

Abstract
The CD117 (KIT) protein is overexpressed in many human neoplasms including adenoid cystic carcinoma of salivary glands. To evaluate the function of c-kit-activating mutations in adenoid cystic carcinoma of the salivary gland, we studied 14 cases (13 primary, 1 cervical lymph node metastasis) from our institution. KIT protein expression was evaluated by immunohistochemistry using formalin-fixed paraffin-embedded tissue. Mutational analyses of c-kit extracellular (exon 9), juxtamembrane (exon 11) and tyrosine kinase domains (exons 13 and 17) were performed by polymerase chain reaction, clonal selection and DNA sequencing. All 14 cases demonstrated strong KIT expression by immunohistochemistry. Molecular analysis was successful in 8 of 14 cases, and c-kit missense point mutations were detected in seven of eight cases (88%) including seven in exon 11, two in exon 9, two in exon 13 and two in exon 17. Eight silent point mutations were detected in five cases. Two cases contained missense mutations in more than one exon. Different mutations were found in the primary tumor and the cervical lymph node metastasis of one patient. Point mutations in domains similar to those described in gastrointestinal stromal tumors were detected, including Pro551Leu and Lys558Glu (5′ end of exon 11), Leu576Phe (3′ end of exon 11), Val643Ala (exon 13) and Asn822Ser (exon 17). Additional novel point mutations in exons 9, 11, 13 and 17 were also identified. This study is the first to report c-kit gene mutations in primary adenoid cystic carcinoma of the salivary gland. Identification of such potential gain-of-function mutations in exon 11, and less frequently in exons 9, 13 and 17, suggests that KIT may be involved in the pathogenesis of adenoid cystic carcinoma of salivary glands. Our study raises a prospect of correlation of c-kit mutation and a potential treatment of adenoid cystic carcinoma with tyrosine kinase inhibitor (imatinib).
Keywords: adenoid cystic carcinoma, c-kit gene mutations, KIT protein, salivary gland
CD117 (KIT) is a type III receptor tyrosine kinase that is activated by binding of its ligand, stem cell factor. In humans, the c-kit gene has been mapped to chromosome 4q12 adjacent to the highly homologous platelet-derived growth factor receptor (PDGFR).1-3 Binding of stem cell factor initiates a phosphorylation cascade that ultimately leads to activation of various transcription factors that regulate apoptosis, cell differentiation and proliferation. KIT protein expression is important in the development of normal human tissues and in many human neoplasms including mastocytosis, gastrointestinal stromal cell tumors, melanoma, breast cancer, both small- cell and non-small-cell lung cancers, gynecologic cancers, thyroid neoplasms, myeloid leukemia and seminomas.4,5 Recently, KIT expression has been identified in seven types of salivary gland tumors: adenoid cystic carcinomas, epithelial–myoepithelial carcinomas, carcinosarcomas, basal cell adenocarcinomas, acinic cell carcinomas, polymorphous low-grade adenocarcinomas, lymphoepithelioma-like carcinomas and in the lumina of pleomorphic adenomas.5,6 This is in contrast to other salivary gland carcinomas and normal salivary lobules. Of these seven tumors, KIT expression has been most extensively documented in adenoid cystic carcinomas with a frequency of expression ranging from 67 to 100%.7-9 In addition, using fluorescent in situ hybridization analysis, copy number gains have been reported in 6.1% of cases.
Pathologic activation of the KIT protein through gain-of-function mutations in the c-kit gene has been demonstrated in hematopoietic malignancies (mastocytosis, acute myeloid leukemia), germ cell tumors (seminomas) and tumors arising from interstitial cells of Cajal (gastrointestinal stromal tumors). A great majority of c-kit mutations cluster in the juxtamembrane domain (exon 11) and tyrosine kinase 2 domain (exon 17), and less frequently in extracellular domains (exons 2, 8 and 9) and tyrosine kinase 1 domain (exons 13 and 14). Some c-kit mutations have specific clinicopathologic significance and differ in response to tyrosine kinase inhibitor treatment/inhibitor sensitivity. For instance in gastrointestinal stromal tumors, mutations in the juxtamembrane domain (exon 11) or the extracellular domain (exon 9) are responsive to tyrosine kinase inhibitor (imatinib) therapy, whereas mutations in exon 13 (that is, V654A and W670I) and exon 17 (D816V and T823D) are predominately imatinib resistant.10-12 Moreover, resistance often develops within a few years in gastrointestinal stromal tumors treated with imatinib due to acquisition of secondary c-kit mutations and c-kit gene amplifications.13
The genetic explanation for KIT overexpression in adenoid cystic carcinoma of the salivary gland remains to be elucidated. To the best of our knowledge, only a few studies to date have looked for activating mutations in the c-kit gene in adenoid cystic carcinomas.4,5,14,15 Holst et al4 and Jeng et al5 demonstrated KIT protein expression in 27 of 30 (90%) and 20 of 25 (80%) cases, respectively, of adenoid cystic carcinoma of the salivary gland; however they did not find c-kit exon 11 or 17 mutations in any of the tumors examined. Lin et al looked at five cases of metastatic adenoid cystic carcinoma of the salivary gland and found no c-kit or PDGFR-α mutations in the tumors analyzed.15 Similarly, Aubry et al16 found no c-kit mutations in 12 of 34 cases of primary adenoid cystic carcinoma of the lung.
To examine the function of c-kit tumorigenesis in adenoid cystic carcinoma, we examined 14 cases of primary adenoid cystic carcinoma of the salivary gland for KIT protein expression and for c-kit gene mutations in exons 9, 11, 13 and 17 by polymerase chain reaction (PCR) amplification, clonal selection and DNA sequencing. This more sensitive methodology allowed us to identify multiple mutations in the c-kit gene.
Materials and methods
Case Selection
Fourteen cases of primary adenoid cystic carcinoma of the salivary gland were identified through a medical record search of patients who underwent biopsy or surgery for salivary gland tumors between 2000 and 2007 at the University of Florida and Shands Hospital. All cases were reviewed by a head and neck pathologist to assure accuracy of the diagnosis of adenoid cystic carcinoma. Adenoid cystic carcinomas of the salivary gland were graded as low grade and high grade based on the predominant histologic pattern observed. Tumors with predominantly tubular and/or cribriform pattern and with solid areas <30% were considered low grade (favorable histology). Tumors with ≥30% solid component were classified as high grade (unfavorable histology). This study was approved by the University of Florida Institutional Review Board.
Immunohistochemistry
Paraffin slides (4 μm) were cut and placed on plus slides and were dried for 2 h in a 601C oven. The slides were then placed on the Ventana Benchmark automated immunostainer where they were dewaxed and heat-induced epitope retrieval was performed with Ventana’s CC1 retrieval solution. Primary antibody anti-CD117, c-kit (C-Term) (YR145; Cell Marque, Rocklin, CA, USA) prediluted rabbit monoclonal antibody, was applied to sections at 421C for 16 min. Presence of the antigen was visualized by using the ultraView DAB detection kit (Ventana Medical Systems Inc., Tucson, AZ, USA). Slides were counterstained with Ventana hematoxylin taken off the stainer and then were dehydrated, cleared and mounted with permanent mounting media.
Mutation Analysis
Tumor-rich areas of unstained, 10 μm paraffin slides were microdissected for each case. Genomic DNA was extracted using TrimGen DNA extraction kit (Trim Gen, Sparks, MD, USA) in accordance with the manufacturer’s instructions.
All cases were subjected to a PCR reaction using primers designed to amplify exons 9, 11, 13 and 17 of the c-kit gene, as outlined by Antonescu et al.17 These products were amplified for 38 cycles on a DNA Thermal cycler (Applied Biosystems, Foster City, CA, USA) as follows: (1) 94°C for 4 min; (2) 94°C for 30 s, 53°C for 30 s, 72°C for 30 s (38 cycles) and (3) 72°C for 3 min. The amplification reactions were performed in a total volume of 25 μl. The reaction mixture contained 1 μl of DNA, 0.5 μl of the forward and reverse primers (10 μM), 0.25 μl of Taq polymerase (Applied Biosystems), 0.5 μl of dNTP (10 mM), 0.5 μl of MgCl2 (25 mM), 5 μl of 5× PCR buffer and 17.25 μl of PCR grade sterile water.
The PCR products were isolated through electrophoresis on a 1% agarose gel stained with ethidium bromide. PCR products were purified using the Qiaex II Gel Extraction kit (Qiagen Inc., Valencia, CA, USA) before cloning. Cloning was performed using the Promega T/A Cloning kit (Promega, Madison, WI, USA). Two to five colonies were selected per exon from each tumor for DNA sequencing. DNA sequencing was performed at the University of Florida Interdisciplinary Center for Biotechnology Research using a 96-well plate format and M13 forward primer. DNA sequences were analyzed using the National Center for Biotechnology Information human c-kit gene sequence (mRNA X06182) and BLAST software to determine the location and type of mutation within each exon.
Results
The clinicopathologic data for the 14 cases of adenoid cystic carcinoma of the salivary gland are illustrated in Table 1. The mean patient age was 59 years (range 45–86 years) and the male to female ratio was 1:1.5. Tumor sites included the parotid gland (n = 4), sublingual gland (n = 2), minor salivary glands of the palate (n = 2), tongue (n = 1), maxillary and ethmoid sinuses (n = 2), nasopharynx (n = 1) and orbit (n = 1). In addition, metastasis to an ipsilateral Level 2A neck lymph node was included for study in one patient with primary adenoid cystic carcinoma of the soft palate (cases 7a and 7b in Table 1). The average tumor size was 2.5 cm. Histologically, nine cases contained ‘favorable’ histology with tubular and/or cribriform growth pattern and five cases contained ‘unfavorable’ histology with greater ≥30% solid pattern. All 14 cases of adenoid cystic carcinoma of the salivary gland demonstrated strong KIT protein expression by immunohistochemistry in the tumor cells (Figure 1) with no immunoreactivity in the adjacent normal salivary gland tissue. Extraction of genomic DNA was successful in 8 out of 14 cases. In five cases, the PCR product was too weak to allow for successful DNA extraction/purification. One case was discarded due to possible crosscontamination with another tumor sample.
Table 1.
Clinicopathologic characteristics of 14 cases of primary adenoid cystic carcinoma of the salivary gland
| Case | Age/sex | Tumor site | Tumor size (cm) | Histology | Grade | KIT-positive |
|---|---|---|---|---|---|---|
| 1 | 45/M | Sublingual gland | 2.8 | Cribriform | Low | + |
| 2 | 73/F | Ethmoid sinus | 4.2 | Cribriform/tubular and solid (60%) | High | + |
| 3 | 53/M | Parotid | 2.6 | Cribriform/tubular | Low | + |
| 4 | 65/M | Maxillary sinus | 5.5 | Solid | High | + |
| 5 | 43/F | Orbital | 3.0 | Cribriform | Low | + |
| 6 | 64/F | Nasal | 4.6 | Cribriform/tubular and solid (75%) | High | + |
| 7a | 59/F | Glossotonsillar sulcus | 1.9 | Cribriform/tubular | Low | + |
| 7b | Level 2A cervical lymph node | 1.8 | Cribriform/tubular and solid (5%) | Low | + | |
| 8 | 59/F | Parotid | 1.4 | Cribriform/ tubular | Low | + |
| 9 | 64/M | Palate | 2.5 | Cribriform | Low | + |
| 10 | 86/F | Sublingual gland | 2.4 | Cribriform/tubular and solid (50%) | High | + |
| 11 | 72/F | Tongue | 1.1 | Solid | High | + |
| 12 | 45/F | Parotid | 1.0 | Cribriform | Low | + |
| 13 | 37/M | Parotid | 0.9 | Cribriform | Low | + |
Figure 1.

KIT immunohistochemical expression in primary adenoid cystic carcinoma of the salivary gland: (a) low-grade adenoid cystic carcinoma of the salivary gland with predominant cribriform/tubular pattern and (c) high-grade adenoid cystic carcinoma of the salivary gland with greater than 30% solid pattern with strong c-kit immunoreactivity in tumor cells (b, d). (a, b) Original magnification ×100; (c, d) magnification ×200.
The results of the mutational analyses are summarized in Table 2. Overall, c-kit missense point mutations, resulting in the substitution of one amino acid for a different amino acid, were detected in seven of eight cases (88%) as follows: seven in exon 11, two in exon 9, two in exon 13 and two in exon 17. In addition, eight silent mutations resulting in no change in amino-acid sequence were detected in five cases. The types of point mutations detected were heterogeneous, but the majority were clustered in exon 11 near the 5′ and 3′ ends (Table 3). A 1673C>T substitution leading to Pro551Leu mutation, 1747C>T leading to Leu576Phe and 1759C>T leading to His580Tyr were detected in one tumor (case 3), illustrated in Figure 2. A second tumor (case 7a) also showed multiple exon 11 mutations including 1693A>G leading to Lys558Glu, 1706A>G leading to Glu562Val and 1792T>C resulting in Phe591Leu.
Table 2.
Summary of KIT exon 9, 11, 13 and 17 mutations detected in adenoid cystic carcinoma of the salivary gland and comparison with KIT mutations in gastrointestinal stromal tumors reported in major studies
| Case | Exon | Substitution | Type | Mutation | KIT mutations reported in gastrointestinal stromal tumors | |
|---|---|---|---|---|---|---|
| 3 | Exon 11 | 1673C>T | Missense | Pro551Leu | Pro551Leu Met552del | Antonescu et al17 |
| 3 | Exon 11 | 1747C>T | Missense | Leu576Phe | Leu576Phe | Zamo et al20, Gomes et al21 |
| 3 | Exon 11 | 1755T>C | Silent | Tyr578 | ||
| 3 | Exon 11 | 1759C>T | Missense | His580Tyr | ||
| 5 | Exon 17 | 2458C>T | Silent | Leu813 | ||
| 7a | Exon 11 | 1693A>G | Missense | Lys558Glu | Lys558Ile Val559del | Zamo et al20 |
| Lys558Gln | Antonescu et al17 | |||||
| 7a | Exon 11 | 1706A>T | Missense | Glu562Val | ||
| 7a | Exon 11 | 1792T>C | Missense | Phe591Leu | ||
| 7a | Exon 17 | 2459T>C | Missense | Leu813Pro | ||
| 7b | Exon 9 | 1508A>T | Missense | Asp496Val | ||
| 7b | Exon 9 | 1476C>T | Silent | His485 | ||
| 7b | Exon 17 | 2430T>C | Silent | Gly803 | ||
| 8 | Exon 9 | 1470C>T | Silent | Phe483 | ||
| 8 | Exon 11 | 1731C>T | Silent | Tyr570 | ||
| 8 | Exon 13 | 1936T>C | Missense | Ser639Pro | ||
| 8 | Exon 17 | 2415C>T | Silent | Ile798 | ||
| 10 | Exon 9 | 1512G>A | Silent | Val497 | ||
| 10 | Exon 9 | 1555A>G | Missense | Asn512Asp | ||
| 10 | Exon 11 | 1771T>C | Missense | Phe584Leu | ||
| 11 | Exon 13 | 1949T>C | Missense | Val643Ala | Val643Ala | Lasota et al23 |
| 12 | Exon 17 | 2486A>G | Missense | Asn822Ser | Asn822Lys, Asn822His Asn822Tyr | Lasota et al23 |
Table 3.
Summary of KIT exon 11 point mutations found in adenoid cystic carcinoma of the salivary gland
| 550 | 560 | 570 | 580 | 590 | |
|---|---|---|---|---|---|
| Wild type | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SFG |
| Case no. | |||||
| 3 | KLMYEVQWKV | VEEINGNNYV | YIDPTQFPYD | YKWEFPRNRL | SFG |
| 5 | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SFG |
| 7a | KPMYEVQWEV | VEVINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SLG |
| 7b | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SFG |
| 8 | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SFG |
| 10 | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWELPRNRL | SFG |
| 11 | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SFG |
| 12 | KPMYEVQWKV | VEEINGNNYV | YIDPTQLPYD | HKWEFPRNRL | SFG |
The first row depicts the wild-type sequence of c-kit exon 11 (codons 550–592) from the 5′ to 3′ ends (left to right). Missense point mutations resulting in amino-acid substitutions are in bold, whereas silent mutations resulting in no amino-acid change are in italics.
Figure 2.

KIT exon 11 mutational analyses in case of adenoid cystic carcinoma of the salivary gland. Multiple point mutations in exon 11 were detected in this case including Leu576Phe, similar to that described in gastrointestinal stromal tumors, novel mutations Pro551Leu and His580Tyr and a silent mutation in codon 578.
Exon 9 sequence alterations consisted of 1508A>T (Asp>Val) substitution in codon 496 and 1555A>G (Asn>Asp) in codon 512. Exon 13 mutations included 1936T>C (Ser>Pro) in codon 639 and 1949T>C (Val>Ala) in codon 643 (Figure 3). Mutations in exon 17 consisted of 2486A>G (Asn>Ser) substitution in the classically altered region of codon 822 (Figure 4), and 2459T>C (Leu>Pro) in codon 813. No duplications or insertions were detected.
Figure 3.
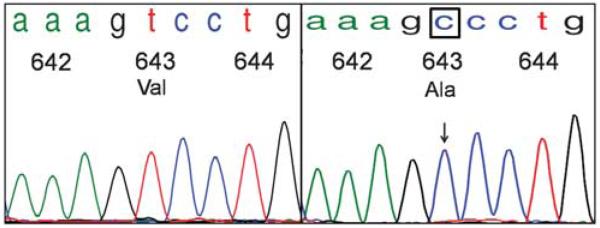
Adenoid cystic carcinoma of the salivary gland with KIT exon 13 point mutation Val643Ala, similar to a variant mutation found in gastrointestinal stromal tumors. The chromatogram tracing on the left depicts a wild-type sequence of KIT exon 13, codons 642–644. On the right is a case of adenoid cystic carcinoma of the salivary gland with a point mutation 1949T>C in codon 643, resulting in substitution of Valine for Alanine.
Figure 4.
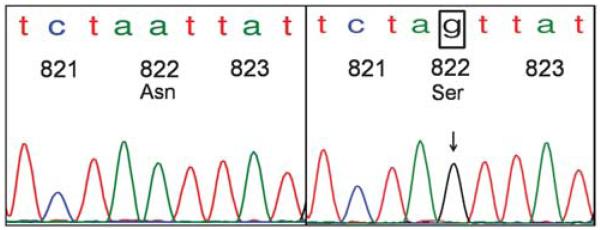
Adenoid cystic carcinoma of the salivary gland with KIT exon 17 point mutation Asn822Ser in hotspot region of codon 822. The wild-type KIT exon 17 sequence depicting codons 821–823 is illustrated on the left. On the right is a case of adenoid cystic carcinoma of the salivary gland with 2486A>G mutation, resulting in substitution of Asparagine for Serine in the frequently mutated c-kit region of codon 822.
Analyses of the cervical lymph node metastasis of one patient (case 7b) revealed different mutations in the metastatic tumor (Asp496Val) vs primary tumor (Lys558Glu, Glu562Val, Phe591Leu and Leu813Pro). Two tumors contained mutations in more than one exon (cases 7a and 10). Mutations in exons 9, 11 and 13 were found in both low- and high-grade tumors. No differences were seen in the types of mutations found between low- and high-grade tumors in this study.
Discussion
This study is the first to report c-kit gene mutations in primary adenoid cystic carcinoma of the salivary gland. Activating mutations in the c-kit gene have been shown in a spectrum of human tumors including kinase domain mutations in mast cell neoplasms and seminomas and extracellular and transmembrane mutations in acute myeloid leukemia and myeloproliferative disorders. In gastrointestinal stromal tumors, 65–92% of tumors are reported to harbor KIT-activating mutations, the majority of which are localized to the juxtamembrane region involving exon 11.18,19 The majority of exon 11 mutations are clustered within the classic hotspot region of the 5′ end involving codons 550–560, however, a second hot spot at the 3′ end involving codons 576–590 has been described by Antonescu et al.17 These include frame deletions of one to several codons (typically involving codons 557–560), point mutations and internal tandem duplications (typically involving the 3′ end). In our study, point mutations in exon 11 were most frequently detected and were clustered in the 5′ and 3′ regions of exon 11. The types of point mutations were heterogeneous; however, substitutions similar to those described in gastrointestinal stromal tumors were detected including Leu576Phe in exon 11 and substitutions involving codons 551 and 558 (Pro551-Leu and Lys 558Glu).17,20,21 Novel exon 11 mutations detected in our study included Glu562Val, His580-Tyr, Phe584Leu and Phe591Leu.
Mutations in exons 9, 13 and 17 were less frequently detected than exon 11 mutations. These are considered rare in gastrointestinal stromal tumors with a reported frequency of less than 10%, but are seen more commonly in hematopoietic malignancies and germ cell neoplasms.22-24 Exon 13 mutations tend to occur in the vicinity of codon 642, with the 1945 A>G substitution leading to Lys642-Glu mutation being the most common.22 We detected the variant Val643Ala mutation observed in gastrointestinal stromal tumors. In addition, exon 17 mutations preferentially involve codon 822 with the 2487T>A substitution being most frequent.22 We detected a variant mutation in adenoid cystic carcinoma consisting of 2486T>A substitution leading to Asn822Ser. Additional novel mutations included Asp496Val and Asn512Asp in exon 9, Ser639Pro in exon 13, Leu813Pro in exon 17. We did not detect any insertions or duplications in exon 9, as have been reported involving codons 502–503 AY and 506–508 FAF.17,23-25
In two patients, we detected more than one mutation in exon 11. In addition, in two cases with exon 11 mutations, we detected additional mutations in exons 9 and 17, respectively. Interestingly, in one patient whose primary tumor contained multiple point mutations in exons 11 and 17 (Lys558Glu, Glu562Val, Phe591Leu and Leu813Pro), we detected a different mutation in exon 9 (Asp496Val) in the cervical level IIA lymph node metastasis. In general, involvement of different c-kit domains is mutually exclusive; however, our results are in keeping with a few studies, which have reported multiple c-kit mutations within the same tumor.26-28 Similar to our study, Andersson et al27 found multiple KIT mutations involving different exons and also loss or addition of mutations in the tumor metastases compared with the primary tumors. Sakurai et al28 likewise identified different mutations in different samples from the same patient. These findings demonstrate the heterogeneity of KIT mutations found within cells of the same tumor population.
Our results differ from four previous studies that did not detect KIT mutations in adenoid cystic carcinomas of the salivary gland and lung.4,5,14-16 These studies used direct PCR sequencing techniques, which is by far, the most widely used; however, direct PCR sequencing lacks significant sensitivity compared to subcloning of the DNA products, because of the heterogeneous DNA from both normal and tumor samples. The high percentage of mutations detected in our study may be attributed to the increased sensitivity obtained through cloning of PCR products and selection of multiple colonies per each exon for DNA sequencing. The cloning selection allowed us to sequence the single species of DNA fragment for each sequencing reaction.
Our study demonstrates the presence of c-kit gene mutations in adenoid cystic carcinomas. The functional significance of these mutations for KIT protein remains to be determined through many cell biology studies. Even less is known about how these mutations are related to potential treatment. There is conflicting data regarding the function of tyrosine kinase inhibitor treatment in adenoid cystic carcinoma. The results of Hotte et al29 showed no evidence of objective response among 15 patients treated with imatinib mesylate. Pfeffer et al30 and Ochel et al31 similarly showed no therapeutic response of locally advanced or metastatic adenoid cystic carcinomas to imatinib. These studies assessed response to tyrosine kinase inhibitor treatment using tumors with strong c-kit immunohistochemical expression; however, they did not assess genetic alterations of the c-kit gene. Lin et al15 observed an unexpected rate of progression of metastatic adenoid cystic carcinoma during 2–3 weeks of treatment with imatinib in five tumors that contained no detectable c-kit or PDGFR-α mutations. Based on their results, the authors concluded that use of imatinib to treat cancers without c-kit or PDGFR-α mutations should be approached with caution. However, preliminary results of a phase II multicenter trial by Faivre et al32 have demonstrated that imatinib may have, at least in a subset of patients, antitumor activity in adenoid cystic carcinoma. Alcedo et al33 also reported two successfully treated cases of adenoid cystic carcinoma of the salivary gland with imatinib. Further studies are needed to investigate the type of KIT mutations harbored by responders compared to nonresponders, as this may be of prognostic value in assessing response to tyrosine kinase inhibitor treatment.
In summary, this study is first to report the presence of c-kit gene mutations in primary adenoid cystic carcinoma. These potential gain-of-function mutations in exon 11, and less frequently in exons 9, 13 and 17 may be involved in KIT overexpression in adenoid cystic carcinomas. Future studies in a larger series may be of value to determine the prognostic implications and benefits of KIT-directed therapy in tumors harboring such mutations.
Acknowledgements
We thank Dr Christine Orlando for her helpful contribution to this study. This study was supported by a grant from Clinical Research Committee, Department of Pathology, University of Florida, Gainesville, FL, and partially supported by a grant (DK02958) from NIH.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
References
- 1.Vliagoftis H, Worobec AS, Metcalfe DD. The proto-oncogene c-kit and c-kit ligand in human disease. J Allergy Clin Immunol. 1997;100:435–440. doi: 10.1016/s0091-6749(97)70131-3. [DOI] [PubMed] [Google Scholar]
- 2.Vandenbark GR, deCastro CM, Taylor H, et al. Cloning and structural analysis of the human c-kit gene. Oncogene. 1992;7:1259–1266. [PubMed] [Google Scholar]
- 3.Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6:3341–3351. doi: 10.1002/j.1460-2075.1987.tb02655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holst VA, Marshall CE, Moskaluk CA, et al. KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol. 1999;12:956–960. [PubMed] [Google Scholar]
- 5.Jeng YM, Lin CY, Hsu HC. Expression of the c-kit protein is associated with certain subtypes of salivary gland carcinoma. Cancer Lett. 2000;154:107–111. doi: 10.1016/s0304-3835(00)00387-6. [DOI] [PubMed] [Google Scholar]
- 6.Andreadis D, Epivatianos A, Poulopoulos A, et al. Detection of C-KIT (CD117) molecule in benign and malignant salivary gland tumours. Oral Oncol. 2006;42:57–65. doi: 10.1016/j.oraloncology.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Azoulay S, Lae M, Freneaux P, et al. KIT is highly expressed in adenoid cystic carcinoma of the breast, a basal-like carcinoma associated with a favorable outcome. Mod Pathol. 2005;18:1623–1631. doi: 10.1038/modpathol.3800483. [DOI] [PubMed] [Google Scholar]
- 8.Crisi GM, Marconi SA, Makari-Judson G, et al. Expression of c-kit in adenoid cystic carcinoma of the breast. Am J Clin Pathol. 2005;124:733–739. doi: 10.1309/61MV-ENEK-5EJ7-JKGF. [DOI] [PubMed] [Google Scholar]
- 9.Freier K, Flechtenmacher C, Walch A, et al. Differential KIT expression in histological subtypes of adenoid cystic carcinoma (ACC) of the salivary gland. Oral Oncol. 2005;41:934–939. doi: 10.1016/j.oraloncology.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–695. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743–1749. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 12.Frost MJ, Ferrao PT, Hughes TP, et al. Juxtamembrane mutant V560GKit is more sensitive to imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther. 2002;1:1115–1124. [PubMed] [Google Scholar]
- 13.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Sorensen KB, Godballe C, de Stricker K, et al. Parotid carcinoma: expression of kit protein and epidermal growth factor receptor. J Oral Pathol Med. 2006;35:286–291. doi: 10.1111/j.1600-0714.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- 15.Lin CH, Yen RF, Jeng YM, et al. Unexpected rapid progression of metastatic adenoid cystic carcinoma during treatment with imatinib mesylate. Head Neck. 2005;27:1022–1027. doi: 10.1002/hed.20274. [DOI] [PubMed] [Google Scholar]
- 16.Aubry MC, Heinrich MC, Molina J, et al. Primary adenoid cystic carcinoma of the lung: absence of KIT mutations. Cancer. 2007;110:2507–2510. doi: 10.1002/cncr.23075. [DOI] [PubMed] [Google Scholar]
- 17.Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: kit mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9:3329–3337. [PubMed] [Google Scholar]
- 18.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- 19.Lasota J, Jasinski M, Sarlomo-Rikala M, et al. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol. 1999;154:53–60. doi: 10.1016/S0002-9440(10)65250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zamo A, Bertolaso A, Franceschetti I, et al. Microfluidic deletion/insertion analysis for rapid screening of KIT and PDGFRA mutations in CD117-positive gastrointestinal stromal tumors: diagnostic applications and report of a new KIT mutation. J Mol Diagn. 2007;9:151–157. doi: 10.2353/jmoldx.2007.060041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomes AL, Bardales RH, Milanezi F, et al. Molecular analysis of c-Kit and PDGFRA in GISTs diagnosed by EUS. Am J Clin Pathol. 2007;127:89–96. doi: 10.1309/M1EC8JE9ACAMJACU. [DOI] [PubMed] [Google Scholar]
- 22.Lasota J, Corless CL, Heinrich MC, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008;21:476–484. doi: 10.1038/modpathol.2008.2. [DOI] [PubMed] [Google Scholar]
- 23.Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol. 2000;157:1091–1095. doi: 10.1016/S0002-9440(10)64623-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156:791–795. doi: 10.1016/S0002-9440(10)64946-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–3825. doi: 10.1200/JCO.2004.05.140. [DOI] [PubMed] [Google Scholar]
- 26.Kitamura Y, Hirotab S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci. 2004;61:2924–2931. doi: 10.1007/s00018-004-4273-y. [DOI] [PubMed] [Google Scholar]
- 27.Andersson J, Sjogren H, Meis-Kindblom JM, et al. The complexity of KIT gene mutations and chromosome rearrangements and their clinical correlation in gastrointestinal stromal (pacemaker cell) tumors. Am J Pathol. 2002;160:15–22. doi: 10.1016/S0002-9440(10)64343-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakurai S, Oguni S, Hironaka M, et al. Mutations in c-kit gene exons 9 and 13 in gastrointestinal stromal tumors among Japanese. Jpn J Cancer Res. 2001;92:494–498. doi: 10.1111/j.1349-7006.2001.tb01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hotte SJ, Winquist EW, Lamont E, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol. 2005;23:585–590. doi: 10.1200/JCO.2005.06.125. [DOI] [PubMed] [Google Scholar]
- 30.Pfeffer MR, Talmi Y, Catane R, et al. A phase II study of imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral Oncol. 2007;43:33–36. doi: 10.1016/j.oraloncology.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 31.Ochel HJ, Gademann G, Rocken C, et al. Effects of imatinib mesylate on adenoid cystic carcinomas. Anticancer Res. 2005;25:3659–3664. [PubMed] [Google Scholar]
- 32.Faivre S, Raymond E, Casiraghi O, et al. Imatinib mesylate can induce objective response in progressing, highly expressing KIT adenoid cystic carcinoma of the salivary glands. J Clin Oncol. 2005;23:6271–6273. doi: 10.1200/JCO.2005.01.6055. author reply 6273-4. [DOI] [PubMed] [Google Scholar]
- 33.Alcedo JC, Fabrega JM, Arosemena JR, et al. Imatinib mesylate as treatment for adenoid cystic carcinoma of the salivary glands: report of two successfully treated cases. Head Neck. 2004;26:829–831. doi: 10.1002/hed.20094. [DOI] [PubMed] [Google Scholar]