

Abstract
Here we report an approach to the design and production of antibody/ligand pairs, to achieve functional affinity far greater than avidin/biotin. Using fundamental chemical principles, we have developed antibody/ligand pairs that retain the binding specificity of the antibody, but do not dissociate. Choosing a structurally characterized antibody/ligand pair as an example, we engineered complementary reactive groups in the antibody binding pocket and the ligand, so that they would be in close proximity in the antibody/ligand complex. Cross-reactions with other molecules in the medium are averted because of the low reactivity of these groups; however, in the antibody/ligand complex the effective local concentrations of the complementary reactive groups are very large, allowing a covalent reaction to link the two together. By eliminating the dissociation of the ligand from the antibody, we have made the affinity functionally infinite. This chemical manipulation of affinity is applicable to other biological binding pairs.
The specific recognition and binding of biological molecules by antibodies is fundamental to many phenomena in biology and medicine. Antibodies associate with their ligands in a reversible manner, to form complexes held together by noncovalent molecular interactions. Natural antibodies are multivalent, having at least two identical ligand-binding sites; this provides a substantial increase in their ability to bind preferentially at sites, such as cell or virus surfaces, that present multiple copies of ligand attached to a single particle (1). Natural antibodies that bind to soluble monovalent ligands, such as most small molecules, do not share this multivalent advantage. Nor do engineered fragments of antibodies, such as single-chain Fv proteins or Fab fragments, which generally possess only a single ligand-binding site.
The association and binding of a ligand L to a single binding site on its antibody Ab to form a complex C can be written as
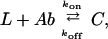 |
where kon (M−1⋅s−1) and koff (s−1) are rate constants. For practical applications it is desirable to produce antibodies that bind very tightly to their ligands, because this permits the assay of minute concentrations or extends the residence of antibodies on the surfaces of targeted cells. Approaches based on molecular biology have been developed to increase the binding affinity (KA = kon/koff) of an engineered antibody fragment for its ligand (2–8). They generally succeed in reducing koff, while having little effect on kon, functionally increasing the affinity. Antibody-ligand complexes having KA ≥ 1010 can be achieved in this way (9).
An alternative binding pair involves association of the small molecule biotin with the protein avidin. The biotin-avidin complex is unusually stable (KA ≥ 1014) and does not dissociate into its components at a significant rate under normal circumstances (10). This has led to the widespread use of the biotin-avidin (or biotin-streptavidin) linkage to immobilize target molecules for applications in vitro and in vivo; highly promising results for cancer therapy have recently been reported by Axworthy et al. (11). Recently, a tight-binding trivalent vancomycin/d-Ala-d-Ala complex also has been characterized (12). In contrast to avidin, which binds only biotin, and vancomycin, which binds only the dipeptide d-Ala-d-Ala (or close analogs), antibodies can be prepared to bind specifically to a staggering variety of ligands.
A slow rate of dissociation is particularly important for in vivo targeting applications, where a targeted therapeutic drug (or enzyme or radionuclide) requires a long period on the target to be effective (11), but slow dissociation is also important for in vitro procedures such as immunoassays. In typical cases, a (monovalent) engineered antibody fragment against a small molecule will remain bound to its ligand for an average period of a few minutes to a few hours (13, 14), whereas a biotin molecule will remain bound to streptavidin with a half-life of about 35 h (10).
The surest way to prolong the lifetime of a complex is to make a covalent bond between its components. Covalent attachment of protein to ligand can prevent dissociation entirely, extending the life of the complex infinitely (koff = 0 ∴ KA → ∞). Other workers have attached cofactors covalently near the active site of a catalytic antibody (e.g., ref. 15).
We reasoned that it should be possible to prepare antibody/ligand pairs that possess the binding specificity of antibodies, but do not dissociate. This might be achieved by taking advantage of the slow dissociation of the correct ligand from the antibody combining site, to form a covalent bond during the lifetime of the complex (Fig. 1). To explore the practical aspects of this concept, we chose the anti-chelate antibody CHA255 (Fig. 2), which possesses high affinity for (S)-benzyl-EDTA-indium chelates (KA ≈ 4 × 109) and exquisite specificity for these small molecules (16). Using the gene for CHA255 and the crystal structure of the antibody-ligand complex (17), we engineered chemically reactive sites near the ligand-binding site of the antibody by substituting cysteine at either position 95 or 96 (Kabat position 93 or 94) of the light chain (Fig. 2). These residues, which are in complementarity determining region 3 of the light chain, were chosen because their side chains (i) are not exposed on the outer surface of the antibody, (ii) do not have any direct contacts with the bound ligand, and (iii) lie within a few angstroms of the para substituent of the ligand in the complex. Thus we expected to produce stable mutant proteins that retain the binding selectivity of the antibody.
Figure 1.

Requirements for an antibody with infinite affinity. (a) When the antibody and ligand are apart, their complementary reactive groups do not react significantly with other molecules in the medium. (b) When the ligand binds to the antibody, the effective concentrations of their complementary reactive groups are sharply elevated, and a covalent link is formed. (c) The covalently linked antibody/ligand complex cannot dissociate.
Figure 2.

Crystal structure of antibody/ligand complex, adapted from Love et al. (17). Two residues in the wild-type antibody that are not directly involved in ligand binding but are favorably located close to the para-substituent of the ligand (yellow) are light-chain residues S95 and N96 (Kabat positions 93 and 94).
As reactive ligands, we prepared a small set of chelates bearing electrophilic para substituents (Fig. 3); these were designed to form stable thioether bonds on reaction with the cysteine side chain, either by Michael addition or by nucleophilic substitution. To be most useful, the electrophilic ligands should be stable in biological media; it is important that they not react prematurely with common biological nucleophiles such as the cysteine on glutathione or in albumin (18). Because of the high local concentrations of cysteine (nucleophile) and ligand (electrophile) expected in the antibody-ligand complex, weak electrophiles were chosen. The effect of local concentration can be appreciated by comparing rate constants for the same chemical reaction between two separate reactants (bimolecular) and between two reactive groups joined by a link (unimolecular) (19) ![]() :
:
Figure 3.

Ligands containing electrophilic substituents, tested with engineered Fab fragments S95C and N96C. BABE, bromoacetamidobenzyl-EDTA; CABE, chloroacetamidobenzyl-EDTA; CpABE, (S)-1-p-chloropropionamidobenzyl-EDTA.
The effective local concentration of A in the presence of B in the unimolecular reaction may be defined = k1/k2 (units: moles/liter, M). The effective local concentration defined in this way can be enormous (>105 M).
Materials and Methods
Materials.
CHA255 hybridoma cells were a gift from David Goodwin, Palo Alto Veterans Administration Hospital, Palo Alto, CA. Primers were synthesized by Operon Technologies, Alameda, CA. Oligotex Direct mRNA Extraction kit was purchased from Qiagen, Chatsworth, CA. Ig Prime kit was purchased from Novagen. S2 cells (CRL-1963) were supplied by the American Type Culture Collection. FCS (heat inactivated) and Excell 420 serum-free media were purchased from JRH Biosciences, Lenexa, KS. Schneider cell media were purchased from Sigma. The vectors pMT/Bip/V5/His and pCoHygro, anti-V5-horseradish peroxidase conjugate, and hygromycin B were purchased from Invitrogen. NpC3tt was a gift from Carlos Barbas, Scripps Research Institute, La Jolla, CA. Restriction enzymes and T4 ligase were purchased from New England Biolabs. Goat anti-human-Kappa antibody was purchased from Southern Biotechnology Associates. Aminolink resin and Slow-3,3′,5,5′ tetramethyl benzidine were supplied by Pierce. Carrier-free 111InCl3 was purchased from Nordion, Vancouver, BC. Pure water (18 MΩ-cm) was used throughout.
Molecular Modeling.
The crystal structure for the native CHA255 Fab domain in complex with indium p-(N-2-hydroxyethyl thioureido)benzyl-EDTA (PDB ID code: 1IND) was used as the starting structure for all manipulations. Using insight ii (Molecular Simulations, San Diego) the structure was manipulated by using the biopolymer and modeler modules to introduce cysteine mutations in the protein and for structural manipulation of the ligand.
Cloning and Expression of Chimeric CHA255 Mutants.
CHA255 hybridoma cells were grown in RPMI 1640 medium supplemented with 10% FCS and used as a source of genetic template. Messenger RNA was extracted with the Oligotex Direct mRNA Extraction kit according to the manufacturer's protocol. Complementary DNA synthesis and PCR amplification of the variable domain genes were done by using the Ig Prime kit and ligated into pT7Blue per manufacturer's protocol. Site-directed substitution of cysteine at positions 95 (S95C) and 96 (N96C) of the light chain was done via the method of Ito et al. (20) using the T7 and U19 primers of the pT7 vector system and the primer KxbaI (CTGCAGGTCGACTGTAGAGGATCTACTAGT) and the mutagenesis primers S95C (ATACCCAGAGGTTGCAGTACCATAGAGCAC) and N96C (ATACCCAGAGGCAGCTGTACCATAGAGCAC). Thus, plasmids pTVlS95CCha255 and pTVlN96CCha255 encoding the variable light-chain domains of CHA255 with the mutations at positions 95 and 96 were produced. For expression of Fab molecules, chimeric constructs containing the variable regions of CHA255 with the constant regions of human antitetanus toxoid were constructed by two-step overlap extension (21) from the vectors pTVHCha255, pTVlCha255, pTVlS95CCha255, pTVlN96CCha255, and npC3tt. The primers used to amplify the full chimeric gene contained BglII and XbaI restriction sites for introduction into the expression cassette of pMT/Bip/V5His for expression of the chimeric Fab molecules in Drosophila S2 cells. All chimeric constructs were sequenced completely. The resulting plasmids pMTBipVlCha/tt, pMTBipVlS95CCha/tt, and pMTBipVlN96CCha/tt, encoding the native and mutant chimeric light-chain domains and pMTBipVHCha/tt/V5His, encoding the chimeric heavy-chain domain, were cotransfected in equimolar ratio into exponentially growing cultures of S2 cells by using the calcium phosphate coprecipitation method. Protein expression was induced by addition of CuSO4 to a final concentration of 500 μM. For production of stable cell lines additional cotransfections with the selection vector pCohygro encoding the hygromycin B phosphotransferase gene were carried out, followed by 3–4 weeks of selection with 300 μg/ml of hygromycin B in complete medium (22).
Synthesis of Aminobenzyl-EDTA Derivatives.
Electrophilic derivatives were synthesized by acylation of t-butyl-aminobenzyl-EDTA in CH2Cl2 with the appropriate acid halide. The resulting t-butyl protected chelate was purified by flash chromatography on silica, unmasked by deprotection with neat trifluoroacetic acid, and characterized by MS, NMR, and metal binding assay. Details will be reported elsewhere.
Labeling of Mutant Fabs with 111In Chelates.
For initial surveys of protein properties, electrophilic benzyl-EDTA chelates such as (S)-1-p-acrylamidobenzyl-EDTA (AABE) (Fig. 3) were loaded with carrier-free 111In and mixed with complete media from the respective S2 cultures expressing each Fab. For kinetic studies, aliquots taken at various times were applied to gel filtration columns, and the protein eluant was analyzed by reducing SDS/PAGE, with quantification of labeled protein by PhosphorImager. Competition for binding sites was studied in the same fashion except that competitor nonradioactive indium-, iron-, cadmium-, scandium- or gallium-(S)-1-p-aminobenzyl-EDTA (ABE) chelates were added to a final concentration of 10 μM and then a trace amount of 111In-AABE was added.
Competitive ELISA.
Benzyl-EDTA-indium was immobilized in 96-well microtiter plates by conjugation of isothiocyanatobenzyl-EDTA to human serum albumin at a ratio of 2:1 as determined by 57Co metal binding assay (23) and loaded with indium. The conjugate was adsorbed to the plates in coating buffer (50 mM carbonate, pH 9.6) followed by blocking of remaining adsorption sites with 1% (wt/vol) BSA solution. ABE was loaded with indium, iron, cadmium, scandium, and gallium by addition of 1.5 eq of metal in 0.05 M HCl to ABE in water, analyzed for completeness of chelation by competition with standardized 57Co and adjusted to pH 6. The respective metal-ABE solutions were diluted over a range of 6 orders of magnitude and applied to the prepared plates followed by S95C Cha Fab in 20 mM Tris (pH 7.4), 50 mM NaCl, 1 mM EDTA to a final concentration of 10 nM. The competition between the solution phase chelates and those immobilized took place over a 1-h incubation at which time the plates were washed extensively with phosphate buffer saline with 0.05% Tween (PBST). Bound Fab was detected with anti-V5-horseradish peroxidase conjugate and 3,3′,5,5′ tetramethyl benzidine was used as the colorimetric substrate and quantified after oxidation with 1 M H2SO4 at 450 nM.
Results and Discussion
To confirm the desired lack of reactivity with molecules normally found in biological media, we labeled para-substituted derivatives of benzyl-EDTA bearing chloroacetamido, chloropropionamido, acrylamido, or amino groups (Fig. 3) with 111In and incubated them with mouse ascites fluid containing native mAb CHA255. The acrylamidobenzyl-EDTA(111In) (AABE) chelate showed practically no reaction with any of the proteins in this medium, as did the nonreactive aminobenzyl-EDTA(111In) (ABE) chelate, whereas the more reactive chloroacetamidobenzyl-EDTA(111In) and bromoacetamidobenzyl-EDTA(111In) chelate reacted significantly with several proteins during a 2-hr incubation at 37°C.
To investigate whether either the S95C or the N96C Fab would bind irreversibly to its target, we incubated 111In-labeled chelates bearing electrophilic groups at physiological pH and temperature with raw tissue culture medium from the cells expressing these recombinant proteins. The culture medium contained 10% FCS, representative of typical biological media. This specific covalent attachment of an 111In-chelate to a mutant Fab—under conditions where it does not covalently attach to other molecules such as albumin present in the culture medium, or to the native Fab (it binds reversibly to the native Fab)—shows the potential value of this procedure.
The samples were analyzed by denaturing gel electrophoresis (SDS/PAGE). Separation under reducing and denaturing conditions on SDS/PAGE separates the light chain from the heavy chain of each Fab, functionally destroying the antibody-binding pocket. Chelates bound to the Fab but not covalently linked will dissociate because the antibody-binding pocket is no longer folded. Unbound chelates do not migrate with the antibody chains. However, a chelate that not only bound to a Fab but also covalently linked will be attached to the Fab light chain and migrate with it on SDS/PAGE. We saw this result with the Fab S95C; nucleophilic Fab S95C reacted equally well with the very electrophilic 111In-chloroacetamidobenzyl-EDTA chelate and with the weakly electrophilic 111In-AABE chelate, but not with electrophilic 111In-(S)-1-p-chloropropionamidobenzyl-EDTA or nonelectrophilic 111In-ABE (Fig. 4). We did not observe any covalent cross-linking between the nonnucleophilic native Fab and any of the electrophilic chelates. Nor did we observe cross-links when the nucleophilic Fab N96C was studied; apparently the orientation of the Cys in position 96 is inferior to position 95 for reaction with these ligands.
Figure 4.

Conjugation of electrophilic 111In-benzyl-EDTA derivatives with engineered Fab S95C. Phosphorimage of 10–20% SDS/PAGE gel of samples of complete culture media incubated with 111In labeled (A) chloroacetamidobenzyl-EDTA, (B) (S)-1-p-chloropropionamidobenzyl-EDTA, (C) AABE, and (D) ABE.
We conclude that an appropriately structured electrophilic ligand and an appropriately placed cysteine side chain are required for efficient covalent attachment. Examination of the protein-ligand structures in the Protein Data Bank suggests that a similar approach can be applied in a number of other cases.
The mAb CHA255 has exquisite selectivity for binding to benzyl-EDTA chelates bearing different chelated metals (In>Fe>Cd>Sc>Ga). To be assured that mutation of residue 95 in the Fab binding site had no deleterious effects on the selectivity of the Fab for benzyl-EDTA complexes we analyzed the metal selectivity of the S95C mutant Fab by competitive ELISA (Fig. 5a). The concentration of each competitor that is effective at blocking the attachment of S95C Fab to the plate is related to the competitor's affinity for binding to the antibody and is practically identical to the native antibody under the same conditions. The present ELISA results show that either wild-type or S95C mutant antibody favors binding to indium chelates relative to others by free energy differences (ΔΔG, kJ/mol) of ≈7.6 (FeIII), 30 (CdII), 34 (ScIII), or 35 (GdIII), in reasonable agreement with previous results determined by less precise methods (16). To further confirm that S95C Fab retained the ligand-binding specificity, we performed a competition assay to see whether nonradioactive ABE-indium, -iron, -cadmium, -scandium, or -gallium could block the covalent attachment of 111In-AABE. As shown in Fig. 5b, excess ABE-indium effectively blocks the reaction, whereas the other metal chelates are less effective, in order of their relative affinities.
Figure 5.

Demonstration that engineered Fab S95C retains the ligand-binding selectivity of the parent antibody. (a) Competition ELISA curves. Different metal-ABE chelates compete with an immobilized indium chelate for binding Fab S95C. (b) Competitive binding of a series of metal-ABE chelates, measured by blocking the covalent attachment of 111In-AABE to Fab S95C. SDS/PAGE gel bands show increased labeling of the light chain with decreasing competition.
We explored the rate of covalent attachment of the AABE-111In chelate to the S95C mutant, by monitoring the extent of reaction as a function of time. The covalent attachment was 50% complete in ≈10 min at 22°C (Fig. 6). This is shorter than the typical 50- to 100-min bound half-lives of complexes between the native CHA255 antibody and nonelectrophilic ligands of similar structure (13), implying that when AABE-indium binds to S95C, it should form a covalent bond with high efficiency.
Figure 6.

(a) Phosphorimage of a representative SDS/PAGE assay of extent of conjugation of 111In-AABE to Fab S95C. (b) Kinetics of formation of the covalent bond between bound 111In-AABE and Fab S95C, at 22°C, pH 7.4.
These results provide the foundation for a generation of engineered binding pairs possessing the specificity characteristic of mAbs along with unprecedented strength of binding to their specific targets. Besides the pretargeted delivery of radiation (11), these molecules could find many applications in biology and medicine, in vitro and in vivo. They should lead to more stable biosensors, more sensitive detection methods, improved targeting of effector molecules to specific cells for imaging or therapy, and other innovations.
Acknowledgments
We thank Dr. David Goodwin and Chung Song for gifts of material and helpful discussions. This work was supported by National Institutes of Health Research Grant CA16861 to C.F.M. The communicating member is a member of the Scientific Advisory Board of Lexrite Labs.
Abbreviations
- AABE
(S)-1-p-acrylamidobenzyl-EDTA
- ABE
(S)-1-p-aminobenzyl-EDTA
References
- 1.Pluckthun A, Pack P. Immunotechnology. 1997;3:83–105. doi: 10.1016/s1380-2933(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 2.Hoogenboom H R. Trends Biotechnol. 1997;15:62–70. doi: 10.1016/S0167-7799(97)84205-9. [DOI] [PubMed] [Google Scholar]
- 3.Rader C, Barbas C F., 3rd Curr Opin Biotechnol. 1997;8:503–508. doi: 10.1016/s0958-1669(97)80075-4. [DOI] [PubMed] [Google Scholar]
- 4.Crameri A, Cwirla S, Stemmer W P C. Nat Med. 1996;2:100–102. doi: 10.1038/nm0196-100. [DOI] [PubMed] [Google Scholar]
- 5.Schier R, Bye J, Apell G, McCall A, Adams G P, Malmqvist M, Weiner L M, Marks J D. J Mol Biol. 1996;255:28–43. doi: 10.1006/jmbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Wiesmann C, Fuh G, Li B, Christinger H W, McKay P, de Vos A M, Lowman H B. J Mol Biol. 1999;293:865–881. doi: 10.1006/jmbi.1999.3192. [DOI] [PubMed] [Google Scholar]
- 7.Yang W-P, Green K, Pinz-Sweeney S, Briones A T, Burton D R, Barbas C F I. J Mol Biol. 1995;254:392–403. doi: 10.1006/jmbi.1995.0626. [DOI] [PubMed] [Google Scholar]
- 8.Boder E T, Midelfort K S, Wittrup K D. Proc Natl Acad Sci USA. 2000;97:10701–10705. doi: 10.1073/pnas.170297297. . (First Published September 12, 2000; 10.1073/pnas.170297297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shreder K. Methods. 2000;20:372–379. doi: 10.1006/meth.1999.0929. [DOI] [PubMed] [Google Scholar]
- 10.Chilkoti A, Stayton P S. J Am Chem Soc. 1995;117:10622–10628. [Google Scholar]
- 11.Axworthy D B, Reno J M, Hylarides M D, Mallett R W, Theodore L J, Gustavson L M, Su F-M, Hobson L J, Beaumier P L, Fritzberg A R. Proc Natl Acad Sci USA. 2000;97:1802–1807. doi: 10.1073/pnas.97.4.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao J, Lahiri J, Isaacs L, Weis R M, Whitesides G M. Science. 1998;280:708–711. doi: 10.1126/science.280.5364.708. [DOI] [PubMed] [Google Scholar]
- 13.Meyer D L, Fineman M, Unger B W, Frincke J M. Bioconjugate Chem. 1990;1:278–284. doi: 10.1021/bc00004a009. [DOI] [PubMed] [Google Scholar]
- 14.Winklmair M, Schuetz A J, Weller M G, Niessner R. Fresenius J Anal Chem. 1999;363:619–624. [Google Scholar]
- 15.Tanaka F, Lerner R A, Barbas C F., III . Chem. Commun. 1999. , 1383–1384. [Google Scholar]
- 16.Reardan D T, Meares C F, Goodwin D A, McTigue M, David G S, Stone M R, Leung J P, Bartholomew R M, Frincke J M. Nature (London) 1985;316:265–268. doi: 10.1038/316265a0. [DOI] [PubMed] [Google Scholar]
- 17.Love R, Villafranca J E, Aust R, Nakamura K, Jue R, Jr, J. M, Radharkrishnan R, Butler W. Biochemistry. 1993;32:10950–10959. doi: 10.1021/bi00092a004. [DOI] [PubMed] [Google Scholar]
- 18.Lenter C, editor. Geigy Scientific Tables. Basel: Ciba-Geigy; 1984. [Google Scholar]
- 19.Fersht A. Enzyme Structure and Mechanism. 2nd Ed. New York: Freeman; 1985. pp. 56–63. [Google Scholar]
- 20.Ito W, Ishiguro H, Kurosawa Y. Gene. 1991;102:67–70. doi: 10.1016/0378-1119(91)90539-n. [DOI] [PubMed] [Google Scholar]
- 21.Pont-Kingdon G. BioTechniques. 1994;16:1010–1011. and erratum (1995) 18, 636. [PubMed] [Google Scholar]
- 22.van der Straten A, Johansen H, Rosenberg M, Sweet R. Methods Mol Cell Biol. 1989;1:1–8. [Google Scholar]
- 23.Meares C F, McCall M J, Reardan D T, Goodwin D A, McTigue M. Anal Biochem. 1984;142:68–78. doi: 10.1016/0003-2697(84)90517-7. [DOI] [PubMed] [Google Scholar]