon AChE cells = 7.109 + (6.816 * # of alleles), R=0.748, R2=0.559, regression p<0.001) consistent with a role for all three RALDH enzymes in determining submucosal neuron density. A similar linear regression analysis of myenteric neuron density as a function of the number of alleles had a much lower R2 value (average colon Hu+ cells = 141.249 + (35.231 * # of alleles), R=0.574, R2=0.330, regression p=0.004), consistent with the observation that the Raldh2 mutation alone accounts for the reduced density of myenteric neurons in the colon of the Raldh mutant mice evaluated.
Figure 2. Colon myenteric and submucosal neuron density are differentially affected by Raldh1, Raldh2 and Raldh3 mutations.
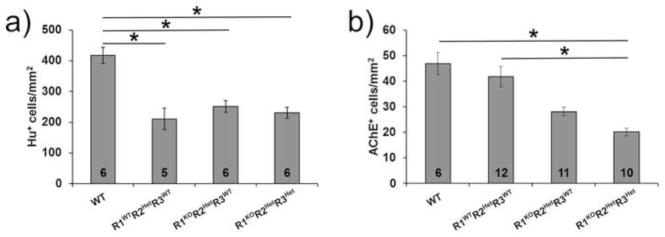
a) Quantification of colon myenteric neuron density (HuC/HuD+ cells) (*, p<0.001 versus WT). b) Quantification of colon submucosal neuron density (*, p<0.05 for indicated comparisons). Number of animals analyzed is indicated on each bar. All pairwise comparisons were done. Unlabeled comparisons were not significant (p>0.05).
Neuron fiber density is altered in Raldh mutants
Previous studies showed that unlike other neuron types, cultured enteric neuron progenitors had longer neurites when RA deprived than when cultured with RA (Sato and Heuckeroth, 2008). Because individual neurites are challenging to trace in the intact ENS, we hypothesized that longer neurites in Raldh mutant mice might be detectable in vivo via an increased ratio of neurites to neuronal cell bodies. Neuronal fiber density in the myenteric plexus visualized with TuJ1 antibody was equal in WT and R1KOR2HetR3Het mice in the small intestine (Bundles: WT: 19.4±0.6 bundles/mm2, n=5 vs. R1KOR2HetR3Het: 18.6±1.3 bundles/mm2, n=5, p=0.597; Fibers: WT: 227.6±20.7 fibers/mm2, n=5 vs. R1KOR2HetR3Het: 206.8±23.6 fibers/mm2, n=5, p=0.526) and in the colon (Bundles: WT: 24.9±0.7 bundles/mm2, n=5 vs. R1KOR2HetR3Het: 26.3±0.8 bundles/mm2, n=4, p=0.241; Fibers: WT: 369.3±28.3 fibers/mm2, n=5 vs. R1KOR2HetR3Het: 376.5±8.2 fibers/mm2, n=4, p=0.413) (Fig. 3a, b). However, the density of TuJ1+ bundles and fibers per Hu+ neuron in the myenteric plexus was significantly decreased in the small intestine (Bundles: WT: 0.06±0.003 bundles/neuron, n=4 vs. R1KOR2HetR3Het: 0.04±0.006 bundles/neuron, n=4, p=0.028; Fibers: WT: 0.63±0.03 fibers/neuron, n=4 vs. R1KOR2HetR3Het: 0.38±0.05 fibers/neuron, n=4, p=0.001) and significantly increased in the colon (Bundles: WT: 0.06±0.004 bundles/neuron, n=5 vs. R1KOR2HetR3Het: 0.12±0.014 bundles/neuron, n=4, p=0.002; Fibers: WT: 0.76±0.1 fibers/neuron, n=5 vs. R1KOR2HetR3Het: 1.16±0.16 fibers/neuron, n=4, p=0.002) of R1KOR2HetR3Het mice (Fig. 3c, d). These data suggest regional differences in the effect of Raldh mutations on neurite growth from enteric neurons.
Figure 3. Ratio of nerve fibers to neuron cell bodies is altered in Raldh mutants.

a) Representative whole mount immunohistochemistry from adult WT and R1KOR2HetR3Het mice show neuronal fibers (TuJ1+, red) in the small intestine (SI) and colon (CO) myenteric plexus. Scale bar: 100 μm. b) Quantification of myenteric plexus neuronal fiber density in adult mice. c) Ratio of nerve fiber density to neuron cell body density (*, p<0.01). d) Ratio of nerve bundle density to neuron cell body density (*, p<0.05). Number of animals analyzed is indicated on each bar.
Nitrergic neurons appear less affected than total neurons by Raldh mutations
To determine if all types of enteric neuron are equally affected by Raldh mutations, we examined myenteric nitric oxide producing neurons using NADPH diaphorase (NADPH-d) staining. Surprisingly, we found no statistically significant differences in nitrergic neuron density between WT and R1KOR2HetR3Het mice in either the small intestine (WT: 40.3±1.6 cells/mm2, n=5 vs. R1KOR2HetR3Het: 42.0±0.7 cells/mm2, n=5, p=0.358) or the colon (WT: 124.8±3.5 cells/mm2, n=5 vs. R1KOR2HetR3Het: 96.7±16.9 cells/mm2, n=4, p=0.11) (Fig. 4a, b), although the mean number of colon NADPH-d neurons was 22.5% lower than WT. In addition, we found no statistically significant differences in nitrergic fiber density between WT and R1KOR2HetR3Het mice in either the small intestine (Bundles: WT: 15.6±0.4 bundles/mm2, n=5 vs. R1KOR2HetR3Het: 16.5±0.5 bundles/mm2, n=5, p=0.189; Fibers: WT: 238.3±10.9 fibers/mm2, n=5 vs. R1KOR2HetR3Het: 217.0±17.0 fibers/mm2, n=5, p=0.325) or the colon (Bundles: WT: 25.1±1.0 bundles/mm2, n=5 vs. R1KOR2HetR3Het: 22.8±0.6 bundles/mm2, n=4, p=0.091; Fibers: WT: 447.2±30.9 fibers/mm2, n=5 vs. R1KOR2HetR3Het: 429.4±17.1 fibers/mm2, n=4, p=0.653) (Fig. 4a, c). Furthermore, when we calculated the number of nitrergic fibers per neuron we found no significant differences between WT and R1KOR2HetR3Het in both the small intestine (WT: 5.9±0.2 fibers/neuron, n=5 vs. R1KOR2HetR3Het: 5.2±0.4 fibers/neuron, n=5, p=0.122) and the colon (WT: 3.6±0.3 fibers/neuron, n=5 vs. R1KOR2HetR3Het: 4.9±0.9 fibers/neuron, n=4, p=0.18) (Fig. 4d). However, a direct comparison also failed to show a significant difference in the ratio of NADPH-d to total neuron density between WT and R1KOR2HetR3Het in the small intestine (WT: 0.14±0.01 NADPH-d/Hu neurons, n=4 vs. R1KOR2HetR3Het: 0.99±0.02 NADPH-d/Hu neurons, n=4, p=0.103) or the colon (WT: 0.30±0.01 NADPH-d/Hu neurons, n=5 vs. R1KOR2HetR3Het: 0.44±0.07 NADPH-d/Hu neurons, n=4, p=0.190) (Fig. 4e) as might be expected if NADPH-d neuron density was unchanged. These data are consistent with the hypothesis that enteric neuron subtypes are not uniformly affected by Raldh mutations and that NADPH-d expressing neurons are better preserved than the other neuronal populations.
Figure 4. Nitrergic neuron and nerve fiber density is similar to WT in the myenteric plexus of R1KOR2HetR3Het mice.

a) Representative NADPH diaphorase (NADPH-d) stained whole mount preparations from WT and R1KOR2HetR3Het mice show nitrergic neurons and fibers in the small intestine and colon. Scale bar: 100 μm. b) Quantification of myenteric plexus nitrergic neuron density c) Quantification of myenteric nitrergic fiber density. d) Ratio of fiber density to neuron cell body density. e) Ratio of nitrergic neuron (NADPH-d) density to total neuron (Hu) density. Number of animals analyzed is indicated on each bar. R1KOR2HetR3Het measurements were not statistically different from WT for any comparisons (p>0.05).
Raldh mutants have altered bowel motility
To determine if bowel function was altered by Raldh mutations, we examined the physiologic response to mucosal stimulus using a three chamber organ bath. In response to mucosal stroking, WT colon had significantly greater ascending contraction when compared to R1WTR2HetR3WT, R1KOR2HetR3WT, and R1KOR2HetR3Het for all stroke numbers examined (WT: n=6; R1WTR2HetR3WT: n=4; R1KOR2HetR3WT: n=5; R1KOR2HetR3Het: n=5) (Fig. 5a, b, Table 2). In addition, R1WTR2HetR3WT had significantly greater ascending contraction compared to R1KOR2HetR3Het with 6 strokes and 8 strokes and more ascending contraction compared to R1KOR2HetR3WT after 8 strokes. Furthermore R1KOR2HetR3WT had stronger ascending contraction compared to R1KOR2HetR3Het using 8 strokes stimulation. Similarly, colon descending relaxation in response to mucosal stroking was greater in WT than in R1KOR2HetR3Het with 6 or 8 strokes and R1WTR2HetR3WT had greater descending relaxation when compared to R1KOR2HetR3Het with 6 strokes (Fig. 5a, b, Table 3). Collectively these data suggest that Raldh1, Raldh2 and Raldh3 each influence colon contractility in response to mucosal stimulation.
Figure 5. Raldh mutants have altered colon contractility.
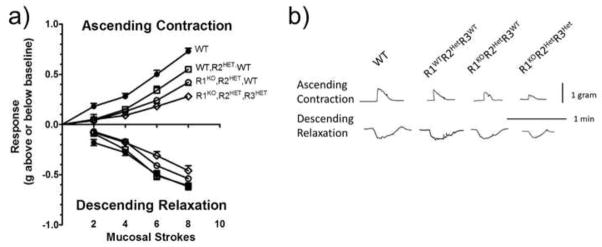
a) Quantification of ascending contraction and descending relaxation in response to mucosal stimulus. b) Representative tracings from flat-sheet preparations of mouse colon from each of the experimental groups. The upper tracings are representative of ascending contraction responses recorded by the force-displacement transducer attached to the circular muscle of the section of colon in the orad compartment and the lower tracings are representative of descending relaxation responses recorded by the force-displacement transducer attached to the circular muscle of the caudad compartment elicited by 8 brush strokes applied to the mucosa of the colonic segment in the central compartment. P values comparing different genotypes to each other are presented in Tables 2 and 3.
Table 2. Ascending contraction p values.
One way ANOVA with pairwise comparisons using the Holm-Sidak method.
| Strokes | WT vs. R1WTR2HetR3WT | WT vs. R1KOR2HetR3WT | WT vs. R1KOR2HetR3Het | R1WTR2HetR3WT vs. R1KOR2HetR3WT | R1WTR2HetR3WT vs. R1KOR2HetR3Het | R1KOR2HetR3WT vs. R1KOR2HetR3Het |
|---|---|---|---|---|---|---|
| 2 | 0.010* | 0.003* | 0.008* | 0.836 | 1.000 | 0.922 |
| 4 | 0.004* | <0.001* | <0.001* | 0.575 | 0.283 | 0.425 |
| 6 | 0.014* | <0.001* | <0.001* | 0.128 | 0.017* | 0.216 |
| 8 | 0.001* | <0.001* | <0.001* | 0.006* | <0.001* | 0.005* |
Asterisks denote statistically significant comparisons.
Table 3. Descending relaxation p values.
One way ANOVA with pairwise comparisons using the Holm-Sidak method.
| Strokes | WT vs. R1WTR2HetR3WT | WT vs. R1KOR2HetR3WT | WT vs. R1KOR2HetR3Het | R1WTR2HetR3WT vs. R1KOR2HetR3WT | R1WTR2HetR3WT vs. R1KOR2HetR3Het | R1KOR2HetR3WT vs. R1KOR2HetR3Het |
|---|---|---|---|---|---|---|
| 2 | NS | NS | NS | NS | NS | NS |
| 4 | 0.784 | 0.069 | 0.099 | 0.248 | 0.288 | 0.799 |
| 6 | 0.807 | 0.207 | 0.009* | 0.243 | 0.010* | 0.165 |
| 8 | 0.762 | 0.479 | 0.042* | 0.512 | 0.115 | 0.387 |
NS, non-significant. Asterisks denote statistically significant comparisons.
Colon colonization by enteric progenitors is enhanced by Raldh mutations
One possible explanation for the reduced enteric neuron density in the colon of R1KOR2HetR3Het mice would be that reduced RA impaired enteric neuron precursor colonization of the colon. To test this hypothesis, we analyzed the position of the most distal TuJ1+ cells in the colon at E13.5 in WT, R1WTR2HetR3WT, R1KOR2HetR3WT, R1KOR2HetR3Het, and R1KOR2HetR3KO mice. Unexpectedly, these analyses demonstrated a statistically significant increase in colonization in the embryos with fewer functional Raldh alleles (WT: 82.4±1.2% colonized, n=18 vs. R1WTR2HetR3WT: 82.5±2.1% colonized, n=7 vs. R1KOR2HetR3WT: 84.3±1.3% colonized, n=11 vs. R1KOR2HetR3Het: 87.7±1.5% colonized, n=18 vs. R1KOR2HetR3KO: 95.3±3.6% colonized, n=4) (Fig. 6a, b). These data suggest that RALDH1, RALDH2 and RALDH3 each affect colon colonization efficiency by ENS precursors.
Figure 6. Colon colonization by enteric progenitors is enhanced by Raldh mutations.
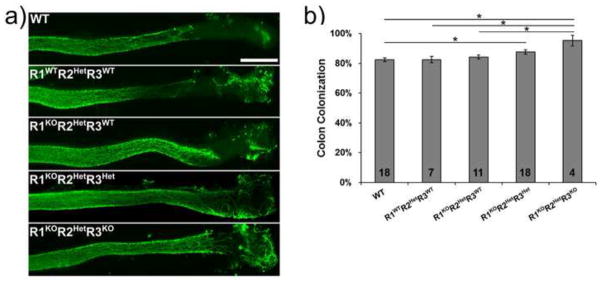
a) Representative images from WT, R1WTR2HetR3WT, R1KOR2HetR3WT, R1KOR2HetR3Het, and R1KOR2HetR3KO mice show enteric neuron precursor cells (TuJ1+ cells, green) that colonized distal colon at E13.5. Scale bar: 500 μm. b) Quantification of extent of colon colonization by enteric progenitors (TuJ1+ cells, green) (*, p<0.05). Number of animals analyzed is indicated on each bar. All pairwise comparisons were done and all unlabeled comparisons were not significant (p>0.05).
Discussion
The well-known developmental morphogen vitamin A and its active metabolite RA, have been studied extensively in many regions of the body, however, there is little information about how retinoid metabolism affects ENS development. Although Raldh2 has been previously shown to be essential for ENS development (Niederreither et al., 2003), we now demonstrate that Raldh1 and Raldh3 also influence ENS structure and function and that heterozygosity for Raldh2 causes ENS defects. Raldh mutations reduced the density of colon myenteric neurons and glia and increased the neuron-to-glia ratio in adult mice. Interestingly, not all enteric neuron subtypes were affected equally since NADPH diaphorase expressing (i.e., NO producing) neuron density was not statistically different in WT and R1KOR2HetR3Het mice. Similarly, there was no statistically significant effect of R1KOR2HetR3Het mutations on measured anatomic parameters for the ENS in the small bowel although the relatively large variance between mice with a mixed genetic background might make it challenging to detect small differences in some parameters (e.g. Hu+ neuron number).
The colon ENS phenotype of Raldh mutant mice fits well with prior observations since RA increases differentiation of many precursor cells into neurons, including enteric neurons (Guan et al., 2001; Sato and Heuckeroth, 2008). Furthermore, RA enhances proliferation of ENS precursors at some stages of development, an observation that might explain the reduced neuron and glia density in the colon of R1KOR2HetR3Het mutant mice. The normal density of enteric neurons and glia in the small bowel of Raldh mutants, however, was not expected. We initially hypothesized that this might occur because RA is needed for efficient migration of ENS precursors into distal colon, but R1KOR2HetR3Het mice do not have delayed colonization of fetal bowel by ENS precursors. Additional mechanistic studies to determine why colon and small bowel myenteric plexus structure are differentially affected in Raldh mutant mice, however, will be challenging since the relevant time frame for RA induced effects on cell number is not known. It is also likely that Raldh mutations will affect the ENS indirectly by altering the expression of one or more of the mesenchymal or epithelial proteins that control ENS development. For example, RA treatment of fetal mice can cause severe caudal regression, gut looping anomalies, and delayed enteric progenitor migration into the hindgut with ectopic Ret and Hoxb5 expression (Pitera et al., 2001). Reduced RA in Raldh2−/− embryos also results in caudal patterning defects, in association with altered sonic hedgehog (Shh) signaling (Ribes et al., 2009). Shh is expressed in the mucosa of the developing bowel and regulates enteric progenitor cell proliferation, differentiation, and migration (Fu et al., 2004). Collectively these data demonstrate important roles for RA, RALDH1, RALDH2, and RALDH3 in colonic ENS development, and defects in the colon ENS even without the severe intestinal patterning abnormalities previously reported with excess RA or complete loss or RALDH2 activity.
Our data also suggest different roles for the various RALDH proteins in the developing myenteric and the submucosal plexus. In the myenteric plexus, loss of a single Raldh2 allele reduced neuron density by 45%, but the additional loss of both Raldh1 alleles and one Raldh3 allele did not cause any additional reduction in myenteric neuron density. In contrast, in the submucosal plexus, Raldh1, Raldh2 and Raldh3 each appear to have a role, with neuron density declining as the number of functional Raldh alleles is reduced. A possible explanation could be the pattern of expression of various RALDH enzymes during development. Our prior studies demonstrated that at E14.5, Raldh1 is primarily expressed within the layer of the developing myenteric plexus, whereas Raldh2 is intensely expressed in the mesenchyme surrounding the ENS, and Raldh3 is expressed within the ENS, mesenchyme, and the intestinal epithelium (Sato and Heuckeroth, 2008). The location of RALDH2 in the developing bowel is ideally suited to paracrine RA signaling to developing myenteric neurons, but apparently not adequate to support developing submucosal neurons. The importance of RALDH1 and RALDH3 in the submucosal plexus may suggest direct signaling within crest-derived cells that form the ENS or important roles for RA in the gut submucosa or epithelium that secondarily supports and influences submucosal neuron development. It is also not known if the reduced number of submucosal neurons occurs because of reduced precursor proliferation or a defect in secondary migration from the myenteric plexus (Gershon et al., 1993).
The effect of Raldh mutations on the ratio of neurites to neuronal cell bodies in the colon is consistent with our prior observations in vitro. In contrast to the neurite elongating effects of RA on other neuronal cell types (Clagett-Dame et al., 2006; Corcoran et al., 2000; So et al., 2006), cultured E12.5 ENS precursors treated with RA had reduced neurite length possibly due to reduced levels of SMURF1, a ubiquitin ligase that degrades RhoA in growth cones (Sato and Heuckeroth, 2008). As a surrogate for neurite length, which we cannot easily measure directly in the intact ENS, we measured neuronal fiber density, a parameter likely to be affected by neurite length. As might be predicted based on our in vitro studies, the ratio of neurites per neuron in the colon was elevated in R1KOR2HetR3Het mice. In contrast, in the small intestine there was a reduction in the ratio of the density of neurites to neuronal cell bodies. One caveat to these studies is that TuJ1 immunohistochemistry does not distinguish intrinsic from extrinsic nerve fibers. To do this would require a single neuron labeling method in the ENS. One elegant approach has recently been described using conditional mutant animals, but this challenging technique would require breeding two additional gene mutations to the Raldh mutant strains (i.e., 5 different interbred genotypes) and is currently impractical (Sasselli et al., 2013).
The mechanisms that might explain differences in the effect of Raldh mutations on colon versus small bowel neuronal cell body to neurite ratio are unknown, but could reflect differences in expression patterns for RA generating or degrading enzymes in different bowel regions, differential sensitivity of these neuron types to RA, or regional differences in RA sensitive gene expression. Our prior work, for example showed differential sensitivity of colon versus small bowel myenteric neurons to loss of mitochondria (Viader et al., 2011) and effects of RA on ENS precursor proliferation that vary by stage of development (Sato and Heuckeroth, 2008). Determining the mechanism underlying these differences, will therefore require detailed information about how RA affects gene expression within the ENS and in surrounding bowel, and at this time the critical time frame for Raldh mutation induced changes is unclear making these studies challenging.
The effect of Raldh mutations on enteric precursor colonization of the distal bowel was unexpected. Our prior study using a maternal vitamin A deficiency model showed decreased colonization of the distal bowel when RA signaling or RA levels were reduced (Fu et al., 2010). Interestingly, the effect was even more profound in mice with heterozygous mutations in Ret, the gene most commonly associated with Hirschsprung disease in humans (Amiel et al., 2008). In contrast, our current data demonstrate that the Raldh mutations studied enhance enteric precursor colonization of the distal colon. The basis for these differing results is unclear, but may be due to the systemic nature of maternal vitamin A deficiency, as opposed to the local effects of RALDH enzyme loss. Maternal vitamin A deficiency reduces the amount of retinol crossing the placenta and therefore should globally reduce RA synthesis. Loss of RALDH enzymes, in contrast, can cause specific structural defects related to the location and timing of enzyme expression (Martín et al., 2005; Mic et al., 2004, 2002; Molotkova et al., 2007; Niederreither et al., 2003; Zhao et al., 2009). The surprising increase in colon colonization by ENS precursors might, for example, result from enhanced neurite growth in migrating ENS precursors (Sato and Heuckeroth, 2008), since neurites from early differentiating enteric neurons can provide a scaffold upon which less differentiated cells migrate, resulting in apparent improvements in colonization. Recent evidence supports this theory of enteric precursor migration using single-cell labeling (Nishiyama et al., 2012).
In addition to the anatomic defects in the colon enteric nervous system demonstrated in Raldh mutant mice, functional analysis of the peristaltic response to mucosal stroking shows that Raldh1, Raldh2 and Raldh3 each support intestinal motility. Raldh2 heterozygosity alone significantly reduced ascending contraction compared to WT animals. The strength of contractions was further reduced in mice heterozygous for Raldh2 and deficient in Raldh1. Additional loss of a single Raldh3 allele again reduced contractile strength in the context of Raldh1 and Raldh2 mutations. It is noteworthy that the peristaltic reflex is dependent on intrinsic primary sensory neurons of both the myenteric and submucosal plexus. Thus, the progressive reduction in functional response with progressively greater loss of RALDH alleles may reflect in part the increasing loss of submucosal neurons as noted in our measurements of neuronal number and density. Interestingly, the effect of these Raldh mutations on descending relaxation was less severe than the effect on ascending contraction, consistent with our observation that nitrergic, inhibitory neurons were unaffected (or at least less affected) relative to total (HuC/D+ neurons). It is also possible that the observed effects of Raldh mutations on the peristaltic response occur because of RA activity in other cell types within the bowel since the Rarα, Rarβ and Rarγ receptors are expressed in all layers of the bowel during fetal development and retinoids are known to affect many cell types (Plateroti et al., 1997; Sato and Heuckeroth, 2008). Thus, although none of the adult Raldh mutant mice tested appear ill, it seems likely that genetic polymorphisms that alter retinoid metabolism may predispose to intestinal motility disorders even without causing distal bowel aganglionosis that occurs in human Hirschsprung disease. Interestingly, the many effects of Raldh mutations identified occurred in mice on a mixed genetic background, increasing the likelihood that polymorphisms in these genes may be relevant for human disease.
We are not aware of any data regarding the role of RALDH enzymes in human enteric nervous system structure or function. Human mutations in ALDH1a2 (RALDH2) have been associated with cardiac outflow tract defects (Pavan et al., 2009), suggesting that other neural crest derived cell populations may be affected by such mutations. These studies lend additional credence to the idea that alterations in retinoid biosynthesis, defects in RA signaling, or suboptimal maternal and post-natal retinoid levels may cause long-term problems with intestinal motility.
Supplementary Material
Neuronal fiber and bundle density were quantified using a counting grid eyepiece in twenty randomly selected fields per region. Only fibers or fiber bundles crossing the top or left side of the grid were counted. Primary fiber structures were considered “bundles” (black arrowheads). Secondary and tertiary fiber structures were considered “fibers” (black arrows). Neuron density was quantified by counting all neuronal cell bodies within the counting grid including those that crossed the top or left side of the grid. Neurons that crossed the bottom or right side of the grid were not counted (white arrowhead). Note that not all counted fibers are marked on this image.
Highlights.
RALDH enzymes are essential for maintaining colon neuron and glial cell density
RALDH mutations alter the density of enteric neuronal fibers per neuron
RALDH mutations enhance enteric neuron precursor migration
RALDH mutations alter bowel motility
Acknowledgments
We would like to thank Hongtao Wang, Ming Fu, Jonathan Lake, Ellen Merrick, and Marina Avetisyan for technical advice and many helpful comments during the course of this work. Funding was provided by the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital CH-II-1008-123 and CH-II-2010-390 (R.O.H.), NIH RO1 DK087715 (R.O.H.), NIH RO1 DK057038 (R.O.H.), a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research 1008525 (R.O.H.) and from the Pediatric Gastroenterology Research Training Program T32 DK077653-18 (E.C.W.). G.D. was supported by NIH grant R01 GM062848. JRG was supported by NIH grant R01 DK34153.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KKY, Lyonnet S, Chakravarti A, Tam PKH, Ceccherini I, Hofstra RMW, Fernandez R. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- Clagett-Dame M, McNeill EM, Muley PD. Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J Neurobiol. 2006;66:739–756. doi: 10.1002/neu.20241. [DOI] [PubMed] [Google Scholar]
- Corcoran J, Shroot B, Pizzey J, Maden M. The role of retinoic acid receptors in neurite outgrowth from different populations of embryonic mouse dorsal root ganglia. J Cell Sci. 2000;113 (Pt 14):2567–2574. doi: 10.1242/jcs.113.14.2567. [DOI] [PubMed] [Google Scholar]
- Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupé V, Matt N, Garnier JM, Chambon P, Mark M, Ghyselinck NB. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci USA. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM, Jr, Milbrandt J. GFR alpha1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998;21:317–324. doi: 10.1016/s0896-6273(00)80541-3. [DOI] [PubMed] [Google Scholar]
- Fan X, Molotkov A, Manabe SI, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, Duester G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol Cell Biol. 2003;23:4637–4648. doi: 10.1128/MCB.23.13.4637-4648.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure C. Pediatric neurogastroenterology gastrointestinal motility and functional disorders in children. Humana Press; Totowa, NJ: 2013. Humana Press: Imprint. [Google Scholar]
- Fu M, Lui VCH, Sham MH, Pachnis V, Tam PKH. Sonic hedgehog regulates the proliferation, differentiation, and migration of enteric neural crest cells in gut. J Cell Biol. 2004;166:673–684. doi: 10.1083/jcb.200401077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Sato Y, Lyons-Warren A, Zhang B, Kane MA, Napoli JL, Heuckeroth RO. Vitamin A facilitates enteric nervous system precursor migration by reducing Pten accumulation. Development. 2010;137:631–640. doi: 10.1242/dev.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness JB. The enteric nervous system. Blackwell Pub; Malden, Mass: 2006. [Google Scholar]
- Gershon MD, Chalazonitis A, Rothman TP. From neural crest to bowel: development of the enteric nervous system. J Neurobiol. 1993;24:199–214. doi: 10.1002/neu.480240207. [DOI] [PubMed] [Google Scholar]
- Grider JR. Neurotransmitters mediating the intestinal peristaltic reflex in the mouse. J Pharmacol Exp Ther. 2003;307:460–467. doi: 10.1124/jpet.103.053512. [DOI] [PubMed] [Google Scholar]
- Guan K, Chang H, Rolletschek A, Wobus AM. Embryonic stem cell-derived neurogenesis. Retinoic acid induction and lineage selection of neuronal cells. Cell Tissue Res. 2001;305:171–176. doi: 10.1007/s004410100416. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sandell LL, Trainor PA, Koentgen F, Duester G. Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim Biophys Acta. 2012;1821:198–205. doi: 10.1016/j.bbalip.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín M, Gallego-Llamas J, Ribes V, Kedinger M, Niederreither K, Chambon P, Dollé P, Gradwohl G. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol. 2005;284:399–411. doi: 10.1016/j.ydbio.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Mic FA, Haselbeck RJ, Cuenca AE, Duester G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development. 2002;129:2271–2282. doi: 10.1242/dev.129.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mic FA, Molotkov A, Molotkova N, Duester G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Dev Dyn. 2004;231:270–277. doi: 10.1002/dvdy.20128. [DOI] [PubMed] [Google Scholar]
- Molotkov A, Molotkova N, Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006;133:1901–1910. doi: 10.1242/dev.02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkova N, Molotkov A, Duester G. Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev Biol. 2007;303:601–610. doi: 10.1016/j.ydbio.2006.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhuber WL, Wörl J, Berthoud HR, Conte B. NADPH-diaphorase-positive nerve fibers associated with motor endplates in the rat esophagus: new evidence for co-innervation of striated muscle by enteric neurons. Cell Tissue Res. 1994;276:23–30. doi: 10.1007/BF00354780. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Fraulob V, Garnier JM, Chambon P, Dollé P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech Dev. 2002;110:165–171. doi: 10.1016/s0925-4773(01)00561-5. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Le Roux I, Schuhbaur B, Chambon P, Dollé P. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development. 2003;130:2525–2534. doi: 10.1242/dev.00463. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Messaddeq N, Schuhbaur B, Chambon P, Dollé P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development. 2001;128:1019–1031. doi: 10.1242/dev.128.7.1019. [DOI] [PubMed] [Google Scholar]
- Nishiyama C, Uesaka T, Manabe T, Yonekura Y, Nagasawa T, Newgreen DF, Young HM, Enomoto H. Trans-mesenteric neural crest cells are the principal source of the colonic enteric nervous system. Nat Neurosci. 2012;15:1211–1218. doi: 10.1038/nn.3184. [DOI] [PubMed] [Google Scholar]
- Obermayr F, Hotta R, Enomoto H, Young HM. Development and developmental disorders of the enteric nervous system. Nat Rev Gastroenterol Hepatol. 2012 doi: 10.1038/nrgastro.2012.234. [DOI] [PubMed] [Google Scholar]
- Pavan M, Ruiz VF, Silva FA, Sobreira TJ, Cravo RM, Vasconcelos M, Marques LP, Mesquita SMF, Krieger JE, Lopes AAB, Oliveira PS, Pereira AC, Xavier-Neto J. ALDH1A2 (RALDH2) genetic variation in human congenital heart disease. BMC Med Genet. 2009;10:113. doi: 10.1186/1471-2350-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitera JE, Smith VV, Woolf AS, Milla PJ. Embryonic gut anomalies in a mouse model of retinoic Acid-induced caudal regression syndrome: delayed gut looping, rudimentary cecum, and anorectal anomalies. Am J Pathol. 2001;159:2321–2329. doi: 10.1016/S0002-9440(10)63082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plateroti M, Freund JN, Leberquier C, Kedinger M. Mesenchyme-mediated effects of retinoic acid during rat intestinal development. J Cell Sci. 1997;110 (Pt 10):1227–1238. doi: 10.1242/jcs.110.10.1227. [DOI] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes V, Roux IL, Rhinn M, Schuhbaur B, Dollé P. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signalling pathways. Development. 2009;136:665–676. doi: 10.1242/dev.016204. [DOI] [PubMed] [Google Scholar]
- Sasselli V, Boesmans W, Vanden Berghe P, Tissir F, Goffinet AM, Pachnis V. Planar cell polarity genes control the connectivity of enteric neurons. J Clin Invest. 2013;123:1763–1772. doi: 10.1172/JCI66759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Heuckeroth RO. Retinoic acid regulates murine enteric nervous system precursor proliferation, enhances neuronal precursor differentiation, and reduces neurite growth in vitro. Developmental Biology. 2008;320:185–198. doi: 10.1016/j.ydbio.2008.05.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So PL, Yip PK, Bunting S, Wong LF, Mazarakis ND, Hall S, McMahon S, Maden M, Corcoran JPT. Interactions between retinoic acid, nerve growth factor and sonic hedgehog signalling pathways in neurite outgrowth. Dev Biol. 2006;298:167–175. doi: 10.1016/j.ydbio.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Viader A, Wright-Jin EC, Vohra BPS, Heuckeroth RO, Milbrandt J. Differential regional and subtype-specific vulnerability of enteric neurons to mitochondrial dysfunction. PLoS ONE. 2011;6:e27727. doi: 10.1371/journal.pone.0027727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hughes I, Planer W, Parsadanian A, Grider JR, Vohra BPS, Keller-Peck C, Heuckeroth RO. The timing and location of glial cell line-derived neurotrophic factor expression determine enteric nervous system structure and function. J Neurosci. 2010;30:1523–1538. doi: 10.1523/JNEUROSCI.3861-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JD, Alpers DH, Andrews PL. Fundamentals of neurogastroenterology. Gut. 1999;45(Suppl 2):II6–II16. doi: 10.1136/gut.45.2008.ii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Sirbu IO, Mic FA, Molotkova N, Molotkov A, Kumar S, Duester G. Retinoic acid promotes limb induction through effects on body axis extension but is unnecessary for limb patterning. Curr Biol. 2009;19:1050–1057. doi: 10.1016/j.cub.2009.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Neuronal fiber and bundle density were quantified using a counting grid eyepiece in twenty randomly selected fields per region. Only fibers or fiber bundles crossing the top or left side of the grid were counted. Primary fiber structures were considered “bundles” (black arrowheads). Secondary and tertiary fiber structures were considered “fibers” (black arrows). Neuron density was quantified by counting all neuronal cell bodies within the counting grid including those that crossed the top or left side of the grid. Neurons that crossed the bottom or right side of the grid were not counted (white arrowhead). Note that not all counted fibers are marked on this image.