

Abstract
Objective:
To examine cardioprotective effects of Ρ-terminal fragment of adipokine apelin-12 (A12), its novel structural analogue [MeArg1, NLe10]-A12 (I), and [d-Ala12]-A12 (II), a putative antagonist of APJ receptor, employing in vivo model of ischemia/reperfusion (I/R) injury.
Materials and Methods:
Peptides were synthesized by the automatic solid phase method using Fmoc technology. Anesthetized open-chest male Wistar rats were subjected to left anterior descending (LAD) coronary artery occlusion and coronary reperfusion. Hemodynamic variables and electrocardiogram (ECG) were monitored throughout the experiment. Myocardial injury was assessed by infarct size (IS), activity of necrosis markers in plasma, and metabolic state of the area at risk (AAR).
Results:
Intravenous injection of A12, I, or II at the onset of reperfusion led to a transient reduction of the mean arterial pressure. A12 or I administration decreased the percent ratio of IS/AAR by 40% and 30%, respectively, compared with control animals which received saline. Both peptides improved preservation of high-energy phosphates, reduced lactate accumulation in the AAR, and lowered CK-MB and LDH activities in plasma at the end of reperfusion compared with these indices in control. Treatment with II did not significantly affect either the IS/AAR, % ratio, or activities of both markers of necrosis compared with control. The overall metabolic protection of the AAR in the treated groups increased in the following rank: II < A12 < I.
Conclusions:
The structural analogue of apelin-12 [MeArg1, NLe10]-A12 may be a promising basis to create a new drug for the treatment of acute coronary syndrome.
Keywords: Analogues of apelin-12, markers of necrosis, metabolism of area at risk, myocardial infarction
INTRODUCTION
Ischemia, which is caused by coronary artery occlusion followed by reperfusion evokes significant myocardial damage, leading to cardiac dysfunction and cell death. A variety of pharmacological agents have been investigated in different experimental models to protect the heart from ischemia/reperfusion (I/R) injury. These include antioxidants, calcium channel blockers, inhibitors of neutrophils, NO donors, inhibitors of the renin–angiotensin system, metabolic protectors, Na+/H+ exchange inhibitors, and antiapoptotic agents. Despite intensive research, treatment for I/R injury remains insufficiently effective. Therefore, development of drugs to improve cardiac function, delay the onset of necrosis, or limit development of infarction during ischemia and reperfusion is of great clinical importance.[1] A promising strategy to solve this issue is the use of natural substances or their structural analogues that trigger signaling pathways associated with endogenous cardioprotection. One of them is the adipocytokine apelin, the endogenous ligand for the G-protein-coupled APJ receptor.[2] To date, apelin and APJ receptor have been shown to play an important role in the maintenance of cardiovascular homeostasis and protection against I/R injury.[3]
Apelin is produced as a 77 amino acid prepropeptide which is cleaved to shorter biologically active C-terminal fragments.[4] Apelin-12 (A12), apelin-13, and, to a lesser extent, apelin-36 are capable to reduce infarct size (IS) and to augment contractile function recovery in the heart of rodents after regional or global ischemia.[3,5,6,7] In cultured cardiomyocytes, apelin-13 suppressed apoptosis.[6,8] The beneficial effects of apelin are attributed to mobilization of the PI3K–Akt and MEK1/2–ERK1/2 salvage kinases, and inhibition of the mitochondrial permeability transition pore (mPTP).[9,10] Phosphorylation and activation of endothelial nitric oxide synthase (eNOS) are also implicated in myocardial protection afforded by apelin.[6,11,12] Recent clinical study revealed that plasma apelin concentration is reduced early after acute myocardial infarction and remains significantly low baseline at 24 weeks.[13] These facts strongly suggest that apelin administration may be promising in the treatment of coronary heart disease. A12 is one of the most potent C-terminal fragments of the polypeptide that possesses a high affinity to APJ receptor and bioactivity in vivo.[7,14,15] However, apelin peptides rapidly cleared from the circulation with a half life of no longer than 5 min.[16] It is believed that this is due to their rapid hydrolysis by various peptidases including angiotensin-converting enzyme 2 (ACE2).[17] Therefore, it is necessary to modify the chemical structure of A12 to improve its physiological stability as a potential drug. This study was designed to evaluate efficacy of a novel structural analogue of A12 (I), potentially more resistant to enzymatic degradation, in model of myocardial I/R injury in rats. Protective action of this peptide was compared with effects of A12 and [d-Ala12]-A12 (II), a putative antagonist of APJ receptor.[18]
MATERIALS AND METHODS
Synthesis of A12 and its analogues
Peptides A12, I, and II were synthesized by the automatic solid phase method using an Applied BioSystems 431 A peptide synthesizer (Germany) and Fmoc technology. To increase the chemical stability of A12 and its resistance to aminopeptidase cleavage, we replaced easily oxidized methionine by norleucine and included Nα-methylarginine moiety in N-terminus of the peptide (analogue I). We replaced the carboxyl-terminal phenylalanine by d-alanine in A12 to produce a putative functional APJ antagonist (analogue II) in accordance with the work of Lee, et al.[18] The synthesized peptides [Table 1] were purified by preparative HPLC and identified by 1H-NMR spectroscopy and mass spectrometry.
Table 1.
Structure of apelin-12 and its analogues

Animals
Adult male Wistar rats weighing 290-340 g were used in this study. All animals were housed in cages in groups of three, maintained at 20-30°C with a natural light-dark cycle, and had free access to standard pelleted diet (Aller Petfood, St. Petersburg, Russia) and tap water ad libitum. The care and use of the animals were conducted in accordance with the European Convention for the Protection of Vertebrate Animals Used for Experimental and other Scientific Purposes (No. 123 of 18 March 1986).
General preparation
Rats were anesthetized with 20% urethane (Aldrich, 120 mg/kg body wt. ip) and artificially ventilated with a KTR-5 animal respirator (Hugo Sacks Electronik) with a volume of 2-3 ml at a rate of 70-75 breaths/min. The right jugular vein was catheterized for drug administration. The left carotid artery was cannulated for monitoring arterial blood pressure and arterial blood sampling. Electrocardiogram (ECG) leads were placed to record heart rate (HR). The chest was opened by a left thoracotomy in the fifth intercostal space, and the heart was exposed by removing the pericardium. After pericardiotomy, a 5-0 prolene ligature was placed under the left anterior descending (LAD) coronary artery where it emerges from beneath the left atrial appendage and the ends were threaded through a small plastic tube to form a snare for reversible LAD coronary artery occlusion. Complete LAD coronary artery occlusion was confirmed by observing cyanosis of the myocardium as well as the ST-segment elevation and immediate fall in mean arterial pressure (MAP) by 15-30 mmHg. Arterial blood pressure was recorded with a pressure transducer (Statham p23Db, Oxnard, USA) using a polygraph Biograph-4 (St. Petersburg, Russia). The MAP, HR, and standard lead II ECG were recorded on a computer using a LabView 7.1 data acquisition system ((National Instruments, USA). Arterial blood pH, partial CO2 pressure, and O2 saturation values were monitored with an ABL-30 acid–base gas analyzer (Radiometer, Denmark) and maintained at the physiological level throughout the experiment.
Experimental design
Acute myocardial infarction was induced as described previously.[19] After 30 min stabilization of hemodynamic parameters (initial state), LAD coronary artery was occluded for 40 min to simulate regional ischemia; the duration of subsequent reperfusion was 1 h. The prepared animals were randomly assigned onto one of four groups: Control, A12, analogue I or analogue II. After the period of LAD coronary artery occlusion, 0.5 ml of saline was administrated by i.v. bolus injection at the onset of reperfusion in control. A12 or its analogues were administrated by i.v. bolus injection at the onset of reperfusion at a dose of 0.35 μmol/kg. A preliminary assessment of IS limitation by peptides was carried out in the dose range from 0.07 to 0.70 μmol/kg. The peptides were dissolved in saline before administration; the volume of injected solution was 0.5 ml. It was found that a dose of 0.35 μmol/kg is optimal for A12 and both analogues. To minimize effects of collateral blood flow on IS, a relatively large number of animals (n = 12) were used in each group.
At the end of the reperfusion, LAD coronary artery was reoccluded and 2 ml of 2% Evans Blue (Sigma, USA) solution was injected through the jugular vein to distinguish the myocardial nonischemic area from area at risk (AAR). In separate series of experiments, the myocardial AAR was freeze-clamped in liquid nitrogen after steady state or at the end of reperfusion for metabolite analysis.
Determination of infarct size
After staining with Evans Blue, the heart was excised, and the left ventricle (LV) was transversely cut into 1.5-mm-thick slices which were incubated in 0.1 M sodium phosphate buffer pH 7.40, containing 1% 2,3,5-triphenyl-tetrazolium chloride (TTC, Sigma, USA), for 10 min at 37°C. The noninfarcted AAR was stained to deep red, infarct tissue grey white, and nonischemic area blue. The slices were fixed in 10% formalin for 20 min. Then, they were placed between two transparent glasses and captured using a scanner at 600 dpi resolution; the saved images were analyzed by computerized planimetry using Imagecall software. The slices were then weighed for determination of LV weight. The AAR was expressed as a percentage of LV weight; the IS was expressed as a percentage of the AAR in each group.
Analysis of metabolites
Frozen AAR samples were quickly homogenized in cooled 6% HClO4 (10 ml/g) using an Ultra-Turrax T-25 homogenizer (IKA-Labortechnik, Staufen, Germany), and the homogenates were centrifuged at 2800 × g for 10 min at 4oC. The supernatants were then neutralized with 5 M K2 CO3 to pH 7.40, and the extracts were centrifuged after cooling to remove KClO4 precipitate. Tissue dry weights were determined by weighing a portion of the pellets after extraction with 6% HClO4 and drying overnight at 110°C. Concentrations of ATP, ADP, AMP, phosphocreatine (PCr), creatine (Cr), and lactate in neutralized tissue extracts were determined specrtophotometrically by enzymatic methods.[20] Enzymes and chemicals were purchased from Sigma Chemical Co. (St Louis, MO, USA). Solutions were prepared using deionized water (Milli Ro-4; Milli-Q, Millipore Corp. Bedford, MA, USA).
Determination of cardiac biomarkers
At the end of the steady state and reperfusion, blood samples were collected for plasma separation and stored at −70oC for further analysis. Plasma lactate dehydrogenase (LDH) was determined enzymatically with pyruvate as substrate by using standard kits from BioSystems S.A. (Barcelona, Spain). Plasma CK-MB activity was assessed by an immunoinhibition method using standard kits from BioSystems S.A. (Barcelona, Spain) from the rate of NADPH formation by means of the hexokinase and glucose-6-phosphate dehydrogenase coupled reactions.
Statistical analysis
All data are presented as mean ± SEM. Results were analyzed by one-way ANOVA followed by Student–Newman–Keuls test for evaluation differences between more than two groups. Comparisons between two groups involved use of the Student's unpaired t-test. A P < 0.05 was considered statistically significant.
RESULTS
Hemodynamic data
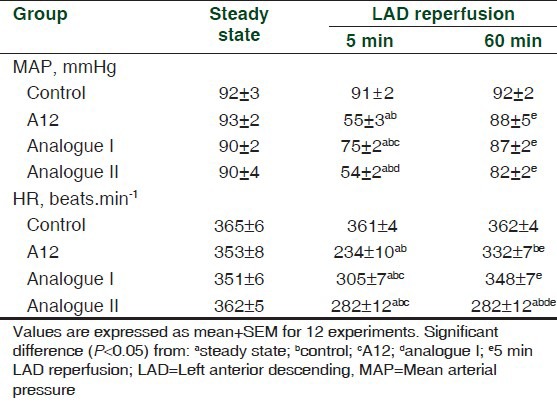
There were no differences in the MAP or HR between the groups in the steady state [Table 2]. In the control, there were no significant changes in either MAP or HR between the steady state and the reperfusion period. Bolus injection of A12 after the period of LAD coronary artery occlusion resulted in a transient fall in MAP (to 59 ± 2% of the initial value at the 5th min of reperfusion). By the end of reperfusion, MAP recovered to near baseline (95 ± 6%). Similar changes in MAP occurred after the administration of analogue II. Injection of analogue I led to significantly less fall in MAP (to 83 ± 2% of baseline) which was accompanied by its rapid recovery. Administration of all peptides induced a short-term reduction in HR lasting 10-15 s, which was quickly replaced by its complete restoration. The exception was the analogue II group that exhibited reduced HR at the end of reperfusion.
Table 2.
Effects of administration of A12 or its analogues on systemic hemodynamic variables
Myocardial Infarction
In control, the percentage ratios of AAR/LV and IS/AAR were 38.6 ± 1.60 and 40.5 ± 2.1%, correspondently [Figure 1]. The AAR of the LV was similar among all treated groups and did not differ significantly from the value in control. After treatment with A12 or analogue I, the percentage ratio of IS/AAR was substantially reduced (by 40% and 30% compared with control, correspondently, P < 0.01) thus indicating limitation of IS. There was no difference in the infarcted area between A12 and analogue I groups. Treatment with analogue II did not limit the IS compared with control (34.2 ± 3.6%) but significantly increased this index compared with A12 administration.
Figure 1.
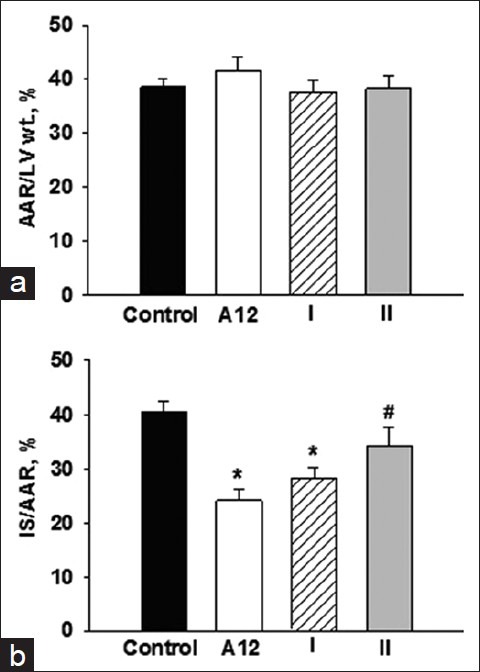
The area at risk (AAR/LV wt., %, a) and myocardial infarct size (IS/AAR,%, b) in the control and experimental groups. Values are expressed as mean ± SEM of 12 experiments. Significant difference: *P < 0.01 vs. control; #P < 0.05 vs. A12 and analogue I
Plasma CK-MB and lactate dehydrogenase activity
In the steady state, a plasma CK-MB activity of 274.3 ± 27.2 IU/l was observed [Figure 2a]. In the control animals, the plasma CK-MB activity increased by 7.5 times by the end of reperfusion. Administration of A12 or analogue I markedly reduced the CK-MB activity (915.1 ± 131.2 and 1200.1 ± 110.1 IU/l, respectively, P < 0.05) at the end of reperfusion compared with control. Treatment with analogue II did not alter significantly the plasma CK-MB activity compared with the control group of animals (1624.5 ± 131.2 IU/l, P = 0.07) thus indicating a marked damage to myocardial tissue. This index was higher than in A12 or analogue I groups (P < 0.05).
Figure 2.
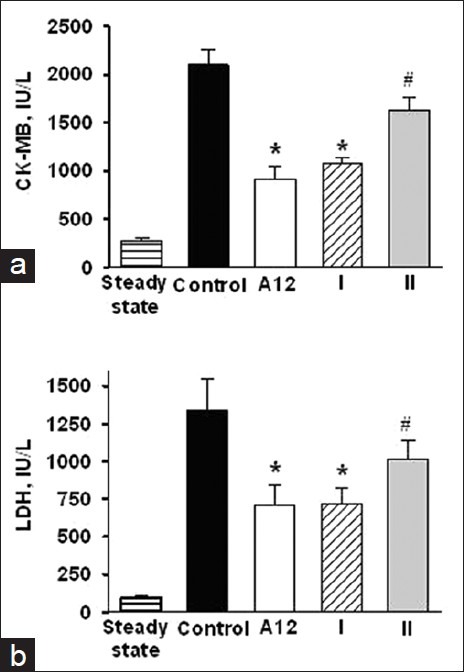
Effects of i/v injection of A12 or its analogues on plasma CK-MB (a) and LDH (b) activities at the end of reperfusion. Values are expressed as mean ± SEM of 6 experiments. Significant difference: *P < 0.01 vs. control; #P < 0.05 vs. A12 and analogue I
In the steady state, a plasma LDH activity of 91.7 ± 20.5 IU/l was observed [Figure 2b]. By the end of reperfusion, the plasma LDH activity increased by more than ten times in control. Administration of A-12 or analogue I substantially reduced the LDH activity (on average by 47% compared with control). Treatment with analogue II did not cause significant change in plasma LDH activity compared with control. The plasma LDH activity in this group was significantly higher than after administration of A-12 or analogue I.
Metabolic state of the area at risk
Myocardial metabolite contents in the AAR are shown in Table 3. By the end of reperfusion, ATP and ∑AN pool in the control animals were reduced by 4 and 2 times as compared with the steady state values, respectively. The control group exhibited only 30% recovery of PCr, almost 40% loss of ∑Cr and a substantial increase in lactate content in comparison with the indices before LAD coronary artery occlusion. Treatment with A12 did not affect preservation of adenine nucleotides in the AAR compared with the values in control. Administration of analogue I or II slightly improved ATP preservation but did not change ADP or AMP contents comparing with the values in control. Myocardial ∑AN pool in analogue I group was significantly higher than in control. Additionally, application of analogue I significantly increased the energy charge of postischemic cardiomyocytes in the AAR compared with this index in A12 group.
Table 3.
Effects of A12 and its analogues on metabolic state of the AAR
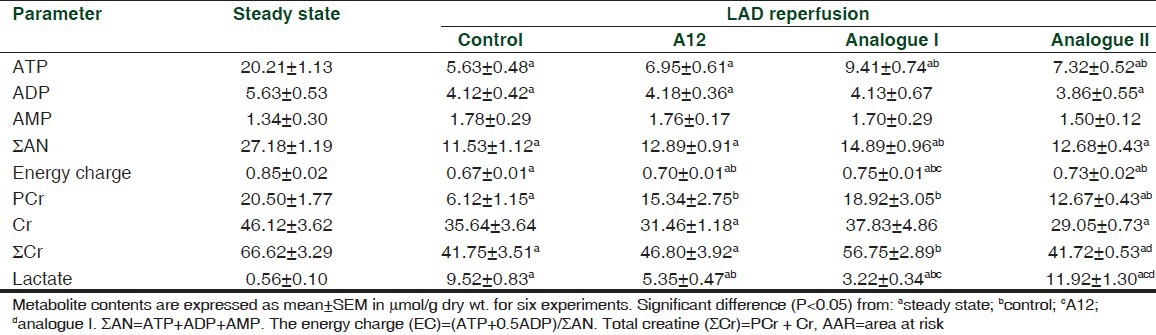
Administration of A-12 and analogue I markedly enhanced preservation of PCr and ∑Cr in the AAR compared with control. This effect was most obvious after treatment with analogue I. In this case, PCr and ∑Cr did not differ significantly from the steady state values. Administration of A12 or analogue I markedly reduced lactate accumulation in the AAR compared with control. Lactate level was significantly lower in analogue I group compared with the value in the A12-treated animals. Treatment with analogue II did not change lactate content compared with that in the control animals. Therefore efficiency of metabolic protection of the AAR induced by peptides increased in the following order: Analogue II < A12 < analogue I.
DISCUSSION
The present study demonstrates the protective effects of a novel A12 analogue I in myocardial regional ischemia and reperfusion in narcotized rats in vivo. They are manifested by a significant limitation of IS [Figure 1b] and enhanced restoration of aerobic metabolism in the AAR [Table 3]. Substitution of methionine to norleucine and modification of amino-terminal arginine residue in A12 provides myocardial protection comparable to that of natural template, A12. Certain advantages of analogue I are enhanced chemical and proteolytic stability, less MAP drop after administration, and a clear trend towards better metabolic preservation of postischemic cardiomyocytes. In contrast, mutation of the carboxy-terminal phenylalanine in analogue II significantly reduced overall efficiency of this peptide in I/R injury.
Previously, Hamada and colleagues attempted to synthesize cyclic analogues of A12 and to evaluate their bioactivities in recombinant human APJ expressed cell line.[21] These peptides were cyclo-A12 in combination with amino-terminal to carboxy-terminal, cyclourea-A12 in combination with amino-terminal and amino acid side chain at positions 7, and cyclo-A12 in combination with amino acid side chain at positions 7 to carboxy-terminal. All three compounds inhibited forskolin-induced cyclic AMP formation, which was almost abolished by pertussis toxin (PTx) treatment. Moreover, they induced the activation of Akt and extracellular signal-regulated kinase (ERK1/2) in PTx-sensitive manner. Thus, the cyclo-A12 analogues might act as the effective agonist at APJ receptors in cell-based in vitro assays. The results of our work testified successfully that modified A12 peptide retains high functional potency in the physiologically relevant model. They are principally consistent with an earlier study which showed that preischemic administration of analogue I attenuated cardiac dysfunction in isolated perfused rat heart subjected to global ischemia and reperfusion.[22]
Cardioprotective effects of exogenous C-terminal fragments of apelin are mediated in part by activation of components of the reperfusion injury salvage kinase (RISK) pathway, PI3K/Akt and MEK1/2–ERK1/2.[10] This is confirmed by abolishing reduction of IS induced by apelin-13 in the presence of the inhibitors of PI3K-Akt and p44/42 phosphorylation, LY294002 and UO126, respectively.[3] Similarly, addition of wortmannin and PD098059, the inhibitors of PI3K/Akt and ERK1/2, to culture medium during hypoxia/reoxygenation, suppressed cardiomyocyte viability provided by apelin-13.[6] The activation of the pro-survival PI3K/Akt cascade results in phosphorylation and activation of eNOS with further involvement of NO in diverse mechanisms of cellular protection. In our study, participation of NO-dependent mechanisms in beneficial action of apelin and its structural analogue I is testified by a transient fall in MAP after peptide administration [Table 2]. However, abolishing or attenuating protective effects of apelin-13 and A12 after coadministration with NG-nitro-L-arginine methyl ester (L-NAME), the NOS inhibitor, directly indicate the principal role of NO.[11,12] Since NO prevents mitochondrial oxygen damage and lipid peroxidation,[23] we may assume that reduction of I/R injury by A12 and analogue I might be related to antioxidant properties of these peptides. Such a possibility was recently confirmed by several authors for apelin-13. In various types of experimental preparations, apelin-13 decreased production of reactive oxygen species (ROS) and malonic dialdehyde (MDA) with a simultaneous increase in superoxide dismutase (SOD) activity.[6,24]
We have previously demonstrated an important role of myocardial bioenergetics in postischemic functional recovery of isolated rat heart treated with A12 or its analogues.[22] The results of the present study indicated that improved metabolic state of the AAR is also implicated in amelioration of I/R injury by analogue I. In fact, a better preservation of myocardial adenine nucleotides and PCr may be of critical importance for maintaining cell membrane integrity and ion homeostasis, thus preventing irreversible damage of cardiomyocytes during reperfusion.[25] Various studies point out an emerging role of apelin in stimulation of glucose utilization in normal and insulin-resistant mice.[26] In the heart and skeletal muscle, apelin effects on glucose uptake and oxidation are attributed to the activation of eNOS, AMP-activated protein kinase (AMPK) and Akt-dependent pathways.[27,28] It is known that under ischemic conditions, the detrimental effects of free fatty acids (FFA) on myocardial metabolism are: accumulation of long chain acyl-CoA thioesters and long chain acylcarnitines inhibit glycolytic flux, and uncoupling of oxidative phosphorylation from electron transfer.[29] Therefore, a shift in myocardial substrate utilization from FFA to glucose should facilitate recovery of energy metabolism during reperfusion. Probably apelin-stimulated alterations in glucose utilization may represent a physiologically significant mechanism for correcting metabolic disorders induced by ischemia and reperfusion.
In conclusion, the present study revealed the ability of pharmacological agonist of APJ receptor to reduce myocardial I/R injury in vivo. Further delineation of cardioprotective effects of this peptide requires the use of spin traps for ROS and NO detection, and selective inhibitors of NOS isoforms. Synthesis of this peptide by using a solid phase method and Fmoc technology with a high yield, and increased chemical stability of the modified molecule in comparison with natural A12 have been demonstrated in our recent paper.[30] Therefore, we believe that modification of C-terminal fragments of apelin may be a useful approach to design new therapeutic tools for the treatment of acute coronary syndrome.
ACKNOWLEDGMENT
This study was supported in part by a grant from The Russian Foundation for Basic Research No. 11-04-00078a.
Footnotes
Source of Support: The Russian Foundation for Basic Research No. 11-04-00078a
Conflict of Interest: None declared.
REFERENCES
- 1.Ferdinandy P, Schulz R, Baxter G. Interaction of Cardiovascular Risk Factors with Myocardial Ischemia/Reperfusion Injury, Preconditioning, and Postconditioning. Pharmacol Rev. 2007;9:418–58. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 2.Kleinz MJ, Davenport AP. Emerging roles of apelin in biology and medicine. Pharmacol Therap. 2005;107:198–211. doi: 10.1016/j.pharmthera.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Simpkin JC, Yellon DM, Davidson SM, Lim SY, Wynne AM, Smith CC. Apelin-13 and apelin-36 exhibit direct cardioprotective activity against ischemia-reperfusion injury. Basic Res Cardiol. 2007;102:518–28. doi: 10.1007/s00395-007-0671-2. [DOI] [PubMed] [Google Scholar]
- 4.Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–6. doi: 10.1006/bbrc.1998.9489. [DOI] [PubMed] [Google Scholar]
- 5.Kleinz MJ, Baxter GF. Apelin reduces myocardial reperfusion injury independently of PI3K/Akt and P70S6 kinase. Regul Pept. 2008;46:271–7. doi: 10.1016/j.regpep.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Zeng XJ, Zhang LK, Wang HX, Lu LQ, Ma LQ, Tang CS. Apelin protects heart against ischemia/reperfusion injury in rat. Peptides. 2009;30:1144–52. doi: 10.1016/j.peptides.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Pisarenko OI, Serebryakova LI, Pelogeykina YA, Studneva IM, Khatri DN, Tskitishvili OV, et al. In vivo reduction of reperfusion injury to the heart with apelin-12 peptide in rats. Bull Exp Biol Med. 2011;152:79–82. doi: 10.1007/s10517-011-1459-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Yu B, Tao G. Apelin protects against cardiomyocyte apoptosis induced by glucose deprivation. Chin Med J (Engl) 2009;122:2360–5. [PubMed] [Google Scholar]
- 9.Smith CC, Mocanu MM, Bowen J, Wynne AM, Simpkin JC, Dixon RA, et al. Temporal changes in myocardial salvage kinases during reperfusion following ischemia: Studies involving the cardioprotective adipocytokine apelin. Cardiovasc Drugs Ther. 2007;21:409–14. doi: 10.1007/s10557-007-6054-y. [DOI] [PubMed] [Google Scholar]
- 10.Smith CC, Yellon DM. Adipocytokines, cardiovascular pathophysiology and myocardial protection. Pharmacol Ther. 2011;129:206–19. doi: 10.1016/j.pharmthera.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Rastaldo R, Cappello S, Folino A, Berta GN, Sprio AE, Losano G, et al. Apelin-13 limits infarct size and improves cardiac postischemic mechanical recovery only if given after ischemia. Am J Physiol Heart Circ Physiol. 2011;300:H2308–15. doi: 10.1152/ajpheart.01177.2010. [DOI] [PubMed] [Google Scholar]
- 12.Pisarenko OI, Pelogeykina YA, Shulzhenko VS, Studneva IM. Nitric oxide synthase mediates the apelin-induced improvement of myocardial postischemic metabolic and functional recovery. Open J Mol Integ Physiol. 2012;2:1–7. [Google Scholar]
- 13.Weir RA, Chong KS, Dalzell JR, Petrie CJ, Murphy CA, Steedman T, et al. Plasma apelin concentration is depressed following acute myocardial infarction in man. Eur J Heart Fail. 2009;11:551–8. doi: 10.1093/eurjhf/hfp043. [DOI] [PubMed] [Google Scholar]
- 14.Langelaan DN, Rainey JK. Headgroup-dependent membrane catalysis of apelin-receptor interactions is likely. J Phys Chem B. 2009;113:10465–71. doi: 10.1021/jp904562q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohno S, Yakabi K, Ro S, Ochiai M, Onouchi T, Sakurada T, et al. Apelin-12 stimulates acid secretion through an increase of histamine release in rat stomachs. Regul Pept. 2012;174:71–8. doi: 10.1016/j.regpep.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, et al. Acute cardiovascular effects of apelin in humans: Potential role in patients with chronic heart failure. Circulation. 2010;121:1818–27. doi: 10.1161/CIRCULATIONAHA.109.911339. [DOI] [PubMed] [Google Scholar]
- 17.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 18.Lee DK, Saldivia VR, Nguyen T, Cheng R, George SR, O’Dowd BF. Modification of the terminal residue of apelin-13 antagonizes its hypertensive action. Endocrinology. 2005;146:231–6. doi: 10.1210/en.2004-0359. [DOI] [PubMed] [Google Scholar]
- 19.Fishbein MC, Maclean D, Maroko PR. Experimental myocardial infarction in the rat. Am J Pathol. 1978;90:57–70. [PMC free article] [PubMed] [Google Scholar]
- 20.Bergmeyer HU, editor. New York: Academic Press; 1974. Methods of enzymatic analysis; pp. 1464–7. [Google Scholar]
- 21.Hamada J, Kimura J, Ishida J, Kohda T, Morishita S, Ichihara S, et al. Evaluation of novel cyclic analogues of apelin. Int J Mol Med. 2008;22:547–52. [PubMed] [Google Scholar]
- 22.Pisarenko OI, Shulzhenko VS, Pelogeykina YA, Studneva IM. Attenuation of myocardial ischemia and reperfusion injury by novel analogues of apelin-12. Int J Pharm Biomed Res. 2012;3:16–21. [Google Scholar]
- 23.Shultz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:402–13. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 24.Jia YX, Lu ZF, Zhang J, Pan CS, Yang JH, Zhao J, et al. Apelin activates L-arginine/nitric oxide synthase/nitric oxide pathway in rat aortas. Peptides. 2007;28:2023–9. doi: 10.1016/j.peptides.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 25.Portman MA, Zhang J. Myocardial energy transport and heart failure. Curr Cardiol Rev. 2005;1:17–27. [Google Scholar]
- 26.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–53. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Hu X, Selvakumar P, Russell RR, Cushman SW, Holman GD, et al. Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab. 2004;287:E834–41. doi: 10.1152/ajpendo.00234.2004. [DOI] [PubMed] [Google Scholar]
- 28.Yue P, Jin H, Aillaud M, Deng AC, Azuma J, Asagami T, et al. Apelin is necessary for the maintenance of insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E59–67. doi: 10.1152/ajpendo.00385.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hendrickson S, Louis J, Lowe J, Abdel-aleem S. Free fatty acid metabolism during myocardial ischemia and reperfusion. Mol Cell Biochem. 1997;166:85–94. doi: 10.1023/a:1006886601825. [DOI] [PubMed] [Google Scholar]
- 30.Sidorova MV, Az’muko AA, Pal’keeva ME, Molokoedov AS, Bushuev VN, Dvoriantsev SN, et al. Synthesis and cardioprotective properties of apelin-12 and its structural analogues. Russ J Bioorganic Chem. 2012;38:40–51. doi: 10.1134/s1068162012010177. [DOI] [PubMed] [Google Scholar]