

Abstract
Numerous studies have employed mathematical modeling to quantitatively understand release of Ca2+ from the sarcoplasmic reticulum (SR) in heart. Models have been used to investigate physiologically important phenomena such as triggering of SR Ca2+ release by Ca2+ entry across the cell membrane and spontaneous leak of Ca2+ from the SR in quiescent heart cells. In this review we summarize studies that have modeled myocardial Ca2+ at different spatial scales: the sub-cellular level, the cellular level, and the multicellular level. We discuss each category of models from the standpoint of parameter sensitivity analysis, a common simulation procedure that can generate quantitative, comprehensive predictions about how changes in conditions influence model output. We propose that this is a useful perspective for conceptualizing models, in part because a sensitivity analysis requires the investigator to define the relevant parameters and model outputs. This procedure therefore helps to illustrate the capabilities and limitations of each model. We further suggest that in future studies, sensitivity analyses will aid in simplifying complex models and in suggesting experiments to differentiate between competing models built with different assumptions. We conclude with a discussion of unresolved questions that are likely to be addressed over the next several years.
Keywords: Ca2+ spark, Ca2+ wave, Ca2+ transient, arrhythmia, triggered activity, ventricular myocyte, mathematical modeling
1. Introduction
1.1 The multiscale nature of Ca2+ release in heart
In ventricular tissue, release of Ca2+ from the sarcoplasmic reticulum (SR) is important both physiologically and pathologically, as described in several contributions to this special issue. With each heart beat, entry of Ca2+ through L-type Ca2+ channels triggers the release of a larger amount of Ca2+ through ryanodine receptors (RyRs) in the SR membrane, a process known as Ca2+-induced Ca2+ release (CICR). The resulting increase in intracellular [Ca2+], called a Ca2+ transient, enables contraction. Under pathological conditions, however, spontaneous SR Ca2+ release can occur in the form of propagating intracellular Ca2+ waves. These events depolarize the cell membrane and can potentially initiate arrhythmias.
Mathematical models of cardiac Ca2+ release have generated important predictions that have subsequently been verified experimentally (see [1, 2] for recent reviews). Modeling cardiac Ca2+ release is challenging, however, because of the multiscale nature of this process. At the cellular level, for instance, movements of Ca2+ into and out of the cell, and between the cytosol and SR, can alter the quantity of released Ca2+ and modulate the strength of cellular contraction [3]. At a smaller spatial scale, however, each Ca2+ transient reflects the probabilistic triggering of thousands of individual events, Ca2+ sparks [4]. Sparks represent the stochastic gating of small clusters of RyRs, and these events are triggered by local [Ca2+] in the immediate vicinity of the RyRs rather than by average cellular [Ca2+]. To describe cellular Ca2+ transients in quantitative terms, then, one must understand the triggering of Ca2+ release at the sub-cellular level. At a spatial scale larger than the individual myocyte, electrical connections between cells must be considered to understand how changes in cellular CICR manifest themselves at the tissue and organ levels. The interactions between processes at these different spatial scales must be simulated to predict the physiological consequences of changes in Ca2+ release [5].
1.2 Challenges in integrating Ca2+ models across scales
In this review, we discuss mathematical models of cardiac Ca2+ signaling that have been developed at three spatial scales (Figure 1): (1) the sub-cellular level, at which RyR clusters produce Ca2+ sparks; (2) the cellular level, at which Ca2+ fluxes across the sarcolemmal and SR membranes produce cellular Ca2+ transients; and (3) the multicellular level, at which the sub-cellular and cellular level changes may alter muscle contraction and tissue electrical activity. The development of models at these three levels has largely proceeded independently. In recent years, however, effort has been made to integrate mathematical representations across the different spatial scales to produce more mechanistic models. Although this is a welcome development that will undoubtedly lead to more powerful and accurate models, it comes with drawbacks. First, models at larger spatial scales frequently simplify the representations of smaller-scale events. Sometimes, it is not clear how to relate the predictions of the larger scale, simplified model with those of the smaller scale, detailed model. Second, two models built by different groups may represent the same process, but with different underlying assumptions, and these may be equally successful at reproducing important physiological behaviors. In these cases it is generally not clear how to differentiate between the two representations. Finally, as models become increasingly multiscale and increasingly integrative, they also usually become larger, with additional parameters and state variables, and it is sometimes easy to lose track of which quantities should be considered important. We argue in this review that sensitivity analysis can play an important role in helping to address these concerns.
Figure 1. The multiscale nature of Ca2+ regulation in heart.
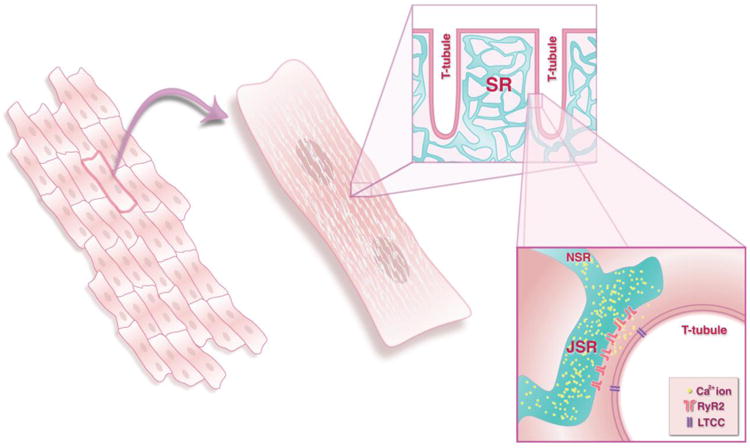
The figure illustrates schematically the different spatial scales at which the effects of cardiac SR Ca2+ release must be considered. Considerable research has been performed to investigate electrophysiology and Ca2+ release at the level of the individual cardiac myocyte (middle). The physiological effects of changes to cellular Ca2+ release, however, occur at the tissue level, when cells are connected to one another through gap junctions (left). At a smaller spatial scale, thousand of individual units of SR Ca2+ release, Ca2+ sparks, are triggered so-called Ca2+ release units (right). These are locations where junctional SR, containing clusters of RyRs, sits in close proximity to T-tubular membranes.
1.3 A role for parameter sensitivity analysis
Parameter sensitivity analysis is a valuable technique for probing and investigating mathematical models of physiological processes. Traditionally, sensitivity analyses are performed by systematically varying each parameter, running simulations, and recording in quantitative terms the effect on model output of each parameter alteration [6-8]. More recently, methods have been developed to compute parameter sensitivities by generating a population of models with variable properties, then using statistical methods to analyze the results generated by the population [9-13]. Regardless of the specific method that is employed, the value of this approach is that it generates a comprehensive set of predictions, potentially experimentally testable, indicating how changes in model parameters influence model outputs.
Here, we review modeling studies of cardiac Ca2+ signaling at different spatial scales, considering each category of models from the standpoint of sensitivity analyses that either have been performed or could be performed with the models. We argue that this is a useful perspective for conceptualizing and understanding models, for several reasons. First, sensitivity analyses provide a direct, quantitative comparison between parameters, and these comparisons allow investigators to assess the relative importance of various model parameters in determining physiologically-important outputs. Second, because these analyses generate comprehensive predictions, the approach can suggest straightforward experimental tests to differentiate between competing models that have different mechanistic assumptions. We believe that this strategy can help to address unresolved questions. Third, we highlight cases in which the same physiological process may be described by both a simple, phenomenological model with few parameters and a complex, mechanistic model with many parameters. Sensitivity analyses can help to relate one type of model with the other, thereby providing guidance in how to simplify models when this is required.
Fourth, the most important benefit of sensitivity analysis is obvious but easily overlooked: namely, the process of performing the analysis requires the investigator to define what he or she considers the relevant parameters and the pertinent model outputs. Even with relatively simple models, these choices may not always be apparent. For instance, if a model calculates intracellular [Ca2+] as a function of time, this does not indicate which aspects of the Ca2+ signal are considered most important: amplitude, frequency, time to peak, decay time, changes with different experimental conditions? As models become increasingly complex and integrative, these questions become more challenging, and therefore more important to define. Once the important parameters and outputs have been identified, however, sensitivity analysis allows for a more sophisticated, higher-level means of conceptualizing a mathematical model (Fig. 2). By itself, a mathematical model may be viewed as simply a machine that converts a set of equations into simulated results (Fig. 2A). Although the output may be experimentally measurable and/or biologically meaningful, the insight that can be gained by a single simulation is rather limited. After a sensitivity analysis is performed, however, the model can be conceptualized as a tool that maps changes in model parameters to changes in model outputs (Fig. 2B). In other words, the model can now predict phenotypes from molecular level changes.
Figure 2. The novel viewpoint provided by sensitivity analysis.
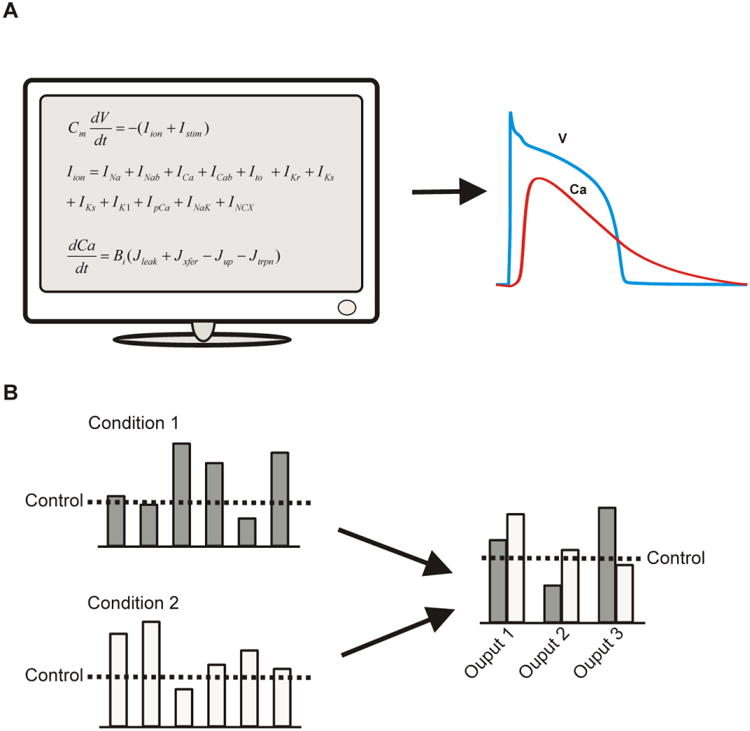
(A) A simulation may be conceived as simply a transformation from a set of equations into meaningful output, such as cellular transmembrane potential (V) and cytosolic Ca2+ concentration. The questions of greater interest, however, often concern the predicted changes in physiological outputs due to alterations caused by drugs or disease states. These questions generally cannot be addressed by the model equations themselves, but they can be addressed by systematically perturbing the model, i.e. performing a sensitivity analysis. (B) Once this has been performed, the investigator has a higher-level viewpoint of the model. It can now be seen as transforming changes in model parameters into altered physiological phenotypes.
2. A review of recent and historically-important studies
We discuss how sensitivity analysis can be helpful for understanding mathematical models of cardiac Ca2+ signaling at different spatial scales, and we propose that these methods can help to resolve controversies and to assist in integrating models across scales. We focus on models of Ca2+ movements and Ca2+ release at three levels: (1) sub-cellular models of Ca2+ sparks and Ca2+ movements within microdomains; (2) cellular models of beat-to-beat regulation and Ca2+ cycling within the cardiac myocyte; and (3) multicellular models that explore how cellular Ca2+ regulation affects tissue-level physiology.
2.1 Studies at the sub-cellular level
Studies at the microscopic, or sub-cellular, level can be grouped into two general categories. In the first, a number of studies have examined how sub-cellular geometric details influence movements and concentrations of [Ca2+] in cellular microdomains [14-21]. These investigations use ordinary differential equations (ODEs) and partial differential equations (PDEs), respectively, to simulate binding of Ca2+ to intracellular buffers and diffusion of Ca2+ within cellular compartments. Many of these studies have also examined how processes such as binding of Ca2+ to fluorescent indicators, indicator diffusion, and blurring by imaging instrumentation influence the properties of measurable Ca2+ signals such as Ca2+ sparks [22-25]. These models may incorporate subtle geometric details, but Ca2+ fluxes through membrane channels or RyRs are generally simulated in a simplified manner. In many models, for instance, RyRs are assumed to open for a fixed period of time [16, 19, 22, 23].
A recent study by Hake et al illustrates the state of the art in this category [25]. These investigators used electron tomography images to build a three-dimensional model of a cardiac release unit, or couplon. The model included spatially realistic representations of transverse tubules, junctional SR, and network SR, as these structures in the model were obtained directly from the electron tomography images. Because these investigators simulated movements of Ca2+ in both the myoplasm and the SR, the model could provide inferences about both Ca2+ sparks and local SR depletion signals, so-called Ca2+ blinks [26]. Two important predictions of this study were: (1) the apparent local SR depletion implied by the Ca2+ blink signal can be considerably less than the true depletion, and (2) the distribution of calsequestrin within the SR can significantly affect the time-to-nadir of the Ca2+ blink.
The second category consists of models that simulate stochastic gating of RyRs to predict the probabilistic triggering and termination of Ca2+ sparks [27-34]. This category originated with the seminal work of Stern [27], who used Monte-Carlo simulations demonstrate the feasibility of his hypothesis that Ca2+ release in heart cells was triggered by local increases in [Ca2+] rather than by average cytosolic [Ca2+]. Subsequent studies have used these models to examine phenomena such as termination of Ca2+ sparks [30], the importance of allosteric interactions between RyRs [28, 32, 33], the functional consequences of local depletion of JSR [Ca2+] [30, 31], and the mechanisms controlling refractoriness of RyR clusters after Ca2+ sparks [34]. These models include schemes representing the details of RyR gating, but they generally use simplified representations of the sub-cellular geometry. For instance, the sub-space between sarcolemmal and SR membranes is frequently assumed to be a single, well-mixed compartment.
We can understand the differences between the two categories of models by considering the type of parameter sensitivity analysis one might perform in either. In the first category, the relevant input parameters would be physical and geometrical variables such as subspace dimensions, diffusion constants for both Ca2+ and indicator, plus the abundances, locations, and affinities of intracellular Ca2+ buffers. In models that explicitly simulate Ca2+ spark formation, the relevant outputs would be quantities such as Ca2+ spark amplitude, duration, and width. In the second category, the parameters considered in an analysis would include additional variables such as the number of RyRs per cluster, the permeability of each RyR, and rate constants that describe RyR gating. Because these models are stochastic, the outputs could include probabilistic variables such as the frequency of spontaneous sparks or the probability that a particular trigger (e.g. L-type channel or RyR opening) will induce a spark. In general, one is also interested in quantities that reflect the characteristics of local SR Ca2+ release, such as release duration, the amount of Ca2+ released per event, and the extent of local JSR depletion. However, since most of these models do not explicitly simulate the processes of spark formation and detection, these outputs do not necessarily correspond to experimentally-measurable variables.
In a recent study, we attempted to both bridge the divide between the two categories of models and to analyze a Ca2+ spark model comprehensively [35]. We performed Monte-Carlo simulations with a stochastic model that simulates the random opening and closing of RyRs as Ca2+ sparks are triggered and terminate [30, 34], then coupled the results of these simulations to a model of buffering and diffusion that converts a local SR Ca2+ release flux into a measurable Ca2+ spark [22]. The geometry was idealized, however, as spherically-symmetric diffusion was assumed. We analyzed parameter sensitivities in this model using recently-developed techniques [9, 13]. This process involved generating a population of models with random parameters, running a single simulation with each parameter set, and using statistical regression methods to relate the random parameters to the model outputs. Ca2+ spark amplitude and duration were analyzed with multivariable linear regression whereas logistic regression was used to analyze the probability that a single RyR opening triggered a spark. Interestingly, spark amplitude and duration depended on physical and structural parameters such as the JSR volume and the quantity of calsequestrin in the JSR whereas triggering probability depended also on RyR gating parameters. Importantly, in this study [35] predictions of the sensitivity analysis were tested experimentally by measuring Ca2+ sparks in cells from genetically-modified mice [36], and the data were found to be consistent with the model predictions.
The results of these recent studies suggest unresolved questions that can be addressed through modeling studies at the Ca2+ spark level over the next few years. First, it will be important to continue to integrate spatially-detailed deterministic models with spatially-simplified stochastic models. In this way the models can provide insight into both the factors that control probabilistic triggering of Ca2+ sparks and the variables that affect the measurable signals. Thorough parameter sensitivity analyses will allow investigators to directly compare the quantitative effects of changes in RyR gating versus changes in structural quantities. Second, it is important to note that many of the models cited above were built using different assumptions about the underlying mechanisms. For instance, some models postulate an important role for Ca2+-dependent inactivation of RyRs in terminating local Ca2+ release [28, 33, 37], whereas others do not [30, 34]. At present, most experimental data support the view that termination occurs because of local depletion of JSR [Ca2+] rather than Ca2+-dependent inactivation [34, 38-40] (reviewed in [41, 42]). Studies that use parameter sensitivity analysis to compare competing models, however, may suggest new experiments that can differentiate between models and potentially uncover a role for inactivation. Third, it will be important to correlate behaviors observed at the Ca2+ spark level with cellular-level phenomena such as Ca2+ transients and SR Ca2+ leak. The next section discusses several recent studies that have initiated these efforts.
2.2 Studies at the cellular level
Mathematical modeling of cardiac cellular physiology has a long history, beginning with the groundbreaking work of Noble [43]. The development of this field has been documented recently in excellent reviews [44-46], and we will not attempt to repeat that effort here. Instead we will discuss selected studies that have focused more specifically on regulation of Ca2+ release from the SR. We focus on simulations of triggered cellular Ca2+ transients and spontaneous SR Ca2+ leak rather than simulations of spontaneous Ca2+ waves. Waves have been modeled in many studies [47-50], but these are discussed more fully in another contribution to this special issue [51]. In our discussion of cellular Ca2+ transients and SR Ca2+ leak, we aim to illustrate the challenges involved in integrating sub-cellular and cellular level models and to highlight the potentially important role of sensitivity analysis for understanding the similarities and differences between alternative mathematical representations.
Given that the importance of Ca2+ sparks has been appreciated for several years, one might expect that stochastic spark triggering would be included in all cellular models. In fact, however, for historical and practical reasons, most cellular models have lumped CICR into a few ODEs. Historically, since the development of cellular models predated the discovery of Ca2+ sparks, it originally made sense to model CICR with the lumped rate constants that had already proved valuable for simulating membrane ionic currents. Moreover, as a practical matter, the stochastic nature of Ca2+ spark triggering makes it difficult and computationally expensive to embed stochastic simulations within cellular models, which usually consist of between 10 and 50 ODEs. As a result, mathematical models of Ca2+ release at the cellular level can be grouped into two categories: (1) models that attempt to represent CICR with a few ODEs, and (2) more recently, models that explicitly incorporate the stochastic triggering of Ca2+ sparks. It is useful to contrast the two types of models from the standpoint of parameter sensitivity analysis. This illustrates the challenges involved in relating predictions of the two types of models and in developing simplified representations of CICR that are appropriate for incorporation into tissue-level and organ-level models.
Cellular models that lump CICR into a few ODEs have a difficult time reproducing the phenomenon of “graded release,” the well-established experimental result that CICR does not exhibit a threshold. Instead, small changes in the L-type Ca2+ current trigger produce small changes in the quantity of released Ca2+ [52, 53]. Cellular models in this category either produce all-or-none release [54], introduce non-physiological corrections, such as CICR that depends explicitly on L-type current rather than on local [Ca2+] [55, 56], or they must tune parameters carefully to reproduce graded release [57]. Despite this limitation, these lumped cellular models have been valuable for understanding phenomena such as changes in Ca2+ transient amplitude with pacing rate [54], altered Ca2+ transients and action potentials resulting from heart failure [58, 59], and the development at rapid pacing rates of alternans, or Ca2+ transients that alternate in magnitude from one beat to the next [60, 61]. From the standpoint of sensitivity analysis, the inputs that can be tested with these cellular models include parameters controlling the magnitude of cellular L-type Ca2+ current, maximal activities of Na+-Ca2+ exchangers and SERCA pumps, and the lumped parameters that control RyR permeability and gating. Relevant simulation outputs include not only Ca2+ transient amplitude at a particular rate, but also the change in amplitude with pacing rate, plus measurable quantities such as diastolic [Ca2+] and SR Ca2+ content.
In the second category are mathematical models of cells that explicitly simulate the stochastic triggering of Ca2+ sparks. The randomness inherent in spark triggering and termination, however, makes this a daunting computational challenge. The first study to simulate both stochastic spark triggering and cellular action potentials (APs) was published by Greenstein and Winslow in 2002 [62]. Several studies published more recently have used either elegant algorithms [63-68], or improvements in computational power [69-74], to develop new models. The biological questions that can be asked with these models, however, are somewhat different from those that can be addressed with the lumped models. For instance, in lumped models leak of Ca2+ from the SR is merely a passive flux that is included to balance SERCA pump activity at rest. In these newer models, however, SR Ca2+ leak via stochastic gating of RyRs is simulated explicitly. Several studies published in the past few years have computed leak in a resting myocyte as a function of SR [Ca2+] and have used the simulations to quantify the relative importance of visible leak in the form of Ca2+ sparks, versus “invisible” leak that would be difficult to detect with standard recording techniques [68, 70, 73, 74]. Sensitivity analyses with these newer models can therefore consider additional model outputs, such as the resting spark rate or the number of Ca2+ sparks triggered by specific voltage-clamp depolarizations. Parameters analyzed can include the explicit RyR gating rate constants, the number of RyRs per cluster, and geometrical parameters such as the volume of each sub-space or the average distances between RyRs and L-type channels.
Unresolved questions that can be addressed with cellular models include the following. First and most fundamentally, how can we relate the lumped RyR gating rate constants in the simplified models to the parameters of the Ca2+ spark triggering models? Despite recent advances in computing power, particularly the advent of graphical processing units (GPUs), there remains a need for simplified representations of CICR for use in tissue level models. We propose that if explicit Ca2+ spark triggering models and lumped models are subjected to sensitivity analyses with identical protocols, this comparison will help to illustrate the strengths and weaknesses of the various lumped representation.
Second, sensitivity analyses can help to potentially identify a role for Ca2+-dependent inactivation of the RyR. Although evidence strongly suggests that Ca2+ dependent inactivation does not terminate Ca2+ release on a millisecond time scale (see [41, 42] for a reviews of relevant studies), these results do not rule out a role for inactivation over a time scale of seconds to minutes. A comparison of models built with different assumptions may suggest experiments to demonstrate such a role. Third, sensitivity analyses will be required to determine the quantitative contributions of different changes during disease states such as heart failure. For instance, how important are structural changes such as T-tubule derangement [75-77] compared with changes in SERCA and Na+-Ca2+ exchange function? Sensitivity analyses performed on cellular models will help to address such questions.
2.3 Studies at the multicellular level
Although studies at the sub-cellular and cellular levels provide critical quantitative data, the physiological effects of myocyte Ca2+ signaling occur when cells are embedded within an intact organ. Modeling studies must therefore consider how sub-cellular and cellular phenomena are translated into tissue-level behaviors. Cardiac tissue-level and organ-level modeling has a long history that has been discussed in recent reviews [78, 79], but most studies have focused on how changes in membrane ionic currents influence electrical and mechanical activity at a larger scale. Comparatively less research has been performed to understand the tissue-level consequences of cellular Ca2+ release. An example of sub-cellular Ca2+ release influencing electrical behavior at the tissue level is the phenomenon known as “alternans,” beat-to-beat alternation in Ca2+ transient or AP characteristics [80-82]. However, since this topic is covered in another contribution to this special issue [83], we do not discuss these important studies in detail. Instead we review recent work that has examined a relevant question: what factors determine whether spontaneous Ca2+ release in a group of cells initiates a propagating AP in tissue? By examining the studies that have looked at these questions we can appreciate the model simplifications that have been employed to allow for multicellular simulations, and thereby understand the challenges that must be overcome to make multiscale modeling truly mechanistic.
At the cellular level it is well-established that spontaneous Ca2+ release from the SR, in the form of a propagating Ca2+ wave, can trigger membrane depolarization. A fraction of released Ca2+ is extruded from the cell by the Na+-Ca2+ exchanger, which depolarizes the membrane as it removes Ca2+. In tissue, however, cells are electrically connected to one another through gap junctions. Any depolarizing current in a cell will spread to its neighbors, thereby blunting the current's depolarizing effect. Thus, whether or not a Ca2+ wave induces an AP depends on tissue characteristics in addition to the Ca2+ release event itself.
In an important recent modeling study [84], Xie et al asked the question: how many myocytes must spontaneously release Ca2+ simultaneously for a propagating AP to be induced? In one, two, and three-dimensional ventricular models, these investigators simulated spontaneous release in a group of cells by imposing a release flux (Jspon), then quantifying how many cells needed to receive Jspon simultaneously for the event to induce a propagating AP. The results showed that the critical number of cells (Ncritical) was considerably greater in multidimensional tissue than in unidimensional tissue because depolarizing current in multidimensional tissue can spread in several directions. Moreover, Ncritical depended not only on the magnitude and timing of Jspon, but also on factors such as the strength of electrical coupling between adjacent myocytes, the presence of fibroblasts between myocytes, and the electrical remodeling that occurs during heart failure. More recently, Chen et al examined the same question in a simplified model that did not include explicit ionic currents and treated AP initiation as a threshold phenomenon [85]. The advantage of this simplified representation, however, is that it allowed the authors the authors to derive analytical expressions for the expected waiting time until the occurrence of a spontaneous, potentially arrhythmogenic AP in tissue. These expressions allow for a straightforward understanding of which factors promote, and which suppress, the development of spontaneous propagating APs.
It is useful to consider the parameter sensitivity analyses that could be performed with model presented by Xie et al [84], as this illustrates the hurdles that must be surmounted to mechanistically integrate such a tissue level model with cellular and subcellular models. The primary output in such an analysis would be Ncritical, the required number of cells with spontaneous Ca2+ release to induce a propagating AP. The potentially important parameters would include the magnitude and kinetics of Jspon, expression of Na+-Ca2+ exchangers, the inward rectifier conductance (GK1), the strength of electrical coupling between myocytes, the degree of electrical anisotropy, and coupling between myocytes and fibroblasts. This analysis could therefore provide a quantitative comparison of the relative importance of Ca2+ release itself, ionic currents, and structural features of the tissue. It is important to note, however, that in these simulations Jspon was imposed on a group of myocytes, whereas in real cells Ca2+ waves originate stochastically when spontaneous Ca2+ sparks trigger additional sparks (see review in this special issue [51]). At present, then, the tissue-level simulations cannot directly provide insight into the importance of factors such as changes in RyR gating, the number of RyRs per cluster, spacing between RyR clusters, and the SR Ca2+ load. The tissue-level models must be integrated with cellular and sub-cellular models in order for the simulations to provide a comparison of the relative importance of the different parameters. Once these integrative models are developed, sensitivity analyses with these models can address important unresolved questions concerning potential anti-arrhythmic targets. For instance, if the goal is to prevent spontaneous propagating APs, is it better to target sub-cellular characteristics of RyR clusters, cellular ionic currents such as L-type channels, or tissue level structural parameters? Equally important, can sensitivity analyses identify targets that inhibit spontaneous APs without causing undesirable effects that may be pro-arrhythmic, such as a decrease in propagation velocity?
3. Current and future challenges
To conclude, we briefly describe some challenges in cardiac Ca2+ modeling that will be encountered by researchers over the next several years. In these areas we believe sensitivity analysis will play an important role in helping to address unresolved questions.
3.1 Integrative models of CICR and additional cellular processes
At the cellular and subcellular scales, a recent trend has been the development of integrative models that simulate both SR Ca2+ release and additional Ca2+-dependent cellular processes. This development provides exciting new opportunities, since the integrated models allow for quantitative predictions regarding mechanisms and phenomena that could not previously be simulated. As these models become increasingly complex, however, we believe that sensitivity analyses will be required to define the scope and capabilities of each model.
Mitochondrial respiration is a vital cellular process that is Ca2+ dependent and closely linked to SR Ca2+ release. The rate of ATP production has long been known to depend on mitochondrial [Ca2+], and experimental studies have established that these organelles take up and extrude Ca2+, although the magnitude and kinetics of changes in mitochondrial [Ca2+] remain controversial [86]. In addition, structural studies demonstrate that mitochondria tend to be located in close proximity to Ca2+ release units [87], which implies that interactions between SR release and mitochondria take place within local microdomains rather than in the bulk cytoplasm. Several published models simulate the Ca2+-dependence of mitochondrial bioenergetics [88, 89]. In these models, important outputs are measures of mitochondrial function such as ATP production, oxygen consumption, or mitochondrial membrane potential. The relevant inputs include both enzyme activities and variables describing Ca2+ transport pathways across the mitochondrial membranes, such as the Ca2+ uniporter and the mitochondrial Na+-Ca2+ exchanger. In these models, however, changes in cytosolic Ca2+ were simulated separately and imposed on the mitochondrial models as an input [88, 89]. More recently, integrative models have been developed that simulate SR Ca2+ release and mitochondrial function together within the same computational framework [90]. From a sensitivity analysis standpoint, then, one can examine how changes in L-type Ca2+ current or SR Ca2+ uptake can influence outputs such as oxygen consumption or ATP production. Conversely, one can also investigate how changes in mitochondrial respiration might affect cellular Ca2+ transients. These models, however, have to date used lumped representations of cellular Ca2+ release [54]. To understand more fully how released Ca2+ interacts with mitochondria in microdomains, the mitochondrial models must be integrated with stochastic models of SR Ca2+ release at the level of the Ca2+ spark. Once these models are built, sensitivity analyses can investigate the relative importance for mitochondrial function of cellular Ca2+ fluxes versus sub-cellular changes in RyR function or release unit structure. In addition, as pathways for mitochondrial Ca2+ entry and efflux become more clearly defined [91], sensitivity analyses of models can be used to predict the quantitative prominence of the different pathways.
A second opportunity for integration is the development of models that simulate both SR Ca2+ release, occurring on a millisecond time scale, and activation of biochemical signaling pathways, which occurs on a time scale of seconds to minutes. Important early work by Saucerman et al investigated how stimulation of β-adrenergic receptors affected the characteristics of Ca2+ release [92]. These models, however, were largely unidirectional, since β-adrenergic stimulation influenced myocyte Ca2+ transport, but Ca2+ had only minimal effects on the biochemical signaling. More recent models have examined pathways that are more explicitly Ca2+-dependent, such as CaMKII (Ca2+/calmodulin-dependent protein kinase) and calcineurin [70, 93, 94]. These models can be bidirectional, since Ca2+ influences the biochemical reactions, and the signaling proteins phosphorylate or dephosphorylate targets that affect Ca2+ cycling. Sensitivity analyses on such integrative models can therefore consider as inputs both variables controlling Ca2+ transport pathways and variables describing enzyme abundances and activities. Outputs can include not only cellular Ca2+ transients, but also measurable biochemical signals such as phosphorylated CaMKII. These analyses can help to address questions that have to date remained unresolved. For instance, activated CaMKII phosphorylates multiple targets that can influence Ca2+ cycling. What is the quantitative importance of each of these targets in determining myocyte Ca2+ regulation and electrophysiology? Similarly, analyzes with these models can address whether enzyme abundances, enzyme affinities, or quantities of Ca2+ transport proteins are most important in determining readouts of myocyte physiology such as Ca2+ transients. Finally, as signaling models become more closely integrated with sub-cellular models of Ca2+ sparks, the importance of enzyme localization, which has been addressed in a few studies [94, 95], can be explored more systematically.
3.2 Scaling from the tissue level to the organ level
Above we have highlighted the challenges in integrating models of CICR from the Ca2+ spark level to the multicellular level. These challenges become even more acute when cellular models are embedded in models of the whole organ. Although it is now possible, with advances in computing power, to simulate stochastic SR Ca2+ release in a model comprising several cells, simplified representations of CICR will continue to be required for organ-level simulations. Sensitivity analyses that compare mechanistic with simplified representations at the cellular level will play an important role by helping to define which simplified representations generate the most realistic output, not only under baseline conditions, but also under conditions simulating pathology. Moreover, as organ-level models become further developed sensitivity analyses of these models can be used to predict the relative importance of cellular CICR characteristics versus structural features such as muscle mechanics and fiber directions in determining physiological outputs such as ejection fraction.
3.3 Conclusions
In this review we have discussed mathematical models of Ca2+ regulation in heart, and we have highlighted the challenges presented by the multiscale nature of this process. The majority of modeling studies published to date have focused on Ca2+ signaling at a particular spatial scale, but lately efforts have been made to both integrate across spatial scales and to combine CICR models with representations of other cellular processes. Besides the mathematical and computational challenges posed by this process of integration, this increase in complexity makes it more difficult to understand the relevant inputs and outputs of each model. As the field moves forward, parameter sensitivity analysis is likely to play an important role in helping to: (1) define the capabilities and limitations of each model; (2) predict the relative importance of different factors in determining physiological responses; (3) generate predictions that can be tested experimentally to discriminate between alternative models.
Footnotes
Disclosures: None to declare
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greenstein JL, Winslow RL. Integrative systems models of cardiac excitation-contraction coupling. Circ Res. 2011;108:70–84. doi: 10.1161/CIRCRESAHA.110.223578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams GS, Smith GD, Sobie EA, Jafri MS. Models of cardiac excitation-contraction coupling in ventricular myocytes. Math Biosci. 2010;226:1–15. doi: 10.1016/j.mbs.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisner DA, Choi HS, Diaz ME, O'Neill SC, Trafford AW. Integrative analysis of calcium cycling in cardiac muscle. Circ Res. 2000;87:1087–94. doi: 10.1161/01.res.87.12.1087. [DOI] [PubMed] [Google Scholar]
- 4.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 5.Qu Z, Garfinkel A, Weiss JN, Nivala M. Multi-scale modeling in biology: how to bridge the gaps between scales? Prog Biophys Mol Biol. 2011;107:21–31. doi: 10.1016/j.pbiomolbio.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nygren A, Fiset C, Firek L, Clark JW, Lindblad DS, Clark RB, et al. Mathematical model of an adult human atrial cell: the role of K+ currents in repolarization. Circ Res. 1998;82:63–81. doi: 10.1161/01.res.82.1.63. [DOI] [PubMed] [Google Scholar]
- 7.Weaver CM, Wearne SL. Neuronal firing sensitivity to morphologic and active membrane parameters. PLoS Comput Biol. 2008;4:e11. doi: 10.1371/journal.pcbi.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero L, Pueyo E, Fink M, Rodriguez B. Impact of ionic current variability on human ventricular cellular electrophysiology. Am J Physiol Heart Circ Physiol. 2009;297:H1436–H1445. doi: 10.1152/ajpheart.00263.2009. [DOI] [PubMed] [Google Scholar]
- 9.Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J. 2009;96:1264–74. doi: 10.1016/j.bpj.2008.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor AL, Goaillard JM, Marder E. How multiple conductances determine electrophysiological properties in a multicompartment model. J Neurosci. 2009;29:5573–86. doi: 10.1523/JNEUROSCI.4438-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarkar AX, Sobie EA. Regression analysis for constraining free parameters in electrophysiological models of cardiac cells. PLoS Comput Biol. 2010;6:e1000914. doi: 10.1371/journal.pcbi.1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkar AX, Sobie EA. Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: A computational analysis. Heart Rhythm. 2011;8:1749–55. doi: 10.1016/j.hrthm.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkar AX, Christini DJ, Sobie EA. Exploiting mathematical models to illuminate electrophysiological variability between individuals. J Physiol. 2012;590:2555–67. doi: 10.1113/jphysiol.2011.223313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langer GA, Peskoff A. Calcium concentration and movement in the diadic cleft space of the cardiac ventricular cell. Biophys J. 1996;70:1169–82. doi: 10.1016/S0006-3495(96)79677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valent I, Zahradnikova A, Pavelkova J, Zahradnik I. Spatial and temporal Ca2+, Mg2+, and ATP2− dynamics in cardiac dyads during calcium release. Biochimica et Biophysica Acta-Biomembranes. 2007;1768:155–66. doi: 10.1016/j.bbamem.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 17.Yu Z, Holst MJ, Hayashi T, Bajaj CL, Ellisman MH, McCammon JA, et al. Three-dimensional geometric modeling of membrane-bound organelles in ventricular myocytes: bridging the gap between microscopic imaging and mathematical simulation. J Struct Biol. 2008;164:304–13. doi: 10.1016/j.jsb.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y, Yu Z, Hoshijima M, Holst MJ, McCulloch AD, McCammon JA, et al. Numerical analysis of Ca2+ signaling in rat ventricular myocytes with realistic transverse-axial tubular geometry and inhibited sarcoplasmic reticulum. PLoS Comput Biol. 2010;6:e1000972. doi: 10.1371/journal.pcbi.1000972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramay HR, Jafri MS, Lederer WJ, Sobie EA. Predicting local SR Ca2+ dynamics during Ca2+ wave propagation in ventricular myocytes. Biophys J. 2010;98:2515–23. doi: 10.1016/j.bpj.2010.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatano A, Okada J, Washio T, Hisada T, Sugiura S. A three-dimensional simulation model of cardiomyocyte integrating excitation-contraction coupling and metabolism. Biophys J. 2011;101:2601–10. doi: 10.1016/j.bpj.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picht E, Zima AV, Shannon TR, Duncan AM, Blatter LA, Bers DM. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108:847–56. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith GD, Keizer JE, Stern MD, Lederer WJ, Cheng H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys J. 1998;75:15–32. doi: 10.1016/S0006-3495(98)77491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izu LT, Mauban JR, Balke CW, Wier WG. Large currents generate cardiac Ca2+ sparks. Biophys J. 2001;80:88–102. doi: 10.1016/S0006-3495(01)75997-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soeller C, Cannell MB. Estimation of the sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks. Biophys J. 2002;82:2396–414. doi: 10.1016/S0006-3495(02)75584-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hake J, Edwards AG, Yu Z, Kekenes-Huskey PM, Michailova AP, McCammon JA, et al. Modelling cardiac calcium sparks in a three-dimensional reconstruction of a calcium release unit. J Physiol. 2012;590:4403–22. doi: 10.1113/jphysiol.2012.227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci U S A. 2005;102:3099–104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, et al. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol. 1999;113:469–89. doi: 10.1085/jgp.113.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rice JJ, Jafri MS, Winslow RL. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys J. 1999;77:1871–84. doi: 10.1016/s0006-3495(99)77030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sobie EA, Dilly KW, Dos Santos CJ, Lederer WJ, Jafri MS. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83:59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huertas MA, Smith GD. The dynamics of luminal depletion and the stochastic gating of Ca2+-activated Ca2+ channels and release sites. Journal of Theoretical Biology. 2007;246:332–54. doi: 10.1016/j.jtbi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Groff JR, Smith GD. Ryanodine receptor allosteric coupling and the dynamics of calcium sparks. Biophys J. 2008;95:135–54. doi: 10.1529/biophysj.107.119982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang X, Hu XF, Hu J. Dynamic interreceptor coupling contributes to the consistent open duration of ryanodine receptors. Biophys J. 2009;96:4826–33. doi: 10.1016/j.bpj.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. doi: 10.1093/cvr/cvr143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee YS, Liu OZ, Hwang HS, Knollmann BC, Sobie EA. Parameter sensitivity analysis of stochastic models provides insights into cardiac calcium sparks. Biophys J. 2012 doi: 10.1016/j.bpj.2012.12.055. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:7636–41. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang SQ, Stern MD, Rios E, Cheng H. The quantal nature of Ca2+ sparks and in situ operation of the ryanodine receptor array in cardiac cells. Proc Natl Acad Sci U S A. 2004;101:3979–84. doi: 10.1073/pnas.0306157101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL. Calsequestrin 2 deletion shortens the refractoriness of Ca2+ release and reduces rate-dependent Ca2+-alternans in intact mouse hearts. J Mol Cell Cardiol. 2012;52:21–31. doi: 10.1016/j.yjmcc.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens SC, Terentyev D, Kalyanasundaram A, Periasamy M, Gyorke S. Intra-sarcoplasmic reticulum Ca2+ oscillations are driven by dynamic regulation of ryanodine receptor function by luminal Ca2+ in cardiomyocytes. J Physiol. 2009;587:4863–72. doi: 10.1113/jphysiol.2009.175547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu OZ, Lederer WJ, Sobie EA. Does the Goldilocks Principle apply to calcium release restitution in heart cells? J Mol Cell Cardiol. 2012;52:3–6. doi: 10.1016/j.yjmcc.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sobie EA, Lederer WJ. Dynamic local changes in sarcoplasmic reticulum calcium: physiological and pathophysiological roles. J Mol Cell Cardiol. 2012;52:304–11. doi: 10.1016/j.yjmcc.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noble D. A modification of the Hodgkin--Huxley equations applicable to Purkinje fibre action and pacemaker potentials. J Physiol. 1962;160:317–52. doi: 10.1113/jphysiol.1962.sp006849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fink M, Niederer SA, Cherry EM, Fenton FH, Koivumaki JT, Seemann G, et al. Cardiac cell modelling: observations from the heart of the cardiac physiome project. Prog Biophys Mol Biol. 2011;104:2–21. doi: 10.1016/j.pbiomolbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Noble D, Garny A, Noble PJ. How the Hodgkin-Huxley equations inspired the Cardiac Physiome Project. J Physiol. 2012;590:2613–28. doi: 10.1113/jphysiol.2011.224238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winslow RL, Cortassa S, O'Rourke B, Hashambhoy YL, Rice JJ, Greenstein JL. Integrative modeling of the cardiac ventricular myocyte. Wiley Interdiscip Rev Syst Biol Med. 2011;3:392–413. doi: 10.1002/wsbm.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Izu LT, Means SA, Shadid JN, Chen-Izu Y, Balke CW. Interplay of ryanodine receptor distribution and calcium dynamics. Biophys J. 2006;91:95–112. doi: 10.1529/biophysj.105.077214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tao T, O'Neill SC, Diaz ME, Li YT, Eisner DA, Zhang H. Alternans of cardiac calcium cycling in a cluster of ryanodine receptors: a simulation study. Am J Physiol Heart Circ Physiol. 2008;295:H598–H609. doi: 10.1152/ajpheart.01086.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen W, Aistrup G, Wasserstrom JA, Shiferaw Y. A mathematical model of spontaneous calcium release in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;300:H1794–H1805. doi: 10.1152/ajpheart.01121.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nivala M, Ko CY, Nivala M, Weiss JN, Qu Z. Criticality in intracellular calcium signaling in cardiac myocytes. Biophys J. 2012;102:2433–42. doi: 10.1016/j.bpj.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Izu LT, Xie Y, Sato D, Banyasz T, Chen-Izu Y. Ca2+ waves in the heart. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2012.11.014. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wier WG, Egan TM, Lopez-Lopez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. J Physiol (Lond) 1994;474:463–71. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–9. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 54.Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J. 1998;74:1149–68. doi: 10.1016/S0006-3495(98)77832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, et al. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol. 2008;45:420–8. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bondarenko VE, Rasmusson RL. Transmural heterogeneity of repolarization and Ca2+ handling in a model of mouse ventricular tissue. Am J Physiol Heart Circ Physiol. 2010;299:H454–H469. doi: 10.1152/ajpheart.00907.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–71. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Winslow RL, Rice J, Jafri S, Marban E, O'Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84:571–86. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]
- 59.Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J. 2005;89:4096–110. doi: 10.1529/biophysj.105.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shiferaw Y, Watanabe MA, Garfinkel A, Weiss JN, Karma A. Model of intracellular calcium cycling in ventricular myocytes. Biophys J. 2003;85:3666–86. doi: 10.1016/S0006-3495(03)74784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie LH, et al. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Greenstein JL, Winslow RL. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys J. 2002;83:2918–45. doi: 10.1016/S0006-3495(02)75301-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hinch R, Greenstein JL, Tanskanen AJ, Xu L, Winslow RL. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys J. 2004;87:3723–36. doi: 10.1529/biophysj.104.049973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J. 2006;90:77–91. doi: 10.1529/biophysj.105.065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams GS, Huertas MA, Sobie EA, Jafri MS, Smith GD. A probability density approach to modeling local control of calcium-induced calcium release in cardiac myocytes. Biophys J. 2007;92:2311–28. doi: 10.1529/biophysj.106.099861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Williams GS, Huertas MA, Sobie EA, Jafri MS, Smith GD. Moment closure for local control models of calcium-induced calcium release in cardiac myocytes. Biophys J. 2008;95:1689–703. doi: 10.1529/biophysj.107.125948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huertas MA, Smith GD, Gyorke S. Ca2+ alternans in a cardiac myocyte model that uses moment equations to represent heterogeneous junctional SR Ca2+ Biophys J. 2010;99:377–87. doi: 10.1016/j.bpj.2010.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hartman JM, Sobie EA, Smith GD. Spontaneous Ca2+ sparks and Ca2+ homeostasis in a minimal model of permeabilized ventricular myocytes. Am J Physiol Heart Circ Physiol. 2010;299:H1996–H2008. doi: 10.1152/ajpheart.00293.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Restrepo JG, Weiss JN, Karma A. Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys J. 2008;95:3767–89. doi: 10.1529/biophysj.108.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hashambhoy YL, Greenstein JL, Winslow RL. Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J Mol Cell Cardiol. 2010;49:617–24. doi: 10.1016/j.yjmcc.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rovetti R, Cui X, Garfinkel A, Weiss JN, Qu Z. Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ Res. 2010;106:1582–91. doi: 10.1161/CIRCRESAHA.109.213975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaur N, Rudy Y. Multiscale modeling of calcium cycling in cardiac ventricular myocyte: macroscopic consequences of microscopic dyadic function. Biophys J. 2011;100:2904–12. doi: 10.1016/j.bpj.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Williams GS, Chikando AC, Tuan HT, Sobie EA, Lederer WJ, Jafri MS. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101:1287–96. doi: 10.1016/j.bpj.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sato D, Bers DM. How does stochastic ryanodine receptor-mediated ca leak fail to initiate a Ca spark? Biophys J. 2011;101:2370–9. doi: 10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–10. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, et al. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. Journal of Physiology-London. 2006;574:519–33. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heinzel FR, MacQuaide N, Biesmans L, Sipido K. Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J Mol Cell Cardiol. 2011;50:390–400. doi: 10.1016/j.yjmcc.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 78.Clayton RH, Bernus O, Cherry EM, Dierckx H, Fenton FH, Mirabella L, et al. Models of cardiac tissue electrophysiology: progress, challenges and open questions. Prog Biophys Mol Biol. 2011;104:22–48. doi: 10.1016/j.pbiomolbio.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 79.Trayanova NA. Whole-heart modeling: applications to cardiac electrophysiology and electromechanics. Circ Res. 2011;108:113–28. doi: 10.1161/CIRCRESAHA.110.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato D, Shiferaw Y, Garfinkel A, Weiss JN, Qu Z, Karma A. Spatially discordant alternans in cardiac tissue: role of calcium cycling. Circ Res. 2006;99:520–7. doi: 10.1161/01.RES.0000240542.03986.e7. [DOI] [PubMed] [Google Scholar]
- 81.Gaeta SA, Bub G, Abbott GW, Christini DJ. Dynamical mechanism for subcellular alternans in cardiac myocytes. Circ Res. 2009;105:335–42. doi: 10.1161/CIRCRESAHA.109.197590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jia Z, Bien H, Shiferaw Y, Entcheva E. Cardiac cellular coupling and the spread of early instabilities in intracellular Ca2+ Biophys J. 2012;102:1294–302. doi: 10.1016/j.bpj.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qu Z, Nivala M, Weiss JN. Calcium alternans in cardiac myocytes: order from disorder. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2012.10.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen W, Asfaw M, Shiferaw Y. The statistics of calcium-mediated focal excitations on a one-dimensional cable. Biophys J. 2012;102:461–71. doi: 10.1016/j.bpj.2011.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O'Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46:767–74. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lukyanenko V, Chikando A, Lederer WJ. Mitochondria in cardiomyocyte Ca2+ signaling. Int J Biochem Cell Biol. 2009;41:1957–71. doi: 10.1016/j.biocel.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cortassa S, Aon MA, Marban E, Winslow RL, O'Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003;84:2734–55. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nguyen MH, Dudycha SJ, Jafri MS. Effect of Ca2+ on cardiac mitochondrial energy production is modulated by Na+ and H+ dynamics. Am J Physiol Cell Physiol. 2007;292:C2004–C2020. doi: 10.1152/ajpcell.00271.2006. [DOI] [PubMed] [Google Scholar]
- 90.Cortassa S, O'Rourke B, Winslow RL, Aon MA. Control and regulation of mitochondrial energetics in an integrated model of cardiomyocyte function. Biophys J. 2009;96:2466–78. doi: 10.1016/j.bpj.2008.12.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chikando AC, Kettlewell S, Williams GS, Smith G, Lederer WJ. Ca2+ dynamics in the mitochondria - state of the art. J Mol Cell Cardiol. 2011;51:627–31. doi: 10.1016/j.yjmcc.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem. 2003;278:47997–8003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
- 93.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys J. 2010;99:2038–47. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heijman J, Volders PG, Westra RL, Rudy Y. Local control of beta-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca2+-transient. J Mol Cell Cardiol. 2011;50:863–71. doi: 10.1016/j.yjmcc.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys J. 2008;95:4597–612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]