

Abstract
Type 1 diabetes (T1D) is an autoimmune disease that results from the destruction of insulin-producing β cells by autoreactive T cells, leading to lifelong dependency on insulin therapy and increased risk of long-term cardiovascular complications. Here we take the opportunity of the 20th anniversary of the generation of the BDC2.5 TCR transgenic non-obese diabetic (NOD) mouse model, to provide a brief overview of the significant progress that has been made in understanding the role of T cells in the disease pathogenesis period. This included development of hundreds of reagents that block or even reverse new-onset disease by directly or indirectly controlling T cells. We also reflect on the sobering fact that none of these strategies has shown significant efficacy in clinical trials and discuss potential reasons hindering translation of the preclinical findings into successful therapeutic strategies and potential ways forward.
Keywords: Autoimmune diabetes, Immunotherapy, T cells, BDC2.5 T cells, Anti-CD3, Immunosuppression
Core tip: Our understanding of type 1 diabetes pathogenesis has significantly improved over the last three decades. We went from not knowing very little to acquisition of significant details about the role of the immune system and different T cell subsets in the disease process. The non-obese diabetic mouse model contributed and continues to contribute to our understanding of the disease process. This article pays tributes to the major role T-cells bearing-cell-specific T-cell receptors transgenic mouse played in shaping of our understanding of the disease process. We also divulge to briefly discuss current challenges facing development of a safe immunotherapy for the disease.
COMMENTARY ON HOT TOPICS
Diabetes is a heterogeneous metabolic disease caused by glucose intolerance and manifested clinically as hyperglycemia. Based on the underlying cause of the hyperglycemia, diabetes is divided into type 1 (T1D) and 2 (T2D). T1D is autoimmune in nature and results from the destruction of insulin-producing β cells by autoreactive T cells, leading to insulin deficiency and dependency on exogenous insulin to maintain glucose homeostasis. In contrast, T2D is a complex metabolic disorder associated with insulin resistance in peripheral tissues. Currently, there is no cure for either type of diabetes. In the interim, T1D is managed by multiple daily injections of insulin, whereas T2D is controlled by medications that improve insulin sensitivity and/or reduce glucose production by the liver. Maintenance of glucose homeostasis, however, is challenging and most patients eventually develop fatal cardiovascular complications. Intensive efforts are therefore being directed toward development of cure or prevention strategies. Small animal models play profoundly important roles in these efforts, particularly in T1D research.
Small animal research in T1D began in earnest with the development and use of spontaneous and induced disease models in 1970s and 1980s. Among several T1D models, the non-obese diabetic (NOD) mouse became the most commonly used and favorite model soon after its development about 33 years ago[1]. The value of the NOD mouse in understanding the disease mechanism increased exponentially in the late 80s and early 90s following development of technologies that allowed engineering of the genome to generate mice bearing particular transgenes or lacking specific molecules to interrogate their roles in the disease process[2]. Consequently, more than 250 different genetically modified NOD mice were produced and characterized (http://jaxmice.jax.org/findmice/index.html). Results of these efforts uncovered a wealth of information about the roles of various cell types and molecules in modulating T cells and established key cellular and molecular events in the disease process.
Of interest is that uncovering the role of T cells in autoimmune diabetes traversed several key steps that culminated in the generation of the NOD mouse bearing TCR transgenic T cells [reviewed in detail in by Haskins[3]. Considerable evidence accumulated in the early 1990s indicating a central role for T cells in mediating T1D in mice. These included demonstration that the disease development can be prevented by immunosuppressive agents that target T cells[4], and by anti-CD4 and anti-CD8 antibody treatments[5,6]. Furthermore, the disease was shown to be transferrable to neonatal NOD mice and immunodeficient NOD-severe combined immunodeficiency mice (NOD-SCID) by adoptive transfer of T cells from spontaneously diabetic NOD donors[7]. A clearer picture of the role of T cells began to emerge with the generation of islet antigen-specific T cell clones. Several groups independently generated islet antigen-specific T cell clones capable of transferring the disease to susceptible recipients[4]. It was found that different T cell clones expressed different T-cell receptors (TCRs), suggesting for the first time that islet-specific T cells recognize several different islet antigens and pointing to the complexity of the disease. Among the well-characterized clones is the BDC2.5 clone, the TCR that was later used to generate the T-cells bearing-cell-specific T-cell receptors (BDC 2.5 TCR) transgenic (tg) mouse in 1993[8]. Thus, generation of T cell clones was crucial in cementing the role of T cell in the disease pathogenesis and the existence of diabetogenic T cells in autoimmune-prone hosts. Yet clones have limited value in providing details regarding the nature and in vivo action mechanisms of diabetogenic T cells. Among the pressing questions (some of which are still incompletely understood) are how autoreactive T cells escape negative selection, where they reside in the periphery, what triggers them to become diabetogenic, and how they cause the disease. Diabetogenic T cells among the peripheral T cell repertoire are rare and the lack of appropriate reagents that permit their identification in vivo precluded addressing these questions directly in vivo in unmanipulated NOD mice. To overcome this problem, researchers generated TCR tg mice by using TCRs derived from generated clones. Among the widely used TCR transgenic mice in autoimmune diabetes is the BDC2.5 TCR tg mouse generated in 1993 by Katz et al[8], in which all T cells express the TCRα (Vα1) and β (Vβ4) chain genes from the BDC2.5 TCR CD4 T cell clone[9]. Unlike in wild type NOD mice, which harbor a diverse repertoire where autoreactive T cells are very rare and are difficult to track in vivo, all T cells in BDC2.5 tg mice recognize and respond uniformly to an elusive islet autoantigen [It was recently reported by two groups[10,11] that BDC2.5 T cells recognize peptides from chromogranin A (ChgA)]. Therefore, by studying T cells in BDC2.5 tg mice, the authors were able to track the behavior and fate of diabetogenic T cells in vivo and test hypotheses pertaining to roles of thymic selection, site of priming and peripheral activation of diabetogenic T cells, trafficking, and timing of response to islet autoantigens. Results showed that diabetogenic TCR can be produced in a large proportion of thymocytes in the TCR αβ tg mice, are positively selected without undergoing massive clonal deletion, and migrate to the periphery where they constitute the majority of the T cell repertoire. The model is still providing an important platform for in vivo dissecting of diabetogenic T cells, including roles of various molecules and cell types in modulating their pathogenicity. It has not only resulted in a wealth of information regarding pathogenesis of autoimmune diabetes, but also shed light on the immune system and autoimmunity.
Tracking disease development in BDC2.5 TCR tg mice showed that initiation of the disease is highly regulated with two important checkpoints controlling the diabetogenic process. These two checkpoints are especially evident and synchronous in BDC2.5 tg mice. The autoreactive T cells appear to ignore the β cells for the first 2 wk of life. Soon after, BDC2.5 T cells abruptly invade the pancreatic islets resulting in insulitis that progresses rapidly, with almost all islets heavily infiltrated by the age of 3 to 4 wk. Surprising at the time, however, was the observation that insulitis in most BDC2.5 tg mice never progresses to full-blown diabetes. But when the BDC2.5 transgene is introduced into NOD-Rag-1 knockout mice, they do develop aggressive disease at a very early age. Failure of BDC2.5 TCR tg mice to develop full-blown disease in Rag-1-sufficient background was due to incomplete allelic exclusion of endogenous TCRβ chains, resulting in developing thymocytes that differentiate into regulatory T cells that oppose the pathogenic effect of diabetogenic T cells leading to standstill insulitis. On the other hand, in the absence of the Rag-1 gene all developing T cells bear the BDC2.5 TCR transgene, resulting in a pathogenic repertoire devoid of regulatory cells, inducing a rapid onset of aggressive disease. The results provide critical hints of a major role for regulatory T cells in opposing the disease development. The synchronous development of the disease in BDC2.5 mice combined with other studies, including adoptive transfer of BDC2.5 T cells, led to the concept that immunoregulatory mechanisms exist at two check points, at the pancreatic draining lymph nodes and the islet itself, respectively. Breach of these checkpoints by diabetogenic T cells is clearly visualized in NOD mice by using adoptive transfer of BDC2.5 in appropriate hosts[12,13]. This paradigm is depicted in Figure 1. Subsequent studies revealed critical roles for regulatory T and B cells and various molecules involved in controlling the major checkpoints, and prevention and cure of the disease in the NOD mouse. Over the last two decades, vast numbers of molecules necessary for maintaining immunoregulatory mechanisms and others that facilitate their subversion have been identified. Targeting these molecules identified more than 250 interventions capable of preventing the disease in the NOD mouse. Some, like treatment with anti-CD3[14] and anti-CD20[15] reversed the disease in as many as 30%-50% of new-onset cases, raising hope of developing strategies to reverse disease in newly diabetic patients. Consequently, in the last few years, clinical trials have been conducted to test efficacy of several molecules including anti-CD3 and anti-CD20.
Figure 1.
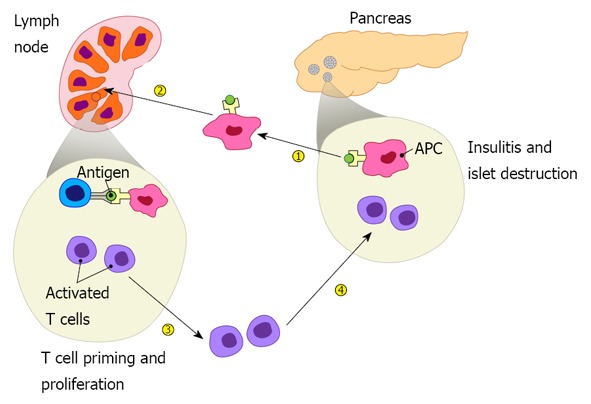
Pathogenesis of type 1 diabetes includes four major steps: islet autoantigens are picked up by antigen presenting cells from the pancreas, which then migrate to draining lymph nodes and present the autoantigens to autoreactive T cells, leading to their priming. Activated autoreactive T cells undergo proliferation, differentiation, and acquire homing molecules that direct them to the pancreas where they infiltrate the islets resulting in insulitis and β cell destruction.
Sobering reality facing translation of preclinical data into effective immunotherapeutics and ways forward
Translating immunotherapies found effective in preclinical studies into human therapies is proving challenging[16], at least for now. Several high profile clinical trials including phase III have failed to demonstrate significant efficacy for all those tested[17,18]. The disappointing results in the clinic are forcing a retreat to drawing boards and generating second thoughts about whether the NOD mouse has surpassed its life expectancy as a research model and even the value of NOD mice in predicting and evaluating immunotherapy for T1D. It is easy to lay the blame on biologic differences between humans and mice, accentuated by more than 60 million years since their divergence into two species that differ in size, lifespan, and lifestyle (habitat/environment). The immune system in humans and mice, however, are generally quite similar, and with few notable exceptions, most paradigms translate well between them. Thus, the intangible efficacy of modalities such as anti-CD3 in humans is not entirely justified by biologic differences between the two species.
We argue that environmental factors play a dominant, if not the dominant role, in subverting therapeutic efficacy of modulators acting alone or in synergy with genetic factors[19,20]. This is acutely evident in the NOD mouse itself. For instance, the variability of anti-CD3 efficacy in reversing new-onset hyperglycemia ranges from about 30%-80% in newly diabetic NOD mice housed in the same facility[14,21] and mostly likely mice in the same cage responded differently. The low efficacy in NOD mice given the extremely small variations in their genetic makeup and exogenous influence of the environment suggests that treating the same mice under virtually identical conditions, the treatment would be successful only once out of at least two attempts. Applying the comparison to patients with markedly different genetic backgrounds, types of food, environment, and microbiota, the odds of success would be extremely low. Therefore, there is still much to be learned in the NOD mouse to uncover causes of variability on rate of disease onset, timing and response to treatment. In addition, understanding why females are more susceptible to disease than males[22-24] and why NOD mice housed in conventional facilities do not develop disease remains unclear[25]. It will also be important to understand why inactivation of molecules such as Fas death receptor or its ligand prevents disease in NOD mice[13,16,26-29]. Understanding mechanisms underlying these observations would provide important clues that could potentially facilitate the development of therapeutic strategies with high efficacy rates that are effective in both mice and men.
ACKNOWLEDGMENTS
We apologize for not having the opportunity to cite all seminal work describing islet reactive clones and transgenic mice expressing islet antigens. We thank Catherine E. Kiefe, MLA, at The Department of Art as Applied to Medicine at Johns Hopkins University for illustration of Figure 1.
Footnotes
Supported by The NIH (1R56AI099027 and 1R01AI099027-01); and American Heart Association (10GRNT4200003)
P- Reviewer Ilangumaran S S- Editor Zhai HH L- Editor A E- Editor Lu YJ
References
- 1.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken Dobutsu. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 2.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 3.Haskins K. Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol. 2005;87:123–162. doi: 10.1016/S0065-2776(05)87004-X. [DOI] [PubMed] [Google Scholar]
- 4.Haskins K, Wegmann D. Diabetogenic T-cell clones. Diabetes. 1996;45:1299–1305. doi: 10.2337/diab.45.10.1299. [DOI] [PubMed] [Google Scholar]
- 5.Miller BJ, Appel MC, O’Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol. 1988;140:52–58. [PubMed] [Google Scholar]
- 6.Phillips JM, Harach SZ, Parish NM, Fehervari Z, Haskins K, Cooke A. Nondepleting anti-CD4 has an immediate action on diabetogenic effector cells, halting their destruction of pancreatic beta cells. J Immunol. 2000;165:1949–1955. doi: 10.4049/jimmunol.165.4.1949. [DOI] [PubMed] [Google Scholar]
- 7.Bendelac A, Carnaud C, Boitard C, Bach JF. Syngeneic transfer of autoimmune diabetes from diabetic NOD mice to healthy neonates. Requirement for both L3T4+ and Lyt-2+ T cells. J Exp Med. 1987;166:823–832. doi: 10.1084/jem.166.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 9.Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. 1988;37:1444–1448. doi: 10.2337/diab.37.10.1444. [DOI] [PubMed] [Google Scholar]
- 10.Nikoopour E, Sandrock C, Huszarik K, Krougly O, Lee-Chan E, Masteller EL, Bluestone JA, Singh B. Cutting edge: vasostatin-1-derived peptide ChgA29-42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol. 2011;186:3831–3835. doi: 10.4049/jimmunol.1003617. [DOI] [PubMed] [Google Scholar]
- 11.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.André I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohamood AS, Guler ML, Xiao Z, Zheng D, Hess A, Wang Y, Yagita H, Schneck JP, Hamad AR. Protection from autoimmune diabetes and T-cell lymphoproliferation induced by FasL mutation are differentially regulated and can be uncoupled pharmacologically. Am J Pathol. 2007;171:97–106. doi: 10.2353/ajpath.2007.070148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutfreund R, Hamad AR Immunotherapy for Type 1 Diabetes: Necessity, Challenges and Unconventional Opportunities. Type 1 Diabetes-Pathogenesis, Genetics and Immunotherapy. In: Wager D, editor. InTech: 409-424 [Google Scholar]
- 17.Staeva TP, Chatenoud L, Insel R, Atkinson MA. Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes. 2013;62:9–17. doi: 10.2337/db12-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michels AW, von Herrath M. 2011 Update: antigen-specific therapy in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2011;18:235–240. doi: 10.1097/MED.0b013e32834803ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wicker LS, Todd JA, Peterson LB. Genetic control of autoimmune diabetes in the NOD mouse. Annu Rev Immunol. 1995;13:179–200. doi: 10.1146/annurev.iy.13.040195.001143. [DOI] [PubMed] [Google Scholar]
- 20.Chervonsky A. Innate receptors and microbes in induction of autoimmunity. Curr Opin Immunol. 2009;21:641–647. doi: 10.1016/j.coi.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, von Herrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–1381. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivakine EA, Fox CJ, Paterson AD, Mortin-Toth SM, Canty A, Walton DS, Aleksa K, Ito S, Danska JS. Sex-specific effect of insulin-dependent diabetes 4 on regulation of diabetes pathogenesis in the nonobese diabetic mouse. J Immunol. 2005;174:7129–7140. doi: 10.4049/jimmunol.174.11.7129. [DOI] [PubMed] [Google Scholar]
- 23.Ivakine EA, Mortin-Toth SM, Gulban OM, Valova A, Canty A, Scott C, Danska JS. The idd4 locus displays sex-specific epistatic effects on type 1 diabetes susceptibility in nonobese diabetic mice. Diabetes. 2006;55:3611–3619. doi: 10.2337/db06-0758. [DOI] [PubMed] [Google Scholar]
- 24.Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339:1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- 25.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–444. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- 27.Desbarats J, Duke RC, Newell MK. Newly discovered role for Fas ligand in the cell-cycle arrest of CD4+ T cells. Nat Med. 1998;4:1377–1382. doi: 10.1038/3965. [DOI] [PubMed] [Google Scholar]
- 28.Atkinson MA, Bluestone JA, Eisenbarth GS, Hebrok M, Herold KC, Accili D, Pietropaolo M, Arvan PR, Von Herrath M, Markel DS, et al. How does type 1 diabetes develop?: the notion of homicide or β-cell suicide revisited. Diabetes. 2011;60:1370–1379. doi: 10.2337/db10-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Herrath M. Combination therapies for type 1 diabetes: why not now? Immunotherapy. 2010;2:289–291. doi: 10.2217/imt.10.23. [DOI] [PubMed] [Google Scholar]