

Abstract
To date, two maternal-effect genes have been shown to have causative roles in recurrent hydatidiform moles (RHMs); NLRP7 that is mutated in 48–60% of patients with RHMs and C6orf221 (HUGO-approved nomenclature is now KHDC3L), a recently identified gene, that is mutated in 14% of patients with RHMs who are negative for NLRP7 mutations. We sequenced KHDC3L in 97 patients with RHMs and reproductive loss who are mostly negative for NLRP7 mutations. We identified three unrelated patients, each homozygous for one of the two protein-truncating mutations, a novel 4-bp deletion resulting in a frameshift, c.299_302delTCAA, p.Ile100Argfs*2, and a previously described 4-bp deletion, c.322_325delGACT, p.Asp108Ilefs*30, transmitted on a shared haplotype to three patients from different populations. We show that five HM tissues from one of these patients are diploid and biparental similar to HMs from patients with two defective NLRP7 mutations. Using immunofluorescence, we show that KHDC3L protein displays a juxta perinuclear signal and colocalizes with NLRP7 in lymphoblastoid cell lines from normal subjects. Using cell lines from patients, we demonstrate that the KHDC3L mutations do not change the subcellular localization of the protein in hematopoietic cells. Our data highlight the similarities between the two causative genes for RHMs, KHDC3L and NLRP7, in their subcellular localization, the parental contribution to the HM tissues caused by them, and the presence of several founder mutations and variants in both of them indicating positive selection and adaptation.
Keywords: KHDC3L, 4-bp deletion, diploid biparental hydatidiform mole, NLRP7, colocalization
Introduction
Hydatidiform mole (HM) is an aberrant human pregnancy with absence of, or abnormal embryonic development, hydropic degeneration of chorionic villi, and excessive proliferation of the trophoblast.1 Sporadic HMs are common, not recurrent, and affect 1 in 600 pregnancies (all forms combined) in western countries2 but have higher frequencies in developing countries.3, 4 Recurrent HMs (RHMs) affect 1–6% of women with sporadic HMs5, 6, 7, 8 and occur either in patients with no family history of HMs (singleton cases) or occasionally in sisters or related females from the same family (familial cases). Histologically, HMs can be categorized into two types, complete HM (CHM) and partial HM (PHM). CHMs are characterized by excessive trophoblastic proliferation and absence of fetal tissues beside the chorionic villi. PHMs are characterized by less pronounced trophoblastic proliferation and may contain embryonic tissues.1
Two maternal-effect genes responsible for RHMs are known, NLRP7 and KHDC3L. NLRP7 is mutated in 88% and 60% of analyzed familial and singleton cases of RHMs, respectively.9, 10, 11, 12, 13, 14 Recently, we demonstrated that ex vivo lipopolysaccharides (LPS)-stimulated peripheral blood mononuclear cells (PBMCs) from patients with mutations and rare variants in NLRP7 have defective interleukin-1 beta (IL-1β) and tumor necrosis factor secretion but normal to higher intracellular levels of pro- and mature-IL-1β.13, 15 The requirement of NLRP7 for normal IL-1β secretion by macrophages was also confirmed in in vitro studies after NLRP7 silencing using small interfering RNA (siRNA).16 Within hematopoietic cells, we showed that NLRP7 colocalizes with the Golgi apparatus and the microtubule organizing center (MTOC) and associates with microtubules suggesting that mutations in this gene may decrease cytokine secretion by affecting microtubule structure and consequently intracellular cytokine trafficking.15
Recently, mutations in a second maternal-effect gene, KHDC3L, were shown to be responsible for RHMs.17 Mutations in this gene were found in 1 familial case and in 2 out of 14 analyzed singleton cases of RHMs (14.2%) that are negative for NLRP7 mutations by conventional Sanger sequencing.17 Among the identified KHDC3L mutations, a 4-bp deletion in exon 2, c.322_325delGACT, p.Asp108Ilefs*30, which was found in a homozygous and heterozygous state in one Tunisian and one Iranian patient, respectively.17
Here, we report two KHDC3L mutations, including one novel 4-bp deletion, in two new singleton cases and one familial case of RHMs. We confirm the causality of KHDC3L mutations in 10% of our patients with RHMs who are negative for NLRP7 mutations and exclude the causative role of KHDC3L in our cohorts of other forms of reproductive losses. We show that five HM tissues from one patient with two defective alleles in KHDC3L are diploid and have biparental contribution to their genomes similar to HMs from patients with two defective alleles in NLRP7. We demonstrate that KHDC3L protein colocalizes with NLRP7 in lymphoblastoid cell lines and that this localization is not affected by the two protein-truncating mutations. Our data shed new lights on our understanding of HMs and indicate positive selective pressures underlying their increased incidences from North to South and from West to East.
Materials and methods
Patients
The study was approved by the McGill Institutional Review Board. Patients were ascertained through various collaborative hospitals and written consents were obtained. DNA was isolated from whole blood cells using Flexigene DNA Kit (Qiagen, Toronto, ON, Canada). Mutation analysis was performed by conventional Sanger sequencing on DNAs from 97 patients with no mutations in NLRP7 with the exception of 8 patients who have one NLRP7 defective allele each. Patients were divided into three categories according to their reproductive histories, (i) 29 patients with at least 2 HMs (⩾2 HMs) (Supplementary Table S1), (ii) 27 patients with at least two reproductive losses including 1 HM (⩾2 RL including 1 HM) (Supplementary Table S2) and (iii) 41 patients with at least 3 spontaneous abortions and no HMs (⩾3 SAs) (Supplementary Table S3). DNA from a total of 142 subjects of African origin were used as controls, 50 of African-American origin obtained from the Corriell Institute from the Human Variation Panel (HD50AA) and 92 of from the general Senegalese population. Both cohorts of controls included males and females and no data about their reproductive outcomes were available.
Mutation and haplotype analyses
The three exons of KHDC3L were PCR amplified on genomic DNA as previously described.17 Sequence analysis was performed using DNASTAR and the sequences were compared with reference sequence NM_001017361. DNA mutation numbering is based on cDNA sequence with a ‘c.' symbol before the number and uses the A of the ATG translation initiation start site as nucleotide +1. Protein numbering starts from the initiation codon (codon 1).We refer to non-synonymous variants (NSVs) for DNA changes leading to amino acids changes that are present in the general population. Two additional primers, Ex2del_forward, 5′-CGTGAATCGATTGGACCCTA-3′ and Ex2del_reverse, 5′-GCTCCAGGTAGCCCTATTCC-3′ that amplify a 156-bp PCR fragment spanning the 4-bp deletion in exon 2 were used. A total of 24 single-nucleotide polymorphisms (SNPs) spanning a 22-kb interval surrounding KHDC3L were selected from the NCBI database of SNPs and used to determine the haplotypes carrying mutation c.322_325delGACT.
Flow cytometry and fluorescent microsatellite genotyping of HM tissues
Flow cytometry was performed on one paraffin-embedded tissue containing substantial amount of chorionic villi from each available product of conception (POC) according to standard methods.18 Five serial 8 μm sections were performed on one paraffin block from each POC and stained with hematoxylin and eosin. Pinpoint solution (Zymo Research, Orange, CA, USA) was applied to the areas containing only chorionic villi defined by stereomicroscopy. The tissues were then removed and used to extract DNA that was used in multiplex PCR to amplify 16 highly polymorphic microsatellite loci in a single PCR reaction using PowerPlex 16 HS System (Promega Corporation, Fitchburg, WI, USA). Amplified PCR products were resolved by capillary electrophoresis using an ABI 3130 Genetic Analyzer (Life Science technologies, Carlsbad, CA, USA). The genotype of the HM tissue was compared with that of the patient to determine the parental contribution to the HMs.
Constructs, cell culture and transfection
GFP (PS100010, Origene, Rockville, MD, USA), GFP wild-type wt-KHDC3L (RG215631, Origene) and FLAG-tagged wild-type wt-NLRP7 constructs15 were used for transfections. One day before transfection, HEK293T cells were seeded at a density of 1 × 105 cells per well using four-well chamber slides (Ultident, Montreal, QC, Canada) in 500 μl of Dulbecco's modified Eagle's medium (Invitrogen, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA). The cells were transfected as previously described.15
Immunofluorescence
Immunofluorescence was performed as previously described.15 The following primary antibodies were used: rabbit anti-KHDC3L (1:100; HPA043699, Sigma (Oakville, ON, Canada) prestige raised against the last 70 amino acids at the C-terminus of the protein (C-term)), rabbit anti-KHDC3L from Abgent (Brockville, ON, Canada) (AP11238a) raised against amino acids 27–56 at the N-terminal (N-term) part of the protein, goat anti-NLRP7 (1:100; sc-50642 from Santa Cruz, Santa Cruz, CA, USA) and rabbit anti-NLRP7 (1:100; IMG-6357A from Imgenex, San Diego, CA, USA). The secondary antibodies were Alexa Fluor 568 conjugated donkey anti-rabbit (1:500; A10042, Invitrogen) and Alexa Fluor 488 donkey anti-goat (1:500; A11055, Invitrogen). Finally, the slides were mounted using Vectashield hard-set mounting medium with 4,6-diamidino-2-phenylindol-2-HCl (DAPI; Vector Laboratories, Burlington, ON, Canada). Fluorescence images were captured on an Axioskop 2 plus microscope (Carl Zeiss, Thornwood, NY, USA).
Results
KHDC3L mutations in three unrelated patients and a novel protein-truncating mutation
KHDC3L mutation analysis in a cohort of 29 unrelated patients, 26 singleton and 3 familial cases, each with at least 2 HMs revealed 2 mutations in 3 patients (Table 1). The first mutation was a novel 4-bp deletion, c.299_302delTCAA, p.Ile100Argfs*2, in a homozygous state in patient 481 of African-American origin, who had 7 HMs with 3 partners (Figure 1a). This 4-bp deletion was not found in 142 control subjects, 50 of African-American origin who were screened by PCR amplification and electrophoresis on Nusieve:agarose (3:1) gel and 92 of Senegalese origin who were screened by DNA sequencing. The second mutation was another 4-bp deletion, c.322_325delGACT, p.Asp108Ilefs*30, previously reported to be causative of RHMs17 and was seen in a homozygous state in two unrelated patients, 654 and 1096 (Figure 2). Patient 654 is of Turkish origin and had 5 HMs from natural conceptions and two failed cycles of in vitro fertilization followed by pre-implantation genetic screening for aneuploidies. In the two cycles, a total of 11 embryos were analyzed. One embryo showed a monosomy for chromosome 16, another displayed monosomies for chromosomes 1 and 16, and all the other embryos had complex aneuploidies except for one that was transferred to the patient but did not result in a positive pregnancy test. Screening of this mutation in available family members of patient 654 using Nusieve:agarose (3:1)gel electrophoresis and DNA sequencing showed that both her parents and a sister with two normal pregnancies were heterozygous for the mutation while her brother has two normal copies of the KHDC3L gene (Figure 2). Patient 1096 is of Indian origin and had 10 RHMs and is from a familial case of RHMs. Screening the other family members revealed the same mutation, c.322_325delGACT, in a homozygous state in her sister with three RHMs and in a heterozygous state in her two parents, but not in her brother who does not carry any copy of the mutation (Figure 2). The two mutations were not reported in the 1000 Genomes database (www.1000genomes.org), in which DNA variants found in 1488 individuals from 5 different populations, including 245 from sub-Saharan Africa, are listed.
Table 1. KHDC3L mutation analysis in a cohort of 97 patients with recurrent reproductive losses.
| Categories of patients | ⩾2 HMs | ⩾2 RL including 1 HM | ⩾3 SAs |
|---|---|---|---|
| Number of analyzed unrelated patients | 29a | 27 | 41 |
| Number of patients with mutations in KHDC3L | 3 | 0 | 0 |
Indicates that 3 of the analyzed 29 patients were from familial cases, all the others were from singleton cases.
Figure 1.

Pedigree structure of patient 481, reproductive outcomes, mutation analysis, histopathology and parental contribution to five HMs. (a) Mutation analysis showing a novel 4-bp deletion in a homozygous state in patient 481. (b) The upper row shows the ploidy analysis of five of her POCs showing a single diploid peak in each. Microphotographs showing the histopathology of the five HMs and the diagnosis based on independent evaluations performed by two pathologists. From each POC, 4–9 different slides were available. HM, indicates hydatidiform mole and is used when the two pathologists did not reach a conclusion as to whether the HM is partial or complete (CHM). Arrows, indicate abnormal circumferential trophoblastic proliferation around chorionic villi (CV). We note that, in general, trophoblastic proliferation in the five HMs was milder than that seen in common sporadic HMs. (c) Genotypes of the five POCs using the PowerPlex 16 HS System (Promega Corporation) demonstrated the biparental contribution to the five analyzed HMs. Informative alleles showing biparental contribution are shown in bold.
Figure 2.

Pedigree structure, reproductive outcomes and KHDC3L mutation analysis of one singleton (case MoTu98, patient 654) and one familial case of RHMs (MoIn355, patients 1094 and 1096). In both cases, a previously described mutation was identified. IVF, in vitro fertilization; ET, embryo transfer; PGS, pre-implantation genetic screen, HD indicates heteroduplexes seen in heterozygous subjects.
Before the identification of the KHDC3L, a SNP microarray, Illumina 610 Quad (San Diego, CA, USA), had been performed on DNAs from patient 654, her two parents, and unaffected sisters. This analysis revealed a large homozygous region encompassing KHDC3L and spanning a total of 44-Mb, from rs9267522 to rs3777505 in patient 654. Altogether, the SNP microarray data combined with the homozygous state of the mutation are in favor of the causality of KHDC3L mutation in the recurrent HMs in this patient.
A shared small haplotype carrying c.322_325delGACT in KHDC3L in patients of different countries of origin
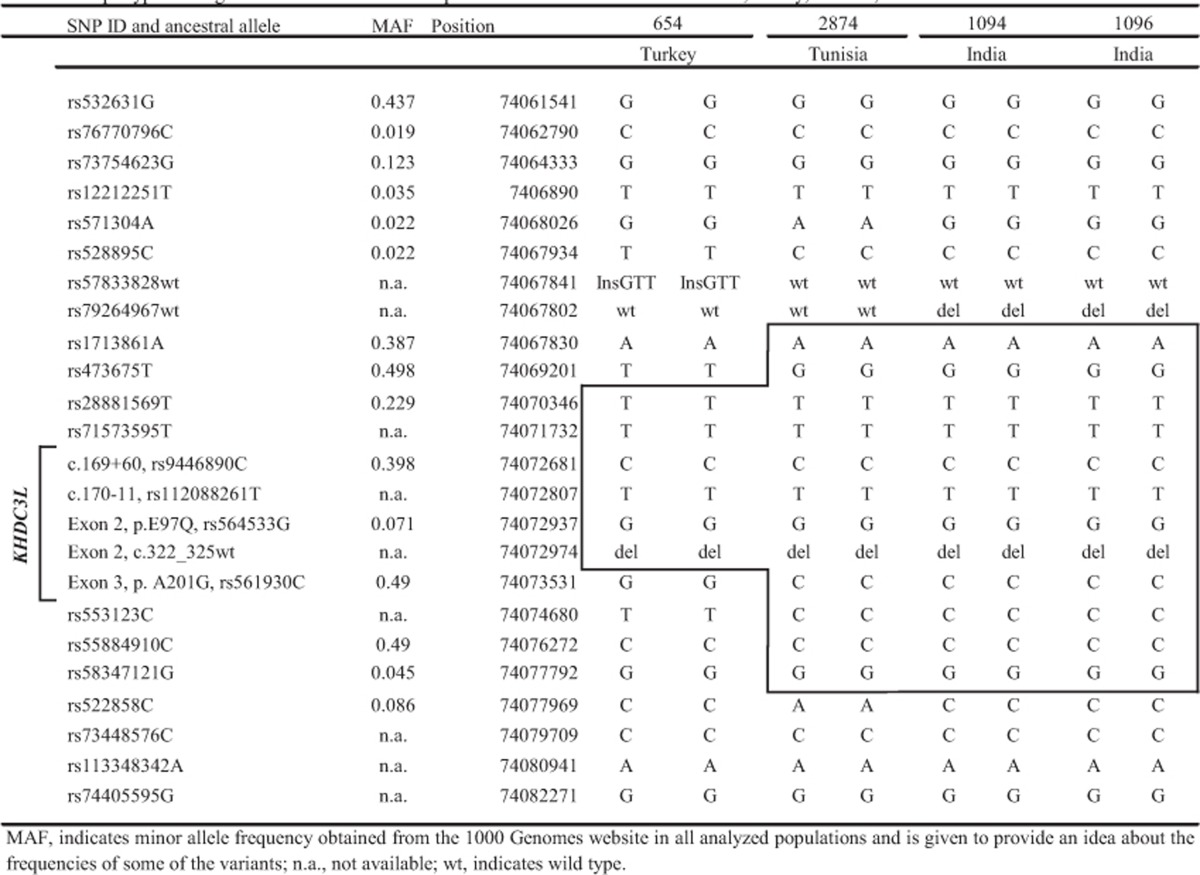
The presence of the same mutation, c.322_325delGACT, in two patients of Tunisian and Iranian origins17 and in two of our patients from different countries, Turkey and India, prompted us to look for shared alleles at markers around the common mutation in our patients and in the previously reported Tunisian patient17 whose DNA was available to us. The analysis of a total of 24 SNPs spanning 21-kb around KHDC3L revealed shared alleles at 5–11 adjacent SNPs around the mutation (Table 2). These data indicate the presence of an old founder effect for the c.322_325delGACT mutation and its inheritance from a common ancestor to the Tunisian, Turkish and Indian patients.
Table 2. Haplotype sharing around KHDC3L between patients from three different countries, Turkey, Tunisia and India.
Absence of KHDC3L mutations or associated variants in patients with sporadic HMs and recurrent spontaneous abortions
To investigate the potential involvement of KHDC3L in sporadic HMs and recurrent spontaneous abortions, we screened this gene in 27 unrelated patients with at least two reproductive losses including one HM (⩾2 RL including 1 HM; Supplementary Table S2), and 41 unrelated patients with at least three spontaneous abortions (⩾3 SAs; Supplementary Table S3). However, we did not find any mutation in these 68 patients.
In the three analyzed categories of patients, only two NSVs in KHDC3L that are reported in public databases were observed, c.289G>C leading to p.E97Q and c.602C>G leading to p.A201G, and were found in the same frequencies in patients and controls indicating that these two NSVs do not predispose our patients for reproductive loss (Table 3).
Table 3. Frequencies of KHDC3L NSVs in the three categories of European patients.
|
rs ID(NSVs)/number of minor allele |
|||
|---|---|---|---|
| Total number of alleles | rs564533C (E97Q) | rs561930G (A201G) | |
| Patients with ⩾2 HMs | 18 | 2 | 9 |
| Patients with ⩾2 RL including 1 HM | 22 | 0 | 13 |
| Patients with ⩾3 SAs | 56 | 6 | 23 |
| Total | 96 | 8 | 45 |
| Frequency in our patients | 0.083 | 0.469 | |
| Frequency in European subjects from the 1000 genomes | 762 | 0.15 | 0.58 |
Abbreviations: HM, hydatidiform mole; NSVs, non-synonymous variants; RL, reproductive loss; SA, spontaneous abortion.
Among the 97 patients included in this study, 8 had one defective allele in NLRP7 and these patients did not have KHDC3L mutations. We note that the four patients with KHDC3L mutations do not have any of the rare NLRP7 NSVs that confer susceptibility for reproductive loss.13
HM tissues from one patient with two defective alleles in KHDC3L are diploid biparental
Flow cytometry analysis on five different POCs from patient 481 (Figure 1b) showed the presence of a single diploid peak demonstrating that the five POCs are diploid (Figure 1b). Fluorescent microsatellite genotyping on the five POCs demonstrated that the analyzed tissues are biparental based on one to seven informative markers, in each POC, where a second allele of non-maternal origin was identified (Figure 1c). These data demonstrate that the five HM tissues from patient 481 with two KHDC3L defective alleles are diploid biparental similar to HMs from patients with two defective NLRP7 alleles.
KHDC3L expression and colocalization with NLRP7 in hematopoietic cells
KHDC3L is reported to be transcribed in a wide range of human tissues and cell lines including hematopoietic cells (http://biogps.org/). PCR after reverse transcription on RNA extracted from EBV-transformed cells from the two patients, 481 and 654, and two controls was performed using exonic primers located in exons 2 and 3. This analysis revealed the amplification of a 284-bp cDNA fragment of the expected size (Supplementary Figure S1) whose identity was also confirmed by DNA sequencing. This demonstrates the transcription of KHDC3L in EBV-transformed cells and the presence of KHDC3L transcripts in cells from the two patients with homozygous protein-truncating mutations. Also, these data indicated that EBV-transformed cells can be used to perform subcellular localization of KHDC3L protein.
Before investigating the subcellular localization of KHDC3L, we first validated a commercially available antibody (Sigma, prestige HPA043699) that detects the C-terminal part of the protein (C-term) using transfected HEK293T cells with a GFP-tagged wild-type KHDC3L cDNA (GFP-wtKHDC3L) construct and simultaneous revelation of the transfected protein with GFP immunofluorescence and the KHDC3L C-term antibody (Figures 3a–d). Using the GFP-wtKHDC3L, the C-term antibody signal overlapped exactly with that of GFP and both revealed a perinuclear staining that was not obtained with the transfected empty vector carrying only GFP (Figures 3e and f). This analysis demonstrated that C-term antibody recognizes the exogenous transfected wild-type KHDC3L protein. We next used the C-term antibody to detect endogenous KHDC3L. This antibody revealed a juxta perinuclear signal that colocalized with NLRP7 protein in EBV-transformed cells from controls (Figures 3g–i). The validation of one NLRP7 antibody (Santa-Cruz, sc-50642) was previously reported15 and additional validations of this antibody (Santa Cruz, sc-50642) and of another one (Imgenex, IMG-6357A) were performed during this study (Supplementary Figure S2). The juxta and perinuclear signal obtained with the C-term KHDC3L antibody on EBV-transformed cells from controls was also confirmed on the same cells with another antibody that detects the N-terminal (N-term) part of KHDC3L (Abgent, AP11238a) (Figures 3j–l). Analysis of EBV-transformed cells from the two patients, 481 and 654, revealed their truncated proteins with the KHDC3L N-term antibody but not with the C-term antibody. With the N-term antibody, the frequency of the signal in patients' cells was lower than in control cells and many cells displayed diffuse signal. In both patients, when present, the signal with the N-term antibody overlapped with that of NLRP7 (Figures 3m and o and s-u). Altogether, our immunofluorescence data demonstrate first that KHDC3L colocalizes with NLRP7 in EBV-transformed cells and second that the two KHDC3L 4-bp deletions identified in our patients do not change the subcellular localization of the protein in hematopoietic cells.
Figure 3.

Sub-cellular localization of KHDC3L and colocalization with NLRP7. (a–d) Simultaneous revelation of GFP-tagged wild-type KHDC3L (GFP-wtKHDC3L) construct with GFP signal (green) and KHDC3L C-terminal (C-term) antibody (Sigma, prestige) (red) in transfected HEK293T cells showing that the antibody recognizes the exogeneous protein found close to the nucleus. DNA was counterstained with DAPI (blue). (e, f) Absence of perinuclear staining in HEK293T cells transfected with the empty vector carrying GFP. (g–i) KHDC3L C-term antibody detected a juxta and perinuclear signal that colocalizes with NLRP7 (green) in EBV-transformed cells from a control subject. (j–l) A similar signal that colocalizes with NLRP7 was also detected by another N-terminal (N-term) KHDC3L antibody (Abgent). (m–x) Presence of KHDC3L signals with the N-term antibody and absence of signals with the KHDC3L C-term antibody in EBV-transformed cells from patients 481 and 654 with protein truncations at positions 100 and 108 amino acids, respectively. The truncated proteins in both patients colocalize with NLRP7 (m–o and s–u).
Discussion
Here we report a novel 4-bp deletion resulting in a frameshift in exon 2 of KHDC3L, c.299_302delTCAA, p.Ile100Argfs*2, in a patient with seven HMs. We also report the identification of a previously reported 4-bp deletion in exon 2, c.322_325delGACT, p.Asp108Ilefs*30, in two sisters and in another unrelated singleton patient with RHMs.
To date and including this study, six unrelated patients with mutations in KHDC3L have been described.17 Of these patients, two have two different DNA substitutions leading to a shift in the initiation codon and four have two different 4-bp deletions leading to frameshift and premature termination of the protein. In the reported patients, the presence of small deletions (20-bp or less) in KHDC3L in four out of six patients is comparatively higher than the average observed in human diseases (15.6%) (http://www.hgmd.org/). In addition, the fact that both deletions occurred in exon 2 and one of them was found in four unrelated patients from different populations, Iranian, Tunisian, Turkish and Indian, prompted us to search for direct repeat DNA, palindromes and mirror repeat sequences that could lead to hotspots of deletions and promote such mutations using publicly available softwares.19 However, we did not find any such elements. On the contrary, haplotype analysis revealed that our Turkish and Indian patients and the previously reported Tunisian patient17 share the same haplotype around the mutation indicating its inheritance from a common ancestor. Although this finding was surprising, it is not inconceivable because the Ottomans conquered Tunisia in 1574 and assimilated it to their Empire for >300 years until the establishment of the French colonization in 1881. Also, geneflow and haplotype sharing through maternal and paternal lineages have been reported between the Iranian, Turkish and Indian populations.20, 21
In our study, we found KHDC3L mutations in about 10% of analyzed patients with at least two HMs. Our finding is in agreement with those reported by Parry et al.17 describing KHDC3L mutations in 14% of analyzed patients and also confirms that KHDC3L is a minor gene responsible for RHMs. In addition, our data demonstrate that KHDC3L, at least in our patients, does not have a causative role in other forms of reproductive losses, ⩾2 RL including 1 HM and ⩾3 SAs. Analysis of additional cohorts of patients with these conditions will allow validating our observations in other populations.
KHDC3L is the second identified maternal-effect gene in humans after NLRP7.17 Maternal-effect genes are a subset of genes whose products, mRNA and proteins, are coded by the maternal genome and are stored in the oocyte to sustain normal postzygotic development until the activation of the fetal genome.22 The maternal-effect of KHDC3L is supported by the facts that one of our patients, 481, had HMs with three different partners, and a second one, patient 654, had a high rate of postzygotic aneuploidies after two cycles of IVF performed at the age of 32. These data are in agreement with our previous observations in some patients with NLRP7 mutations11, 23 and suggest that the two genes might have similar or overlapping functions in early development. This suggestion is corroborated by the sub-cellular localization of endogenous KHDC3L and its colocalization with NLRP7 whose strongest signal localizes to the Golgi apparatus and the MTOC. Along with others, we demonstrated the requirement of wild-type NLRP7 for normal IL-1β secretion by ex vivo LPS-stimulated PBMCs from patients with NLRP7 mutations and by macrophages in which NLRP7 was knocked-down using siRNA studies.15, 16 At the present time, we do not know exactly how defects in NLRP7 cause all the features of HM pregnancies, but we believe that at least two independent events that occur at different times during embryonic development are required for HM manifestation. The first event is an oocyte defect that is leading to an early arrest of the pregnancy before the establishment of a functional fetal vascular system.24 The second event is the retention of the arrested pregnancy and its delayed rejection by the mother until the end of the first trimester. At this point in time, we do not know what causes the early pregnancy arrest, but we believe that the impaired cytokine secretion by patients' cells prevents them from mounting appropriate inflammatory reaction to timely reject these early arrested pregnancies. The delayed rejection of these conceptions, in which fetal vessels do not form inside the chorionic villi, leads to their hydropic degeneration and HM manifestation. Our proposal is in agreement with a recent case where a patient with two NLRP7 defective alleles conceived a healthy baby from a donated oocyte.25 The colocalization of KHDC3L with NLRP7 suggests that KHDC3L may also have a role in the intracellular trafficking and secretion of cytokines that will be interesting to assess in PBMCs from the patients, which unfortunately could not be done in this study because of the lack of accessibility to fresh blood samples from our patients.
To date, the parental contribution to four HM tissues from patients with two defective alleles in KHDC3L, three from patient L114, 26 and one from the Tunisian patient, 2874,27 were reported to be biparental. In this analysis, we demonstrated that five HMs from one patient are diploid by flow cytometry and have biparental contribution to their genomes. In line with these findings, the fact that all characterized HMs to date from patients with two defective NLRP7 alleles, approximately a total of 45 HMs from 30 patients, have been found diploid biparental and indicate common mechanisms leading to HMs caused by mutations in KHDC3L and NLRP7.
HMs is a condition known to display wide geographic variations in its frequencies with up to 10-fold increase in the rates of HMs from West to East and a 2-fold increase from North to South (Figure 4a).3, 4 One of the factors that could explain these gradients is the increased frequencies of some mutations and NSVs predisposing to HMs in Eastern and Southern populations. In line with these data are the presence of the same small haplotype carrying the same mutation in KHDC3L in patients of Middle Eastern and Asian origins. Similarly, we previously reported the presence of several founder mutations in NLRP7 in the Indian, Chinese and Pakistani populations based on haplotype analyses.10 Also, along with others, we previously reported the presence of the same rearrangement and missense in unrelated patients from Egypt and Mexico, respectively.23, 28 Re-reviewing SNPs in the sequenced NLRP7 amplicons in our patients and in those found in patients with identical mutations, reported by our collaborators, revealed more shared haplotypes, not only in patients from regions with high rate of consanguinity, but also in patients from different populations. For instance, mutation p.Arg693Trp in NLRP7 was found in seven patients from six different populations, five of which share the same haplotype 5′ of the mutation, and two others share another haplotype (Supplementary Table S4). In addition, we previously showed that four NLRP7 NSVs, associated with recurrent reproductive loss in Europeans, display gradients of increasing frequencies from Europe to sub-Saharan Africa (Figures 4b–e) with the Tunisian population (Mediterranean) having intermediate frequencies.29 Consistent with these data are the global distribution of the countries of origin of patients with mutations in NLRP7 and in KHDC3L with most of them originating from southern, eastern and Asian countries (Figure 4f). This is further corroborated by the presence of several founder mutations in both NLRP7 and KHDC3L, which is surprising for a condition that prevents the reproduction of women with two defective alleles and indicates some selective advantages for males and/or heterozygous females.
Figure 4.

Global distribution of frequencies of HMs, four NLRP7 NSVs, and countries of origin of unrelated patients with mutations in KHDC3L and NLRP7 genes. (a) Epidemiology of HMs. Countries with low incidences are in white, countries with middle incidences (2-fold increase) are in yellow and those with high incidences (up to 10-fold) are in red. (b–e) Distribution of the minor allele frequencies of four NSVs in NLRP7 according to data from the 1000 Genomes database. Ancestral alleles are indicated by their colors. (f) Distribution of the countries of origin of patients with NLRP7 (black dots) and KHDC3L (red dots) mutations.
We previously showed that LPS-stimulated PBMCs from patients with NLRP7 mutations and rare NSVs have low cytokine secretion. We suggested that this will lower the inflammatory response of the patients and make them more tolerant to the growth of aberrant HM conceptions. By the same token, the low inflammatory response of patients with NLRP7 mutations and NSVs may increase their tolerance for various micro-organisms and could confer selective advantages for heterozygous carriers that may explain the persistence of old mutations and NSVs across several populations. Our data are in agreement with previous reports showing that mutations and susceptibility alleles for autoimmune diseases such as celiac disease and type 1 diabetes have been maintained in human populations because of past positive selection and adaptation.30, 31, 32 Immune response and reproduction are believed to be the main selective pressures that have shaped human evolution.30, 33 Exploring further, the geographic distribution of variants in these two genes will provide a unique model to dissect evolutionary constraints on genes with roles in both immune responses and reproduction.
Acknowledgments
We thank the patients and their family for their cooperation. We thank Dr Ertan Kervancioglu for referring one case. We also thank Li Zhang for technical assistance and Sarah-Slim Diwan for the preparation of the world map. The authors wish to acknowledge the use of the Sequencing Platform of the McGill University and Génome Québec Innovation Center and the histopathology services of the ‘Institut d'Immunologie et de Cancerologie' of the University of Montreal. RR was supported by a Trainee Award from the Center for the Study of Reproduction, EA by a Fellowship from the McGill University Health Center Research Institute, NMPN by an RQR CREATE fellowship from the Reseau Quebecois en Reproduction, RS by a Hugh and Helen McPherson Memorial Award. This study was supported by operating grants from the Canadian Institute of Health Research grant numbers MOP102469 and MOP86546 to RS.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Szulman AE, Surti U. The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. Am J Obstet Gynecol. 1978;132:20–27. doi: 10.1016/0002-9378(78)90792-5. [DOI] [PubMed] [Google Scholar]
- Savage P, Williams J, Wong SL, et al. The demographics of molar pregnancies in England and Wales from 2000-2009. J Reprod Med. 2010;55:341–345. [PubMed] [Google Scholar]
- Grimes DA. Epidemiology of gestational trophoblastic disease. Am J Obstet Gynecol. 1984;150:309–318. doi: 10.1016/s0002-9378(84)90370-3. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Brinton LA, Hayashi K. Epidemiology of hydatidiform mole and choriocarcinoma. Epidemiol Rev. 1984;6:52–75. doi: 10.1093/oxfordjournals.epirev.a036275. [DOI] [PubMed] [Google Scholar]
- Kronfol NM, Iliya FA, Hajj SN. Recurrent hydatidiform mole: a report of five cases with review of the literature. J Med Liban. 1969;22:507–520. [PubMed] [Google Scholar]
- Kim JH, Park DC, Bae SN, Namkoong SE, Kim SJ. Subsequent reproductive experience after treatment for gestational trophoblastic disease. Gynecol Oncol. 1998;71:108–112. doi: 10.1006/gyno.1998.5167. [DOI] [PubMed] [Google Scholar]
- Horn LC, Kowalzik J, Bilek K, Richter CE, Einenkel J. Clinicopathologic characteristics and subsequent pregnancy outcome in 139 complete hydatidiform moles. Eur J Obstet Gynecol Reprod Biol. 2006;128:10–14. doi: 10.1016/j.ejogrb.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Berkowitz RS, Im SS, Bernstein MR, Goldstein DP. Gestational trophoblastic disease. Subsequent pregnancy outcome, including repeat molar pregnancy. J Reprod Med. 1998;43:81–86. [PubMed] [Google Scholar]
- Wang CM, Dixon PH, Decordova S, et al. Identification of 13 novel NLRP7 mutations in 20 families with recurrent hydatidiform mole; missense mutations cluster in the leucine-rich region. J Med Genet. 2009;46:569–575. doi: 10.1136/jmg.2008.064196. [DOI] [PubMed] [Google Scholar]
- Slim R, Bagga R, Chebaro W, Srinivasan R, Agarwal N. A strong founder effect for two NLRP7 mutations in the Indian population: an intriguing observation. Clin Genet. 2009;76:292–295. doi: 10.1111/j.1399-0004.2009.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Cheng Q, Murdoch S, et al. The genetics of recurrent hydatidiform moles in China: correlations between NLRP7 mutations, molar genotypes, and reproductive outcomes. Mol Hum Reprod. 2011;17:612–619. doi: 10.1093/molehr/gar027. [DOI] [PubMed] [Google Scholar]
- Murdoch S, Djuric U, Mazhar B, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38:300–302. doi: 10.1038/ng1740. [DOI] [PubMed] [Google Scholar]
- Messaed C, Chebaro W, Roberto RB, et al. NLRP7 in the spectrum of reproductive wastage: rare non-synonymous variants confer genetic susceptibility to recurrent reproductive wastage. J Med Genet. 2011;48:540–548. doi: 10.1136/jmg.2011.089144. [DOI] [PubMed] [Google Scholar]
- Hayward BE, De Vos M, Talati N, et al. Genetic and epigenetic analysis of recurrent hydatidiform mole. Hum Mutat. 2009;30:E629–E639. doi: 10.1002/humu.20993. [DOI] [PubMed] [Google Scholar]
- Messaed C, Akoury E, Djuric U, et al. NLRP7, a NOD-like receptor protein, is required for normal cytokine secretion and co-localizes with the Golgi and the microtubule organizing center. J Biol Chem. 2011;286:43313–433123. doi: 10.1074/jbc.M111.306191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S, Dorfleutner A, Bryan NB, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36:464–476. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, Logan CV, Hayward BE, et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am J Hum Genet. 2011;89:451–458. doi: 10.1016/j.ajhg.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga M, Endo Y, Ushigome S. Flow cytometric and clinicopathologic study of 197 hydatidiform moles with special reference to the significance of cytometric aneuploidy and literature review. Cytometry. 1995;22:135–138. doi: 10.1002/cyto.990220209. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Bacolla A, Ferec C, Vasquez KM, Kehrer-Sawatzki H, Chen JM. On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat. 2011;32:1075–1099. doi: 10.1002/humu.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinnioglu C, King R, Kivisild T, et al. Excavating Y-chromosome haplotype strata in Anatolia. Hum Genet. 2004;114:127–148. doi: 10.1007/s00439-003-1031-4. [DOI] [PubMed] [Google Scholar]
- Eaaswarkhanth M, Haque I, Ravesh Z, et al. Traces of sub-Saharan and Middle Eastern lineages in Indian Muslim populations. Eur J Hum Genet. 2010;18:354–363. doi: 10.1038/ejhg.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurttas P, Morency E, Coonrod SA. Use of proteomics to identify highly abundant maternal factors that drive the egg-to-embryo transition. Reproduction. 2010;139:809–823. doi: 10.1530/REP-09-0538. [DOI] [PubMed] [Google Scholar]
- Deveault C, Qian JH, Chebaro W, et al. NLRP7 mutations in women with diploid androgenetic and triploid moles: a proposed mechanism for mole formation. Hum Mol Genet. 2009;18:888–897. doi: 10.1093/hmg/ddn418. [DOI] [PubMed] [Google Scholar]
- Hertig A, Edmonds H. Genesis of hydatidiform mole. Arch Pathol. 1940;30:260–291. [Google Scholar]
- Fisher RA, Lavery SA, Carby A, et al. What a difference an egg makes. Lancet. 2011;378:1974. doi: 10.1016/S0140-6736(11)61751-0. [DOI] [PubMed] [Google Scholar]
- Judson H, Hayward BE, Sheridan E, Bonthron DT. A global disorder of imprinting in the human female germ line. Nature. 2002;416:539–542. doi: 10.1038/416539a. [DOI] [PubMed] [Google Scholar]
- Landolsi H, Rittore C, Philibert L, et al. Screening for NLRP7 mutations in familial and sporadic recurrent hydatidiform moles: report of 2 Tunisian families. Int J Gynecol Pathol. 2011;30:348–353. doi: 10.1097/PGP.0b013e31820dc3b0. [DOI] [PubMed] [Google Scholar]
- Kou YC, Shao L, Peng HH, et al. A recurrent intragenic genomic duplication, other novel mutations in NLRP7 and imprinting defects in recurrent biparental hydatidiform moles. Mol Hum Reprod. 2008;14:33–40. doi: 10.1093/molehr/gam079. [DOI] [PubMed] [Google Scholar]
- Slim R, Coullin P, Diatta AL, et al. NLRP7 and the genetics of post-molar choriocarcinomas in Senegal. Mol Hum Reprod. 2012;18:52–56. doi: 10.1093/molehr/gar060. [DOI] [PubMed] [Google Scholar]
- Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. 2010;11:17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admetlla A, Pattini L, Nielsen R. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011;7:e1002355. doi: 10.1371/journal.pgen.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga TS, Ioana M, Alonso S, et al. The evolutionary history of TLR4 polymorphisms in Europe. J Innate Immun. 2012;4:168–175. doi: 10.1159/000329492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur E, Patin E, Laval G, et al. The selective footprints of viral pressures at the human RIG-I-like receptor family. Hum Mol Genet. 2011;20:4462–4474. doi: 10.1093/hmg/ddr377. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.