

Abstract
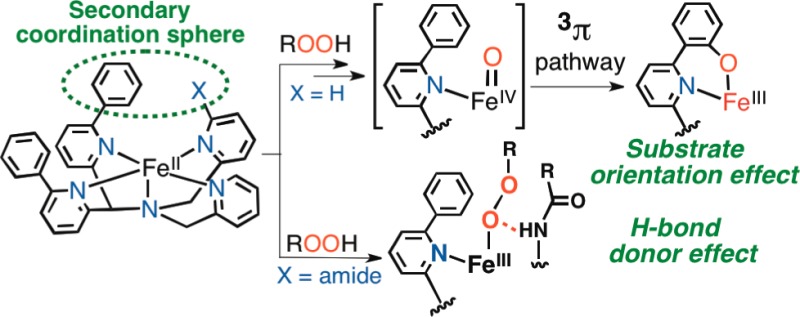
The new biomimetic ligands N4Py2Ph (1) and N4Py2Ph,amide (2) were synthesized and yield the iron(II) complexes [FeII(N4Py2Ph)(NCCH3)](BF4)2 (3) and [FeII(N4Py2Ph,amide)](BF4)2 (5). Controlled orientation of the Ph substituents in 3 leads to facile triplet spin reactivity for a putative FeIV(O) intermediate, resulting in rapid arene hydroxylation. Addition of a peripheral amide substituent within hydrogen-bond distance of the iron first coordination sphere leads to stabilization of a high-spin FeIIIOOR species which decays without arene hydroxylation. These results provide new insights regarding the impact of secondary coordination sphere effects at nonheme iron centers.
Nonheme iron oxygenases are potent and selective catalysts, typically operating through iron-peroxo (FeOO(H/R)) and iron-oxo (Fe(O)) intermediates. For example, arene hydroxylation is mediated by a class of mammalian nonheme iron enzymes known as aromatic amino acid hydroxylases (e.g., Tyr, Phe, and Trp hydroxylases), and both FeIIOOR and FeIV(O) species are postulated as key intermediates in their catalytic cycles.1 Much effort has gone into the preparation of synthetic analogs of FeOO(H/R) and FeIV(O) intermediates, employing ligands designed to stabilize these species through the use of oxidatively inert, biologically relevant donor groups and steric shielding of the metal center. Enzymes, however, also have at their disposal the ability to control the second coordination sphere through the juxtaposition of substrates at appropriate distances from the metal center and through interacting residues that can tune reactivity via hydrogen bonds. In contrast, model complexes that are designed to incorporate these second-coordination sphere effects are less developed.
Herein we report the synthesis of two new polydentate ligands, N4Py2Ph (1) and N4Py2Ph,amide (2) (N4Py = N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine) (Figure 1), which have been designed to examine second-coordination sphere effects in nonheme Fe model complexes. The new ligand 1 incorporates phenyl substituents as constrained substrates for oxidation, while 2 includes an additional amide group as a hydrogen-bond donor for interaction with metal–oxygen intermediates. An iron(II) complex from 1 undergoes rapid, regioselective arene hydroxylation at one Ph ring, and a novel reaction channel appears accessible by the positioning of the phenyl ring in the second coordination sphere. In contrast, the FeII complex from 2 does not undergo arene hydroxylation but rather leads to the formation of a metastable FeIII-OOR complex. The latter result suggests a significant influence of the amide-derived H-bond donor group on the stability of the FeIII-OOR species. Computational studies, in combination with the experimental data, provide key insights regarding the importance of the second-coordination sphere effects introduced by 1 and 2.
Figure 1.
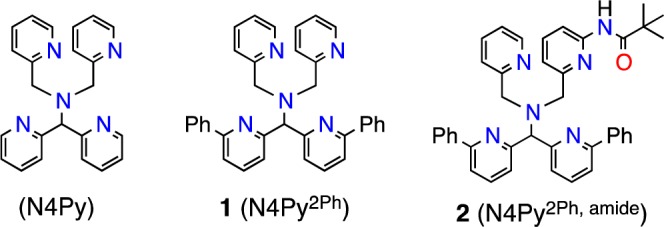
Structures of new ligands 1 and 2.
A key step in the synthesis of diphenyl-substituted 1 was a Suzuki–Miyaura coupling between bis(6-bromo-2-pyridyl) ketone and C6H5B(OH)2, with the final ligand 1 prepared as in Figure S1. Stirring of 1 equiv of Fe(BF4)2 and 1 in CH3CN (Scheme 1) leads to X-ray quality crystals of [FeII(N4Py2Ph)(NCCH3)](BF4)2 (3) (92%) from Et2O/CH3CN. X-ray diffraction reveals the structure of 3 as shown in Figure 2, which has a six-coordinate FeII center bound to the pentadentate N4Py2Ph ligand and one CH3CN molecule. The Fe-Npy distances for 3 (2.165(1)–2.379(1); 2.172(1)–2.442(1) Å) are indicative of high-spin (hs) FeII-Npy bond lengths.2 The paramagnetically shifted 1H NMR spectrum (from 151 to −7 ppm) as well as a magnetic moment measurement by Evan’s method in CD3CN (exptl μeff = 5.2; calcd (spin-only, S = 2) μeff = 4.9) confirmed that 3 is hs-FeII. Thus the addition of phenyl substituents to the N4Py scaffold causes a spin state change from low-spin (ls) FeII for [FeII(N4Py)(CH3CN)]2+ to hs-FeII for 3.3
Scheme 1.
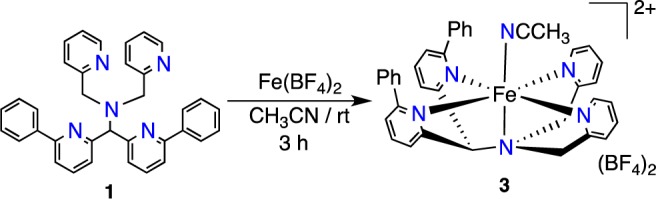
Figure 2.
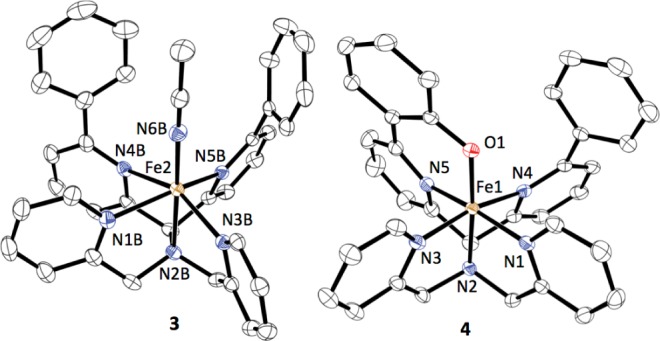
Displacement ellipsoid plots (50% probability) of the cations of 3 and 4. H atoms are omitted for clarity.
Addition of a small excess (4–5 equiv) of tBuOOH to 3 in CH3CN at room temperature results in an immediate color change from yellow to green and a new UV–vis band (λmax = 758 nm, ε = 1880 M–1 cm–1) (Figure 3). Characterization by LDI-MS(+) revealed a parent ion at m/z 590.167, corresponding to [Fe(N4Py2Ph)-H+O]+. Isolation of the modified ligand and analysis by FAB-MS is consistent with ligand monohydroxylation. The product N4Py2PhOH was isolated in 65% yield following purification.
Figure 3.
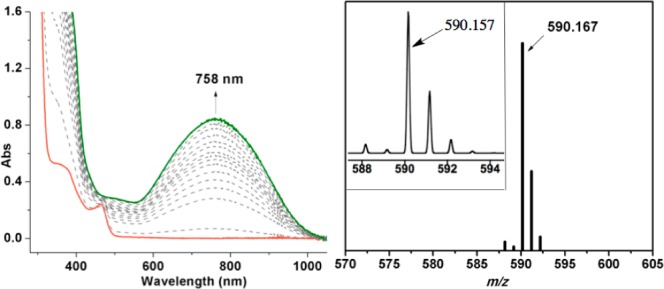
UV–vis spectral changes (0–17 min) (left) and LDIMS(+) (inset: calcd for 4) (right) for 3 + tBuOOH (5 equiv) in CH3CN.
Hydroxylation of the ligand in the reaction of 3 + tBuOOH suggests a reactive oxidant, such as a high-valent FeIV(O) species, is formed as a transient intermediate via O-O bond cleavage from an Fe-OOtBu precursor.4 We turned toward oxygen-atom transfer (OAT) agents in an attempt to generate an FeIV(O) species directly. Addition of PhIO or the more soluble analog isopropyl 2-iodoxybenzoate (IBX-ester)4d,5 (1 equiv) to 3 in CH3CN resulted in the rapid formation of a green species with an almost identical UV–vis signature (λmax = 763 nm, ε = 2190 M–1 cm–1) and LDI-MS spectrum (m/z 589.892) as that seen for the product obtained from tBuOOH. The IBX-ester reaction afforded X-ray quality crystals of [FeIII(N4PyPh,PhO)](BF4)2 (4) from iPr2O/CH3CN (Figure 2). The structure confirmed that arene hydroxylation had occurred to give a phenolato-iron(III) complex. The Fe-N bond lengths (1.9193(15)–2.0358(15) Å) and EPR spectrum (g 2.39, 2.12, 1.90) indicate 4 is an ls-FeIII complex.
The reaction of 3 with tBuOOH, PhIO or IBX-ester results in a regioselective intramolecular arene hydroxylation to give a single ortho-hydroxylated product. This selectivity for the same product points to a common metal-based intermediate for all three oxidants, which is most likely the proposed [FeIV(O)(N4Py2Ph)]2+ species (Scheme 2).4b,4c Further support for the involvement of [FeIV(O)(N4Py2Ph)]2+ comes from UV–vis and NMR spectral titrations, where maximal formation of 4 is clearly seen following the addition of 1 equiv of IBX-ester (Figures S9, S10). These data are consistent with two-electron OAT from IBX-ester to 3 to give an FeIV(O) species that then hydroxylates the phenyl ring. Reaction of PhI18O + 3 leads to 18O incorporation (88%) in 4 as seen by LDI-MS (Figure S16), indicating that the O atom in 4 is derived exclusively from the organic oxidant, and providing additional evidence for the FeIV(O) species as a key intermediate.
Scheme 2.
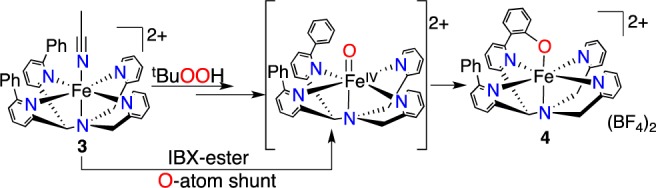
Attempts were made to trap the FeIV(O) intermediate derived from 3. Reaction of 3 with IBX-ester and tBuOOH was examined at low temperature but led only to the slow formation of 4 (−35 °C/CH3CN, Figure S11) or decomposition (−60 °C/CH2Cl2), suggesting that the FeIV(O) species rapidly reacts with the C6H5 substituent even at low temperature. In contrast, the stable [FeIV(O)(N4Py)]2+ 6 does not react with C6H6 (500 equiv) even over prolonged reaction times (>14 h). Thus [FeIV(O)(N4Py)]2+ is not competent to mediate benzene hydroxylation.7
A few examples of aromatic hydroxylation mediated by nonheme Fe complexes are known, and some have implicated the importance of orienting the aromatic substrate near the metal.4,8 However, the mechanism of hydroxylation and identity of the active oxidant in these systems remain poorly understood. In the case of 3, rapid intramolecular phenyl hydroxylation is observed, but there is a complete lack of reactivity between [FeIV(O)(N4Py)]2+ and C6H6. Similar contradictory observations have been discussed for intra- vs intermolecular phenyl hydroxylations in the Fe-TPA (TPA = tris(2-pyridylmethyl)amine) system, but no explanation has been given.4a,4b,8g To gain insight into the mechanism of intra- vs intermolecular arene hydroxylation, we performed density functional theory (DFT) calculations on [FeIV(O)(N4Py2Ph)]2+ and compared the results with those previously obtained for [FeIV(O)(N4Py)]2+ + C6H6.7
The [FeIV(O)(N4Py2Ph)]2+ structure has close lying triplet and quintet spin configurations, with a favorable triplet spin state in the gas phase and almost degenerate spin states at free energy level at 298 K. This spin state splitting is much smaller than that found for [FeIV(O)(N4Py)]2+, where the triplet spin state was found to be at least 8 kcal mol–1 more stable than the quintet.7 Electrophilic attack of the oxo group on the ortho-carbon atom leads to electron transfer from Ph to the π*xz orbital in the triplet spin state (3π-pathway), whereas in the quintet spin state the virtual σ*z2 (5σ-pathway) or π*xz (5π-pathway) are the possible acceptor orbitals. The 3π-pathway generates an intermediate with configuration π*xy2 π*xz2 π*yz↑ ϕL↑, while the 5σ-channel gives π*xy↑ π*xz↑ π*yz↑ σ*z2↑ σ*x2–y2↑ ϕL↓, and the 5π-pathway gives π*xz2 π*xy↑ π*yz↑ σ*x2–y2↑ ϕL↑.
As seen in Figure 4, the gas-phase energies predict that the 3π channel is the lowest in energy, although the quintet transition states are not that far above 3TSπ. The 5σ-pathway is slightly favored over the 3π-pathway at the free-energy level. These results differ dramatically from calculations on [FeIV(O)(N4Py)]2+, which show that the 3π-pathway is highly destabilized compared to the 5σ-pathway (δΔG‡ > 15 kcal/mol).7
Figure 4.
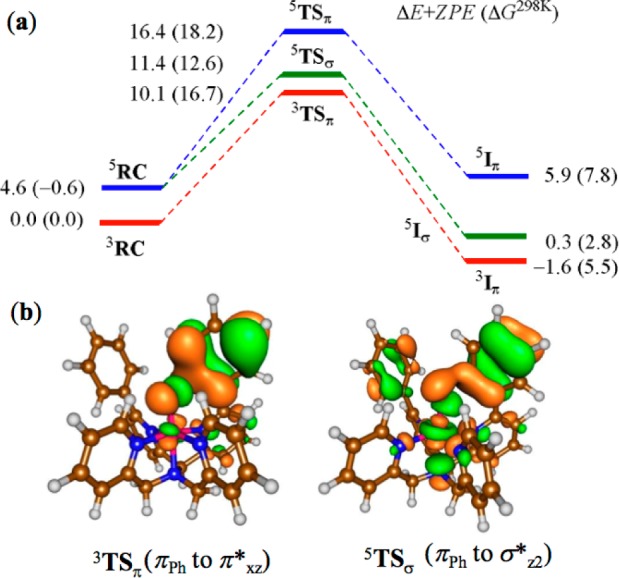
Potential energy landscape (kcal/mol) for arene hydroxylation of [FeIV(O)(N4Py2Ph)]2+ (a) and molecular orbitals for the electron transfer pathways (b).
Much effort has gone into determining the factors that control the reactivity of FeIV(O) species. For both nonheme iron models and enzymes, the triplet (low-spin) state for FeIV(O) is described as generally unreactive, whereas quintet (high-spin) FeIV(O) is considered a powerful oxidant.9 The high reactivity for the quintet species has been attributed to the accessibility of the 5σ-reaction channel, with an approximate collinear approach (Fe–O–C(or H) = 180°) of the substrate donor orbital with the Fe=O unit. The triplet FeIV(O), on the other hand, is limited to the 3π-reaction channel with an approximate perpendicular approach for overlap with the π*xz/yz orbital. The ligands in the equatorial plane of triplet FeIV(O) complexes typically provide a steric barrier to this channel and make it prohibitively high in energy, and a low-lying quintet excited state is often invoked to explain the observed reactivity for triplet FeIV(O).
The DFT calculations for [FeIV(O)(N4Py2Ph)]2+ indicate that the low-spin, 3π-pathway becomes a viable reaction channel by positioning the phenyl substrate in the second coordination sphere. In comparison, triplet [FeIV(O)(N4Py)]2+ is completely unreactive toward C6H6, consistent with the fact that the 3π-pathway is sterically blocked.7,9g,10 Computational investigations have asserted the importance of substrate positioning (σ vs π) in controlling nonheme Fe(O) reactivity,9 but to our knowledge the results herein provide the first combined experimental/theoretical evidence for the importance of σ vs π substrate orientation in a synthetic nonheme iron system.
Addition of another secondary coordination sphere element, in the form of a potential H-bond donor group,11 dramatically changes the reactivity of the nonheme FeII complex. The complex [FeII(N4Py2Ph,amide)](BF4)2 (5) was readily prepared from 2, and an X-ray structure reveals a six-coordinate, hs-FeII complex with the new amide group bound in the open site (Figure 5). Reaction of 5 with tBuOOH (10 equiv) at room temperature does not induce arene hydroxylation as seen for 3(12) but instead gives rise to a transient green intermediate that rapidly decays (∼t1/2 < 30 s). This intermediate can be trapped at −30 °C, revealing a long-lived, dark-blue species with λmax = 606 nm (ε = 2100 M–1 cm–1 based on total Fe), which rapidly decays upon warming without any ligand hydroxylation (Figures S22, S23). Resonance Raman spectroscopy of the 606 nm species (6) shows vibrations at 642 and 876 cm–1, which are typical of hs-FeIIIOOR species and can be assigned to ν(Fe–O) and ν(O–O) (Figure S21), respectively.13 EPR revealed hs-FeIII peaks (g 7.89, 5.55, 4.24), along with a minor, unidentified ls-FeIII component (∼20%). Based on these data, a reasonable structure for the blue species is proposed for complex 6 as shown in Figure 5.
Figure 5.
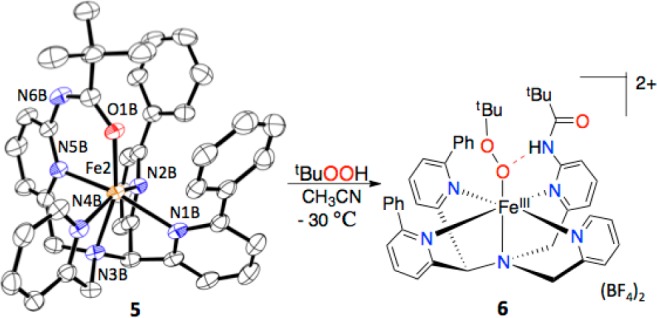
Generation of the FeIII(OOtBu) complex 6.
DFT calculations (see Supporting Information) fully support the proposed, hs-FeIII–OOR structure with an amide N–H···O bond. The influence of H-bond donors on the stability of iron–oxygen species in nonheme Fe systems is of great interest but is still not well-understood.11 We conclude that the amide group in 5 helps to trap an FeIII–OOR complex, in contrast to 3, which likely forms an FeIII–OOR species as a transient intermediate during arene hydroxylation.
We have employed an experimental and computational approach to examine the influence of secondary coordination sphere modifications in nonheme iron model complexes. The results herein give new insights regarding how nonheme Fe enzymes may utilize two critical secondary coordination sphere effects, substrate orientation, and hydrogen bonding, to control reactivity.
Acknowledgments
The NIH (D.P.G., GM62309; P.M.L., GM074785) is gratefully acknowledged for financial support. S.d.V. thanks the NSCCS for CPU time and M.G.Q. thanks BBSRC for a studentship. H.M. acknowledges support from the JSPS.
Supporting Information Available
Experimental and DFT details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Fitzpatrick P. F. Biochemistry 2003, 42, 14083–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bruijnincx P. C. A.; van Koten G.; Klein Gebbink R. J. M. Chem. Soc. Rev. 2008, 37, 2716–2744. [DOI] [PubMed] [Google Scholar]; c Iron-containing enzymes: Versatile catalysts of hydroxylation reaction in nature; de Visser S. P., Kumar D., Eds.; RSC Publishing: Cambridge (U.K.), 2011. [Google Scholar]
- a Krishnamurthy D.; Sarjeant A. N.; Goldberg D. P.; Caneschi A.; Totti F.; Zakharov L. N.; Rheingold A. L. Chem.—Eur. J. 2005, 11, 7328–7341 and reference therein. [DOI] [PubMed] [Google Scholar]; b Complex 3 is hs-FeII from 4–300 K in the solid state as seen by magnetic susceptibility measurements.
- a Lubben M.; Meetsma A.; Wilkinson E. C.; Feringa B.; Que L. Jr. Angew. Chem., Int. Ed. 1995, 34, 1512–1514. [Google Scholar]; b We have confirmed that [FeII(N4Py)(CH3CN)](BF4)2, which has the same counter ion as 3, is also low-spin FeII.
- a Makhlynets O. V.; Das P.; Taktak S.; Flook M.; Mas-Ballesté R.; Rybak-Akimova E. V.; Que L. Jr. Chem.—Eur. J. 2009, 15, 13171–13180. [DOI] [PubMed] [Google Scholar]; b Jensen M. P.; Lange S. J.; Mehn M. P.; Que E. L.; Que L. Jr. J. Am. Chem. Soc. 2003, 125, 2113–2128. [DOI] [PubMed] [Google Scholar]; c Mekmouche Y.; Ménage S.; Toia-Duboc C.; Fontecave M.; Galey J. B.; Lebrun C.; Pécaut J. Angew. Chem., Int. Ed. 2001, 40, 949–952. [PubMed] [Google Scholar]; d Makhlynets O. V.; Rybak-Akimova E. V. Chem.—Eur. J. 2010, 16, 13995–14006. [DOI] [PubMed] [Google Scholar]; e Ansari A.; Kaushik A.; Rajaraman G. J. Am. Chem. Soc. 2013, 135, 4235–4249. [DOI] [PubMed] [Google Scholar]
- a Ye W.; Staples R. J.; Rybak-Akimova E. V. J. Inorg. Biochem. 2012, 115, 1–12. [DOI] [PubMed] [Google Scholar]; b Ye W.; Ho D. M.; Friedle S.; Palluccio T. D.; Rybak-Akimova E. V. Inorg. Chem. 2012, 51, 5006–5021. [DOI] [PubMed] [Google Scholar]
- a Kaizer J.; Klinker E. J.; Oh N. Y.; Rohde J.-U.; Song W. J.; Stubna A.; Kim J.; Münck E.; Nam W.; Que L. Jr. J. Am. Chem. Soc. 2004, 126, 472–473. [DOI] [PubMed] [Google Scholar]; b Klinker E. J.; Kaizer J.; Brennessel W. W.; Woodrum N. L.; Cramer C. J.; Que L. Jr. Angew. Chem., Int. Ed. 2005, 44, 3690–3694. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Oh K.; Han A.-R.; Nam W. Inorg. Chem. 2007, 46, 4632–4641. [DOI] [PubMed] [Google Scholar]
- a Bigi J. P.; Harman W. H.; Lassalle-Kaiser B.; Robles D. M.; Stich T. A.; Yano J.; Britt R. D.; Chang C. J. J. Am. Chem. Soc. 2012, 134, 1536–1542. [DOI] [PubMed] [Google Scholar]; b Mehn M. P.; Fujisawa K.; Hegg E. L.; Que L. Jr. J. Am. Chem. Soc. 2003, 125, 7828–7842. [DOI] [PubMed] [Google Scholar]; c Nielsen A.; Larsen F. B.; Bond A. D.; McKenzie C. J. Angew. Chem., Int. Ed. 2006, 45, 1602–1606. [DOI] [PubMed] [Google Scholar]; d Ménage S.; Galey J.-B.; Dumats J.; Hussler G.; Seité M.; Luneau I. G.; Chottard G.; Fontecave M. J. Am. Chem. Soc. 1998, 120, 13370–13382. [Google Scholar]; e Avenier F.; Dubois L.; Latour J.-M. New J. Chem. 2004, 28, 782–784. [Google Scholar]; f Yamashita M.; Furutachi H.; Tosha T.; Fujinami S.; Saito W.; Maeda Y.; Takahashi K.; Tanaka K.; Kitagawa T.; Suzuki M. J. Am. Chem. Soc. 2007, 129, 2–3. [DOI] [PubMed] [Google Scholar]; g Oh N. Y.; Seo M. S.; Lim M. H.; Consugar M. B.; Park M. J.; Rohde J.-U.; Han J.; Kim K. M.; Kim J.; Que L. Jr.; Nam W. Chem. Commun. 2005, 5644–5646. [DOI] [PubMed] [Google Scholar]
- a Neidig M. L.; Decker A.; Choroba O. W.; Huang F.; Kavana M.; Moran G. R.; Spencer J. B.; Solomon E. I. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12966–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]; b de Visser S. P. J. Am. Chem. Soc. 2006, 128, 9813–9824. [DOI] [PubMed] [Google Scholar]; c de Visser S. P. J. Am. Chem. Soc. 2006, 128, 15809–15818. [DOI] [PubMed] [Google Scholar]; d Hirao H.; Kumar D.; Que L. Jr.; Shaik S. J. Am. Chem. Soc. 2006, 128, 8590–8606. [DOI] [PubMed] [Google Scholar]; e Geng C.; Ye S. F.; Neese F. Angew. Chem., Int. Ed. 2010, 49, 5717–5720. [DOI] [PubMed] [Google Scholar]; f Ye S.; Neese F. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 1228–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Srnec M.; Wong S. D.; England J.; Que L. Jr.; Solomon E. I. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Wilson S. A.; Chen J.; Hong S.; Lee Y.-M.; Clémancey M.; Garcia-Serres R.; Nomura T.; Ogura T.; Latour J.-M.; Hedman B.; Hodgson K. O.; Nam W.; Solomon E. I. J. Am. Chem. Soc. 2012, 134, 11791–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald A. R.; Guo Y.; Vu V. V.; Bominaar E. L.; Münck E.; Que L. Jr. Chem. Sci. 2012, 3, 1680–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shook R. L.; Borovik A. S. Inorg. Chem. 2010, 49, 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Berreau L. M.; Makowska-Grzyska M. M.; Arif A. M. Inorg. Chem. 2001, 40, 2212–2213. [DOI] [PubMed] [Google Scholar]; c Yeh C.-Y.; Chang C. J.; Nocera D. G. J. Am. Chem. Soc. 2001, 123, 1513–1514. [DOI] [PubMed] [Google Scholar]; d Wada A.; Harata M.; Hasegawa K.; Jitsukawa K.; Masuda H.; Mukai M.; Kitagawa T.; Einaga H. Angew. Chem., Int. Ed. 1998, 37, 798–799. [DOI] [PubMed] [Google Scholar]; e Latifi R.; Sainna M. A.; Rybak-Akimova E. V.; de Visser S. P. Chem.—Eur. J. 2013, 19, 4058–4068. [DOI] [PubMed] [Google Scholar]
- Reaction of 5 with PhIO or IBX-ester also does not result in arene hydroxylation (Figure S24).
- Namuswe F.; Hayashi T.; Jiang Y.; Kasper G. D.; Sarjeant A. A. N.; Moënne-Loccoz P.; Goldberg D. P. J. Am. Chem. Soc. 2010, 132, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.