

Abstract
Discovering the genetic origin of aging-related traits could greatly advance strategies aiming to extend health span. The results of genome-wide association studies (GWAS) addressing this problem are controversial, and new genetic concepts have been fostered to advance the progress in the field. A limitation of GWAS and new genetic concepts is that they do not thoroughly address specifics of aging-related traits. Integration of theoretical concepts in genetics and aging research with empirical evidence from different disciplines highlights the conceptual problems in studies of genetic origin of aging-related traits. To address these problems, novel approaches of systemic nature are required. These approaches should adopt the non-deterministic nature of linkage of genes with aging-related traits and, consequently, reinforce research strategies for improving our understanding of mechanisms shaping genetic effects on these traits. Investigation of mechanisms will help determine conditions that activate specific genetic variants or profiles and explore to what extent these conditions that shape genetic effects are conserved across human lives and generations.
Introduction
The increases in life expectancy in humans worldwide require effective strategies to reduce the burdens of morbidity and extend years of healthy life.1–3 Studies of health in long-living people indicate that it is possible to avoid major diseases for long periods of life.4–9 Studies of heritability also show that health at old ages and life span can have a genetic origin.10–18 Accordingly, yielding insights into genetic predisposition to aging-related traits could be a major breakthrough in addressing the problem of extending health span. Aging-related traits are referred to phenotypes that are characteristic for the post-reproductive period (e.g., diseases of heart, cancer, type 2 diabetes, Alzheimer disease).
Genome-wide association studies (GWAS) have been thought as a breakthrough compared to candidate gene studies to foster progress in the field. Although GWAS indeed have pinpointed genes associated with aging-related traits, the views on GWAS progress19 range from exciting20 to disappointing,21,22 making the picture on the role of genes in health span elusive.
Controversial discussion of GWAS findings is motivated by two major concerns. One is that, unlike the original expectations, GWAS suggest that complex traits are likely controlled by a large number of genes (see, e.g., refs. 20, 21), many of which are of tiny effect.23 The other is that even a large number of genetic variants discovered by GWAS explain only a small portion of the genetic variance in a complex trait.24,25 Taken together, both of these concerns constitute a serious problem of “missing genetic variance.”26
Different genetic strategies to address this problem are fostered including a more prominent role of rare variants compared to common variants, epigenetics, structural diversity of genome, epistasis, pleiotropy, interactions through different layers of genomic complexity, etc.19,23,26–28 It is also suggested that the problem of missing genetic variance can be overblown because standard GWAS strategies have important limitations.26,27
These genetic strategies, however, do not typically address a fundamental problem in genetic susceptibility to aging-related traits—the lack of direct, evolutionary programmed mechanisms linking genes to such traits. This problem implies that genetic determinism (in evolutionary context) in the case of aging-related traits is an implausible concept. The latter becomes particularly important given numerous GWAS evidence on the highly polygenic origin of complex traits, i.e., that such traits can be influenced by a large number of common and/or rare variants.29 This evidence questions the original GWAS hypothesis of common disease–common variants (CDCV)30,31 and strengthen the “risk” hypothesis. Unlike the CDCV hypothesis, the risk hypothesis emphasizes the quantitative nature of common traits in the sense that genes rather confer the risks of common traits than cause them32 (originally proposed by Fisher33).
Paired together, the risk hypothesis and the lack of direct, evolutionary programmed mechanisms highlight four inter-related aspects of the problem of genetic susceptibility to aging-related traits not commonly discussed in genetic literature, including (1) inheritance, (2) evolutionary selection, (3) life-course-related processes, and (4) etiologic complexity.
Inheritance of the Aging-Related Traits
The most plausible explanation of inheritance of common aging-related traits within the framework of Mendelian genetics is when these traits are controlled by a few common variants. In this case, these traits will segregate as in an ordinary case of Mendelian genetics. Having a large number of unlinked disease alleles poses theoretical challenges. Fisher33 provided a theoretical basis for inheritance of polygenic phenotypes, suggesting that they could be inherited through a mechanism of inheritance of quantitative traits. A key of this mechanism is the concept of allelic equivalence, i.e., that different alleles confer risk of, rather than cause, the same trait.33,34 In this sense, common traits are considered as non-Mendelian, whereas the risk alleles still follow Mendelian segregation.33
The problem is that this basis is plausible mostly for common genetic variants.32 The CDCV hypothesis, however, becomes discouraging 21 whereas the role of rare variants is promoted.35–37 For rare variants there is a fundamental problem of explaining individualized risks of aging-related traits in the context of their inheritance.
Rare variants and individualized risks of aging-related traits
Individualized risks of aging-related traits in a population can be very high. For example, the risk of cardiovascular disease (CVD) in men with an unfavorable risk profile from 45 to 80 years is about 50%,38 i.e., every other man in such a population will develop CVD over 35 years. Similarly, according to the Surveillance Epidemiology and End Results (SEER) Cancer Statistics Review, 1975–2009 (http://seer.cancer.gov), men have a 45% lifetime risk of cancer. High-risk groups can have 16% risk of developing type 2 diabetes.39 High individualized risks imply that the person's genome has an adequate number of the rare risk variants. This number will depend on how strong the effect of an individual allele is; the weaker effect is, the larger number of the risk alleles is required to collectively explain high disease risk in a given person.35,39–41
Analyses of biomolecular mechanisms involved in regulation of aging-related traits suggest that these mechanisms are likely associated with genome-wide networks.42,43 This implies that the risks of such traits in a given individual should be explained by multiple variants spread throughout the entire genome. Indirectly, this conclusion is supported by the results of recent GWAS. For example, Teslovich et al.20 showed that single-nucleotide polymorphisms (SNPs) at 95 loci spread throughout the entire genome can be involved in regulation of blood lipids and, potentially, diseases of heart.
If individualized risks are conferred by a large number of rare variants spread throughout the entire genome, then we face a principal problem in the framework of Mendelian genetics provided that these variants are not in either genetic or functional linkage. Indeed, suppose we have n rare risk alleles with minor allele frequency (MAF) of 1%. Following the Hardy–Weinberg principle, the frequency of the minor allele homozygotes in a population is MAF-squared, i.e., 1 of 10,000 individuals will carry both risk alleles. One copy of this allele is carried by 198 of 10,000 heterozygous individuals. Therefore, the most common mode of transmittance of rare risk alleles to progeny in a population is through mating of major allele homozygous and heterozygous parents. In the case of Mendelian segregation, this crossing implies that the number of the risk (minor) alleles in progeny declines in a power law fashion, i.e., 0.5n, where the base is the Mendelian expectation in this type of crossing. For example, in the case of four alleles, only 6.25% of children of major allele homozygous and heterozygous parents inherit the parental rare risk alleles. This implies that if a large number of rare alleles confer risks of aging-related traits, heritability of these traits should be tiny. This does not comply either with clustering of aging-related traits in families or with large narrow-sense heritability estimates for such traits (e.g., 40% for type 2 diabetes39).44
Do rare variants really matter for common aging-related traits?
Thus, transmittance of rare variants in families implies that unless (1) there is a large pool of rare variants in a population with exceptionally strong effects when one or two variants from this pool explain high individualized risks of highly prevalent (e.g., about 30% of all deaths in the United States in 2008 were attributed to diseases of heart and stroke combined and about 23% to cancers45) aging-related traits (that essentially resembles the case of Mendelian traits) or (2) the rare variants do not demonstrate “clear Mendelian segregation,”40 the leading role of rare variants in common aging-related traits appears to be elusive. Accordingly, common genetic variants should play a more fundamental role in the etiology of aging-related traits. Then, the question is: Why have GWAS results not been so encouraging? The following three sections help in addressing this question.
Genes, Aging-Related Traits, and Evolutionary Selection
Evolutionary constraints are a major theoretical challenge for explaining linkage of genes with aging-related traits. For example, classical evolutionary hypothesis assumes that aging processes and the related traits are a result of a decline in the force of natural selection with age that results in accumulation of mutations.46 According to this hypothesis, aging-related traits should be non-adaptive and subject to stochastic variation.47 Another widespread hypothesis views aging and its related traits as a result of antagonistic pleiotropy, where the same gene can be favorable for fitness but can confer risks of traits in late life.48,49 Aging and the related traits are also viewed as a side effect of the evolutionary programmed mechanism of an organism's development50 when genes that are optimized for development become deleterious or fade in the post-maturational period. It is also hypothesized that parents' life span can be an evolutionary factor improving the fitness of children of reproductive age (the so-called grandmother hypothesis51). Kirkwood and Austad47 review other evolutionary hypotheses of aging-related processes.
Neither one of these hypotheses supports deterministic linkage of genes with aging-related traits, including longevity, which could be established by direct evolutionary selection against or in favor of these traits as, e.g., in the case of the program of an organism's development.17 The word “direct” in this context means that aging-related traits are irrelevant for reproductive success of the same organism. This linkage can, however, be a side effect of evolutionary programmed mechanisms, for example, when fitness factors coincide with the risk factors for diseases. Furthermore, although longevity can indirectly be relevant to the evolutionary advantage of children of long-living parents, life span constraints in the history of humans are an important factor to consider. Indeed, the world record life expectancy (i.e., essentially mean life span) in 1840 was about 45 years for women.52 Accordingly, aging-related traits could not even theoretically be a major contributor to mortality, even relatively recently; a major non-violent player during those times was infectious diseases. This poses theoretical constraints on aging-related traits as the driving force of evolutionary programming of their mechanisms. The lack of evolutionary programmed linkage of genes with aging-related traits is a source of issues discussed in the next two sections.
Life Course and Genetics of Aging-Related Traits
A fundamental problem of genetic susceptibility to aging-related traits is two-fold. First, a genetic profile is transmitted from parents to offspring at inception, whereas the risks of these traits sharply increase in late life, i.e., genes and aging-related traits are separated by a large portion of life of an organism (called life course). Second, evolutionary selection did not directly program deterministic mechanisms linking genes with those traits. This two-fold problem, which is typically not in the focus of genetic strategies, implies that processes associated with life course appear to be a key in understanding the effect of genes on aging-related traits (Fig. 1). These processes are a superposition of two major inter-related processes, i.e., the process of intrinsic biological aging (senescence) and dynamic environmental exposures (Fig. 1B). Both of these processes act at the level of individual genes and the level of phenotypes, which are eventually defined by sets of genes.
FIG. 1.

The role of genes in aging-related traits: Deterministic and dynamic concepts. Currently prevailing strategies traditionally consider genetic susceptibility to aging-related traits in framework of a concept of heritability. This concept assumes that a phenotype (P) can be represented as an additive superposition of genetic (G) and environmental (E) effects (A). Linkage of genes to a phenotype within this concept is implicitly assumed to be of deterministic nature. This concept, however, was developed for reproduction-related phenotypes used in breeding experiments with plants and referred to “the genetic contribution to variance within a population and in a specific environment.” 112 Extension of this concept to aging-related human traits is, therefore, at best problematic because of: (1) Lack of directly programmed deterministic mechanisms linking genes with those traits and (2) uncontrolled changes in environmental exposures for humans. Given these constraints, B illustrates the dynamic concept when the effects of genes on aging-related traits have to be inevitably shaped by aging-related processes (senescence) in dynamic environment. The role of environment in this concept is through activation of genes at different periods of life and modulation of gene actions over the life course and across generations. Color images available online at www.liebertpub.com/rej
Senescence primarily contributes to intra-individual variability of genetic effects. This contribution is indirectly seen as a variation of phenotypes with aging, i.e., during an organism's life. For example, decades of biodemographic and gerontological studies provided evidence on changes in various endophenotypes (e.g., the levels of physiological markers,53–55 bone mineral density56), and risks of diseases57 and death with age. As long as one believes that aging-related traits have a genetic basis, the observed changes in phenotypic expression over the life course imply that the effects of genes on these phenotypes inevitably have to change with aging. Candidate gene studies support this conclusion by showing the differential role of genes in complex traits at different periods of life.58–63
An organism's life is accompanied by varying environmental exposures that primarily contribute to inter-individual variability, diversifying genetic heterogeneity at different ages. However, known exposures may not necessarily explain all heterogeneity in genetic susceptibility to aging-related traits. For example, life span of even genetically identical species in an environment controlled for known exposures (e.g., diet, collective behavior, temperature) can still vary dramatically (e.g., see refs. 64, 65). Such evidence is a basis of a hypothesis on a stochastic origin of aging-related traits.66 However, stochasticity in this context should be interpreted with great caution because unless we know its fundamental laws (such as, e.g., the Heisenberg uncertainty principle in physics), it can be simply a measure of insufficient knowledge about the mechanisms linking genes to aging-related traits.
The complex role of genes in aging-related traits is one of sources of missing genetic variance. A classical example is antagonistic pleiotropy (postulated by Williams49; examples are provided in refs. 48, 67–71), which can result in underestimation of effects because gene function can be different at different ages (see also the Pleiotropy section, below).
Dynamic concept of genetic susceptibility to aging-related traits and its clinical relevance
The clinical relevance of the dynamic concept (Fig. 1B) is supported by epidemiological and clinical studies showing that aging-related traits develop during a substantial period of life.72 This evidence implies that the dynamic component in the effect of genes on aging-related traits should be relatively sustainable, i.e., it should be on a long timescale, comparable with aging-related changes in an organism. Long-term dynamics make this concept feasible for practical implications.
Analysis of the role of life course in genetic susceptibility to aging-related traits addresses an important problem of personalization of medicine and diminishing iatrogenesis (i.e., unintentional harm resulting from medical treatment or advice). Iatrogenesis is a serious problem that can result in losing up to about 100,000 lives each year only in the United States.73
De novo mutations and genetics of aging-related traits
Although new mutations may play a substantial role in genetic diseases,74 they are unlikely to play a pivotal role in aging-related traits. This conclusion is based on empirical observations of the extent of recent secular changes in incidence of aging-related traits,27 e.g., incidence of type 2 diabetes doubled from the 1970s to 1990s in the United States,75 that are not accompanied by adequate extensions in life span.76 Such extensive changes in health can unlikely be explained by the modest rate of de novo mutations.77 Accordingly, the observed secular changes strengthen the dynamic concept, i.e., that the existing genetic variants with different roles at different periods of life in changing environment should primarily explain susceptibility to aging-related traits across human generations.
Etiologic Complexity of Aging-Related Traits
Trait-specific and systemic mechanisms of gene actions on aging-related traits
The lack of mechanisms of development of aging-related traits directly programmed by evolutionary selection implies that genes can confer risks of such traits through different mechanisms. There are at least two fundamentally different etiologic groups of such mechanisms.16 One of them is associated with the biochemical genetic basis of a specific aging-related trait (trait-specific mechanisms) and the other with systemic processes of decline in functioning of an organism with age (systemic or aging-related mechanisms).16,18,78–81
Currently prevailing GWAS mostly focus on a group of trait-specific mechanisms. The inherent heterogeneity of aging-related traits is one of the key problems in these studies.26,40,82 GWAS typically follow a reductionist approach to overcome this problem, and the basis of this approach is to select more homogeneous sub-phenotypes. For example, one common strategy is to focus on endophenotypes that can be in a causative pathway to a trait in question.83,84 The limitation of this strategy is that endophenotypes can be even more heterogeneous than the trait itself. Accordingly, alleles involved in regulation of endophenotypes may not necessarily be involved in regulation of the downstream trait. Epidemiology explicitly illustrates this limitation. For example, 30.7% of women who have two and more major physiological risk factors for CVD develop this disease between the ages of 45 and 90 years.38 Accordingly, 69.3% of women do not develop this disease at those ages, even though they have those risk factors (these estimates address the problem of death as a competing risk38). Therefore, genes that regulate these risk factors in 69.3% of women in this sample will not affect CVD within their current life span.
Another common GWAS strategy is to more precisely define a trait in question or select its more homogeneous components.82 A clear limitation of this strategy is that it does not guarantee etiologic homogeneity because of limited knowledge about etiology of aging-related traits.82 Current GWAS following trait-specific mechanisms do not typically address the role of life course in aging-related traits.
Existence of a group of systemic mechanisms is supported by extensive evidence from epidemiology and gerontology studies that highlight health deterioration with aging across not just one but multiple health domains, regardless of population specifics (provided the same levels of populations' development are used). Clearly, such systemic processes should have a genetic basis associated with fundamental changes in intrinsic biology with aging.16,18,80,81 This group of mechanisms is not commonly considered in GWAS, although genetic studies of long-living individuals (e.g., centenarians) may reveal such mechanisms. Contrary to the reductionist approach adopted in the trait-specific mechanism, the aging-related mechanism requires systemic approaches to embrace the problem of heterogeneity by focusing on multiple traits. The systemic mechanism naturally accommodates the dynamic concept of genetic susceptibility to aging-related traits (Fig. 1B).
Pleiotropy
The diversity of mechanisms of gene actions on aging-related traits implies that pleiotropy (i.e., the same allele affects multiple traits) should play a fundamental role in genetic susceptibility to such traits. Due to confinement of early GWAS to a specific trait, studies did not typically focus on pleiotropy—for example, quoting Frazer et al.29 from a 2009 Nature paper: “A surprising finding of genome-wide association (GWA) studies is that over 15 loci are associated with the risk of developing two or more diseases” (italic is by A.K.). Currently, the fundamental role of pleiotropy is more commonly recognized.23 However, pleiotropy in most GWAS is still considered in terms of the trait-specific mechanisms for causally related traits.85 Systemic mechanisms of genetic susceptibility to aging-related traits suggest a more fundamental role of pleiotropy.42,43,67,86,87 Pleiotropy is further diversified by the life course processes shaping actions of genes on aging-related traits (Fig. 1B).
As a result of diversity of mechanisms of pleiotropy, the same direction of the effects of the same alleles on different traits may not be generally plausible. Accordingly, the same allele can confer risks to some traits but protect against the others exhibiting genetic trade-offs.29,87–98 Trade-off is a broader concept than antagonistic pleiotropy49 because it may not necessarily include fitness traits. Trade-off can be one of the sources of weak genetic signals. For example, trade-offs among upstream traits can result in tiny or no overall effect on a downstream trait, e.g., the same apolipoprotein e4 allele can confer risk for CVD but show a protective effect against cancer that significantly alters estimates for life span.87 Clearly, mishandling genetic trade-offs may contribute to iatrogenesis. This is a particularly important issue to consider in newly emerging methods of multi-variate genotype–phenotype analyses.99,100 Abundance of antagonism in gene actions on aging-related traits is supported by evidence of antagonistic relationships at the phenotypic level (e.g., see refs. 93, 101–104).
The problem of replication
Considering the CDCV hypothesis, GWAS assumed that the true effect of the same genetic variant should be replicated in different populations. However, numerous recent GWAS reports challenge the CDCV hypothesis and strengthen the risk hypothesis.32,33 A key concept of the risk hypothesis is a concept of allelic equivalence, i.e., that different alleles confer risk of the same trait. Therefore, a basic GWAS concept of replication of the association of the same allele with the same trait becomes questioned, even in framework of the risk hypothesis. Evolutionary constraints and the derivative problem of the diversity of etiologic mechanisms of aging-related traits further challenge the traditional concept of replication of association at the same variant, with the same allele, in the same direction with the same trait in different populations.
Recognition of this problem22,105–107 suggests that alternative strategies are needed. One of them is to use biological evidence for validation.108,109 Another strategy could be to use pleiotropy. The latter is plausible because evidence on additive associations of the same genetic variant with different traits reduces the probability of false discoveries of stochastic origin, especially, in framework of systemic strategies.
Conclusions and Implications
Integration of theoretical fundamentals with empirical evidence from different disciplines, including biodemography, epidemiology, and gerontology, highlights the inherently complex role of genes in aging-related traits (Fig. 1). Figure 2 summarizes the evolution of conceptual hypotheses of the structure of linkage of genes with such traits based on that integrative view. The complex structure of such a linkage formalized as a dynamic diseasome (Fig. 2E) requires effective strategies beyond those adopted in currently prevailing studies using genome-wide resources that are unable “to fully describe the architecture of phenotypic variation.”26 Straightforward strategies on “increasing the size of human disease cohorts is likely only to scale the heterogeneity in parallel” 82 with unclear chances for success.
FIG. 2.
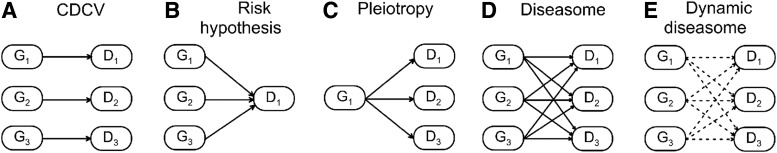
Conceptual hypotheses of structure of linkage of genes (G) with aging-related traits (D). (A) The basic concept, known as the common disease–common variants (CDCV) hypothesis, adopts linear structure when few genes can influence a given trait. (B) The CDCV hypothesis becomes discouraging whereas the risk hypothesis is strengthened; the risk hypothesis emphasizes the concept of polygenicity (many genes–one phenotype). (C) Recognition of fundamental role of pleiotropy (one gene–multiple phenotypes) in aging-related traits implies that the pleiotropy concept should complement the concept of polygenicity. (D) Superposition of these two concepts constitutes qualitative transition from the linear structure to the net structure formalized as the diseasome. (E) Recognition of the dynamic concept of genetic susceptibility to aging-related traits (Fig 1B) leads to the concept of the dynamic diseasome.
A major conclusion from the integrative discussion in this paper is that determinism in the linkage of genes with aging-related traits is not supported either by theory or by empirical evidence as a key in the strategies aiming to unravel genetic origin of these traits. As long as such genetic determinism is implausible, one inevitably concludes that the effects of genes on such traits have to be shaped by processes characteristic for the period of life between inception (when genes are transmitted from parents to their children) and expression of those traits (in post-reproductive period) (Fig. 1B). This implies that the key strategy in studies of genetic origin of aging-related traits becomes understanding the role of aging-related processes (including those in utero110) in changing environment within and across human generations. The key is to unravel mechanisms that shape genetic effects on aging-related traits but not merely report significant hits. In this regard, next-generation sequencing technologies may substantially extend the scope of the research questions about the nature of genetic mechanisms contributing to regulation of health in late life and life span by highlighting new variations in the human genome at different layers of genomic complexity.111
Investigation of mechanisms is aimed at determining conditions that activate specific genetic variants or profiles and exploring to what extent these conditions shaping genetic effects are conserved across specific periods of human lives and across generations. Given the concept of dynamic diseasome (Fig. 2E), these studies require systemic approaches integrating insights not just on one trait but on a major subset of them. Ideally, these studies should focus on life span, a wide array of aging-related diseases, and wide spectrum of endophenotypes characterizing health-related changes occurring over the large portion of individual's life and across different generations. Such data are already available in longitudinal studies of health and aging, including the Framingham Heart Study, the Cardiovascular Health Study, and the Long Life Family Study, among others. Despite the great promise of using rich longitudinal data for systemic analyses of mechanisms of genetic susceptibility to aging-related traits,63 their potential in studies using genome-wide resources is heavily underused. Collection of new longitudinal data for systemic analyses is essential to advancing progress in the field.
Acknowledgments
The research reported in this paper was supported, in part, by Award Number R01AG030612 from the National Institute on Aging. I thank my colleagues, I. Culminskaya, S. Ukraitseva, K. Arbeev, and A. Yashin, for fruitful discussion.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Sierra F. Hadley E. Suzman R. Hodes R. Prospects for life span extension. Annu Rev Med. 2009;60:457–469. doi: 10.1146/annurev.med.60.061607.220533. [DOI] [PubMed] [Google Scholar]
- 2.Olshansky SJ. Perry D. Miller RA. Butler RN. Pursuing the longevity dividend: Scientific goals for an aging world. Ann NY Acad Sci. 2007;1114:11–13. doi: 10.1196/annals.1396.050. [DOI] [PubMed] [Google Scholar]
- 3.Robine J-M. Determining Health Expectancies. Wiley; Chichester, West Sussex, England, Hoboken, NJ: 2003. [Google Scholar]
- 4.Evert J. Lawler E. Bogan H. Perls T. Morbidity profiles of centenarians: Survivors, delayers, and escapers. J Gerontol A Biol Sci Med Sci. 2003;58:232–237. doi: 10.1093/gerona/58.3.m232. [DOI] [PubMed] [Google Scholar]
- 5.Perls TT. The different paths to 100. Am J Clin Nutr. 2006;83:484S–487S. doi: 10.1093/ajcn/83.2.484S. [DOI] [PubMed] [Google Scholar]
- 6.Barzilai N. Atzmon G. Schechter C. Schaefer EJ. Cupples AL. Lipton R. Cheng S. Shuldiner AR. Unique lipoprotein phenotype and genotype associated with exceptional longevity. JAMA. 2003;290:2030–2040. doi: 10.1001/jama.290.15.2030. [DOI] [PubMed] [Google Scholar]
- 7.Willcox DC. Willcox BJ. Wang NC. He Q. Rosenbaum M. Suzuki M. Life at the extreme limit: Phenotypic characteristics of supercentenarians in Okinawa. J Gerontol A Biol Sci Med Sci. 2008;63:1201–1208. doi: 10.1093/gerona/63.11.1201. [DOI] [PubMed] [Google Scholar]
- 8.Willcox BJ. Donlon TA. He Q. Chen R. Grove JS. Yano K. Masaki KH. Willcox DC. Rodriguez B. Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci USA. 2008;16105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulminski AM. Ukraintseva SV. Culminskaya IV. Arbeev KG. Land KC. Akushevich L. Yashin AI. Cumulative deficits and physiological indices as predictors of mortality and long life. J Gerontol A Biol Sci Med Sci. 2008;63:1053–1059. doi: 10.1093/gerona/63.10.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cournil A. Kirkwood TB. If you would live long, choose your parents well. Trends Genet. 2001;17:233–235. doi: 10.1016/s0168-9525(01)02306-x. [DOI] [PubMed] [Google Scholar]
- 11.Sebastiani P. Hadley EC. Province M. Christensen K. Rossi W. Perls TT. Ash AS. A family longevity selection score: Ranking sibships by their longevity, size, and availability for study. Am J Epidemiol. 2009;170:1555–1562. doi: 10.1093/aje/kwp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown WM. Beck SR. Lange EM. Davis CC. Kay CM. Langefeld CD. Rich SS. Age-stratified heritability estimation in the Framingham Heart Study families. BMC Genet. 2003;4(Suppl 1):S32. doi: 10.1186/1471-2156-4-S1-S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herskind AM. McGue M. Holm NV. Sorensen TIA. Harvald B. Vaupel JW. The heritability of human longevity: A population-based study of 2872 Danish twin pairs born 1870–1900. Hum Genet. 1996;97:319–323. doi: 10.1007/BF02185763. [DOI] [PubMed] [Google Scholar]
- 14.Iachine IA. Holm NV. Harris JR. Begun AZ. Iachina MK. Laitinen M. Kaprio J. Yashin AI. How heritable is individual susceptibility to death? The results of an analysis of survival data on Danish, Swedish and Finnish twins. Twin Res. 1998;1:196–205. doi: 10.1375/136905298320566168. [DOI] [PubMed] [Google Scholar]
- 15.Matteini AM. Fallin MD. Kammerer CM. Schupf N. Yashin AI. Christensen K. Arbeev KG. Barr G. Mayeux R. Newman AB. Walston JD. Heritability Estimates of Endophenotypes of Long and Health Life: The Long Life Family Study. J Gerontol A Biol Sci Med Sci. 2010;65A:1375–1379. doi: 10.1093/gerona/glq154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin GM. Bergman A. Barzilai N. Genetic determinants of human health span and life span: Progress and new opportunities. PLoS Genet. 2007;3:e125. doi: 10.1371/journal.pgen.0030125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vijg J. Suh Y. Genetics of longevity and aging. Annu Rev Med. 2005;56:193–212. doi: 10.1146/annurev.med.56.082103.104617. [DOI] [PubMed] [Google Scholar]
- 18.Finch CE. Tanzi RE. Genetics of aging. Science. 1997;278:407–411. doi: 10.1126/science.278.5337.407. [DOI] [PubMed] [Google Scholar]
- 19.Visscher PM. Brown MA. McCarthy MI. Yang J. Five Years of GWAS Discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teslovich TM. Musunuru K. Smith AV. Edmondson AC. Stylianou IM. Koseki M. Pirruccello JP. Ripatti S. Chasman DI. Willer CJ. Johansen CT. Fouchier SW. Isaacs A. Peloso GM. Barbalic M. Ricketts SL. Bis JC. Aulchenko YS. Thorleifsson G. Feitosa MF. Chambers J. Orho-Melander M. Melander O. Johnson T. Li X. Guo X. Li M. Shin Cho Y. Jin Go M. Jin Kim Y. Lee JY. Park T. Kim K. Sim X. Twee-Hee Ong R. Croteau-Chonka DC. Lange LA. Smith JD. Song K. Hua Zhao J. Yuan X. Luan J. Lamina C. Ziegler A. Zhang W. Zee RY. Wright AF. Witteman JC. Wilson JF. Willemsen G. Wichmann HE. Whitfield JB. Waterworth DM. Wareham NJ. Waeber G. Vollenweider P. Voight BF. Vitart V. Uitterlinden AG. Uda M. Tuomilehto J. Thompson JR. Tanaka T. Surakka I. Stringham HM. Spector TD. Soranzo N. Smit JH. Sinisalo J. Silander K. Sijbrands EJ. Scuteri A. Scott J. Schlessinger D. Sanna S. Salomaa V. Saharinen J. Sabatti C. Ruokonen A. Rudan I. Rose LM. Roberts R. Rieder M. Psaty BM. Pramstaller PP. Pichler I. Perola M. Penninx BW. Pedersen NL. Pattaro C. Parker AN. Pare G. Oostra BA. O'Donnell CJ. Nieminen MS. Nickerson DA. Montgomery GW. Meitinger T. McPherson R. McCarthy MI. McArdle W. Masson D. Martin NG. Marroni F. Mangino M. Magnusson PK. Lucas G. Luben R. Loos RJ. Lokki ML. Lettre G. Langenberg C. Launer LJ. Lakatta EG. Laaksonen R. Kyvik KO. Kronenberg F. Konig IR. Khaw KT. Kaprio J. Kaplan LM. Johansson A. Jarvelin MR. Janssens AC. Ingelsson E. Igl W. Kees Hovingh G. Hottenga JJ. Hofman A. Hicks AA. Hengstenberg C. Heid IM. Hayward C. Havulinna AS. Hastie ND. Harris TB. Haritunians T. Hall AS. Gyllensten U. Guiducci C. Groop LC. Gonzalez E. Gieger C. Freimer NB. Ferrucci L. Erdmann J. Elliott P. Ejebe KG. Doring A. Dominiczak AF. Demissie S. Deloukas P. de Geus EJ. de Faire U. Crawford G. Collins FS. Chen YD. Caulfield MJ. Campbell H. Burtt NP. Bonnycastle LL. Boomsma DI. Boekholdt SM. Bergman RN. Barroso I. Bandinelli S. Ballantyne CM. Assimes TL. Quertermous T. Altshuler D. Seielstad M. Wong TY. Tai ES. Feranil AB. Kuzawa CW. Adair LS. Taylor HA., Jr. Borecki IB. Gabriel SB. Wilson JG. Holm H. Thorsteinsdottir U. Gudnason V. Krauss RM. Mohlke KL. Ordovas JM. Munroe PB. Kooner JS. Tall AR. Hegele RA. Kastelein JJ. Schadt EE. Rotter JI. Boerwinkle E. Strachan DP. Mooser V. Stefansson K. Reilly MP. Samani NJ. Schunkert H. Cupples LA. Sandhu MS. Ridker PM. Rader DJ. van Duijn CM. Peltonen L. Abecasis GR. Boehnke M. Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 22.McClellan J. King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 23.Stranger BE. Stahl EA. Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SH. Wray NR. Goddard ME. Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88:294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraja AT. Hunt SC. Rao DC. Davila-Roman VG. Arnett DK. Province MA. Genetics of hypertension and cardiovascular disease and their interconnected pathways: Lessons from large studies. Curr Hypertens Rep. 2011;13:46–54. doi: 10.1007/s11906-010-0174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eichler EE. Flint J. Gibson G. Kong A. Leal SM. Moore JH. Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson G. Rare and common variants: Twenty arguments. Nat Rev Genet. 2011;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeggini E. Next-generation association studies for complex traits. Nat Genet. 2011;43:287–288. doi: 10.1038/ng0411-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frazer KA. Murray SS. Schork NJ. Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 30.Swales JD. London: Keynes Press; 1985. Platt versus pickering: an episode in recent medical history; p. 155. [Google Scholar]
- 31.Schork NJ. Murray SS. Frazer KA. Topol EJ. Common vs. rare allele hypotheses for complex diseases. Curr Opin Genet Dev. 2009;19:212–219. doi: 10.1016/j.gde.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plomin R. Haworth CM. Davis OS. Common disorders are quantitative traits. Nat Rev Genet. 2009;10:872–878. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- 33.Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinburgh. 1918;52:399–433. [Google Scholar]
- 34.Wright S. Genetics of abnormal growth in the guinea pig. Cold Spring Harbor Symp Quant Biol. 1934;2:137–147. [Google Scholar]
- 35.Bodmer W. Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorlov IP. Gorlova OY. Frazier ML. Spitz MR. Amos CI. Evolutionary evidence of the effect of rare variants on disease etiology. Clin Genet. 2011;79:199–206. doi: 10.1111/j.1399-0004.2010.01535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tennessen JA. Bigham AW. O'Connor TD. Fu W. Kenny EE. Gravel S. McGee S. Do R. Liu X. Jun G. Kang HM. Jordan D. Leal SM. Gabriel S. Rieder MJ. Abecasis G. Altshuler D. Nickerson DA. Boerwinkle E. Sunyaev S. Bustamante CD. Bamshad MJ. Akey JM. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berry JD. Dyer A. Cai X. Garside DB. Ning H. Thomas A. Greenland P. Van Horn L. Tracy RP. Lloyd-Jones DM. Lifetime risks of cardiovascular disease. N Engl J Med. 2012;366:321–329. doi: 10.1056/NEJMoa1012848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pawitan Y. Seng KC. Magnusson PKE. How many genetic variants remain to be discovered? PLoS One. 2009;4(12):e7969. doi: 10.1371/journal.pone.0007969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manolio TA. Collins FS. Cox NJ. Goldstein DB. Hindorff LA. Hunter DJ. McCarthy MI. Ramos EM. Cardon LR. Chakravarti A. Cho JH. Guttmacher AE. Kong A. Kruglyak L. Mardis E. Rotimi CN. Slatkin M. Valle D. Whittemore AS. Boehnke M. Clark AG. Eichler EE. Gibson G. Haines JL. Mackay TF. McCarroll SA. Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarthy MI. Abecasis GR. Cardon LR. Goldstein DB. Little J. Ioannidis JP. Hirschhorn JN. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 42.Goh KI. Cusick ME. Valle D. Childs B. Vidal M. Barabasi AL. The human disease network. Proc Natl Acad Sci USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chavali S. Barrenas F. Kanduri K. Benson M. Network properties of human disease genes with pleiotropic effects. BMC Syst Biol. 2010;4:78. doi: 10.1186/1752-0509-4-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weiss KM. Tilting at quixotic trait loci (QTL): An evolutionary perspective on genetic causation. Genetics. 2008;179:1741–1756. doi: 10.1534/genetics.108.094128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miniño AM. Murphy SL. Xu J. Kochanek KD. Deaths: Final Data for 2008. National Vital Statistics Reports. 2011;59:1–127. [PubMed] [Google Scholar]
- 46.Medawar PB. H.K. Lewis and Co.; London: Dec 6, 1951. 1952. An Unsolved problem of biology; an inaugural lecture delivered at University College, London. [Google Scholar]
- 47.Kirkwood TB. Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- 48.Williams PD. Day T. Antagonistic pleiotropy, mortality source interactions, and the evolutionary theory of senescence. Evolution. 2003;57:1478–1488. doi: 10.1111/j.0014-3820.2003.tb00356.x. [DOI] [PubMed] [Google Scholar]
- 49.Williams GC. Pleiotropy, natural-selection, and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 50.de Magalhaes JP. Church GM. Genomes optimize reproduction: Aging as a consequence of the developmental program. Physiology (Bethesda) 2005;20:252–259. doi: 10.1152/physiol.00010.2005. [DOI] [PubMed] [Google Scholar]
- 51.Hawkes K. O'Connell JF. Jones NG. Alvarez H. Charnov EL. Grandmothering, menopause, and the evolution of human life histories. Proc Natl Acad Sci USA. 1998;95:1336–1339. doi: 10.1073/pnas.95.3.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oeppen J. Vaupel JW. Demography. Broken limits to life expectancy. Science. 2002;296:1029–1031. doi: 10.1126/science.1069675. [DOI] [PubMed] [Google Scholar]
- 53.Hershcopf RJ. Elahi D. Andres R. Baldwin HL. Raizes GS. Schocken DD. Tobin JD. Longitudinal changes in serum cholesterol in man: An epidemiologic search for an etiology. J Chronic Dis. 1982;35:101–114. doi: 10.1016/0021-9681(82)90111-4. [DOI] [PubMed] [Google Scholar]
- 54.Scheen AJ. Diabetes mellitus in the elderly: Insulin resistance and/or impaired insulin secretion? Diabetes Metab. 2005;31(Spec No 2):5S27–25S34. doi: 10.1016/s1262-3636(05)73649-1. [DOI] [PubMed] [Google Scholar]
- 55.Yashin AI. Akushevich IV. Arbeev KG. Akushevich L. Ukraintseva SV. Kulminski A. Insights on aging and exceptional longevity from longitudinal data: Novel findings from the Framingham Heart Study. Age. 2006;28:363–374. doi: 10.1007/s11357-006-9023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sheu Y. Cauley JA. Wheeler VW. Patrick AL. Bunker CH. Ensrud KE. Orwoll ES. Zmuda JM. Age-related decline in bone density among ethnically diverse older men. Osteoporos Int. 2011;22:599–605. doi: 10.1007/s00198-010-1330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akushevich I. Kravchenko J. Ukraintseva S. Arbeev K. Yashin AI. Age patterns of incidence of geriatric disease in the U.S. Elderly population: Medicare-based analysis. J Am Geriatr Soc. 2012;60:323–327. doi: 10.1111/j.1532-5415.2011.03786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Benedictis G. Carotenuto L. Carrieri G. De Luca M. Falcone E. Rose G. Yashin AI. Bonafe M. Franceschi C. Age-related changes of the 3′ APOB-VNTR genotype pool in ageing cohorts. Ann Hum Genet. 1998;62(Pt 2):115–122. doi: 10.1046/j.1469-1809.1998.6220115.x. [DOI] [PubMed] [Google Scholar]
- 59.Ilveskoski E. Perola M. Lehtimaki T. Laippala P. Savolainen V. Pajarinen J. Penttila A. Lalu KH. Mannikko A. Liesto KK. Koivula T. Karhunen PJ. Age-dependent association of apolipoprotein E genotype with coronary and aortic atherosclerosis in middle-aged men: An autopsy study. Circulation. 1999;100:608–613. doi: 10.1161/01.cir.100.6.608. [DOI] [PubMed] [Google Scholar]
- 60.Yashin AI. Ukraintseva SV. De Benedictis G. Anisimov VN. Butov AA. Arbeev K. Jdanov DA. Boiko SI. Begun AS. Bonafe M. Franceschi C. Have the oldest old adults ever been frail in the past? A hypothesis that explains modern trends in survival. J Gerontol A Biol Sci Med Sci. 2001;56:B432–B442. doi: 10.1093/gerona/56.10.b432. [DOI] [PubMed] [Google Scholar]
- 61.Jarvik GP. Austin MA. Fabsitz RR. Auwerx J. Reed T. Christian JC. Deeb S. Genetic influences on age-related change in total cholesterol, low density lipoprotein-cholesterol, and triglyceride levels: Longitudinal apolipoprotein E genotype effects. Genet Epidemiol. 1994;11:375–384. doi: 10.1002/gepi.1370110407. [DOI] [PubMed] [Google Scholar]
- 62.Jarvik GP. Goode EL. Austin MA. Auwerx J. Deeb S. Schellenberg GD. Reed T. Evidence that the apolipoprotein E-genotype effects on lipid levels can change with age in males: A longitudinal analysis. Am J Hum Genet. 1997;61:171–181. doi: 10.1086/513902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kulminski AM. Culminskaya I. Arbeev KG. Ukraintseva SV. Stallard E. Arbeeva L. Yashin AI. The role of lipid-related genes, aging-related processes, and environment in healthspan. Aging Cell. 2013;12:237–246. doi: 10.1111/acel.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carey JR. Harshman LG. Liedo P. Muller HG. Wang JL. Zhang Z. Longevity-fertility trade-offs in the tephritid fruit fly, Anastrepha ludens, across dietary-restriction gradients. Aging Cell. 2008;7:470–477. doi: 10.1111/j.1474-9726.2008.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kulminski AM. Molleman F. Culminskaya IV. Arbeev KG. Ukraintseva SV. Carey JR. Yashin AI. Date of eclosion modulates longevity: Insights across dietary-restriction gradients and female reproduction in the mexfly Anastrepha ludens. Exp Gerontol. 2009;44:718–726. doi: 10.1016/j.exger.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kirkwood TB. Cordell HJ. Finch CE. Speed-bumps ahead for the genetics of later-life diseases. Trends Genet. 2011;27:387–388. doi: 10.1016/j.tig.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 67.Martin GM. Modalities of gene action predicted by the classical evolutionary biological theory of aging. Ann NY Acad Sci. 2007;1100:14–20. doi: 10.1196/annals.1395.002. [DOI] [PubMed] [Google Scholar]
- 68.Kulminski AM. Culminskaya I. Ukraintseva SV. Arbeev KG. Land KC. Yashin AI. Beta2-adrenergic receptor gene polymorphisms as systemic determinants of healthy aging in an evolutionary context. Mech Ageing Dev. 2010;131:338–345. doi: 10.1016/j.mad.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Summers K. Crespi BJ. Xmrks the spot: Life history tradeoffs, sexual selection and the evolutionary ecology of oncogenesis. Mol Ecol. 2010;19:3022–3024. doi: 10.1111/j.1365-294x.2010.04739.x. [DOI] [PubMed] [Google Scholar]
- 70.Alexander DM. Williams LM. Gatt JM. Dobson-Stone C. Kuan SA. Todd EG. Schofield PR. Cooper NJ. Gordon E. The contribution of apolipoprotein E alleles on cognitive performance and dynamic neural activity over six decades. Biol Psychol. 2007;75:229–238. doi: 10.1016/j.biopsycho.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 71.Schnebel EM. Grossfield J. Antagonistic pleiotropy—an interspecific Drosophila comparison. Evolution. 1988;42:306–311. doi: 10.1111/j.1558-5646.1988.tb04134.x. [DOI] [PubMed] [Google Scholar]
- 72.Wilson PW. D'Agostino RB. Levy D. Belanger AM. Silbershatz H. Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 73.Weingart SN. Ship AN. Aronson MD. Confidential clinician-reported surveillance of adverse events among medical inpatients. J Gen Intern Med. 2000;15:470–477. doi: 10.1046/j.1525-1497.2000.06269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veltman JA. Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 75.Fox CS. Pencina MJ. Meigs JB. Vasan RS. Levitzky YS. D'Agostino RB., Sr. Trends in the incidence of type 2 diabetes mellitus from the 1970s to the 1990s: The Framingham Heart Study. Circulation. 2006;113:2914–2918. doi: 10.1161/CIRCULATIONAHA.106.613828. [DOI] [PubMed] [Google Scholar]
- 76.Yashin AI. Begun AS. Boiko SI. Ukraintseva SV. Oeppen J. The new trends in survival improvement require a revision of traditional gerontological concepts. Exp Gerontol. 2001;37:157–167. doi: 10.1016/s0531-5565(01)00154-1. [DOI] [PubMed] [Google Scholar]
- 77.Roach JC. Glusman G. Smit AF. Huff CD. Hubley R. Shannon PT. Rowen L. Pant KP. Goodman N. Bamshad M. Shendure J. Drmanac R. Jorde LB. Hood L. Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Franco OH. Karnik K. Osborne G. Ordovas JM. Catt M. van der Ouderaa F. Changing course in ageing research: The healthy ageing phenotype. Maturitas. 2009;63:13–19. doi: 10.1016/j.maturitas.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 79.Martin GM. The biology of aging: 1985–2010 and beyond. FASEB J. 2011;25:3756–3762. doi: 10.1096/fj.11-1102.ufm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sierra F. Hadley E. Suzman R. Hodes R. Prospects for life span extension. Annu Rev Med. 2009;60:457–469. doi: 10.1146/annurev.med.60.061607.220533. [DOI] [PubMed] [Google Scholar]
- 81.Jazwinski SM. Biological aging research today: Potential, peeves, and problems. Exp Gerontol. 2002;37:1141–1146. doi: 10.1016/s0531-5565(02)00171-7. [DOI] [PubMed] [Google Scholar]
- 82.MacRae CA. Vasan RS. Next-generation genome-wide association studies: Time to focus on phenotype? Circ Cardiovasc Genet. 2011;4:334–336. doi: 10.1161/CIRCGENETICS.111.960765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arking DE. Chakravarti A. Understanding cardiovascular disease through the lens of genome-wide association studies. Trends Genet. 2009;25:387–394. doi: 10.1016/j.tig.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 84.Bloss CS. Pawlikowska L. Schork NJ. Contemporary human genetic strategies in aging research. Ageing Res Rev. 2011;10:191–200. doi: 10.1016/j.arr.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sivakumaran S. Agakov F. Theodoratou E. Prendergast JG. Zgaga L. Manolio T. Rudan I. McKeigue P. Wilson JF. Campbell H. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet. 2011;89:607–618. doi: 10.1016/j.ajhg.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koga H. Kaushik S. Cuervo AM. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev. 2011;10:205–215. doi: 10.1016/j.arr.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kulminski AM. Culminskaya I. Ukraintseva SV. Arbeev KG. Arbeeva L. Wu D. Akushevich I. Land KC. Yashin AI. Trade-off in the effects of the apolipoprotein E polymorphism on the ages at onset of CVD and cancer influences human lifespan. Aging Cell. 2011;10:533–541. doi: 10.1111/j.1474-9726.2011.00689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kulminski AM. Ukraintseva SV. Arbeev KG. Manton KG. Oshima J. Martin GM. Il'yasova D. Yashin AI. Health-protective and adverse effects of the apolipoprotein E epsilon2 allele in older men. J Am Geriatr Soc. 2008;56:478–483. doi: 10.1111/j.1532-5415.2007.01574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crespi BJ. The origins and evolution of genetic disease risk in modern humans. Ann NY Acad Sci. 2010;1206:80–109. doi: 10.1111/j.1749-6632.2010.05707.x. [DOI] [PubMed] [Google Scholar]
- 90.Kulminski AM. Culminskaya IV. Ukraintseva SV. Arbeev KG. Akushevich I. Land KC. Yashin AI. Polymorphisms in the ACE and ADRB2 genes and risks of aging-associated phenotypes: The case of myocardial infarction. Rejuvenation Res. 2010;13:13–21. doi: 10.1089/rej.2009.0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang K. Baldassano R. Zhang H. Qu HQ. Imielinski M. Kugathasan S. Annese V. Dubinsky M. Rotter JI. Russell RK. Bradfield JP. Sleiman PM. Glessner JT. Walters T. Hou C. Kim C. Frackelton EC. Garris M. Doran J. Romano C. Catassi C. Van Limbergen J. Guthery SL. Denson L. Piccoli D. Silverberg MS. Stanley CA. Monos D. Wilson DC. Griffiths A. Grant SF. Satsangi J. Polychronakos C. Hakonarson H. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum Mol Genet. 2010;19:2059–2067. doi: 10.1093/hmg/ddq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barnes JA. Dix DJ. Collins BW. Luft C. Allen JW. Expression of inducible Hsp70 enhances the proliferation of MCF-7 breast cancer cells and protects against the cytotoxic effects of hyperthermia. Cell Stress Chaperones. 2001;6:316–325. doi: 10.1379/1466-1268(2001)006<0316:eoihet>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ukraintseva SV. Arbeev KG. Akushevich I. Kulminski A. Arbeeva L. Culminskaya I. Akushevich L. Yashin AI. Trade-offs between cancer and other diseases: Do they exist and influence longevity? Rejuvenation Res. 2010;13:387–396. doi: 10.1089/rej.2009.0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weilbach FX. Toyka KV. Does Down's syndrome protect against multiple sclerosis? Eur Neurol. 2002;47:52–55. doi: 10.1159/000047947. [DOI] [PubMed] [Google Scholar]
- 95.Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci USA. 2010;107(Suppl 1):1718–1724. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Charlesworth B. Evolution of senescence: Alzheimer's disease and evolution. Curr Biol. 1996;6:20–22. doi: 10.1016/s0960-9822(02)00411-6. [DOI] [PubMed] [Google Scholar]
- 97.Martin GM. APOE alleles and lipophylic pathogens. Neurobiol Aging. 1999;20:441–443. doi: 10.1016/s0197-4580(99)00078-0. [DOI] [PubMed] [Google Scholar]
- 98.van Heemst D. Mooijaart SP. Beekman M. Schreuder J. de Craen AJ. Brandt BW. Slagboom PE. Westendorp RG. Variation in the human TP53 gene affects old age survival and cancer mortality. Exp Gerontol. 2005;40:11–15. doi: 10.1016/j.exger.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 99.van der Sluis S. Posthuma D. Dolan CV. TATES: Efficient multivariate genotype-phenotype analysis for genome-wide association studies. PLoS Genet. 2013;9:e1003235. doi: 10.1371/journal.pgen.1003235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Q. Wang Y. Methods for analyzing multivariate phenotypes in genetic association studies. J Prob Statist. 2012;2012:652569. doi: 10.1155/2012/652569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yashin AI. Ukraintseva SV. Akushevich IV. Arbeev KG. Kulminski A. Akushevich L. Trade-off between cancer and aging: What role do other diseases play? Evidence from experimental and human population studies. Mech Ageing Dev. 2009;130:98–104. doi: 10.1016/j.mad.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stallard E. Underlying and multiple cause mortality at advanced ages: United States 1980–1998. N Am Actuarial J. 2002;6:64–87. [Google Scholar]
- 103.Driver JA. Beiser A. Au R. Kreger BE. Splansky GL. Kurth T. Kiel DP. Lu KP. Seshadri S. Wolf PA. Inverse association between cancer and Alzheimer's disease: Results from the Framingham Heart Study. Br Med J. 2012:344. doi: 10.1136/bmj.e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tabares-Seisdedos R. Rubenstein JL. Inverse cancer comorbidity: A serendipitous opportunity to gain insight into CNS disorders. Nat Rev Neurosci. 2013;14:293–304. doi: 10.1038/nrn3464. [DOI] [PubMed] [Google Scholar]
- 105.Yashin AI. Wu D. Arbeev KG. Ukraintseva SV. Polygenic effects of common single-nucleotide polymorphisms on life span: When association meets causality. Rejuvenation Res. 2012;15:381–394. doi: 10.1089/rej.2011.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Day-Williams AG. Zeggini E. The effect of next-generation sequencing technology on complex trait research. Eur J Clin Invest. 2011;41:561–567. doi: 10.1111/j.1365-2362.2010.02437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neale BM. Sham PC. The future of association studies: Gene-based analysis and replication. Am J Hum Genet. 2004;75:353–362. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luo L. Peng G. Zhu Y. Dong H. Amos CI. Xiong M. Genome-wide gene and pathway analysis. Eur J Hum Genet. 2010;18:1045–1053. doi: 10.1038/ejhg.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peng G. Luo L. Siu H. Zhu Y. Hu P. Hong S. Zhao J. Zhou X. Reveille JD. Jin L. Amos CI. Xiong M. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010;18:111–117. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barker DJ. The developmental origins of well-being. Philos Trans R Soc Lond B Biol Sci. 2004;359:1359–1366. doi: 10.1098/rstb.2004.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.de Magalhaes JP. Finch CE. Janssens G. Next-generation sequencing in aging research: Emerging applications, problems, pitfalls and possible solutions. Ageing Res Rev. 2010;9:315–323. doi: 10.1016/j.arr.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rose SPR. Commentary: Heritability estimates—long past their sell-by date. Int J Epidemiol. 2006;35:525–527. doi: 10.1093/ije/dyl064. [DOI] [PubMed] [Google Scholar]