

Abstract
Turneriella parva Levett et al. 2005 is the only species of the genus Turneriella which was established as a result of the reclassification of Leptospira parva Hovind-Hougen et al. 1982. Together with Leptonema and Leptospira, Turneriella constitutes the family Leptospiraceae, within the order Spirochaetales. Here we describe the features of this free-living aerobic spirochete together with the complete genome sequence and annotation. This is the first complete genome sequence of a member of the genus Turneriella and the 13th member of the family Leptospiraceae for which a complete or draft genome sequence is now available. The 4,409,302 bp long genome with its 4,169 protein-coding and 45 RNA genes is part of the Genomic Encyclopedia of Bacteria and Archaea project.
Keywords: Gram-negative, motile, axial filaments, helical, flexible, non-sporulating, aerobic, mesophile, Leptospiraceae, GEBA
Introduction
Strain HT (= DSM 21527 = NCTC 11395 = ATCC BAA-1111) is the type strain of Turneriella parva [1]. The strain was isolated from contaminated Leptospira culture medium [2] and was originally thought to be affiliated with Leptospira [2] because of morphological similarities to other members of the genus. Strain HT was designated as a separate species because of certain morphological and molecular differences: cells were shorter and were more tightly wound, the surface layer formed blebs instead of cross-striated tubules when detached for negative staining preparation and the base composition of DNA differed from that of other Leptospira species [2]. DNA-DNA hybridization [3] and enzyme activity [4] studies revealed sufficient differences between other Leptospira species and L. parva that the ‘Subcommittee on the Taxonomy of Leptospira’ [5] decided to exclude L. parva from the genus Leptospira and assign it as the type strain of a new genus: ‘Turneria’ as ‘Turneria parva’. The genus was named in honor of Leslie Turner, an English microbiologist who made definitive contributions to the knowledge of leptospirosis [1]. However, as the generic name is also in use in botany and zoology, this name was rendered illegitimate and invalidate, but was used in the literature [6,7]. The first 16S rRNA gene-based study (Genbank accession number Z21636), performed on Leptospira parva incertae sedis, confirmed the isolated position of L. parva among Leptonema and Leptospira species [8], a finding later supported by Morey et al. [9]. The reclassification of L. parva as Turneriella parva com. nov. was published by Levett et al. [1], reconfirming the separate position of the type strain [10] and an additional strain (S-308-81, ATCC BAA-1112) from the uterus of a sow from all other leptospiras on the basis of DNA-DNA hybridization and 16S rRNA gene sequence analysis (Genbank accession number AY293856). The strain was selected for genome sequencing because of its deep branching point within the Leptospiraceae lineage.
Here we present a summary classification and a set of features for T. parva HT together with the description of the complete genomic sequencing and annotation.
Classification and features
16S rRNA gene sequence analysis
A representative genomic 16S rDNA sequence of T. parva HT was compared using NCBI BLAST [11,12] under default settings (e.g., considering only the high-scoring segment pairs (HSPs) from the best 250 hits) with the most recent release of the Greengenes database [13] and the relative frequencies of taxa and keywords (reduced to their stem [14]) were determined, weighted by BLAST scores. The most frequently occurring genera were Geobacter (48.7%), Leptospira (19.2%), Pelobacter (13.4%), Spirochaeta (8.1%) and Turneriella (6.4%) (56 hits in total). Regarding the single hit to sequences from members of the species, the average identity within HSPs was 95.8%, whereas the average coverage by HSPs was 89.8%. Among all other species, the one yielding the highest score was Leptonema illini (AY714984), which corresponded to an identity of 85.7% and an HSP coverage of 62.6%. (Note that the Greengenes database uses the INSDC (= EMBL/NCBI/DDBJ) annotation, which is not an authoritative source for nomenclature or classification.) The highest-scoring environmental sequence was DQ017943 (Greengenes short name 'Cntrl Erpn Rnnng Wtrs Exmnd TGGE and uplnd strm cln S-BQ2 83'), which showed an identity of 95.6% and an HSP coverage of 97.8%. The most frequently occurring keywords within the labels of all environmental samples which yielded hits were 'microbi' (5.5%), 'sediment' (2.6%), 'soil' (2.5%), 'industri' (2.1%) and 'anaerob' (1.9%) (194 hits in total). Environmental samples which yielded hits of a higher score than the highest scoring species were not found.
Figure 1 shows the phylogenetic neighborhood of T. parva HT in a 16S rRNA based tree. The sequences of the two identical 16S rRNA gene copies in the genome do not differ from the previously published 16S rRNA sequence (AY293856).
Figure 1.

Phylogenetic tree highlighting the position of T. parva relative to the type strains of the other species within the phylum 'Spirochaetes'. The tree was inferred from 1,318 aligned characters [15,16] of the 16S rRNA gene sequence under the maximum likelihood (ML) criterion [17]. Rooting was done initially using the midpoint method [18] and then checked for its agreement with the current classification (Table 1). The branches are scaled in terms of the expected number of substitutions per site. Numbers adjacent to the branches are support values from 500 ML bootstrap replicates [19] (left) and from 1,000 maximum-parsimony bootstrap replicates [20] (right) if larger than 60%. Lineages with type strain genome sequencing projects registered in GOLD [21] are labeled with one asterisk, those also listed as 'Complete and Published' with two asterisks [22-28]; for Sphaerochaeta pleomorpha see CP003155. The collapsed Treponema subtree contains three species formerly assigned to Spirochaeta that have recently been included in the genus Treponema, even though those names are not yet validly published [27].
Table 1. Classification and general features of T. parva HT according to the MIGS recommendations [29].
| MIGS ID | Property | Term | Evidence code |
|---|---|---|---|
| Domain Bacteria | TAS [30] | ||
| Phylum Spirochaetes | TAS [31] | ||
| Class Spirochaetes | TAS [32,33] | ||
| Current classification | Order Spirochaetales | TAS [34,35] | |
| Family Leptospiraceae | TAS [1,35,36] | ||
| Genus Turneriella | TAS [1] | ||
| Species | Turneriella parva | TAS [1] | |
| MIGS-7 | Subspecific genetic lineage (strain) | Turneriella parva HT | TAS [1] |
| MIGS-12 | Levett at al. 2005 | TAS [1] | |
| Gram stain | negative | TAS [1] | |
| Cell shape | spiral-shaped | TAS [1] | |
| Motility | motile | TAS [1] | |
| Sporulation | non-sporulating | ||
| Temperature range | mesophile | TAS [1] | |
| Optimum temperature | grows between 11 and 37 °C | TAS [1] | |
| Salinity | not reported | ||
| MIGS-22 | Relationship to oxygen | aerobe | TAS [1] |
| Carbon source | long-chain fatty acids and long-chain alcohols | TAS [4] | |
| Energy metabolism | chemoheterotrophic | TAS [4] | |
| MIGS-6 | Habitat | not reported | |
| MIGS-6.2 | pH | not reported | |
| MIGS-15 | Biotic relationship | free living | TAS [1] |
| MIGS-14 | Known pathogenicity | not reported | |
| MIGS-16 | Specific host | not reported | |
| MIGS-18 | Health status of host | unknown | |
| Biosafety level | 1 | TAS [37] | |
| MIGS-19 | Trophic level | unknown | |
| MIGS-23.1 | Isolation | contaminated culture medium | TAS [1] |
| MIGS-4 | Geographic location | Regina, Saskatchewan, Canada | TAS [1] |
| MIGS-5 | Time of sample collection | 1981 | TAS [1] |
| MIGS-4.1 | Latitude | 50.45 | TAS [1] |
| MIGS-4.2 | Longitude | -104.61 | TAS [11] |
| MIGS-4.3 | Depth | ||
| MIGS-4.4 | Altitude |
Evidence codes - TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). Evidence codes are from the Gene Ontology project [38].
Morphology and physiology
Cells of strain HT are Gram-negative, flexible and helical with 0.3 µm in diameter and 3.5-7.5 µm in length and a wavelength of 0.3-0.5 µm (Figure 2). Motility is achieved by means of two axial filaments, similar to those of other leptospiras. The surface of the cells show several blebs with no apparent substructure when prepared for negative staining while under the same conditions, cross-striated tubules are visible in other leptospiras [1,2]. The strain is obligately aerobic and oxidase positive. Slow and limited growth occurs in polysorbate albumin medium [39] at 11, 30 and 37 °C. Growth is inhibited by 8-azaguanine (200 µg ml-1) and 2,6 diaminopurine (µg ml-1). Lipase is produced, long-chain fatty acids and long-chain fatty alcohols are utilized as carbon and energy sources. L-lysine arylamidase, α-L-glutamate arylamidase, glycine arylamidase, leucyl-glycine arylamidase and α-D-galactosidase activities are lacking [4]. The type strain is not pathogenic for hamsters [1].
Figure 2.

Scanning electron micrograph of T. parva HT
Chemotaxonomy
Information on peptidoglycan composition, major cell wall sugars, fatty acids, menaquinones and polar lipids is not available. The mol% G+C of DNA was originally reported to be approximately 48% [3], significantly less than the G+C content inferred from the genome sequence.
Genome sequencing and annotation
Genome project history
This organism was selected for sequencing on the basis of its phylogenetic position [40], and is part of the Genomic Encyclopedia of Bacteria and Archaea project [41]. The genome project is deposited in the Genomes On Line Database [21] and the complete genome sequence is deposited in GenBank. Sequencing, finishing and annotation were performed by the DOE Joint Genome Institute (JGI) using state of the art sequencing technology [42]. A summary of the project information is shown in Table 2.
Table 2. Genome sequencing project information.
| MIGS ID | Property | Term |
|---|---|---|
| MIGS-31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | Five genomic libraries: 454 standard library, 454 PE libraries (3 kb, 4kb and 11 kb insert size), one Illumina library |
| MIGS-29 | Sequencing platforms | Illumina GAii, 454 GS FLX Titanium |
| MIGS-31.2 | Sequencing coverage | 1,675.1 × Illumina; 47.0 × pyrosequence |
| MIGS-30 | Assemblers | Newbler version 2.3-PreRelease-6/30/2009, Velvet 1.0.13, phrap version SPS - 4.24 |
| MIGS-32 | Gene calling method | Prodigal 1.4, GenePRIMP |
| INSDC ID | CP002959 (chromosome) CP002960 (plasmid) |
|
| GenBank Date of Release | June 12, 2012 | |
| GOLD ID | Gc02242 | |
| NCBI project ID | 50821 | |
| Database: IMG | 2506520013 | |
| MIGS-13 | Source material identifier | DSM 21527 |
| Project relevance | Tree of Life, GEBA |
Growth conditions and DNA isolation
T. parva strain HT, DSM 21527, was grown in semisolid DSMZ medium 1113 (Leptospira medium) [43] at 30°C. DNA was isolated from 1-1.5 g of cell paste using MasterPure Gram-positive DNA purification kit (Epicentre MGP04100) following the standard protocol as recommended by the manufacturer with modification st/DL for cell lysis as described in Wu et al. 2009 [41]. DNA is available through the DNA Bank Network [44].
Genome sequencing and assembly
The genome was sequenced using a combination of Illumina and 454 sequencing platforms. All general aspects of library construction and sequencing can be found at the JGI website [45]. Pyrosequencing reads were assembled using the Newbler assembler (Roche). The initial Newbler assembly consisting of 217 contigs in 1 scaffold was converted into a phrap [46] assembly by making fake reads from the consensus, to collect the read pairs in the 454 paired end library. Illumina GAii sequencing data (8,018.4 Mb) was assembled with Velvet [47] and the consensus sequences were shredded into 1.5 kb overlapped fake reads (shreds) and assembled together with the 454 data. The 454 draft assembly was based on 200.6 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are -consed -a 50 -l 350 -g -m -ml 21. The Phred/Phrap/Consed software package [46] was used for sequence assembly and quality assessment in the subsequent finishing process. After the shotgun stage, reads were assembled with parallel phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with gapResolution [45], Dupfinisher [48], or sequencing cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks (J.-F. Chang, unpublished). A total of 361 additional reactions and 11 shatter library were necessary to close some gaps and to raise the quality of the final contigs. Illumina reads were also used to correct potential base errors and increase consensus quality using a software Polisher developed at JGI [49]. The error rate of the final genome sequence is less than 1 in 100,000. Together, the combination of the Illumina and 454 sequencing platforms provided 1,722.1 × coverage of the genome. The final assembly contained 348,698 pyrosequence and 97,925,368 Illumina reads.
Genome annotation
Genes were identified using Prodigal [50] as part of the DOE-JGI annotation pipeline [51], followed by a round of manual curation using the JGI GenePRIMP pipeline [52]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes - Expert Review (IMG-ER) platform [53].
Genome properties
The genome statistics are provided in Table 3 and Figure 3. The genome in its current assembly consists of two linear scaffolds with a total length of 4,384,015 bp and 25,287 bp, respectively, and a G+C content of 53.6%. Of the 4,214 genes predicted, 4,169 were protein-coding genes, and 45 RNAs; 30 pseudogenes were also identified. The majority of the protein-coding genes (57.9%) were assigned a putative function while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COGs functional categories is presented in Table 4.
Table 3. Genome Statistics.
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 4,409,302 | 100.00 |
| DNA coding region (bp) | 4,062,544 | 92.14 |
| DNA G+C content (bp) | 2,364,784 | 53.63 |
| Number of scaffolds | 2 | |
| Extrachromosomal elements | 0 | |
| Total genes | 4,214 | 100.00 |
| RNA genes | 45 | 1.07 |
| rRNA operons | 2 | |
| tRNA genes | 38 | 0.90 |
| Protein-coding genes | 4,169 | 98.93 |
| Pseudo genes | 30 | 0.71 |
| Genes with function prediction | 2,446 | 58.04 |
| Genes in paralog clusters | 1,807 | 42.88 |
| Genes assigned to COGs | 2,698 | 64.02 |
| Genes assigned Pfam domains | 2,897 | 68.75 |
| Genes with signal peptides | 508 | 12.06 |
| Genes with transmembrane helices | 1,034 | 24.54 |
| CRISPR repeats | 0 |
Figure 3.
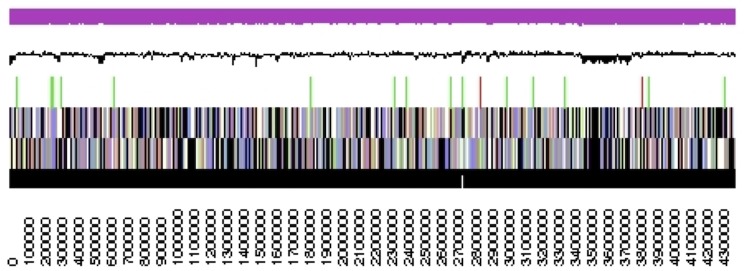
Graphical map of the largest scaffold (smaller scaffold not shown). From bottom to the top: Genes on forward strand (color by COG categories), Genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew (purple/olive).
Table 4. Number of genes associated with the general COG functional categories.
| Code | Value | % age | Description |
|---|---|---|---|
| J | 164 | 5.5 | Translation, ribosomal structure and biogenesis |
| A | 0 | 0.0 | RNA processing and modification |
| K | 169 | 5.7 | Transcription |
| L | 158 | 5.3 | Replication, recombination and repair |
| B | 2 | 0.1 | Chromatin structure and dynamics |
| D | 34 | 1.2 | Cell cycle control, cell division, chromosome partitioning |
| Y | 0 | 0.0 | Nuclear structure |
| V | 49 | 1.7 | Defense mechanisms |
| T | 266 | 9.0 | Signal transduction mechanisms |
| M | 222 | 7.5 | Cell wall/membrane/envelope biogenesis |
| N | 80 | 2.7 | Cell motility |
| Z | 0 | 0.0 | Cytoskeleton |
| W | 0 | 0.0 | Extracellular structures |
| U | 70 | 2.4 | Intracellular trafficking, secretion, and vesicular transport |
| O | 114 | 3.9 | Posttranslational modification, protein turnover, chaperones |
| C | 158 | 5.3 | Energy production and conversion |
| G | 123 | 4.2 | Carbohydrate transport and metabolism |
| E | 154 | 5.2 | Amino acid transport and metabolism |
| F | 73 | 2.5 | Nucleotide transport and metabolism |
| H | 117 | 4.0 | Coenzyme transport and metabolism |
| I | 146 | 4.9 | Lipid transport and metabolism |
| P | 121 | 4.1 | Inorganic ion transport and metabolism |
| Q | 55 | 1.9 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 405 | 13.7 | General function prediction only |
| S | 279 | 9.4 | Function unknown |
| - | 1,516 | 36.0 | Not in COGs |
Emended description of the species Turneriella parva Levett et al. 2005
The description of the species Turneriella parva is the one given by Levett et al. 2005 [1], with the following modification: DNA G+C content is 53.6 mol%.
Acknowledgements
We would like to gratefully acknowledge the help of Sabine Welnitz for growing T. parva cultures, and Evelyne-Marie Brambilla for DNA extraction and quality control (both at DSMZ). This work was performed under the auspices of the US Department of Energy Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396, UT-Battelle and Oak Ridge National Laboratory under contract DE-AC05-00OR22725.
References
- 1.Levett PN, Morey RE, Galloway R, Steigerwalt AG, Ellis WA. Reclassification of Leptospira parva Hovind-Hougen et al. 1982 as Turneriella parva gen. nov., comb. nov. Int J Syst Evol Microbiol 2005; 55:1497-1499 10.1099/ijs.0.63088-0 [DOI] [PubMed] [Google Scholar]
- 2.Hovind-Hougen K, Ellis WA, Birch-Andersen A. Leptospira parva sp.nov.: some morphological and biological characters. Zentralbl Bakteriol Mikrobiol Hyg [A] 1981; 250:343-354 [PubMed] [Google Scholar]
- 3.Yasuda PH, Steigerwalt AG, Sulzer KR, Kaufmann AF, Rogers F, Brenner DJ. Deoxyribonucleic acid relatedness between serogroups and serovars in the family Leptospiraceae with proposals for seven new Leptospira species. Int J Syst Bacteriol 1987; 37:407-415 10.1099/00207713-37-4-407 [DOI] [Google Scholar]
- 4.Saito T, Ono E, Yanagawa R. Enzyme activities of the strains belonging to the family Leptospiraceae detected by the API ZYM system. Zentralbl Bakteriol Mikrobiol Hyg [A] 1987; 266:218-225 [DOI] [PubMed] [Google Scholar]
- 5.Marshall R. International Committee on Systematic Bacteriology Subcommittee on the Taxonomy of Leptospira. Minutes of the meetings, 13 and 15 September 1990, Osaka, Japan. Int J Syst Bacteriol 1992; 42:330-334 10.1099/00207713-42-2-330 [DOI] [Google Scholar]
- 6.Perolat P, Chappel RJ, Adler B, Baranton G, Bulach DM, Billinghurst ML, Letocart M, Merien F, Serrano MS. Leptospira fainei sp. nov., isolated from pigs in Australia. Int J Syst Bacteriol 1998; 48:851-858 10.1099/00207713-48-3-851 [DOI] [PubMed] [Google Scholar]
- 7.Levett PN. Leptospirosis. Clin Microbiol Rev 2001; 14:296-326 10.1128/CMR.14.2.296-326.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hookey JV, Bryden J, Gatehouse L. The use of 16S rDNA sequence analysis to investigate the phylogeny of Leptospiraceae and related spirochaetes. J Gen Microbiol 1993; 139:2585-2590 10.1099/00221287-139-11-2585 [DOI] [PubMed] [Google Scholar]
- 9.Morey RE, Galloway RL, Bragg SL, Steigerwalt AG, Mayer LW, Levett PN. Species-specific identification of Leptospiraceae by 16S rRNA gene sequencing. Can J Clin Microbiol 2006; 44:3510-3516 10.1128/JCM.00670-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hookey JV. Characterization of Leptospiraceae by 16S DNA restriction fragment length polymorphisms. J Gen Microbiol 1993; 139:1681-1689 10.1099/00221287-139-8-1681 [DOI] [PubMed] [Google Scholar]
- 11.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403-410 [DOI] [PubMed] [Google Scholar]
- 12.Korf I, Yandell M, Bedell J. BLAST, O'Reilly, Sebastopol, 2003. [Google Scholar]
- 13.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069-5072 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter MF. An algorithm for suffix stripping. Program: electronic library and information systems 1980; 14:130-137.
- 15.Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics 2002; 18:452-464 10.1093/bioinformatics/18.3.452 [DOI] [PubMed] [Google Scholar]
- 16.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 2000; 17:540-552 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- 17.Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML web-servers. Syst Biol 2008; 57:758-771 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 18.Hess PN, De Moraes Russo CA. An empirical test of the midpoint rooting method. Biol J Linn Soc Lond 2007; 92:669-674 10.1111/j.1095-8312.2007.00864.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pattengale ND, Alipour M, Bininda-Emonds ORP, Moret BME, Stamatakis A. How many bootstrap replicates are necessary? Lect Notes Comput Sci 2009; 5541:184-200 10.1007/978-3-642-02008-7_13 [DOI] [PubMed] [Google Scholar]
- 20.Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4.0 b10. Sinauer Associates, Sunderland, 2002. [Google Scholar]
- 21.Pagani I, Liolios K, Jansson J, Chen IM, Smirnova T, Nosrat B, Markowitz VM, Kyrpides NC. The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 2012; 40:D571-D579 10.1093/nar/gkr1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abt B, Han C, Scheuner C, Lu M, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, et al. Complete genome sequence of the termite hindgut bacterium Spirochaeta coccoides type strain (SPN1T), reclassification in the genus Sphaerochaeta as Sphaerochaeta coccoides comb. nov. and emendations of the family Spirochaetaceae and the genus Sphaerochaeta. Stand Genomic Sci 2012; 6:194-209 10.4056/sigs.2796069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han C, Gronow S, Teshima H, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S, Cheng JF, Zeytun A, et al. Complete genome sequence of Treponema succinifaciens type strain (6091T). Stand Genomic Sci 2011; 4:361-370 10.4056/sigs.1984594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mavromatis K, Yasawong M, Chertkov O, Lapidus A, Lucas S, Nolan M, Glavina del Rio T, Tice H, Cheng JF, Pitluck S, et al. Complete genome sequence of Spirochaeta smaragdinae type strain (SEBR 4228T). Stand Genomic Sci 2010; 3:136-144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pati A, Sikorski J, Gronow S, Lapidus A, Copeland A, Glavina del Rio T, Nolan M, Lucas S, Chen F, Tice H, et al. Complete genome sequence of Brachyspira murdochii type strain (56-150T). Stand Genomic Sci 2010; 2:260-269 10.4056/sigs.831993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton RA, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 1997; 390:580-586 10.1038/37551 [DOI] [PubMed] [Google Scholar]
- 27.Abt B, Göker MG, Scheuner C, Han C, Lu M, Misra M, Lapidus A, Nolan M, Lucas S, Hammon N, et al. Genome sequence of the thermophilic fresh-water bacterium Spirochaeta caldaria type strain (H1T), reclassification of Spirochaeta caldaria and Spirochaeta stenostrepta in the genus Treponema as Treponema caldaria comb. nov. and Treponema stenostrepta comb. nov., revival of the name Treponema zuelzerae comb. nov., and emendation of the genus Treponema. Stand Genomic Sci 2013; 8:88-105 10.4056/sigs.3096473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Picardeau M, Bulach DM, Bouchier C, Zuerner RL, Zidane N, Wilson PJ, Creno S, Kuczek ES, Bommezzadri S, Davis JC, et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLoS ONE 2009; 13:e1607. [DOI] [PMC free article] [PubMed]
- 29.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, Tatusova T, Thomson N, Allen MJ, Angiuoli SV, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 2008; 26:541-547 10.1038/nbt1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA 1990; 87:4576-4579 10.1073/pnas.87.12.4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW (eds), Bergey's Manual of Systematic Bacteriology, Second Edition, Volume 1, Springer, New York, 2001, p. 119-169. [Google Scholar]
- 32.Ludwig W, Euzeby J, Whitman WG. Draft taxonomic outline of the Bacteroidetes, Planctomycetes, Chlamydiae, Spirochaetes, Fibrobacteres, Fusobacteria, Acidobacteria, Verrucomicrobia, Dictyoglomi, and Gemmatimonadetes http://www.bergeys.org/outlines/Bergeys_Vol_4_Outline.pdf Taxonomic Outline 2008
- 33.Judicial Commission of the International Committee on Systematics of Prokaryotes The nomenclatural types of the orders Acholeplasmatales, Halanaerobiales, Halobacteriales, Methanobacteriales, Methanococcales, Methanomicrobiales, Planctomycetales, Prochlorales, Sulfolobales, Thermococcales, Thermoproteales and Verrucomicrobiales are the genera Acholeplasma, Halanaerobium, Halobacterium, Methanobacterium, Methanococcus, Methanomicrobium, Planctomyces, Prochloron, Sulfolobus, Thermococcus, Thermoproteus and Verrucomicrobium, respectively. Opinion 79. Int J Syst Evol Microbiol 2005; 55:517-518 10.1099/ijs.0.63548-0 [DOI] [PubMed] [Google Scholar]
- 34.Buchanan RE. Studies in the nomenclature and classification of bacteria. II. The primary subdivisions of the Schizomycetes. J Bacteriol 1917; 2:155-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skerman VBD, McGowan V, Sneath PHA. Approved Lists of Bacterial Names. Int J Syst Bacteriol 1980; 30:225-420 10.1099/00207713-30-1-225 [DOI] [PubMed] [Google Scholar]
- 36.Hovind-Hougen K. Leptospiraceae, a new family to include Leptospira Noguchi 1917 and Leptonema gen. nov. Int J Syst Bacteriol 1979; 29:245-251 10.1099/00207713-29-3-245 [DOI] [Google Scholar]
- 37.BAuA. 2010, Classification of bacteria and archaea in risk groups. http://www.baua.de TRBA 466, p. 243.
- 38.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nat Genet 2000; 25:25-29 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sulzer CR, Jones WL. Leptospirosis, methods in laboratory diagnosis, revised ed. Centers for Disease Control publication 80-8275. 1980; Centers for Disease Control, Atlanta. [Google Scholar]
- 40.Klenk HP, Göker M. En route to a genome-based classification of Archaea and Bacteria? Syst Appl Microbiol 2010; 33:175-182 10.1016/j.syapm.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 41.Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, Kunin V, Goodwin L, Wu M, Tindall BJ, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009; 462:1056-1060 10.1038/nature08656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mavromatis K, Land ML, Brettin TS, Quest DJ, Copeland A, Clum A, Goodwin L, Woyke T, Lapidus A, Klenk HP, et al. The fast changing landscape of sequencing technologies and their impact on microbial genome assemblies and annotation. PLoS ONE 2012; 7:e48837 10.1371/journal.pone.0048837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.List of growth media used at DSMZ: http://www.dsmz.de/catalogues/catalogue-microorganisms/culture-technology/list-of-media-for-microorganisms.html
- 44.Gemeinholzer B, Dröge G, Zetzsche H, Haszprunar G, Klenk HP, Güntsch A, Berendsohn WG, Wägele JW. The DNA Bank Network: the start from a German initiative. Biopreserv Biobank 2011; 9:51-55 10.1089/bio.2010.0029 [DOI] [PubMed] [Google Scholar]
- 45.JGI website. http://www.jgi.doe.gov/
- 46.The Phred/Phrap/Consed software package. http://www.phrap.com
- 47.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008; 18:821-829 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han C, Chain P. Finishing repeat regions automatically with Dupfinisher. In: Proceeding of the 2006 international conference on bioinformatics & computational biology. Arabnia HR, Valafar H (eds), CSREA Press. June 26-29, 2006:141-146. [Google Scholar]
- 49.Lapidus A, LaButti K, Foster B, Lowry S, Trong S, Goltsman E. POLISHER: An effective tool for using ultra short reads in microbial genome assembly and finishing. AGBT, Marco Island, FL, 2008. [Google Scholar]
- 50.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci 2009; 1:63-67 10.4056/sigs.632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 2010; 7:455-457 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- 53.Markowitz VM, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 2009; 25:2271-2278 10.1093/bioinformatics/btp393 [DOI] [PubMed] [Google Scholar]