

Abstract
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism that results in accumulation of copper primarily in the liver, brain and cornea. Mutations in the WD gene, ATP7B, cause failure of copper excretion from hepatocyte into bile and a defective synthesis of ceruloplasmin. More than 500 mutations are now recognized, scattered throughout the ATP7B gene. Since WD has protean clinical presentations, awareness of WD in clinical practice is important for the early diagnosis and prevention of accumulated copper toxicity. Molecular genetic testing is playing an increasingly important role in the diagnosis of WD in uncertain cases and family screening. Siblings should be screened for WD once an index case has been diagnosed. Discrimination of heterozygotes from asymptomatic patients is essential to avoid inappropriate lifelong therapy for heterozygotes. Genetic testing, either by haplotype analysis or by mutation analysis, is the only definite solution for differentiating heterozygote carriers from affected asymptomatic patients. Routine genetic testing, because of the multitude of documented mutations, has been thought to be impractical until recently. However, genetic testing is now being more actively applied to the diagnosis of WD, particularly in young children in whom conventional biochemical diagnosis has much limitation and only genetic testing is able to confirm WD. Because advancement of modern biochemical technology now allows more rapid, easier, and less expensive mutation detection, direct DNA sequencing could be actively considered as the primary mode of diagnostic investigation rather than a supplementary test to the conventional biochemical tests. This review will focus on the recent advancement of molecular genetics and genetic diagnosis of WD in very young children on the basis of research data of the Seoul National University Children's Hospital and recent literature.
Keywords: Wilson disease, ATP7B, Genetic diagnosis, Mutation, Child
INTRODUCTION
Wilson disease (WD) is an autosomal recessive disorder of copper metabolism that results in accumulation of copper in the liver, brain, cornea, kidney, and other tissues.
WD occurs at a frequency of approximately 1 in 30,000-50,000 worldwide. In a recent nationwide survey for WD in Korea, the estimated prevalence rate of WD in the pediatric population is one in 37,000 [1,2].
Since the description of WD in 1912 by Samuel Alexander Kinnier Wilson as a "familial syndrome of progressive lenticular degeneration in the brain associated with cirrhosis of the liver" [3], there was a major breakthrough in WD research in 1993 when the WD gene ATP7B was first cloned [4-10].
The protein product of ATP7B gene is a copper transporting adenosine triphosphatase (ATPase), which is mainly expressed in hepatocytes. Mutations in ATP7B cause the functional loss as a copper transporter and result in impairment of hepatic biliary copper excretion and also copper incorporation into apo-ceruloplasmin. Accumulation of copper in the body ultimately leads to liver disease, neurologic symptoms, and Kayser-Fleischer corneal rings which have been described as "classical symptom triad" of WD (Fig. 1) [11].
Fig. 1.

(A) Severe cirrhotic liver in a 13 year old girl with Wilson disease who underwent the first liver transplantation in Korea in 1988 [11], (B) Micro and macro-nodular cirrhosis of the removed liver after liver transplantation at Seoul National University Children's Hospital.
In young children, the onset of manifestations is primarily hepatic. Neurologic and psychiatric onset is usually observed in older children and adolescents [12,13].
Asymptomatic elevation of aminotransferases can be observed in children less than 3 years. The first manifestations of WD can be observed in adults greater than 60 years of age.
Children with WD typically present with liver disease including chronic hepatitis with asymptomatic elevations of aminotransferases, cirrhosis and occasionally fulminant hepatic failure (Table 1 and 2) [1]. Neurologic symptoms of WD including tremor, dysphagia, dystonia, rigidity, dysarthria, and choreoathetosis usually occur later in childhood and in adults. The prevalence of neurologic symptoms increases with age. About half of the adult patients with WD present with neurologic symptoms (Table 1) [1].
Table 1.
Clinical Manifestations of 516 Korean Patients with Wilson Disease

Values are presented as number (%).
Table 2.
Presentation Modes of Liver Diseases in Korean Children and Adults with Wilson Disease

Values are presented as number (%).
Early diagnosis and appropriate management can cure WD. If left untreated, however, WD can progress to fatal hepatic failure or severe neurologic deterioration and death.
Because the misdiagnosis and delay in treatment lead to fatal deterioration, applying appropriate diagnostic tests early in life is most important. However, the diagnosis of WD is very difficult particularly in young children who frequently show atypical or insufficient findings of biochemical and clinical tests for WD.
It has recently been documented that molecular genetic testing detects WD earlier and more securely in very young children who frequently do not meet the diagnostic criteria of laboratory tests for WD.
In the past, genetic testing was once considered impractical because of the great numbers of WD causing mutations reported in literature and most of these mutations beging very rare.
In recent years, direct genetic diagnosis including full DNA sequencing has become much easier and is more rapid than before. As the cost of molecular genetic testing has been decreasing, genetic diagnosis is replacing the copper related biochemical tests and is more actively used particularly in very young children as one of the initial diagnostic workup tests for WD.
In this article, the author reviewed recent advancements in the diagnosis of WD in young children and suggested an algorithm for the diagnosis of WD on the basis of the research experience at Seoul National University Children's Hospital and recently published literature.
INTRA-CELLULAR COPPER TRAFFICKING PATHWAY
Dietary copper is absorbed from the intestinal epithelium into the blood. And copper in the blood, which is bound to albumin, is delivered to the liver. In the liver, copper enters the hepatocyte through the copper-transporter 1 (CTR1), and is transported into hepatocytes. Then a specific copper chaperone ATOX1 carries copper to the ATP7B protein (coppertransporting ATPase) located in the trans-Golgi network (TGN). Copper is then transported by ATP7B from the TGN into the apical membrane-trafficked vesicles, and copper is then excreted into bile (Fig. 2 and 3) [14-18].
Fig. 2.

Three types of defects in copper dependent trafficking pathway among various types of missense mutations [15,17,18]. CTR1: copper-transporter 1, ER: endoplasmic reticulum, TGN: trans-Golgi network.
Fig. 3.

The failure of Cu dependent trafficking pathway and functional defects of copper transport to apoenzyme in representative missense mutations [15,17,18]. ER: endoplasmic reticulum, TGN: trans-Golgi network.
The relocation ATP7B depends on copper concentrations. When copper concentrations are low or in the basal state, ATP7B is located in the TGN, where they deliver copper to the apo-ceruloplasmin. When copper concentrations are elevated, ATP7B moves out of the TGN and is relocated to cytoplasmic vesicles. These copper containing vesicles move periphery to the plasma membrane where the accumulated copper is released into the bile canaliculus. After releasing copper, vesicles are recycled and ATP7B returns back to the TGN.
In WD, ATP7B and copper cannot leave the TGN because of a conformational change including misfolding in ATP7B variants. The functional loss of ATP7B as a copper transporter results in copper accumulation in the hepatocyte and copper toxicity related symptoms occur.
The mutated ATP7B is nonfunctional as a copper transporter. In patients with ATP7B mutations, severe impairments of both copper release into the bile and copper incorporation into ceruloplasmin result in high hepatic copper contents, very low levels of copper bound serum ceruloplasmin, and low biliary copper.
There seems to be a difference in the failure of copper trafficking pathway among various mutations at the molecular level (Fig. 3) [15-20]. Mutations of ATP7B may cause three types of localization defects: 1) a normal steady state, constitutive localization within the TGN, but no response to copper [17,19]; 2) mislocalization at the endoplasmic reticulum (ER); 3) constitutive localization at the cell periphery (Fig. 2).
The mislocalization at the ER seems to be the most common type of failure in copper trafficking pathway. It is found in R778L and H1069 mutations [17]. ER mislocalization of ATP7B is often due to misfolding and associated with proteasomal degradation [21-23].
And in patients with other type of mutations, copper and ATP7B binding complex are constitutively localized at the cell periphery and copper is not excreted into bile.
However, clinical correlation with these molecular level differences among various mutations is not clearly documented at the present.
Not all mutations in ATP7B disrupt both the copper transport into bile and the delivery to ceruloplasmin (Fig. 3) [15-20]. Most of the mutation failuretions including R778L and H1069Q have both of these two defects. Some mutations such as G943S and M769V seem to have only one defect. In G943S, the failure of copper trafficking pathway to bile canalicuil was found but intact cuproenzyme biosynthesis was shown in the complementation assay. These types of mutation could explain the normal serum ceruloplasmin level in some patients with WD. In one patient who had compound heterozygote mutations with G943S, serum ceruloplasmin was reported to be normal [17]. Further studies are needed to confirm the correlation of the localization defects in the cell with laboratory findings or clinical severity observed in patients with WD.
MUTATIONS OF ATP7B
The ATP7B is a large gene which has 21 exons varying from 77 to 1,234 bp [4]. ATP7B, a protein product of the gene, is copper-transporting P type ATPase. The ATP7B gene is expressed mainly in the liver and kidney. The highest expression is shown in hepatocytes. The copper delivery to the apo-ceruloplasmin and copper excretion into bile canaliculi from hepatocytes are essential functions of the ATP7B. Mutations cause conformational changes such as misfolding in the ATP7B which result in loss of function as a copper transporter.
Since the discovery of WD gene, more than 500 different mutations have been documented so far, and novel mutations are continuously being reported. Single nucleotide substitutions-missense or nonsense mutations are the most common mutations. Small deletions, small insertions and splicing site mutations are the next common mutation types (Table 3) [10].
Table 3.
Human Mutation Database (2012)

Mutations differ among ethnic groups. Arg778Leu mutation is located on exon 8 in the trans-membrane domain 4. It is the most common mutation in East Asian countries. This mutation has seldom been reported in European populations, so far. An allele frequency of Arg778Leu in Korean children with WD is 37-41% [2,24-27]. The A874V, L1083F, and N1270S are the next common mutations in Korea, and the total allele frequency of these 4 mutations is 64% [2]. A higher frequency of Arg778Leu was also reported in Taiwanese [28,29], Chinese [30], and Japanese [31] patients.
His1069Gln mutation, which is located on exon 14 close to the ATP binding domain, is the most common mutation in western countries. This mutation accounts for about one third of WD mutations in European and American populations, with the highest allele frequency of 73% reported in polish patients [32].
Patients with WD usually have two different mutations rather than two identical mutations. Since most patients with WD are compound heterozygotes and most mutations are very rare, genotype/phenotype correlation studies are difficult to perform.
DIAGNOSIS OF WD AND LIMITATIONS OF LABORATORY TESTS IN YOUNG CHILDREN
Clinical suspicion of WD is the most important first step for the early diagnosis. WD should be considered in the differential diagnosis of children with unknown chronic hepatitis, unusual neurologic or psychiatric symptoms, and hemolytic anemia.
The diagnosis of WD often depends on the detection of low serum ceruloplasmin levels, increased 24 hour urinary copper excretion, Kayser-Fleisher rings in the descemet membrane of the cornea, and increased hepatic copper contents, (Table 4). None of the laboratory parameters alone allows a definite diagnosis of WD. A combination of any two of these four laboratory findings strongly supports for a diagnosis of WD.
Table 4.
Diagnosis of Wilson Disease

In 550 pediatric and adult patients detected on a nation-wide survey of WD in Korea, low serum ceruloplasmin (<20 mg/dL), high 24-hour urine copper (>100 µg), and Kayser-Fleischer rings were found in 96%, 86%, and 73% of the patients (Table 5) [1]. High hepatic copper content (>250 µg/g of dry weight liver) were found in 88% of the tested patients.
Table 5.
Diagnostic Investigations for Wilson Disease

Values are presented as number (%).
The Kayser-Fleischer ring, which is copper deposits in the periphery of the cornea, is very useful particularly for the diagnosis of patients with neurologic symptoms (Table 6). In the neurologic form of WD, the absence of Kayser-Fleischer rings is extremely rare. If slit lamp examinations reveal Kayser-Fleischer rings in children presenting with typical neurologic manifestations, a definite diagnosis of WD can be made without further biochemical tests.
Table 6.
Kayser-Fleisher Rings in 469 Korean Patients with Wilson Disease (WD)

In clinical practice, less invasive investigations including measurements of serum ceruloplasmin levels, urine collections for copper content, and slit lamp examinations for Kayser-Fleischer rings are performed more frequently at the initial stages of WD diagnosis than hepatic copper assays which need invasive liver biopsy. None of the biochemical tests are highly sensitive or specific.
Structural abnormalities of the mitochondria within hepatocytes are regarded as an important and characteristic feature of WD (Fig. 4) [33]. One of the most striking changes is the deformity of the mitochondrial cristae. Cystic dilatation of the cristae results from separation of the outer membrane from the inner membrane of the cristae. The size and shape of the mitochondria are very pleomorphic. Some mitochondria may contain vacuoles and dense inclusion bodies in the cytoplasm.
Fig. 4.

Structural abnormalities of mitochondriae in the hepatocytes from the article of Sternlieb (Hepatology 1992; 16:728-32) [33].
If cholestatic liver disease is not present, these mitochondrial changes could be considered pathognomonic findings of WD. Although the ultra-structural abnormalities of mitochondriae are very characteristic and specific, diagnostic sensitivity is low. Mitochondrial changes are not prominent in most of the children with WD who are diagnosed in early childhood. Children with WD rarely show all these characteristic mitochondrial changes.
Magnetic resonance imaging (MRI) investigations of the brain should be a part of the evaluation in all children with neurologic WD, and should be considered prior to treatment. Liver MRI including multiple hypointense nodules on T2-weighted images, correlates with the severity of hepatic dysfunction, and also demonstrates reversible changes after clinical improvement [34].
In children with the neurologic form of WD, the most frequently identified abnormality on MRI is bilateral symmetrical high signal intensity in the putamen on T2-weighted images. This lesion subsides after chelating therapy (Fig. 5) [35].
Fig. 5.

High signal intensity in the putamen on T2 weighted magnetic resonance image (A) and reversal of the lesion after therapy in children with neurologic WD (B). From a case of Seoul National University Children's Hospital [35].
LIMITATIONS OF DIAGNOSTIC LABORATORY TESTS FOR WD IN VERY YOUNG CHILDREN
Intermediate or equivocal values in the 24 hour urinary copper are more common in young children (Table 7).
Table 7.
24 Hour Urine Copper Excretion (µg) in 114 Children with Wilson Disease (p=0.0035)

Values are presented as number (%). At Seoul National University Children's Hospital.
In patients over 6 years of age, all but 4 children (96%) showed increased 24 hour urinary copper above 100 µg. In contrast, only 73% of children under the age of 6 showed increased 24 hour urinary copper above 100 µg. In addition, 7 of 26 children (27%) did not meet the diagnostic cut off value of urinary copper above 100 µg.
Young children rarely present with Kayser-Fleischer ring. A Kayser-Fleischer ring was detected with a slit lamp examination in 41% of children older than 6 years of age. In contrast, only one child out of 26 children under 6 years old showed a Kayser-Fleischer ring (Table 8).
Table 8.
Presence of a Kayser-Fleischer Ring by Slit Lamp Examinations in 114 Korean Children
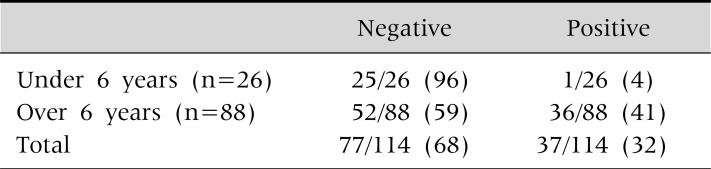
Values are presented as number (%). At Seoul National University Children's Hospital.
In order to increase the diagnostic accuracy of biochemical and clinical investigations, Ferenci diagnostic scoring system (Table 9) was developed at the Eighth International Meeting on WD, in 2001 [36].
Table 9.
Scoring System Developed at the Eighth International Meeting on Wilson Disease, Leipzig, 2003 (Ferenci et al. [36])

*If no quantitative liver copper analysis available. MRI: magnetic resonance imaging, ULN: upper limit of normal.
The Ferenci scoring system has limitations for the diagnosis of WD, particularly in very young children. We evaluated the validity of the Ferenci scoring system using biochemical and clinical parameters, without applying the mutation analysis results, in 114 children with WD. The Ferenci scoring system has excellent diagnostic value and can detect WD in about 95% of the children older than 6 years of age. However, the diagnosis of WD is absent in about 19% of young children under 6 years of age when the scoring system is applied to this very young age group (Table 10).
Table 10.
Age Difference in the Validity of Ferenci Score in 114 Children with WD
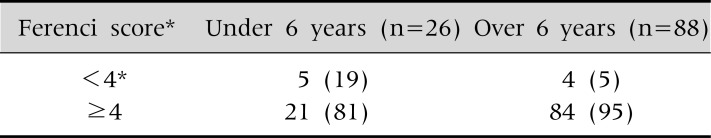
Values are presented as number (%). At Seoul National University Children's Hospital. *Diagnosis of Wilson disease is established.
GENETIC TESTING AND AN ALGORITHM FOR THE DIAGNOSIS OF WD IN INFANTS AND PRESCHOOL CHILDREN
Although majority of patients diagnosed with WD are in the age range between 5 and 35 years old, very young children with WD are being reported more frequently in recent literature. Severe impairments in ATP7B including large deletion, insertion, nonsense mutations and splice site mutations can lead to an earlier onset of WD.
Thomas et al. [14] reported severe liver disease in a 3 year old boy with 54 amino acids missing in ATP7B immediately after the first discovery of WD gene. Early childhood cases of WD were reported in a 13 month old child with hypertransaminasemia, in a 2 year old child with chronic hepatitis, in a 3 year old child with cirrhosis and in a 5 year old child with acute liver failure [37-40].
Caprai et al. [41] reported 3 children who failed to be diagnosed with WD solely on the basis of biochemical diagnostic criteria. These children showed near normal serum ceruloplasmin levels. The 24-hour urinary copper excretions were not high, and hepatic copper contents were below 50 µg/g dry weight, Ferenci scores of these patients (Table 11), were low and the diagnosis of WD was very unlikely. Nevertheless, all 3 children showed two mutations in the ATP7B gene. Only the molecular genetic testing was able to confirm WD in these patients.
Table 11.
Copper Metabolism Parameters of the 3 Patients Described in Details

In our unit, we reported the youngest case of WD in a 9 month-old male infant who visited hospitals because of persistently elevated aminotransferases [42]. His ceruloplasmin level was very low (<7 mg/dL). The 24 hour urine copper concentrations were not high, and Kayser-Fleischer ring was absent. The diagnosis of WD was established through genetic testing in this patient, who was confirmed to be a compound heterozygote of G1186S and 4006delA (Table 12).
Table 12.
Clinical and Biochemical Features of a 9 Month Old Korean Infant with Wilson Disease

AST: aspartate aminotransferase, ALT: alanine aminotransferase. At Seoul National University Children's Hospital.
Molecular genetic testing is playing an increasingly important role in the diagnosis of WD in uncertain cases and family screening. About 20% of heterozygote carriers have low serum ceruloplasmin levels, borderline normal urinary copper concentrations, and moderate elevations of hepatic copper (50-250 µg/g dry weight). Molecular genetic testing is a reliable tool for distinguishing securely healthy heterozygote carriers from affected presymptomatic patients. It could avoid inappropriate lifelong therapy in heterozygotes.
For the screening of siblings, molecular genetic testing either by mutation analysis or haplotype analysis is the only definite solution [43-45]. When the 2 mutations are identified in an index patient, mutation analysis is most efficient in family screening strategy. The cost necessary for checking the presence of already identified 2 mutations is less expensive; about 10% of the cost for a complete DNA sequencing in the index patient. If the mutations of the index patient are unknown, haplotype analysis with closely linked microsatellite markers is useful for family screening.
Because a large number of mutations have been recognized in the ATP7B gene, mutation screening was considered impractical. However, in recent years, direct genetic diagnosis including complete DNA sequencing has become much easier and more rapid than in the past. Therefore, for uncertain cases, genetic diagnosis is now becoming the most specific and sensitive diagnostic test, and is being included among the initial diagnostic workup tests, although the cost of genetic testing is still high to be used extensively in all patients.
Currently reported mutation detection rate is very high.
In 114 children with unexplained hepatitis at our children's hospital, complete gene sequencing of the coding regions of all the exons and flanking regions of the intron was confirmed. In 81% of children, two mutations were found and a definite diagnosis of WD was confirmed. In 17%, one mutation was found and no second mutation was detected after a complete DNA sequencing. No mutations were found in only 2 children, who had a definite clinical diagnosis of WD after observations of their response to therapy and long-term clinical follow-ups (Fig. 6).
Fig. 6.

Genetic diagnosis and copper metabolism associated 3 tests in 114 children with Wilson disease (WD) at Seoul National University Children's Hospital (Low ceruloplasmin+ high urine copper+Kayser-Fleischer ring).
If 2 mutations are found, and one of them was not previously reported, it should be determined whether this mutation is a WD causing novel mutation or a normal variant in the patient's population. A couple of methods are available to determine the functional activity and to predict consequences of the protein structural changes of newly identified variants [17,46,47].
On-line prediction programs such as SIFT (Sorting Intolerant from Tolerant), PolyPhen (Polymorphism Phenotyping), and Align-GVGD (Align-Grantham Variation Grantham Deviation) are available for the evaluation of newly identified unclassified variants. Conservation, structure, biochemical properties, and models of the 3-dimensional protein structure of variants are evaluated by the computational predictive algorithms and compared with original residue at the affected site.
The functional activity of newly identified ATP7B variants can be evaluated in a yeast complementation assay utilizing ATP7B homolog Ccc2p in a strain of Saccharomyces cerevisiae [46,47]. This functional assay helps to distinguish normal from disease causing mutations. With immunofluorescence microscopic examinations, failure of the copper dependent trafficking pathway of the ATP7B variants can be also detected.
The functional assays in the yeast mutant Ccc2p model confirmed 508 WD causing variants including 267 missense mutations in 644 variants which were identified by diagnostic sequencing of ATP7B (http://www.wilsondisease.med.ualberta.ca/database.asp).
An allele frequency study in the normal population for an unknown variant is useful. At least 100 alleles with 50 persons should be included.
When one mutation is detected in a child and the mutation has been previously reported, this child is either a heterozygote carrier coincidentally having unknown hepatitis rather than WD, or a WD patient in whom the second mutation is not identified. In this group of the children with one mutation, it was not difficult to confirm the diagnosis of WD; two of the 3 tests including ceruloplasmin measurement, urinary copper assay, and Kayser-Fleischer ring examination were positive in 60% of this group of children and all 3 tests were positive in 30% of this group of children (Fig. 6). In 10% of children who were found to have one known mutation, one test was positive and other 2 tests were negative.
A long term clinical follow up and evaluation of the treatment response usually confirms the diagnosis of WD in such cases. Therefore, in almost all of the children with unexplained hepatitis, a definite diagnosis of WD could be established with the direct DNA sequencing of ATP7B performed in the initial investigation. Children who have no mutation are extremely rare, and the diagnosis of WD in these patients could be established with long term follow-ups and treatment responses evaluations. Even in patients who have one known mutation and one positive laboratory test, WD is highly likely. Liver biopsy for hepatic copper and mitochondrial structural abnormalities or brain MRI may be recommended to aid in establishing the diagnosis of WD in this group of patients. Long-term evaluation of the response to treatment also can be very helpful in some patients who have no mutations.
Further investigations including Multiplex Ligation-dependent Probe Amplification (MLPA) can be attempted to search for large gene defects such as whole exon deletions which are not easily detected by direct DNA sequencing method. MLPA assay is a recently developed highly sensitive new method for detecting copy number variations in genomic DNA sequences. MLPA can detect large deletions and duplications on whole exon levels, which are not found by direct sequencing. The MLPA assay in combination with a complete DNA sequencing is found to increase the diagnostic accuracy in some pediatric genetic diseases such as Peutz-Jeghers syndrome [48] or Alagille syndrome, which are known to have large gene defects. However, large gene defects in ATP7B are extremely rare in patients with WD, therefore MLPA assay may not be efficient.
The diagnostic algorism of WD in our children's hospital for young children is shown in Fig. 7.
Fig. 7.

Diagnostic approach for Wilson disease (WD) in infants and preschool children - paradigm shift in the era of molecular genetics (at Seoul National University Children's Hospital).
For infants and preschool children with unexplained hepatitis, we test ceruloplasmin levels, 24-hour urinary copper concentrations and Kayser-Fleischer ring with slit lamp examinations. If at least one of these less invasive tests is positive, routine genetic testing is recommended to all children as one of the initial investigational examinations. When ATP7B mutations are detected, we extend mutation analyses to all family members for genetic screening.
If 2 mutations are found, we treat the patient with a definite diagnosis of WD. If one mutation is found and more than 2 laboratory tests are positive, we regard the patient as having a tentative diagnosis of WD and we evaluate the treatment response during follow-ups. If one mutation is found and only one test is positive, we recommend further studies such as liver biopsy for hepatic copper, or brain MRI. And we evaluate the treatment response.
When a detected mutation is not previously reported, allele frequency study in our population is performed to check whether it is a normal variant.
If genetic testing is not performed, many of the very young children may not be diagnosed as having WD because these patients usually have no Kayser-Fleischer rings, equivocal levels of biochemical tests, and low Ferenci scores.
Sequencing of ATP7B could be considered first as an initial test, particularly in young children, for a definitive diagnosis of WD and for secure family screening.
References
- 1.Seo JK, Kim YS, Hahn CJ, Baik SK. A nationwide survey for prevalence and clinical characteristics of Wilson disease in Korea. Korean J Hepatol. 2004;10(Suppl):5–15. [Google Scholar]
- 2.Seo JK. Wilson disease: an update. Korean J Hepatol. 2006;12:333–363. [PubMed] [Google Scholar]
- 3.Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain. 1912;34:295–507. doi: 10.1093/brain/awp193. [DOI] [PubMed] [Google Scholar]
- 4.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 5.Chelly J, Monaco AP. Cloning the Wilson disease gene. Nat Genet. 1993;5:317–318. doi: 10.1038/ng1293-317. [DOI] [PubMed] [Google Scholar]
- 6.Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, et al. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5:338–343. doi: 10.1038/ng1293-338. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi Y, Heiny ME, Gitlin JD. Isolation and characterization of a human liver cDNA as a candidate gene for Wilson disease. Biochem Biophys Res Commun. 1993;197:271–277. doi: 10.1006/bbrc.1993.2471. [DOI] [PubMed] [Google Scholar]
- 9.Petrukhin K, Lutsenko S, Chernov I, Ross BM, Kaplan JH, Gilliam TC. Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum Mol Genet. 1994;3:1647–1656. doi: 10.1093/hmg/3.9.1647. [DOI] [PubMed] [Google Scholar]
- 10.Cox DW, Moore SD. Copper transporting P-type ATPases and human disease. J Bioenerg Biomembr. 2002;34:333–338. doi: 10.1023/a:1021293818125. [DOI] [PubMed] [Google Scholar]
- 11.Kim ST, Park YH, Lee KU, Kum SJ, Youn YK, Seo JK, et al. An experience of first liver transplantation in Korea. J Korean Soc Transplant. 1988;2:27. [Google Scholar]
- 12.Moon JS, Ko JS, Seo JK. Long-term clinical follow-up of Korean children with Wilson disease; twenty years' experience. J Korean Pediatr Soc. 2001;44:127–138. [Google Scholar]
- 13.Seo JK, Moon HR. Hepatitis in childhood as a manifestation of treatable Wilson's disease. Korean J Gastroenterol. 1983;15:55–64. [Google Scholar]
- 14.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- 15.Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev. 2007;87:1011–1046. doi: 10.1152/physrev.00004.2006. [DOI] [PubMed] [Google Scholar]
- 16.Schaefer M, Hopkins RG, Failla ML, Gitlin JD. Hepatocyte-specific localization and copper-dependent trafficking of the Wilson's disease protein in the liver. Am J Physiol. 1999;276:G639–G646. doi: 10.1152/ajpgi.1999.276.3.G639. [DOI] [PubMed] [Google Scholar]
- 17.Forbes JR, Cox DW. Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum Mol Genet. 2000;9:1927–1935. doi: 10.1093/hmg/9.13.1927. [DOI] [PubMed] [Google Scholar]
- 18.de Bie P, Muller P, Wijmenga C, Klomp LW. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673–688. doi: 10.1136/jmg.2007.052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambrosini L, Mercer JF. Defective copper-induced trafficking and localization of the Menkes protein in patients with mild and copper-treated classical Menkes disease. Hum Mol Genet. 1999;8:1547–1555. doi: 10.1093/hmg/8.8.1547. [DOI] [PubMed] [Google Scholar]
- 20.Cater MA, La Fontaine S, Shield K, Deal Y, Mercer JF. ATP7B mediates vesicular sequestration of copper: insight into biliary copper excretion. Gastroenterology. 2006;130:493–506. doi: 10.1053/j.gastro.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 21.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 22.Forbes JR, Hsi G, Cox DW. Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease. J Biol Chem. 1999;274:12408–12413. doi: 10.1074/jbc.274.18.12408. [DOI] [PubMed] [Google Scholar]
- 23.DiDonato M, Hsu HF, Narindrasorasak S, Que L, Jr, Sarkar B. Copper-induced conformational changes in the N-terminal domain of the Wilson disease copper-transporting ATPase. Biochemistry. 2000;39:1890–1896. doi: 10.1021/bi992222j. [DOI] [PubMed] [Google Scholar]
- 24.Seo JK. Molecular genetic testing and diagnosis of Wilson disease. Korean J Pediatr Gastroenterol Nutr. 2008;11(Suppl):72S–82S. doi: 10.5223/pghn.2012.15.4.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seo JK, Kim JW. Mutation analysis of Wilson disease Gene: Arg778Leu mutation in Korean Children. Korean J Pediatr Gastroenterol Nutr. 1999;2:164–168. [Google Scholar]
- 26.Yoo HW. Identification of novel mutations and the three most common mutations in the human ATP7B gene of Korean patients with Wilson disease. Genet Med. 2002;4(6 Suppl):43S–48S. doi: 10.1097/00125817-200211001-00009. [DOI] [PubMed] [Google Scholar]
- 27.Bae SH, Kim JW, Seo JK. Haplotype analysis and possible founder effect at the R778L mutation of the ATP7B gene in Korean patients with Wilson disease. Korean J Hepatol. 2009;15:309–319. doi: 10.3350/kjhep.2009.15.3.309. [DOI] [PubMed] [Google Scholar]
- 28.Tsai CH, Tsai FJ, Wu JY, Chang JG, Lee CC, Lin SP, et al. Mutation analysis of Wilson disease in Taiwan and description of six new mutations. Hum Mutat. 1998;12:370–376. doi: 10.1002/(SICI)1098-1004(1998)12:6<370::AID-HUMU2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 29.Wan L, Tsai CH, Tsai Y, Hsu CM, Lee CC, Tsai FJ. Mutation analysis of Taiwanese Wilson disease patients. Biochem Biophys Res Commun. 2006;345:734–738. doi: 10.1016/j.bbrc.2006.04.136. [DOI] [PubMed] [Google Scholar]
- 30.Xu Y, Fan Y, Yu L, Jiang Y, Yang R, Han Y, et al. Identification of a mutation hotspot in exon 8 of Wilson's disease gene by cycle sequencing. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 1998;15:284–287. [PubMed] [Google Scholar]
- 31.Nanji MS, Nguyen VT, Kawasoe JH, Inui K, Endo F, Nakajima T, et al. Haplotype and mutation analysis in Japanese patients with Wilson disease. Am J Hum Genet. 1997;60:1423–1429. doi: 10.1086/515459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czlonkowska A, Rodo M, Gajda J, Ploos van Amstel HK, Juyn J, Houwen RH. Very high frequency of the His1069Gln mutation in Polish Wilson disease patients. J Neurol. 1997;244:591–592. doi: 10.1007/s004150050149. [DOI] [PubMed] [Google Scholar]
- 33.Sternlieb I. Fraternal concordance of types of abnormal hepatocellular mitochondria in Wilson's disease. Hepatology. 1992;16:728–732. doi: 10.1002/hep.1840160319. [DOI] [PubMed] [Google Scholar]
- 34.Cheon JE, Kim IO, Seo JK, Ko JS, Lee JM, Shin CI, et al. Clinical application of liver MR imaging in Wilson's disease. Korean J Radiol. 2010;11:665–672. doi: 10.3348/kjr.2010.11.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW, et al. MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. AJNR Am J Neuroradiol. 2006;27:1373–1378. [PMC free article] [PubMed] [Google Scholar]
- 36.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 37.Gow PJ, Smallwood RA, Angus PW, Smith AL, Wall AJ, Sewell RB. Diagnosis of Wilson's disease: an experience over three decades. Gut. 2000;46:415–419. doi: 10.1136/gut.46.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iorio R, D'Ambrosi M, Mazzarella G, Varrella F, Vecchione R, Vegnente A. Early occurrence of hypertransaminasemia in a 13-month-old child with Wilson disease. J Pediatr Gastroenterol Nutr. 2003;36:637–638. doi: 10.1097/00005176-200305000-00009. [DOI] [PubMed] [Google Scholar]
- 39.Beyersdorff A, Findeisen A. Morbus Wilson: case report of a two-year-old child as first manifestation. Scand J Gastroenterol. 2006;41:496–497. doi: 10.1080/00365520500389453. [DOI] [PubMed] [Google Scholar]
- 40.Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson's disease in preschool-aged children. J Pediatr. 2000;137:719–722. doi: 10.1067/mpd.2000.108569. [DOI] [PubMed] [Google Scholar]
- 41.Caprai S, Loudianos G, Massei F, Gori L, Lovicu M, Maggiore G. Direct diagnosis of Wilson disease by molecular genetics. J Pediatr. 2006;148:138–140. doi: 10.1016/j.jpeds.2005.07.036. [DOI] [PubMed] [Google Scholar]
- 42.Kim JH, Kim JH, Seo KS, Ko JS, Chang JY, Yang HR, et al. Genetically confirmed Wilson disease in a 9 month-old boy with elevation of aminotransferase. World J Gastroenterol. 2012 doi: 10.4254/wjh.v5.i3.156. [being in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart EA, White A, Tomfohrde J, Osborne-Lawrence S, Prestridge L, Bonne-Tamir B, et al. Polymorphic microsatellites and Wilson disease (WD) Am J Hum Genet. 1993;53:864–873. [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JW, Kim SI, Seo JK. A genetic linkage study of Wilson disease in Korean families. J Korean Pediatr Soc. 1993;36:1596–1612. [Google Scholar]
- 45.Thomas GR, Roberts EA, Walshe JM, Cox DW. Haplotypes and mutations in Wilson disease. Am J Hum Genet. 1995;56:1315–1319. [PMC free article] [PubMed] [Google Scholar]
- 46.Luoma LM, Deeb TM, Macintyre G, Cox DW. Functional analysis of mutations in the ATP loop of the Wilson disease copper transporter, ATP7B. Hum Mutat. 2010;31:569–577. doi: 10.1002/humu.21228. [DOI] [PubMed] [Google Scholar]
- 47.Forbes JR, Cox DW. Functional characterization of missense mutations in ATP7B: Wilson disease mutation or normal variant? Am J Hum Genet. 1998;63:1663–1674. doi: 10.1086/302163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang HR, Ko JS, Seo JK. Germlinemutation analysis of STK11 gene using direct sequencing and multiplex ligation-dependent probe amplification assay in Korean children with Peutz-Jeghers syndrome. Dig Dis Sci. 2010;55:3458–3465. doi: 10.1007/s10620-010-1194-5. [DOI] [PubMed] [Google Scholar]