

Abstract
The incidence of diseases increases rapidly with age, accompanied by progressive deteriorations of physiological functions in organisms. Aging-associated diseases are sporadic but mostly inevitable complications arising from senescence. Senescence is often considered the antithesis of early development, but yet there may be factors and mechanisms in common between these two phenomena over the dynamic process of aging. The association between early development and late-onset disease with advancing age is thought to come from a consequence of developmental plasticity, the phenomenon by which one genotype can give rise to a range of physiologically and/or morphologically adaptive states in response to different environmental or genetic perturbations. On the one hand, we hypothesized that the future aging process can be predictive based on adaptivity during the early developmental period. Modulating the thresholds of adaptive plasticity by chemical genetic approaches, we have been investigating whether any relationship exists between the regulatory mechanisms that function in early development and in senescence using the zebrafish (Danio rerio), a small freshwater fish and a useful model animal for genetic studies. We have successfully conducted experiments to isolate zebrafish mutants expressing apparently altered senescence phenotypes during embryogenesis (“embryonic senescence”), subsequently showing shortened lifespan in adulthoods. We anticipate that previously uncharacterized developmental genes may mediate the aging process and play a pivotal role in senescence. On the other hand, unexpected senescence-related genes might also be involved in the early developmental process and regulation. The ease of manipulation using the zebrafish system allows us to conduct an exhaustive exploration of novel genes and small molecular compounds that can be linked to the senescence phenotype, and thereby facilitates searching for the evolutionary and developmental origins of aging in vertebrates.
Keywords: Aging, Evolution, Development, Disease, Rejuvenation, Zebrafish
1. Introduction
The mechanisms of aging are currently the focus of extensive investigations throughout the world. During the aging process, multiple forms of tissue- and organ-specific damage and pathophysiological change accumulate, and accordingly a number of chronic diseases appear with advancing age. Scientists are now hoping for discovery of the principles governing the ubiquitous process of aging, to ultimately find novel ways to attenuate or delay aging in humans as well as to develop interventions for age-associated diseases. However, understanding the molecular mechanisms of aging in vertebrates is still a major challenge of modern biology and biomedical science. There are various animal models to be considered and utilized in aging studies, and determining the optimal model system(s) remains one of the most important issues. Given certain limitations in each and all the existing models of human aging, calling for an integrative approach that uses diverse species in the hope that each would provide a piece of the puzzle and, together, would help to identify critical elements common to aging in all organisms. One of the essential limitations is availability of genetic and genomic information detailed on relevant species. This provides the rationale for current biomedical investigations into the mechanisms of aging are conducted concurrently in genetically robust species like worms, fruit flies, and mice. In fact, small and prolific invertebrates, such as Caenorhabditis elegans (C. elegans) and Drosophila melanogaster, can provide the strong basis for unbiased genetic screens that identify and determine the precise functions of novel genes that regulate aging and lifespan throughout more levels of evolution. This approach has already led to the successful identification of key genes and pathways evolutionarily conserved in, and associated with, the aging process. Although evolutionally conserved mechanisms of aging could play roles in various organisms, manifestations of aging can differ in different species. For instance, vertebrates from fish through humans often die of cancer or stroke, but invertebrates such as worms and insects unlikely die of either. Thus far, only mice have served as a widely used genetic model system to study the aging process in vertebrates, providing important insights into mammalian aging as an indispensable animal model [1]. However, studying only a single genetic model may be limiting due to diversified complexity of vertebrate aging across species. Some mechanisms of aging could be evolutionarily conserved and publicly common in vertebrates, but there are also more lineage-specific or private mechanisms of aging from a comparatively biological point of view. Even the common notion that caloric (or dietary) restriction extends lifespan has not proven to be universally true, as shown in several cases including humans, some fish species, and honeybees (Apis mellifera) [2-8]. Therefore, a cross-species comparative biology of aging prospective is necessary to understand and obtain a more complete picture for fundamental causes of aging in speciation and biodiversity.
Development is obviously under the precise control of genetic mechanisms programmed in genome having adaptive plasticity through epigenome. However, such precise genetic programming may subsequently be disturbed later on during the aging process that undergoes many deteriorative challenges against regenerative/repair responses and restorations where the reactivation of certain phases of the early developmental program/process contributes and/or are required. Once we consider aging as an extension of the biological process of development, the power of zebrafish development and genetics becomes a monumental and instructive wealth of information. Zebrafish can be extensively utilized in searching for evolutionary and developmental origins of aging common in vertebrates. In fact, some of the genes we identified by an apparent senescence phenotype during embryogenesis (“embryonic senescence”) had already been associated with cellular senescence and chronological aging in other organisms, while many have still waited to be linked to the conventional aging process. Complete loss-of-function of developmentally essential genes induce embryonic (or larval) lethality, whereas it seems like their partial loss-of-function (i.e., decrease-of-function by heterozygote or hypomorphic mutations) still remains sufficient to go through the early developmental process because of its adaptive plasticity or rather heterozygote advantage. On the other hand, in some cases such partial loss-of-function of genes compromise normal homeostasis due to haploinsufficiency later in adult life having a number of environmental or epigenetic challenges which less plasticity can no longer adjust to compensate. By contrast, any heterozygote-advantageous genes might gain a certain benefit(s) (much more fitness) by such partial loss-of-function later in life.
Physiological senescence may evolutionarily arise from both genetic and epigenetic drifts as well as from losing adaptive developmental plasticity in face of stress signals from the external environment that interacts with functions of multiple genes rather than effects of only a single gene mutation or defect. We wish identify a number of such critical genes promptly in a comprehensive manner by utilizing the zebrafish model system. Presumably, certain critical portions of aging phenomena may become readily predictable based on advanced genetic and genomic approaches in zebrafish, and a systems biology approach will allow us to simulate the aging process by modeling potential stochastic values including epistatic and epigenetic effects.
2. Zebrafish as a new vertebrate model of aging
Zebrafish has emerged as a highly promising model for studies of vertebrate aging [9-14]. Combining their optical transparency at embryonic and larval stages [in some cases, even in adults throughout their lives; [15]], the small size and high fecundity of zebrafish has made them a favorite vertebrate of developmental biologists. This has resulted in detailed characterization of the zebrafish genome, the development of multiple mutant and transgenic phenotypes, and the emergence of various molecular genetics techniques (e.g., most recent targeted-gene knockout using nucleases as well as authentic knockdown using morpholino antisense oligos) allowing for investigation of the intrinsic mechanisms underlying normal physiological and pathological events in this organism [16-20]. Consequently, of the vertebrates studied to-date, zebrafish offers the most cost-effective opportunity for high-throughput screens using robotic systems for evaluating mutants, identifying genes, and testing chemicals in vertebrate organisms [21-23].
Zebrafish live for approximately 3 years on average (over 5 years maximally) in laboratory conditions and show gradual senescence that is similar in many aspects to humans [9, 11]. With age, zebrafish often display spinal curvature that is possibly related to muscle abnormalities [11]. In aging zebrafish, we detected senescence-associated β-galactosidase (SA-β-gal) activity in skin (Figure 1) [9, 14, 24]. As zebrafish age, they show increased accrual of lipofuscin (“aging pigment”) in the liver and accumulation of oxidized proteins in muscle, [9, 24], similar to that reported in mice and humans [25-27]. Aged zebrafish also often develop lipofuscin accumulation and drusen-like lesions in retinal pigment epithelium (RPE), which is similar to that seen in age-related macular degeneration (AMD) in humans. Moreover, almost all zebrafish develop cataracts by 4 years of age (unpublished observation), when the majority of the oldest fish show retinal atrophy [24].
Figure 1. Increased senescence-associated β-gal (SA-β-gal) activity in the trunk skin of the adult zebrafish with age [24].
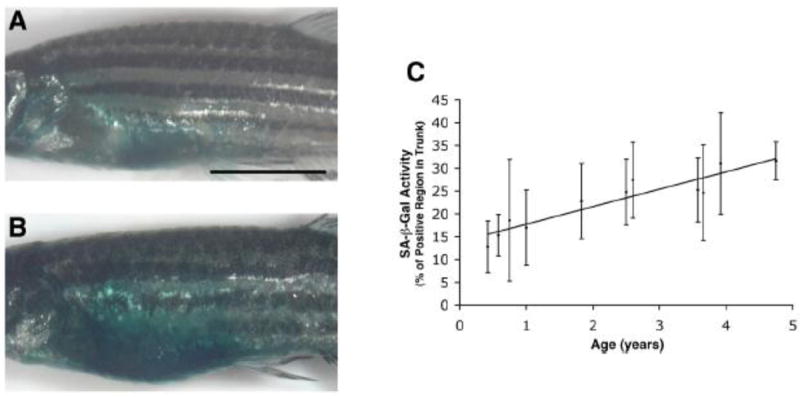
(A, B) Lateral imaging of 5-month (0.42 y) old (A) and 57-month (4.75 y) old (B) whole adult zebrafish stained for SA-β-gal activity. (Scale bar: 0.5 cm.). (C) Quantitative analysis of trunk SA-β-gal staining in fish of various ages showing a near-linear increase in SA-β-gal activity with age. In (C), the numbers of fish at each time point (years) were: 0.42 y (n = 16); 0.58 y (n = 14); 0.75 y (n = 13); 1 y (n = 14); 1.83 y (n = 15); 2.5 y (n = 6); 2.6 y (n = 8); 3.58 y (n = 14); 3.66 y (n = 12); 3.92 y (n = 22); 4.75 y (n = 5). R2 = 0.3027. y; year(s) of age.
Our studies have further revealed several age-related degenerative changes and increases in a variety of pathological lesions in aging zebrafish, as well as an age-dependent decline in both their reproductive and regenerative capacity [9, 14, 24]. In adult zebrafish, fin regeneration is a highly orchestrated processes involving wound healing, establishment of the wound epithelium, recruitment of the blastema from mesenchymal cells underlying the wound epithelium, and differentiation and outgrowth of the regenerated cells [28]. Importantly, such regenerative ability declines with age, leading to impaired morphology and distorted fin shape [14], which may become a valuable hallmark of aging in this animal model.
There have also been observed age-associated alterations in circadian rhythmicity, sleep and cognitive function in zebrafish [12-14, 29]. Taken together, these observations suggest that zebrafish are the great model to investigate the mechanisms underlying gradual senescence in vertebrates, and could serve as a promising model of human aging [9, 24].
3. High-throughput mutant screening for senescence-aging in vertebrates
In addition to studying the function of specific genes that have already been linked to the aging process, it is critically important to conduct unbiased mutant screens for aging phenotypes in vertebrates, in search of additional genes that affect late-onset diseases or age-related dysfunctions. The mechanisms connecting the organismal aging process with multiple chronic age-associated human diseases (‘geriatric diseases’) remain a complex enigma, and scientists sometimes face conflicting arguments about ‘aging phenotypes’ versus ‘diseases’. Both intrinsic gene-gene interactions (‘epistasis’) and environmental factors, as well as gene-environment interactions [30, 31], affect the aging process and disease progression. It is however poorly understood which of the complex genetic elements evolutionarily affect lifespan, how they are correlated with disease susceptibility, and how environmental and epigenetic modulations are responsible for the developmental origins of aging and disease in vertebrates. There may be remarkably complex interactions between unexpected, as of yet unidentified and/or even unrelated genes, requiring unbiased comprehensive methods of detection and systems biology approaches to their identifications. In most vertebrates, unbiased screens for aging mutants are technically difficult and have therefore not yet been done, even in mice. On the other hand, specific phenotype-based and unbiased forward genetic screens in early development for multiple biological and biomedical purposes have already been successfully carried out in zebrafish, and can be readily adapted to current aging research, once any adequate senescence biomarker is identified and can be applied to this model system, particularly in embryos and/or larvae during the developmental process.
The features that distinguish the zebrafish as a high-throughput amenable organism for high-content or large-scale screens include their small body size, transparent embryogenesis, and high reproductive rate, allowing the simultaneous evaluation of many siblings throughout their lives under controlled conditions, with and without certain environmental and pharmacological challenges. Moreover, the existence of various zebrafish mutants and traceable transgenic lines showing particular phenotypes already, including retroviral insertional mutant collections [32], provides a valuable rich resource for the identification of both physiological and pathological aging characteristics.
4. Utility of a biomarker of “embryonic senescence” to predict the chronological aging process
Humans develop and age very gradually, when compared with most of the animals used as biomedical research model systems (some notable exceptions include a certain species of rockfish which can live for over 200 years). This may be a critical limitation of the current study and approach, since some of the aging processes common to humans may, indeed, require a prolonged lifespan to become apparent, and simply short-span models might not be adequate to elucidate the essential determining factors of the gradual nature and mechanisms of aging. On the other hand, the lengthy lifespan of experimental animals represents a practical obstacle for undertaking either cross-sectional or longitudinal aging research, particularly in comprehensive genetic studies and high-throughput mutant screens.
One of the most promising approaches to confronting this dilemma is to assess rather young animals (even embryos and larvae) for reliable and easily measurable biomarkers of senescence that “predict” an actual (both chronological and biological) aging phenotype(s) appeared later in life. In genotypes susceptible to accelerated aging (caused by ‘Heterozygote Disadvantage’) or even delayed aging (caused by ‘Heterozygote Advantage’), such biomarkers might manifest in young animals with certain gene mutations (or alleles), either spontaneously or following stress (Figure 2A). Even if such mutations are lethal in homozygous zebrafish, most heterozygous animals develop normally and survive into adulthood. Under normal conditions, the heterozygous embryos, larvae, or young (adult) fish might not immediately reveal significant vulnerable changes in biomarkers of aging, due to the presence of ‘plasticity’ like partial redundant compensatory mechanisms and/or adaptive responses and upregulations (e.g., hyper activation) mediated by the remaining intact functional allele. However as confirmed by our recent study [24], several homozygous mutant (complete loss-of-function) conditions as well as exposures of the corresponding heterozygote carriers to specific stress challenges can help to amplify the response in animals showing dominant premature senescence (“embryonic senescence”) phenotypes even in early development (in case of senescence “enhancer mutants” of Type I genes by haploinsufficiency; Figure 2B). This applies to cases where gene mutations lead to an augmented response to stress (in Type I genes) or conversely a resistance to it (in Type II genes). Thus, a mutant phenotype in homozygosity as well as a specific stress challenge (instead of an actual aging challenge) in heterozygosity during early development could mimic the actual aging process and help to predict the genotypes that might be susceptible or resistant to homeostatic deteriorations that occur during aging. By using a chemical sensitizer (e.g., hydrogen peroxide), we could indeed identify mutants with an altered response to oxidative stress; that is we expected the chemical sensitizer to induce “heterozygote advantage or disadvantage” impacts in many of the potential target genes. We then denoted this chemical genetic methodology ‘CASH’ (Chemically Assisted Screening in Heterozygotes) [24].
Figure 2. Gene mutations resulting in altered (half) dosages of particular gene functions and leading to altered senescence outcomes through the aging process.
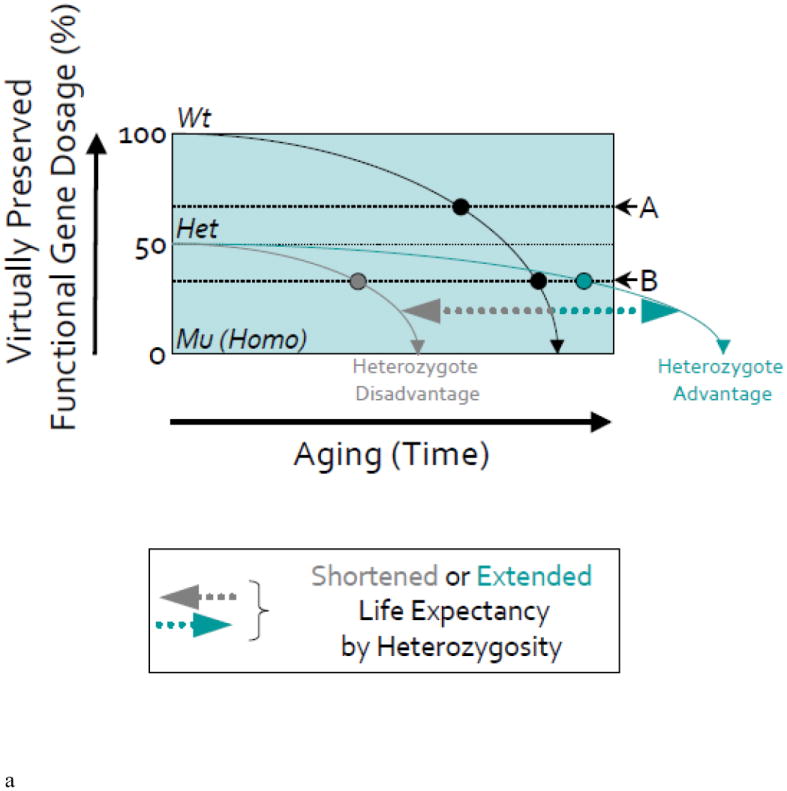
(A) In the schematic, the slope of the solid black line illustrates the age-dependent regression of accuracy in gene function in a wild-type (Wt) organism due to in part to “programmed” senescence, accumulated structural cellular damage following intrinsic and extrinsic (environmental) stochastic events, desynchronization of molecular mechanisms, and so on. The slope of the gray or green line shows the age-dependent decline of accuracy in gene function in a heterozygous (Het) mutant organism identified in our screening i.e., the mutants with a presumed 50% loss of original gene function at birth leading to a shorter (‘heterozygote disadvantage’) or longer (‘heterozygote advantage’) lifespan (on X-axis). Dotted horizontal lines depict the level of gene function at which certain ‘Aging Biomarkers’ can be documented, i.e., aging biomarker thresholds; A and B.
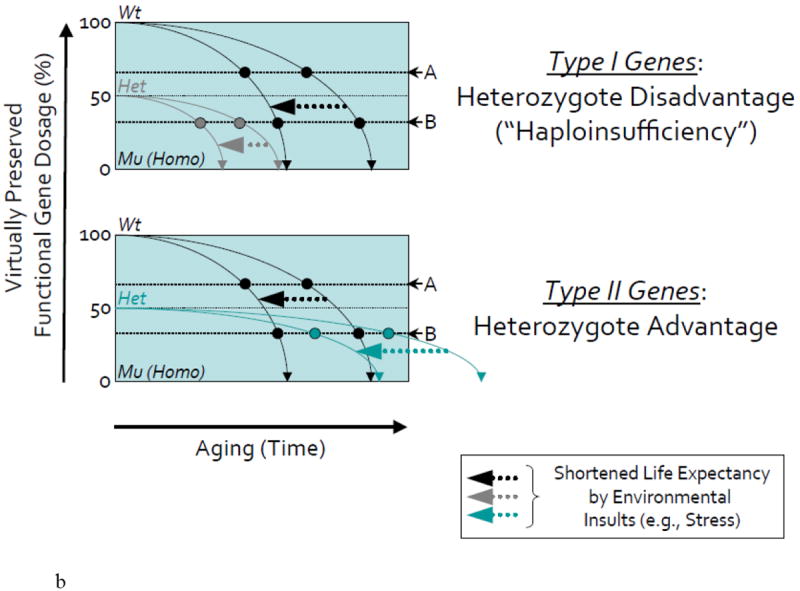
(B) In upper schematic (in mutants of Type I Genes with heterozygote disadvantage or haploinsufficiency), shifted lines to the left by arrows illustrate the more rapid decline of accuracy in the function of the same gene following early exposure to stress (e.g., oxidative stress or gamma-radiation that we used in our studies) in wild-type or heterozygous mutant organisms.
According to this model, in wild-type organisms, Aging Biomarker-A manifests around middle age (Mid-Age). In the heterozygous mutant, this Aging Biomarker-A manifests spontaneously even in early development without stress insult (on Y-axis), since its threshold lies above the 50% level of gene function, thus predicting an accelerated aging phenotype. In contrast, Aging Biomarker-B is characteristic of old age in a wild-type organism and manifests in heterozygous mutants at a relatively earlier period of late adult life. Thus, neither organism would show this biomarker spontaneously during early development.
However, in both organisms, exposure to stress during development or later in life can change the dynamics of gene function with age, presumably through additional structural and functional damage to cells. As a result, after stress, both Aging Biomarkers manifest earlier and the Aging Biomarker-B can now be detected earlier in life of the heterozygous mutant but not the wild-type organism.
In homozygous (Mu; Homo) mutant organism, either Aging Biomarker-A or -B can be detected during early development (i.e., increased SA-β-gal activity) and the mutations are early lethal (on X-axis). Thus, the search for gene mutations that lead to accelerated aging phenotypes can be conducted during early development of homozygous mutants as well as heterozygous mutants with or without the use of environmental stress factors. Note that while this highly-simplified schematic illustrates changes that helped us to predict accelerated aging in zebrafish mutants with altered spns1/nrs and terf2 genes, it does not reflect the more complex relationships between the dynamics of gene functions and age that would be predicted by the “antagonistic pleiotropy” theory of aging [30, 91]. Moreover, the concerted effect of numerous genes functioning in parallel throughout life would predictably cause the overall aging process to exhibit non-linear dynamics, with stochastic environmental factors providing further modifications of age-dependent processes under real-life conditions (e.g., epigenetic drifts).
In the lower schematic (in mutants of Type II Genes with heterozygote advantage’), for the heterozygous mutant, the Aging Biomarker-A may still manifest spontaneously in early development without stress insult (on Y-axis), since its threshold lies above the 50% level of gene function, thus predicting an delayed aging phenotype. In contrast, Aging Biomarker-B is characteristic of old age in the wild-type animals and manifests in heterozygous mutant during relatively late adulthood beyond the age of wild type. Thus, such organisms would not show this biomarker spontaneously during early development.
Exposure to stress during development or even later in life can maintain the robust dynamics of gene function with age, probably due to resistance to structural and functional damage to cells. As a result, even after stress, although Aging Biomarker-A may still manifest relatively earlier but the Aging Biomarker-B can only be detected during Mid-Age of the heterozygous mutant beyond the wild-type age. In the homozygous mutants, certain however Aging Biomarker-A or -B could be “negatively” detected during early development (i.e., decreased SA-β-gal activity) and these mutations are also still early lethal (on X-axis).
In the case of enhancer mutants with mutations in Type I Genes and increased SA-β-gal activity, the normal (Wt) allele (Normal Gene I) can be more beneficial to be against senescence but the heterozygous mutant (Mut: Hetero) allele (Hetero Gene I) could be rather more deleterious and induce accelerated aging and subsequently a shorter lifespan as seen in the spns1/nrs and terf2 mutants (The 11 mutants are categorized as type I). On the other hand, in suppressor mutants with mutations in Type II Genes and decreased SA-β-gal activity, the normal allele (Normal Gene II) could be more deleterious later in life in terms of senescence but the heterozygous mutant allele (Hetero Gene II) may be more beneficial to be against senescence having ‘heterozygote advantage’ in this regard (The 3 mutants are categorized as type II). These heterozygous mutants are anticipated to show slow or delayed aging and a longer lifespan. However, the homozygous (Mu) null mutants have no fitness with lethality.
One obvious biomarker of aging to use in unbiased screens is SA-β-gal, an indicator of cellular senescence in vitro as well as of organismal aging in vertebrates [33-38]. In fact, we detected SA-β-gal activity in the skin as well as oxidized protein accumulation in the muscle of aging zebrafish [9, 14, 24], similar to that demonstrated in humans with age [33]. We employed this marker in a series of screens for embryonic senescence phenotypes using 306 retrovirus-mediated insertional zebrafish mutant lines and more others by N-ethyl-N-nitrosourea (ENU) chemical mutagenesis (approximately total ~500 mutant genomes) [24, 32] (Figure 3). Since all of the 306 insertional mutations screened were ultimately homozygous lethal, we needed to explore the effects of missing just one copy of the genes (‘haploinsufficiency’) in heterozygous adult fish with age. However, instead of characterizing their aging phenotypes throughout their lifespan, we first examined which of these mutants showed increased SA-β-gal activity during embryonic development within 5 days post fertilization (dpf), either spontaneously in homozygote or following oxidative stress in heterozygote [24]. All the mutants showing the altered SA-β-gal activities are currently available at the Zebrafish International Resource Center (ZIRC): <http://zebrafish.org/zirc/fish/lineAll.php?t=Anything&c=aging&searchMenuQuick=Go>.
Figure 3. A scheme of “embryonic/larval senescence” mutant screening in zebrafish.
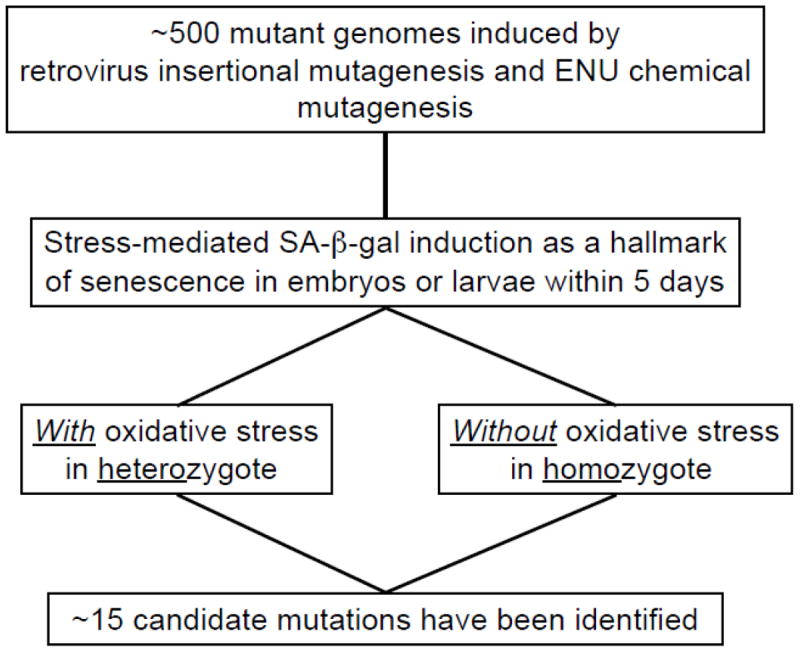
It is possible to hypothetically model our mutant screening of developmentally essential (potentially ‘beneficial’ versus ‘deleterious’) genes for embryonic senescence considering the actual aging process (Figure 2). In enhancer mutants (harboring mutations in Type I Genes) with increased SA-β-gal activity, the normal allele can be more ‘beneficial’ to be against senescence, whereas the heterozygous mutant allele could be rather more ‘deleterious’ in this sense showing accelerated aging and a subsequently shorter lifespan (as evident in the spns1/nrs and terf2 mutants out of the total 11 mutants categorized in this type A group; discussed further below). On the other hand, in suppressor mutants (having mutations in Type II Genes) with decreased SA-β-gal activity, the normal allele could be relatively more ‘deleterious’ in senescence but the heterozygous mutant allele may be more ‘beneficial’ against senescence, having ‘heterozygote advantage’ of fitness (The 3 mutants are categorized as this Type II, as discussed below). These heterozygous mutants are anticipated to show slow or delayed aging and thus a longer lifespan.
From the pool of 306 mutants, we identified 11 mutant genotypes that showed increased higher embryonic SA-β-gal activity and were designated as enhancer mutants (Figure 4), and raised two of these lines to maturity. One of these mutants was found to be null for a homologue of Drosophila spinster (spinster homolog 1/not really started; spns1/nrs), a gene known to regulate lifespan in flies [39], whereas the other harbors a mutation in a homologue of the human telomeric repeat binding factor 2 (terf2) gene, which plays roles in telomere protection and telomere-length regulation [40, 41]. In fact, adult fish that are heterozygous for these mutations show the premature expression of aging biomarkers and the accelerated onset of aging phenotypes including increased SA-β-gal activity, an enhanced accumulation of lipofuscin granules and shorter lifespan, compared with their control siblings. Our current strategy of mutant screening for a senescence-associated biomarker in zebrafish embryos may thus prove to be a useful new approach for the genetic dissection of vertebrate stress response and senescence mechanisms. Importantly, these two genes have been already linked to the organismal aging process (spinster in Drosophila) and cellular senescence (terf2 in mice and human cells), respectively [39, 42, 43]. Thus, this approach allows us to establish the ‘proof-of-principle’ that prominent increases in embryonic SA-β-gal activity correlate with accelerated adult aging phenotypes by haploinsufficiency in the mutants. The remaining other 9 genes linked to increased SA-β-gal activity during embryogenesis await more full characterizations at mature stages (Figure 4), but remain significant interests for their potential involvement and roles in aging. Each gene linked to increased embryonic SA-β-gal activity is listed in the Table 1.
Figure 4. Retrovirus-insertional mutants showing high (increased) SA-β-gal activity [24].
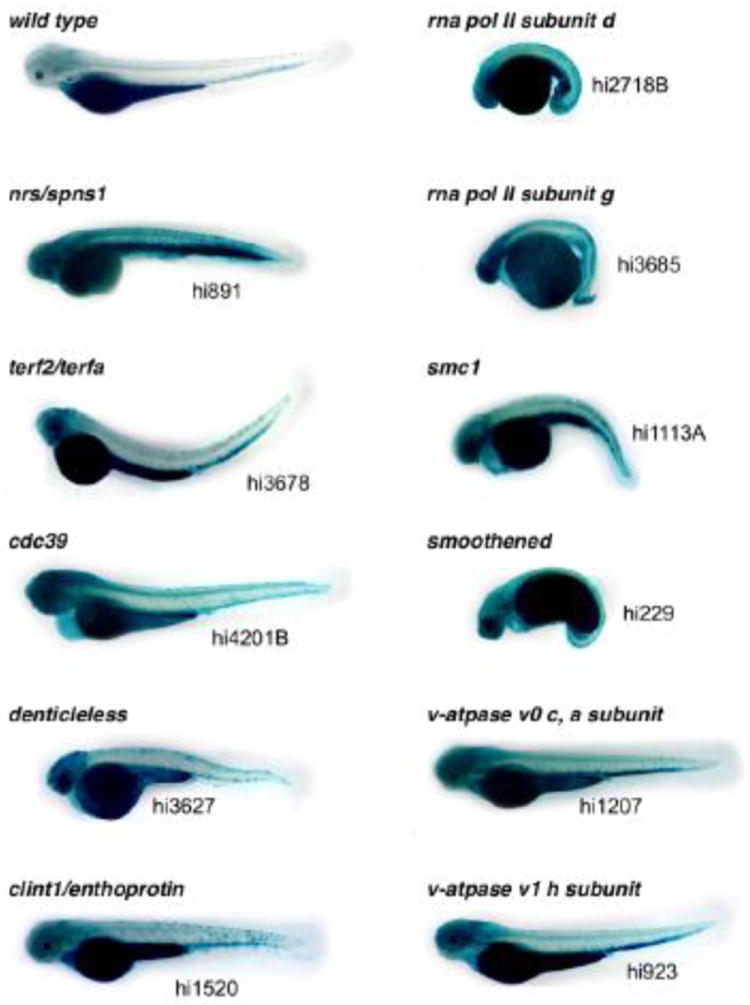
The 11 homozygous 3.5 days post-fertilization (dpf) retrovirus-insertional mutants stained with high SA-β-gal are shown in comparison with a wild-type control (with 1-phenyl-2-thiourea; PTU). All of the original information we obtained regarding these mutants is summarized in the table of our previous publication [24].
Table 1.
Embryonic/Larval Senescence Mutants
| Embryonic SA-β-gal activity |
Gene | GenBank Accession No. |
|---|---|---|
| Enhancer by loss-of-function | spinster homolog 1 (spns1) | NM_153663 |
| telomeric repeat binding factor a (terfa; terf2) | NM_173243 | |
| clathrin interactor 1a (clint1a) | NM_001003412 | |
| ATPase, H+ transporting, lysosomal, V0 subunit c, a (atp6v0ca) | NM_001105136 | |
| ATPase, H+ transporting, lysosomal, V1 subunit H (atp6v1h) | NM_173270 | |
| polymerase (RNA) II (DNA directed) polypeptide D (polr2d) | NM_001002317 | |
| polymerase (RNA) II (DNA directed) polypeptide G-like (polr2gl) | NM_199669 | |
| smoothened homolog (smo) | NM_131027 | |
| CCR4-NOT transcription complex, subunit 1 (cnot1) | NM_001079951 | |
| structural maintenance of chromosomes 1A, like (smc1al) | NM_212810 | |
| denticleless homolog (dtl) | NM_173241 | |
|
| ||
| Suppressor by loss-of-function | eukaryotic translation initiation factor 3 subunit D (eif3d) | NM_200016 |
| ribosomal protein L11 (rpl11) | NM_001002139 | |
| small nuclear ribonucleoprotein D1 polypeptide (snrpd1) | NM_173252 | |
Another intriguing gene identified by increased embryonic SA-β-gal is smoothened (smo) that is originally well known to function prominently in different developmental contexts, as shown from aging studies that have examined the Hedgehog pathway [44, 45]. Smo is a G protein-coupled receptor for Hedgehog signaling [46]. The essential role of Smo in brain development is well established, and the functional presence in adult brain suggests its importance beyond the early developmental stages [47]. Indeed, recent evidence suggests that Hedgehog, acting through Smo, plays a critical role in the expansion and establishment of postnatal hippocampal neural progenitors [48]. Moreover, it has been revealed that there is a potential involvement of Hedgehog signaling in maintaining adult hair follicle stem cells [45], and a certain functional role of the Hedgehog pathway in the retinal aging process has also been highly anticipated [44]. Therefore, further characterizations of zebrafish smo mutants promise to provide crucial information regarding the new function of this gene in aging and senescence beyond early development.
The fact that these interesting mutants were selected based on increased SA-β-gal activity during early development, whilst no apparent abnormalities were found in heterozygous individuals early on, although at least the two of them show the accelerated aging phenotypes later in adult life, underscores the utility of such an approach for identifying early predictors of actual aging phenomena. This suggests that ‘haploinsufficiency’ in some genes might not be detrimental to a developing embryo probably due to adaptive developmental plasticity, but would facilitate alterations in molecular mechanisms and physiological functions characteristic of aging. If biomarkers of such alterations could be documented at an early age, they could help to predict subsequent aging phenotypes later in life.
Furthermore, our mutant screen also revealed three genotypes designated as suppressor mutants with a relative decrease of SA-β-gal activity during early development (Figure 5). Each gene linked to decreased embryonic SA-β-gal activity is also listed in the Table 1. These genes are also of interest for their potential involvement in aging of physiological functions, particularly with regard to the possible existences of long-lived zebrafish mutant animals. The 3 mutants whose SA-β-gal levels were significantly lower harbored the mutations in eukaryotic translation initiation factor 3, subunit D (eif3d), ribosomal protein L11 (rpl11), and small nuclear ribonucleoprotein D1 polypeptide (snrpd1). These genes are involved in protein synthesis, ribosomal functions, and RNA-protein interactions, respectively. The presence of lower SA-β-gal levels in these mutant embryos (‘suppressor mutants by loss-of-function’) is intriguing and makes them attractive for further exploring the possibility of slow or delayed aging regulated by any heterozygote advantage. Identification of longevity assurance genes, which can extend lifespan, should be elucidated to better establish the existence of aging-related genes in the zebrafish model system. Therefore, we currently anticipate being able to identify a real suppressor-type aging mutant, which displays enhanced stress resistance in advanced age and, perhaps, a longer healthy lifespan (“healthspan”). Alternatively, it might also be possible to isolate a “revertant” from the background of an accelerated aging mutant, which would restore the original normal phenotype with rejuvenation by means of any suppressor mutation (i.e., a reversal of the accelerated aging phenotype).
Figure 5. Retrovirus-insertional mutants showing low (decreased) SA-β-gal activity [24].
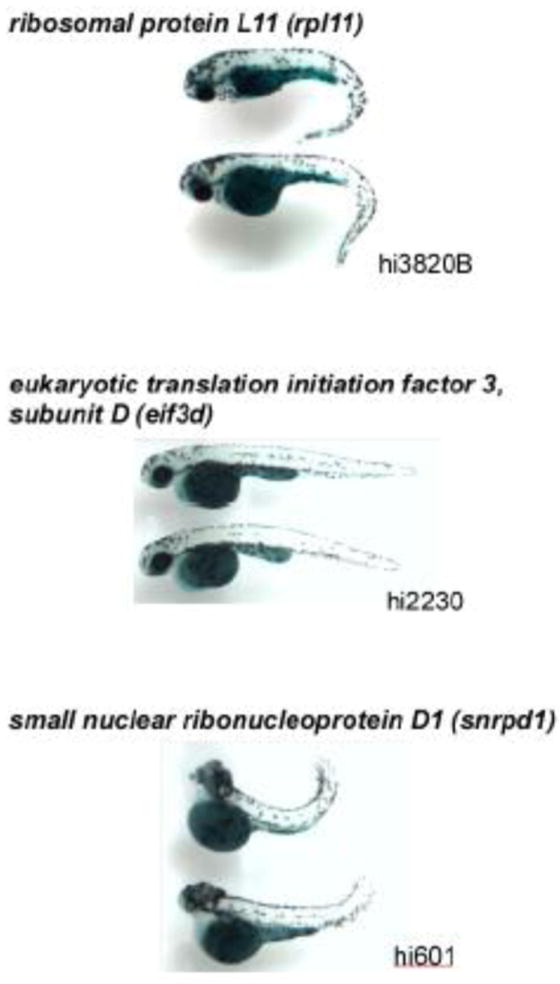
There were the three different mutants (shown by duplications) with low SA-β-gal intensity, hi3820B (60S ribosomal protein L11 gene) (n = 12; 5 dpf), hi2230 (eukaryotic translation initiation factor 3, subunit 7 gene) (n = 15; 4 dpf), and hi601 (small nuclear ribonucleoprotein D1 gene) (n = 14; 3.5 dpf). Representative SA-β-gal stained homozygous embryos (two individuals from each of these three mutant groups) are shown.
Our studies have demonstrated to date that the certain environmental challenges that can accelerate aging in adult zebrafish, e.g., gamma-irradiation or oxidative stress, can be effective in revealing biomarkers of aging during early development. If the coupling of stress responses in zebrafish embryos and/or larvae with aging mechanisms in adult fish works parallel fashion, as has been demonstrated in other invertebrate animal systems and suggested by our results discussed here, this approach will be a useful and powerful tool in the search for aging-related genes more comprehensively in vertebrates. Such speedy identification of genes relevant to vertebrate aging using embryos and/or larvae would partially circumvent the need for time-consuming and lengthy lifespan analyses in any available zebrafish mutants or even natural variants.
5. Developmental origins of aging and disease
Genetic information determines the architecture of individual organisms during development and programs the mechanisms that maintain adequate functions during adult life [49]. There is also substantial evidence which shows that constraints in early life environment are an important determinant of the risk of geriatric diseases including cancer and metabolic disease [50, 51]. One hypothesis is that the increased susceptibility to such diseases has a common origin with the developmental changes induced in the developing tissues and organs under initially exposed particular environmental conditions, as originally proposed by Barker [52]. The developmental and environmental impacts include nutritional status and altered epigenetic regulation of specific genes having each genetic background. The induction of a subsequent disease risk with age is significantly dependent upon the nature of the environmental challenges as well as on the interaction of the original genetic susceptibility with the altered epigenome throughout the life course [53]. However, age-related physiological changes and pathologies may also originally derive from the drifting actions of developmental programs [54]. Age-associated epigenetic drifts in an intrinsic developmental program that tends toward imbalance with advancing age can be a cause of the progressive deteriorative changes in physiological functions and homeostasis, leading to various diseases [55, 56].
With aging, regenerative ability also declines and degenerative potential increases in zebrafish as we discussed above [14]. The loss/decrease of plasticity and/or onset of self-renewal defects in stem cells may be responsible for reduced regenerative capability and inefficient tissue restoration accompanied by aging. On the other hand, inadequate elasticity in aging may cause inappropriate regeneration leading to cancer or degeneration by compromising homeostatic integrity and plasticity. The fundamental aging process is not a disease itself, whereas aging obviously increases vulnerability to many diseases including cancer. In essence, therefore, aging may be considered to be one of the most prominent pathogens and carcinogens. Cancer incidences exponentially increase with age in most vertebrate species as shown in zebrafish as well as humans [57-59]. This age-associated increase of cancer incidence is presumably due to inappropriate developmental drift in aging stem cells or aged somatic cells which are voluntarily trying to reset a program having certain troubles. Most of proto-oncogenes function properly in normal development and growth. Developmentally crucial genes such as oct-3/4 and sox2 and the proto-oncogene c-myc among others can reprogram differentiated somatic cells to stem cells beyond plasticity [60-62]. However, such artificially (or even naturally) induced/reversed stemness is insufficiently robust and often suffer from inevitable carcinogenesis owing to the conflicting relationship between reversing power and forwarding entropy in senescence [63]. Inappropriate fading or unexpectedly excessive actions of developmental functions in the adult stages is conceivably caused by less molecular fidelity with increasing entropy [64, 65]. The reason why adaptive plasticity is restricted to a particular period of early life is possibly that there is a difficulty in reversing the entire developmental processes of adult organisms in vivo [50]. Thus, it might be due to the unaffordable energy cost of assuring reproductive success and fidelity in reconstructing the prior established characteristics of aging organisms.
Segmental progeroid syndromes including Hutchinson-Gilford progeria syndrome (HGPS) (simply referred to as “progeria”) are often accompanied by developmental and growth retardation, but very little is known about the developmental process of such accelerated aging disorders. We have demonstrated the impaired early developmental processes of the mesenchymal lineage in zebrafish embryos harboring the disturbed lamin A expression [66], and other lines of evidence also support the notion that developmental signaling pathways are presumably involved in the lamin A-associated aging mechanisms as well [67]. Because embryonic stem cells appear to express lamin A/C upon differentiation [68], and lamin A-associated stem cell dysfunction has been linked to disruption of the Wnt and Notch signaling pathways [67, 69-71], which are essential for early vertebrate development, our observations of embryonic senescence or apoptosis in the lamin A gene (LMNA)-disturbed zebrafish might be consistent with a dysregulation of somatic stem cells leading to organismal premature aging. In particular, it is plausible that the less adaptive plasticity and insufficient self-renewal ability in mesenchymal stem cells may be responsible for the specificity of accelerated tissue degeneration, as most affected tissues in progeria are of mesenchymal origin [70]. Embryonic senescence and further unforeseen developmental abnormalities may also be occurring in human (or mice) progeria given that severe neonatal lethality has been reported [72-74]. Thus, aging research should therefore extend to the study of early developmental process at least within the context of reproduction and regeneration.
It is reasonable to assume that the developmental theory of aging approach eventually leads to the identification of genes and pathways which are relevant for development alone, for aging alone, and for both development and aging [75]. As a fundamental first task, the pathways that have already been implicated in aging and longevity can be more extensively investigated in early development, as we have been working on the several gene products including lamin A, ataxia telangiectasia mutated (ATM), and telomerase [66, 76, 77].
6. Juvenile protective and rejuvenating factors against aging
Little attention has been devoted to determining ‘physiological’ factors characteristic of early life that could be protective to adult health and contribute to healthy aging. This is presumably because ‘premature aging’ during early life has often been considered pathological rather physiological as a canonical context. Conceivably, however, our identified genes of senescence enhancers by loss-of-function might encode juvenile protective and/or rejuvenating factors that could normally act to delay aging and that when mutated can cause premature aging.
The morphogenic signals mediated by Hedgehog and Wnt are essential to direct pattern formation during embryogenesis and both the signal activities can remain through adulthood. Consistently, it has been reported that both Hedgehog and Wnt pathways function in aging [44, 78-80]. In addition, critical roles of Hedgehog and Wnt signals have been implicated in metabolic regulatory events including insulin sensitivity and fat formation [81, 82]. In lines with these, as we described, our embryonic senescence mutant screens have identified the Hedgehog signal transducer, Smo, as a driver of SA-β-gal activity by the loss of its function [24]. Moreover, haploinsufficient accelerated aging phenotypes in retinal photoreceptors via the heterozygous hedgehog genotype have been reported in zebrafish [44]. Thus, Hedgehog signaling may function as an aging antagonist or rejuvenating factor [83, 84]. The outcome of several plausible candidate mutants in our mutant screening, like smo mutant fish, in addition to spns1/nrs and terf2 mutant fish, has already provided a proof-of-concept for our original approach to assess the rejuvenating connection between developmental and senescence signaling pathways through the common genes during embryogenesis in zebrafish.
The secreted factor Klotho, which acts as a proficient suppressor of aging, may also be classified as a rejuvenating factor. Klotho-deficient mice manifest a syndrome resembling accelerated human aging with multiple deteriorative features [85], whereas Klotho-overexpressed mice extend lifespan displaying anti-aging properties [86]. In the Klotho-deficient animals, increased Wnt signaling activity becomes evident, and in fact Klotho functions as a secreted Wnt antagonist. Klotho binds to various Wnt family members, and Klotho deficiency-induced accelerated aging is due, at least in part, to constitutive Wnt signaling. Hence, an unexpected deleterious role of Wnt in aging has been revealed in a Klotho-deficient mouse model system, while other lines of evidence contradictorily support a beneficial effect(s) of a Wnt signaling against on aging as it can ameliorate multiple age-associated symptoms [87]. A tissue-dependent imbalanced regulation between Klotho and Wnt may be caused by developmental drift in aging stem cells and become a reason of reduced plasticity in tissues and organs with age.
Importantly also, essential involvements of Notch and Wnt signaling pathways in the deterioration of stem cells from progeria patients and model mice have been described, respectively [67, 69-71]. In human progeria, Notch signaling is upregulated in mesenchymal stem cells, leading to activation of the downstream target genes and to increased osteogenesis and decreased adipogenesis by perturbing cellular differentiation. Furthermore, in progeria model mice, the transcriptionally active form of β-catenin, which is the well-characterized regulator of the Wnt signaling pathway, was almost completely absent in hair follicle stem cells.
Moreover, given the recent findings that Wnt signaling and the telomerase catalytic subunit (telomerase reverse transcriptase; TERT) can potentially make a positive regulatory loop: the Wnt pathway component β-catenin can positively regulate the expression of TERT while TERT can positively regulate the expression of Wnt/β-catenin target genes [88, 89], it is now reasonable to propose that the new direct link between theses mechanistic regulations have a crucial impact on balancing regeneration/rejuvenation and cancer during the aging process beyond embryogenesis [90].
Taken together, the cumulative lines of evidence support the notion that developmental signaling pathways and their possible drifting actions may be involved in the aging and rejuvenation mechanisms [54], where we recognize that the zebrafish model system can enormously contribute to the identification of juvenile protective and rejuvenating factors against aging and the study of molecular mechanisms to reverse the aging process that are common amongst vertebrates.
7. Conclusions
Our findings from the recent studies suggest that ‘embryonic senescence’ in zebrafish can be connected to the process of actual aging, which manifests at both the cellular and organismal levels. These include morphological and histological changes in multiple tissues, reduced regeneration and damage to macromolecules (DNA, proteins, and lipids), lipofuscin accumulation possibly resulting in reduced intracellular trafficking, cognitive decline, circadian dysregulation, and sleep alterations among others. These hallmarks of aging, also typical of other vertebrates, have been used in our longitudinal study of aging phenotypes among the zebrafish mutants that were identified based on early-life evaluations. This and other examples indicate that zebrafish can become an ideal system to develop alternative vertebrate models in order to address the many biological questions regarding the relationship between vertebrate development and aging in a more amenable throughput setting.
Aging has been thought of as a stochastically ‘deteriorative/destructive’ process, whereas development is a finely ‘programmed/constructive’ process. However, the genetic robustness of aging may already be defined during early developmental stages. For instance, a certain gene mutation might lead to accelerated decline in gene function that would normally occur only at old age, and this process could be facilitated by challenging of multiple stressors which can mimic various environmental effects. In such cases, robust biomarkers of aging could be potentially revealed at a much earlier age. As we have shown in the previous study, this issue can be fruitfully explored using unbiased genetic mutant screens for senescence-associated biomarkers during early developmental stages [24]. Overall, we propose that the complexity of aging in higher organisms, involving the interaction of multiple genes and signaling pathways, warrants a comprehensive search for the evolutionary developmental origins as early predictors of altered aging phenotypes later in life, where we might be able to reverse the process. The zebrafish animal model, with its well-established genetics and advanced high-throughput technologies, offers an unparalleled opportunity to identify aging-related genes, and to easily analyze their function throughout life. The highly conserved mechanisms of aging on the cellular, tissue and organismal levels can and should be addressed using zebrafish as the most affordable genetic model in vertebrates, comparable to C. elegans and Drosophila in invertebrate.
Highlights.
There may be common factors and mechanisms between early development and senescence.
There are also developmental origins of adult-onset diseases in advanced age.
The aging process can be predictive based on adaptivity during the early development.
Zebrafish mutants showing senescence phenotypes during embryogenesis were identified.
The aging complexity may warrant a search for the evolutionary developmental origins.
Acknowledgments
We are grateful to the current and former members of our laboratory. The work of our laboratory reviewed herein was funded by research grants from The Ellison Medical Foundation, the Glenn Foundation for Medical Research, the A-T Children’s Project, and the NIGMS/NIA/NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 2.Carey JR, Liedo P, Harshman L, Zhang Y, Muller HG, Partridge L, Wang JL. Life history response of Mediterranean fruit flies to dietary restriction. Aging Cell. 2002;1:140–148. doi: 10.1046/j.1474-9728.2002.00019.x. [DOI] [PubMed] [Google Scholar]
- 3.Liao CY, Rikke BA, Johnson TE, Diaz V, Nelson JF. Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell. 2010;9:92–95. doi: 10.1111/j.1474-9726.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper TM, Mockett RJ, Sohal BH, Sohal RS, Orr WC. Effect of caloric restriction on life span of the housefly, Musca domestica. FASEB J. 2004;18:1591–1593. doi: 10.1096/fj.03-1464fje. [DOI] [PubMed] [Google Scholar]
- 5.Phelan JP, Rose MR. Why dietary restriction substantially increases longevity in animal models but won’t in humans. Ageing Res Rev. 2005;4:339–350. doi: 10.1016/j.arr.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Mangel M, Abrahams MV. Age and longevity in fish, with consideration of the ferox trout. Exp Gerontol. 2001;36:765–790. doi: 10.1016/s0531-5565(00)00240-0. [DOI] [PubMed] [Google Scholar]
- 7.Kamakura M. Royalactin induces queen differentiation in honeybees. Nature. 2011;473:478–483. doi: 10.1038/nature10093. [DOI] [PubMed] [Google Scholar]
- 8.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK, de Cabo R. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kishi S, Uchiyama J, Baughman AM, Goto T, Lin MC, Tsai SB. The zebrafish as a vertebrate model of functional aging and very gradual senescence. Exp Gerontol. 2003;38:777–786. doi: 10.1016/s0531-5565(03)00108-6. [DOI] [PubMed] [Google Scholar]
- 10.Keller ET, Murtha JM. The use of mature zebrafish (Danio rerio) as a model for human aging and disease. Comp Biochem Physiol C Toxicol Pharmacol. 2004;138:335–341. doi: 10.1016/j.cca.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Gerhard GS, Kauffman EJ, Wang X, Stewart R, Moore JL, Kasales CJ, Demidenko E, Cheng KC. Life spans and senescent phenotypes in two strains of Zebrafish (Danio rerio) Exp Gerontol. 2002;37:1055–1068. doi: 10.1016/s0531-5565(02)00088-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhdanova IV, Yu L, Lopez-Patino M, Shang E, Kishi S, Guelin E. Aging of the circadian system in zebrafish and the effects of melatonin on sleep and cognitive performance. Brain Res Bull. 2008;75:433–441. doi: 10.1016/j.brainresbull.2007.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu L, Tucci V, Kishi S, Zhdanova IV. Cognitive aging in zebrafish. PLoS ONE. 2006;1:e14. doi: 10.1371/journal.pone.0000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai SB, Tucci V, Uchiyama J, Fabian NJ, Lin MC, Bayliss PE, Neuberg DS, Zhdanova IV, Kishi S. Differential effects of genotoxic stress on both concurrent body growth and gradual senescence in the adult zebrafish. Aging Cell. 2007;6:209–224. doi: 10.1111/j.1474-9726.2007.00278.x. [DOI] [PubMed] [Google Scholar]
- 15.White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, Bourque C, Dovey M, Goessling W, Burns CE, Zon LI. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2008;2:183–189. doi: 10.1016/j.stem.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- 17.Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, Grunwald DJ. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8:e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Oikonomou G, Chiu CN, Niles BJ, Liu J, Lee DA, Antoshechkin I, Prober DA. A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res. 2013 doi: 10.1093/nar/gks1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG, 2nd, Tan W, Penheiter SG, Ma AC, Leung AY, Fahrenkrug SC, Carlson DF, Voytas DF, Clark KJ, Essner JJ, Ekker SC. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta A, Christensen RG, Rayla AL, Lakshmanan A, Stormo GD, Wolfe SA. An optimized two-finger archive for ZFN-mediated gene targeting. Nat Methods. 2012;9:588–590. doi: 10.1038/nmeth.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pardo-Martin C, Chang TY, Koo BK, Gilleland CL, Wasserman SC, Yanik MF. High-throughput in vivo vertebrate screening. Nat Methods. 2010;7:634–636. doi: 10.1038/nmeth.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gehrig J, Reischl M, Kalmar E, Ferg M, Hadzhiev Y, Zaucker A, Song C, Schindler S, Liebel U, Muller F. Automated high-throughput mapping of promoter-enhancer interactions in zebrafish embryos. Nat Methods. 2009;6:911–916. doi: 10.1038/nmeth.1396. [DOI] [PubMed] [Google Scholar]
- 23.Baker M. Screening: the age of fishes. Nat Methods. 2011;8:47–51. doi: 10.1038/nmeth0111-47. [DOI] [PubMed] [Google Scholar]
- 24.Kishi S, Bayliss PE, Uchiyama J, Koshimizu E, Qi J, Nanjappa P, Imamura S, Islam A, Neuberg D, Amsterdam A, Roberts TM. The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet. 2008;4:e1000152. doi: 10.1371/journal.pgen.1000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 26.Schmucker DL. Age-related changes in liver structure and function: Implications for disease? Exp Gerontol. 2005;40:650–659. doi: 10.1016/j.exger.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 28.Poleo G, Brown CW, Laforest L, Akimenko MA. Cell proliferation and movement during early fin regeneration in zebrafish. Dev Dyn. 2001;221:380–390. doi: 10.1002/dvdy.1152. [DOI] [PubMed] [Google Scholar]
- 29.Zhdanova IV. Melatonin as a hypnotic: pro. Sleep Med Rev. 2005;9:51–65. doi: 10.1016/j.smrv.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Martin GM. Modalities of gene action predicted by the classical evolutionary biological theory of aging. Ann N Y Acad Sci. 2007a;1100:14–20. doi: 10.1196/annals.1395.002. [DOI] [PubMed] [Google Scholar]
- 31.Martin GM, Bergman A, Barzilai N. Genetic determinants of human health span and life span: progress and new opportunities. PLoS Genet. 2007b;3:e125. doi: 10.1371/journal.pgen.0030125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci U S A. 2004;101:12792–12797. doi: 10.1073/pnas.0403929101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–213. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005;19:1986–1999. doi: 10.1101/gad.342305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valenzano DR, Terzibasi E, Cattaneo A, Domenici L, Cellerino A. Temperature affects longevity and age-related locomotor and cognitive decay in the short-lived fish Nothobranchius furzeri. Aging Cell. 2006;5:275–278. doi: 10.1111/j.1474-9726.2006.00212.x. [DOI] [PubMed] [Google Scholar]
- 37.Kishi S. Functional aging and gradual senescence in zebrafish. Ann N Y Acad Sci. 2004;1019:521–526. doi: 10.1196/annals.1297.097. [DOI] [PubMed] [Google Scholar]
- 38.Kishi S. Zebrafish as Aging Models. In: Conn M, editor. Handbook of Models for Human Aging. Elsevier Academic Press; 2006. pp. 317–338. [Google Scholar]
- 39.Nakano Y, Fujitani K, Kurihara J, Ragan J, Usui-Aoki K, Shimoda L, Lukacsovich T, Suzuki K, Sezaki M, Sano Y, Ueda R, Awano W, Kaneda M, Umeda M, Yamamoto D. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol Cell Biol. 2001;21:3775–3788. doi: 10.1128/MCB.21.11.3775-3788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21:532–540. doi: 10.1038/sj.onc.1205080. [DOI] [PubMed] [Google Scholar]
- 41.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 42.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF 2 deletion. Nat Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 43.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 44.Stenkamp DL, Satterfield R, Muhunthan K, Sherpa T, Vihtelic TS, Cameron DA. Age-related cone abnormalities in zebrafish with genetic lesions in sonic hedgehog. Invest Ophthalmol Vis Sci. 2008;49:4631–4640. doi: 10.1167/iovs.07-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rittie L, Stoll SW, Kang S, Voorhees JJ, Fisher GJ. Hedgehog signaling maintains hair follicle stem cell phenotype in young and aged human skin. Aging Cell. 2009;8:738–751. doi: 10.1111/j.1474-9726.2009.00526.x. [DOI] [PubMed] [Google Scholar]
- 46.Ayers KL, Therond PP. Evaluating Smoothened as a G-protein-coupled receptor for Hedgehog signalling. Trends Cell Biol. 2010;20:287–298. doi: 10.1016/j.tcb.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 47.Charytoniuk D, Porcel B, Rodriguez Gomez J, Faure H, Ruat M, Traiffort E. Sonic Hedgehog signalling in the developing and adult brain. J Physiol Paris. 2002;96:9–16. doi: 10.1016/s0928-4257(01)00075-4. [DOI] [PubMed] [Google Scholar]
- 48.Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11:277–284. doi: 10.1038/nn2059. [DOI] [PubMed] [Google Scholar]
- 49.Finch CE, Kirkwood TBL. Chance, Development, and Aging. Oxford University Press; USA: 2000. [Google Scholar]
- 50.Bateson P, Barker D, Clutton-Brock T, Deb D, D’Udine B, Foley RA, Gluckman P, Godfrey K, Kirkwood T, Lahr MM, McNamara J, Metcalfe NB, Monaghan P, Spencer HG, Sultan SE. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- 51.Vaiserman A. Early-life origin of adult disease: evidence from natural experiments. Exp Gerontol. 2011;46:189–192. doi: 10.1016/j.exger.2010.08.031. [DOI] [PubMed] [Google Scholar]
- 52.Barker DJ. The developmental origins of adult disease. J Am Coll Nutr. 2004;23:588S–595S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- 53.Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res Rev. 2009;8:268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell. 2008;134:291–303. doi: 10.1016/j.cell.2008.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin GM. Epigenetic drift in aging identical twins. Proc Natl Acad Sci U S A. 2005;102:10413–10414. doi: 10.1073/pnas.0504743102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin GM. Epigenetic gambling and epigenetic drift as an antagonistic pleiotropic mechanism of aging. Aging Cell. 2009;8:761–764. doi: 10.1111/j.1474-9726.2009.00515.x. [DOI] [PubMed] [Google Scholar]
- 57.Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spitsbergen JM, Buhler DR, Peterson TS. Neoplasia and neoplasm-associated lesions in laboratory colonies of zebrafish emphasizing key influences of diet and aquaculture system design. ILAR J. 2012;53:114–125. doi: 10.1093/ilar.53.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DePinho RA. The age of cancer. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 61.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 62.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 63.Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, Vallier L, Gil J. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–2139. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayflick L. Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLoS Genet. 2007;3:e220. doi: 10.1371/journal.pgen.0030220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hershey D. Entropy, Basal Metabolism and Life Expectancy. Gerontologia. 1963;68:245–250. doi: 10.1159/000211200. [DOI] [PubMed] [Google Scholar]
- 66.Koshimizu E, Imamura S, Qi J, Toure J, Valdez DM, Jr, Carr CE, Hanai J, Kishi S. Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS One. 2011;6:e17688. doi: 10.1371/journal.pone.0017688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meshorer E, Gruenbaum Y. Gone with the Wnt/Notch: stem cells in laminopathies, progeria, and aging. J Cell Biol. 2008;181:9–13. doi: 10.1083/jcb.200802155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Constantinescu D, Gray HL, Sammak PJ, Schatten GP, Csoka AB. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24:177–185. doi: 10.1634/stemcells.2004-0159. [DOI] [PubMed] [Google Scholar]
- 69.Espada J, Varela I, Flores I, Ugalde AP, Cadinanos J, Pendas AM, Stewart CL, Tryggvason K, Blasco MA, Freije JM, Lopez-Otin C. Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J Cell Biol. 2008;181:27–35. doi: 10.1083/jcb.200801096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–459. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, Lee HS, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, Stewart CL. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010;19:413–425. doi: 10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Engelen BG, Muchir A, Hutchison CJ, van der Kooi AJ, Bonne G, Lammens M. The lethal phenotype of a homozygous nonsense mutation in the lamin A/C gene. Neurology. 2005;64:374–376. doi: 10.1212/01.WNL.0000149763.15180.00. [DOI] [PubMed] [Google Scholar]
- 73.De Sandre-Giovannoli A, Levy N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog Mol Subcell Biol. 2006;44:199–232. doi: 10.1007/978-3-540-34449-0_9. [DOI] [PubMed] [Google Scholar]
- 74.Rodriguez JI, Perez-Alonso P, Funes R, Perez-Rodriguez J. Lethal neonatal Hutchinson-Gilford progeria syndrome. Am J Med Genet. 1999;82:242–248. [PubMed] [Google Scholar]
- 75.Zwaan BJ. Linking development and aging. Sci Aging Knowledge Environ. 2003;2003:pe32. doi: 10.1126/sageke.2003.47.pe32. [DOI] [PubMed] [Google Scholar]
- 76.Imamura S, Kishi S. Molecular cloning and functional characterization of zebrafish ATM. Int J Biochem Cell Biol. 2005;37:1105–1116. doi: 10.1016/j.biocel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 77.Imamura S, Uchiyama J, Koshimizu E, Hanai J, Raftopoulou C, Murphey RD, Bayliss PE, Imai Y, Burns CE, Masutomi K, Gagos S, Zon LI, Roberts TM, Kishi S. A non-canonical function of zebrafish telomerase reverse transcriptase is required for developmental hematopoiesis. PLoS One. 2008;3:e3364. doi: 10.1371/journal.pone.0003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 79.Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira II, Schimel D, Kuo CJ, Gutkind JS, Hwang PM, Finkel T. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- 80.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 81.Pospisilik JA, Schramek D, Schnidar H, Cronin SJ, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, Bayer M, Haschemi A, Puviindran V, Tar K, Orthofer M, Neely GG, Dietzl G, Manoukian A, Funovics M, Prager G, Wagner O, Ferrandon D, Aberger F, Hui CC, Esterbauer H, Penninger JM. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–160. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 82.Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010;24:1507–1518. doi: 10.1101/gad.1924910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neureiter D. New in Hedgehog signaling: a possible role in aging, and chronic degenerative and inflammatory diseases? Bioessays. 2012;34:828–829. doi: 10.1002/bies.201200049. Comment on. [DOI] [PubMed] [Google Scholar]
- 84.Dashti M, Peppelenbosch MP, Rezaee F. Hedgehog signalling as an antagonist of ageing and its associated diseases. Bioessays. 2012;34:849–856. doi: 10.1002/bies.201200049. [DOI] [PubMed] [Google Scholar]
- 85.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 86.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.DeCarolis NA, Wharton KA, Jr, Eisch AJ. Which way does the Wnt blow? Exploring the duality of canonical Wnt signaling on cellular aging. Bioessays. 2008;30:102–106. doi: 10.1002/bies.20709. [DOI] [PubMed] [Google Scholar]
- 88.Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- 89.Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, McLaughlin M, Veenstra TD, Nusse R, McCrea PD, Artandi SE. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009;460:66–72. doi: 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, Horner JW, Maratos-Flier E, Depinho RA. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102–106. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Williams PD, Day T. Antagonistic pleiotropy, mortality source interactions, and the evolutionary theory of senescence. Evolution. 2003;57:1478–1488. doi: 10.1111/j.0014-3820.2003.tb00356.x. [DOI] [PubMed] [Google Scholar]