

Abstract
Feline immunodeficiency virus (FIV) infection like human immunodeficiency virus (HIV), produces systemic and central nervous system disease in its natural host, the domestic cat, that parallels the pathogenesis seen in HIV-infected humans. The ability to culture feline nervous system tissue affords the unique opportunity to directly examine interactions of infectious virus with CNS cells for the development of models and treatments that can then be translated to a natural infectious model. To explore the therapeutic potential of a new p75 neurotrophin receptor ligand, LM11A-31, we evaluated neuronal survival, neuronal damage and calcium homeostasis in cultured feline neurons following inoculation with FIV. FIV resulted in the gradual appearance of dendritic beading, pruning of processes and shrinkage of neuronal perikarya in the neurons. Astrocytes developed a more activated appearance and there was an enhanced accumulation of microglia, particularly at longer times post-inoculation. Addition of 10 nM LM11A-31, to the cultures greatly reduced or eliminated the neuronal pathology as well as the FIV effects on astrocytes and microglia. LM11A-31 also, prevented the development of delayed calcium deregulation in feline neurons exposed to conditioned medium from FIV treated macrophages. The suppression of calcium accumulation prevented the development of foci of calcium accumulation and beading in the dendrites. FIV replication was unaffected by LM11A-31. The strong neuroprotection afforded by LM11A-31 in an infectious in vitro model indicates that LM11A-31 may have excellent potential for the treatment of HIV-associated neurodegeneration.
Introduction
Human immunodeficiency virus (HIV) infects macrophages and microglia in the central nervous system (CNS) resulting in, inflammation and the gradual development of a range of cognitive-motor deficits. Although combination antiretroviral therapy (CART) has decreased the severity of neurological symptoms, CNS disease continues to progress (2Sacktor et al., 2002; Brew, 2004) and evolve into different types of pathology (Masliah et al., 1997; Langford et al., 2003). Interventions are needed to suppress neuropathogenesis which, if unchecked, is expected to support an increasing neurological disease burden.
To develop therapies that prevent CNS damage in HIV-infected patients we need a better understanding of the underlying neuropathogenesis, particularly during early stages where interventions are likely to have the greatest impact. Animal models have provided essential tools to explore the pathogenesis in both in vivo and in vitro systems. While each of the various animal models allows the opportunity to explore specific contributions to pathogenesis, only two represent natural infections that recapitulate disease progression in HIV-infected humans, simian immunodeficiency virus (SIV) and feline immunodeficiency virus (FIV). Our studies have focused on the use of the FIV model in an effort to develop parallel in vitro and in vivo approaches that identify pathogenic mechanisms and support the testing of interventions in infected cats.
The FIV model recapitulates much of the pathogenesis seen with HIV. It primarily infects CD4+ Tcells and cells of monocyte lineage(Brunner and Pedersen, 1989; Brown et al., 1991; English et al., 1994; Dow et al., 1999) eventually causing immunodeficiency and CNS disease(Pedersen et al., 1987; Sparger et al., 1989; Podell et al., 1993; English et al., 1994; Phillips et al., 1994). FIV rapidly penetrates the brain (Ryan et al., 2003; Liu et al., 2006) where it establishes an infection (Dow et al., 1990) and leads to neuropathogenesis(Dow et al., 1990; Hurtrel et al., 1992; Meeker et al., 1997). Like HIV, interactions with macrophages and microglia result in inflammation and the release of factors that damage neurons(Bragg et al., 2002) resulting in neuropathological changes similar to HIV but typically less severe, including a diffuse gliosis, microglial nodules, meningitis, perivascular infiltrates, white matter lesions and neuronal loss; (Hurtrel et al., 1992; Phillips et al., 1994; Meeker et al., 1997). Cortical atrophy has been demonstrated by MRI(Podell et al., 1993).
Key elements of the neuropathogenesis of FIV can be modeled in vitro. Feline neurons in mixed neural cultures inoculated with FIV have enhanced sensitivity to glutamate-induced calcium accumulation and damage (Meeker et al., 1996; Meeker, 2007). Toxins secreted from feline macrophages induce a delayed calcium deregulation and neural damage(Bragg et al., 2002) due, in part, to the suppression of intracellular calcium recovery (Bragg et al., 2002).
In the present studies we used the FIV model to explore the neuroprotective efficacy of a new neurotrophin ligand, LM11A-31, that targets the p75 neurotrophin receptor (p75NTR)(Longo and Massa, 2008). LM11A-31 was developed as a small molecule ligand to mimic loop 1 of nerve growth factor (Massa, et al, 2006). It competes for binding to p75NTR but not TrkA and increases NFkB phosphorylation and Akt activation in a p75NTR-dependent fashion (Massa, et al, 2006). It does not have the pro-apoptotic actions that can be mediated by pro-NGF(Massa, et al, 2006). The neuroprotective actions are unique from NGF since it offers protection from Aβ-induced neuron death where NGF is ineffective (Knowles, et al, 2008). The potential utility of this ligand for HIV infection was suggested by observations of altered neurotrophin signaling in infected patients. Brain-derived neurotrophic factor (BDNF) and its receptor, TrkB, are increased in HIV-infected brain(Soontornniyomkij et al., 1998) (and BDNF and nerve growth factor (NGF) protect neurons against Tat- (Ramirez et al., 2001) and gp120-mediated apoptosis(Mocchetti and Bachis, 2004; Bachis and Mocchetti, 2005; Nosheny et al., 2005). The neurotrophins may provide broad natural protection by suppressing or counteracting several destructive processes triggered by gp120 and Tat such as pro-apoptotic signaling cascades(Kruman et al., 1998; Garden et al., 2002), interference with NGF signaling(Peruzzi et al., 2002) and activation of p38 MAPK, p53 and glycogen synthase kinase-3β(GSK-3β)(Maggirwar et al., 1999; Kaul et al., 2001). The following studies examined the potential of LM11A-31 to protect cultured neurons from the damaging effects of FIV and associated macrophage toxins.
Materials and Methods
Neural cultures
Neural cultures were prepared from the forebrain of E30–45 fetuses removed during spays of pregnant cats. The protocol for removal and culture of fetal material followed institutional and federal guidelines and was approved by the North Carolina State University College of Veterinary Medicine Institutional Animal Care and Use Committee. After removal, the uterus was placed on ice and transported to the tissue culture facility where the fetuses were removed and placed in ice cold HEPES-buffered Hank’s balanced salt solution (HBSS). The brain was harvested from each fetus, rinsed three times in fresh sterile HBSS and the forebrain containing the cortex, hippocampus and basal ganglia transferred to a 15 ml tube containing calcium-magnesium free HBSS (CMF-HBSS), 1.25 U/ml dispase II + 2 U/ml DNase I. After incubation for 25–30 min at 37° C the cells were gently dissociated by several rounds of trituration and collected in a tube containing 25 mls Minimum Essential Medium (MEM) with glutamine plus 10% fetal bovine serum and 20 μg/ml gentamicin (complete medium). The dissociated cells were seeded into poly-D-lysine-coated culture dishes at a density of 50,000 – 100,000 cells/cm2 for toxicity analysis or 25,000 cells/cm2 on poly-D-lysine-coated coverslips for imaging. Cultures were fed by 50% exchange of complete medium three times per week. Cells were allowed to grow at least 6 days in vitro, at which time they contain an enriched population of neurons (>80%). Over the next week, the cultures slowly become populated with astrocytes. Imaging studies were done prior to extensive outgrowth of the astrocytes at 6–12 days in cultures where fields of isolated neurons could be obtained
Cultures were infected with FIV at times ranging from 6–12 days in vitro for neuronal cultures depending on the experiment. Cultures were not washed following inoculation to facilitate prolonged exposure to the virus with the exception of the infection studies where the cultures were washed five times at 4 hours after inoculation. Viral titers were selected to reflect the high range of virus seen in CSF in vivo (104 – 106 copies/ml).
To assess neurotrophic activity, feline neurons were seeded at very low densities of 2800–4000 cells/cm2. Survival is typically low at this density but can be improved by neurotrophins. Survival over time was monitored under phase contrast and cells with neuronal morphology and distinctive neurite outgrowth were counted. Final survival and elaboration of processes was evaluated by staining with the neuron marker microtubule associated protein-2 (MAP-2).
Sources of FIV
Two strains of FIV were used in these studies. FIVNCSU1 was isolated from a naturally infected cat with lymphopenia and acute enteritis. Peripheral blood mononuclear cells from this cat were used to inoculate 2 purpose bred specific pathogen free cats. Pooled PBMC from these 3 donor cats were co-cultured with a CD4+ lymphocyte cell line (FCD4-E), and high RT activity supernatant collected and frozen as a source of virus stock. Intravenous and intracerebroventricular inoculation of cats results in robust infection, with a persistent decrease in the CD4:CD8 ratio and a gradual loss of CD4+ T cells(English et al., 1994; Meeker et al., 1997; Liu et al., 2006). Both anti-FIV antibodies and FIV provirus are present by 2 to 4 weeks p.i. as detected by ELISA and PCR amplification, respectively. The virus infects both CD4+ T cells and monocyte/macrophages. In addition, an FIV strain with enhanced neurotropism (FIVV1CSF) isolated from the CSF of a cat with neurological disease(Power et al., 1998; Johnston et al., 2002) was used to enhance neurovirulence. To maximize infection of cultures and neurovirulence, a 1:1 mix of viruses was used as the challenge.
Conditioned medium from feline choroid plexus macrophages
Cultures of purified macrophages were prepared from fetal choroid plexus. Choroid plexus was removed from the lateral ventricles of fetal brains at 30–62 days gestation as described above for neural cultures. The choroid plexus was washed extensively and placed into 6-well low adhesion plates (approximately 6/well) containing Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 20 μg/ml gentamicin (complete medium). The cultures were maintained in a 5% CO2 incubator at 36° C. Over a period of approximately one week, macrophages replicate and migrate from the choroid plexus. The macrophages were the only cells that attached to the low adhesion plastic giving rise to a pure population of macrophages. The cultures were initially fed by supplementing the medium with fresh medium (twice) and then were fed three times a week by carefully aspirating medium from the surface (avoiding removal of the choroid plexus explants) and replacing with fresh medium. After 8–12 days in culture, choroid plexus cultures were typically enriched in macrophages at densities ranging from approximately 50 – 170 cells/mm2 (40,000–136,000/well). The choroid plexus was then removed and the macrophages washed with fresh medium. The resulting cultures were conservatively >95% macrophages. These macrophages were then infected with FIV at an MOI of 1 in 1.2 mls of complete DMEM. Medium from these cells was harvested after 3 days and ultrafiltered at 300 kDa to remove virus. Medium collected from the same cells prior to infection was collected as a control. Conditioned medium was aliquoted and stored frozen at −80° until used for toxicity testing.
Real Time PCR quantification of FIV
Viral RNA was extracted from samples using QIAamp Viral RNA Mini Kit (Qiagen, CA). The viral RNA loads were determined by real time RT-PCR with the Taqman onestep RT-PCR master kit (Applied Biosystems, CA). The following specific primers and probe of FIV gag region were used which recognize both NCSU1 and V1CSF variants: FIVNC-491F (5′-GATTAGGAGGTGAGGAA GTTCA GCT–3′), FIVNC-617R (5′-CTTTCATCCAATATTTCTTTATCTGCA–3′) and the labeled probe, FIVNC-555p (5′-FAM- CATGGCCACATTAATAATGG CGCA -TAMRA-3′ (Applied Biosystems, CA). These reactions were performed with a Bio-Rad iCyclerTM iQ and analyzed using the manufacturer’s software. A standard curve was generated within each run from dilutions of an FIV standard of known copy numbers.
Immunocytochemistry
Neurons were identified by microtubule-associated protein-2 (MAP-2) immunostaining. Cells were fixed in ice cold 97% methanol, 3% acetic for 10 min at room temperature and washed in 0.01 M phosphate-buffered saline (PBS, 3 × 5 min). Cells were incubated in blocking buffer containing 3% normal goat serum in 0.01 M PBS for 60 min at room temp. Polyclonal rabbit anti-MAP-2 (Chemicon/Millipore, Bilerica, MA) was then applied at a dilution of 1:500 in blocking buffer and incubated overnight at 4° C. The cells were washed three times in PBS and incubated in goat anti-rabbit Alexa488 (1:500) or mouse anti-rabbit Alexa568 (1:500) for 1 h. The cells were washed three times in PBS. Astrocytes were stained in a similar fashion using 1:500 rabbit anti-GFAP (DAKO). Nuclei were counterstained with bisbenzimide (0.5 μM, Sigma Corp, St Louis, MO) for 20 min in PBS, washed 2 × 5 min and the coverslips were mounted onto slides with Fluoromount (Southern Biotech, Birmingham AL).
Staining was examined with an Olympus IMT-2 or Olympus IX71 inverted microscope and quantified with digital imaging and Metamorph imaging software. Neuron survival was measured by counting the number of viable neurons in five fields. Neuron morphology was evaluated by measuring the microtubule-associated protein-2 (MAP-2) fluorescence stain intensity at magnifications of 134X and 334X and the density of neurons and processes was quantified using the threshold and intensity functions. The total intensity of the MAP-2 stain was recorded and the area occupied by MAP-2 immunoreactive cell bodies and processes was measured and expressed as a percentage of the entire field. Background stain intensity was measured in regions that did not contain neurons and was negligible.
LM11A-31: a non-peptide neurotrophin ligand
The compound LM11A-31 was initially identified by Drs. Longo and Massa using a virtual screen of a small molecule library to identify compounds mimicking the spatial and physical chemical features of the loop 1 region of NGF, a domain known to bind to p75NTR(Massa et al., 2002). Subsequent screening of compounds showed LM11A-31 to have neurotrophic activity, high affinity for p75NTR, and the ability to activate downstream signaling via PI3 kinase and Akt(Massa et al., 2006; Massa et al., 2002) with no direct actions on the nerve growth factor receptor, TrkA or the brain-derived neurotrophic factor (BDNF) receptor, TrkB. LM11A-31, has potent neuroprotective properties in mouse hippocampal cultures under conditions of serum starvation with EC50 values of 200–300 pM and a maximally effective concentration of 5–100 nM in various assays(Massa et al., 2006; Yang et al., 2008). In rat and feline neural cultures, a concentration of 10 nM was shown to be optimal to promote survival under a variety of conditions.
Calcium imaging
Neuronal cultures were washed in HEPES-buffered artificial CSF (aCSF, concentrations in mM: NaCl 137, KCl 5.0, CaCl2 2.3, MgCl2 1.3, glucose 20, HEPES 10, adjusted to pH 7.4 with NaOH) and pre-loaded with the calcium indicator, Fluo-4 AM using the Molecular Probes Fluo-4 NW Calcium Assay Kit (Molecular Probes/Invitrogen, Eugene, OR) at a 1:4 dilution. After 30 minutes the coverslip was mounted in a specialized stage for imaging (Warner Instruments, Hamden, CT). LM11A-31 was added to the coverslips in the chamber to allow approximately 10 min exposure prior to the challenge. The cells were challenged with conditioned medium from feline macrophages infected with FIV. The toxic challenge was added in a volume of 80 μl to 320 μl aCSF in the chamber to provide a 1:5 dilution of the stock. Neural cultures stimulated with aCSF were used to establish the non-toxic reference baseline and the pre-inoculation conditioned medium was used to establish basal activity of the macrophages.
Time lapse digital images were captured automatically by the Metamorph System (Molecular Devices, Union City, CA). Three pre-stimulation measurements were taken to establish basal levels of fluorescence at the beginning of each experiment. Acute changes in calcium were measured at 0.1 min intervals for 6 min. Delayed changes in calcium were measured at 1 min intervals for an additional 60 min. The increase in fluorescence intensity (free calcium) within each cell was then measured relative to the baseline measurements to correct for cell to cell differences in dye loading and intrinsic fluorescence. Responses to each challenge were also compared to runs with aCSF and/or control medium and corrected for non-specific effects if necessary. In most cases the background changes were negligible. Medium that caused a pronounced late destabilization of intracellular calcium was defined as toxic and was used as the challenge against which the actions of LM11A-31 were assessed.
Results
FIV infection of feline neural cultures
Addition of FIVV1CSF and FIVNCSU1 to primary feline neural cultures at biologically relevant levels of 104 – 106 TCID50/ml had no significant acute effect but led to a gradual appearance of dendritic beading, pruning and neuronal shrinkage. Cultured feline neurons inoculated with FIV and stained for MAP-2 after 7 days are illustrated in Figure 1. Untreated neurons (Fig 1A) had excellent outgrowth of dendrites and well stained cell bodies. One week after inoculation with FIV, the dendrites show early signs of beading (small arrows), simplification of the dendrites (open arrow) and shrinkage with very thin or poorly stained cytosol (large arrow). The loss of the dendritic architecture in particular leads to an overall reduction in MAP-2 staining which can be used to assess the extent of damage. The time course for the development of damage based on the mean MAP-2 stain intensity is illustrated in Figure 1C. At 3 days post-inoculation, no changes are seen. At seven days post-inoculation there is a 57% loss of MAP-2 stain intensity (p=0.0049, FIV vs. uninoculated) and by 14 days a 77% loss (p=0.0003 FIV vs. uninoculated).
Figure 1.
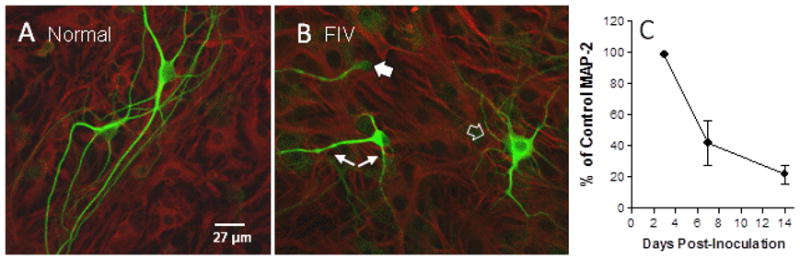
Example of early FIV-induced damage to feline neurons in vitro. A. In the absence of FIV, isolated MAP-2 immunoreactive feline neurons (green) exhibit large cell bodies with elaborate outgrowth of dendrites on a uniform bed of GFAP+ astrocytes (red). B. Seven days after inoculation with 105 TCID50 FIV, neurons often appear shrunken with thin cytosol (large arrow), dendrites show early signs of beading (small arrows) and otherwise healthy neurons show a simplification of processes (open arrow). C. Mean ± sem MAP-2 immunoreactivity at various times after inoculation with FIV. No loss was seen over the first 3 days after which MAP-2 immunoreactivity decreased to 43% of uninfected controls by 7 days and 23% at 14 days (p=0.0049, n=6 and 0.0003, n=4, respectively relative to uninoculated controls).
LM11A-31 provides neurotrophic support to feline neurons
To establish the neurotrophic properties of LM11A-31, cat neurons were seeded at a very low density and half the cultures were treated with 10 nM LM11A-31 in the medium. Initial seeding density was matched within each run at approximately 2800–4000 cells/cm2 and cultures were fed every two days with complete MEM or MEM+LM11A-31. Neuron survival is typically poor at these low densities in the absence of additional neurotrophic support. Three days after seeding, neuron survival was significantly higher in the LM11A-31 treated cultures (Figure 2A). After fourteen days of treatment with LM11A-31, neurons exhibited a 6.6-fold increase in the total MAP-2 stained area (Figure 2B) which reflects both the density of neurons and extent of process outgrowth.
Figure 2.
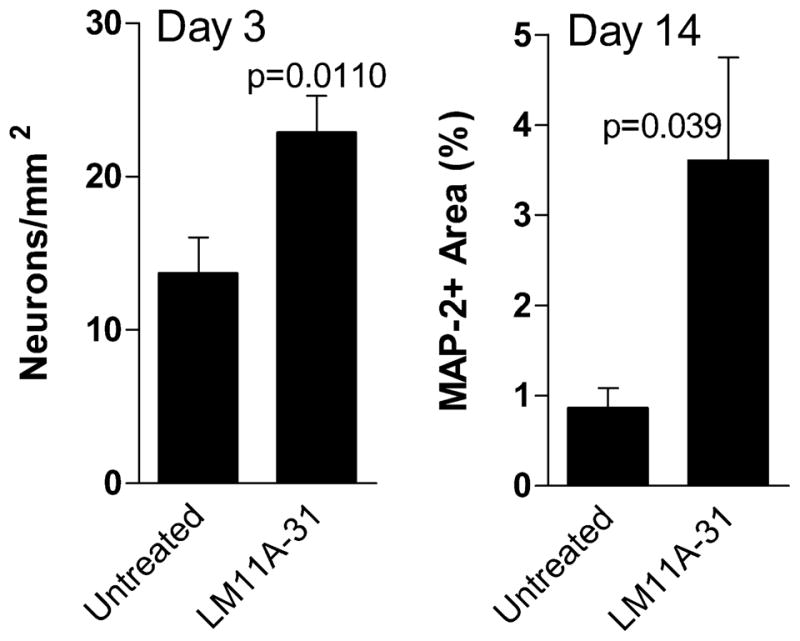
LM11A-31 supports survival of feline neurons in low density cultures. Feline neurons were cultured at a density of 23–40 cells/mm2 in the presence or absence of 10 nM LM11A-31. A. Three days after seeding, untreated neurons were present at an average (± sem) density of 13.7/mm2 (34.2% survival) whereas in the presence of LM11A-31 a significantly higher density of 22.9/mm2 (57.3% survival) was seen (p=0.0110 LM11A-31 vs. untreated, n=12 cultures each). B. A significant effect of LM11A-31 was also seen in the outgrowth of neurons after 14 days of continuous treatment. An average of 3.6% of the culture surface was occupied by MAP-2 immunoreactive cells and processes in the LM11A-31 cultures relative to 0.86% in the untreated cultures (p=0.039 LM11A-31 vs. untreated, n= 6 cultures each).
LM11A-31 reduces FIV-associated damage
To test the effects of LM11A-31 in the presence of FIV, feline neural cultures at 12 days in culture were inoculated with 105 TCID50 FIV mix followed immediately by addition of LM11A-31 at a concentration of 10 nM. LM11A-31 was sustained in the culture throughout the experiment and the cells were fixed at 3 and 7 days post-treatment and stained for MAP-2 and GFAP. The FIV inoculated cultures showed damage after seven days which is illustrated in Figure 3A. Beading and simplification of MAP-2+ processes and neuron shrinkage was seen as in Figure 1 and a co-stain for GFAP stain also showed a retraction of astrocyte processes with development of a more process bearing morphology. Treatment with LM11A-31 reversed much of this damage with most cultures showing healthy neurons with elaborate processes on a more uniform bed of astrocytes (Figure 3B). Quantification of the MAP-2 stain shown in Figure 4, illustrates the mean (± sem) MAP-2 fluorescence intensity relative to untreated control cultures at three days and seven days post-inoculation. At three days no decrease in MAP-2 stain or changes in neuron morphology were seen (94.8 ± 7.1% of control) and by seven days a 58% decrease was seen (41.8 ± 15.7% of control, p=0.0049). In each case, treatment with LM11A-31 resulted in MAP-2 staining that was above the control values (172–193%) and 4-fold greater than the MAP-2 intensity of the neural cultures treated with FIV (FIV+LM11A-31 vs. FIV at 7 days, p=0.0111).
Figure 3.
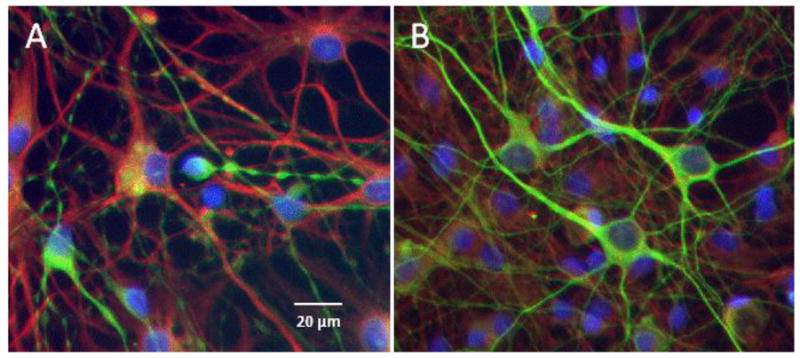
LM11A-31 prevents damage to neurons inoculated with FIV. A. Example of neuronal damage seen after 7 days exposure to FIV. Beading, pruning and shrinkage is evident in MAP-2 stained neurons (green) with a shift toward development of process bearing GFAP-positive astrocytes (red). Nuclei were stained blue with bisbenzimide. B. Concurrent addition of 10 nM LM11A-31 to the FIV-inoculated cultures resulted in a preservation of neuron integrity on a normal bed of astrocytes.
Figure 4.

Quantification of MAP-2 staining relative to untreated controls at 3 and 7 days post-inoculation. A. At 3 days no significant change in average MAP-2 fluorescence intensity from controls was seen whereas by 7 days a 52% decrease was seen (p=0.0049 vs. untreated, n= 6 each). Values are normalized to controls (100%). The FIV-associated decrease in MAP-2 immunoreactivity was completely reversed in the presence of 10 nM LM11A-31 (p=0.0111, FIV vs. FIV+LM11A-31, n=6 each) to a value that was 172% of untreated controls. B. Measurement of the area occupied by MAP-2 stained dendrites at 7 days indicated that the effects of FIV were largely due to a decrease in the dendritic architecture (p=0.00079, n=16) with almost complete prevention of the loss by 10 nM LM11A-31 (p=0.00004, n=11). C. The area of neuronal cell bodies also decreased significantly at 7 days (p=0.0017, n=16) in the FIV-infected cultures but was only partially reversed by LM11A-31 (n=11). D. Characterization of the distribution of neuron cell body areas illustrated that LM11A-31 significantly shifted the distribution toward a broader profile with larger cells (X2 =27.5, df=6, p<0.01, 198–244 neurons), similar to untreated neurons.
To evaluate the relative effect of FIV on dendrites and cell bodies, the MAP-2 stained area was quantified in cultures treated for seven days with FIV (Figure 4B, C). MAP-2 stained dendrites accounted for an average of 82 ± 4% of the stained area or 4.6 times the area of the neuron cell bodies. Figure 4B shows the large reduction in stained dendrites in the presence of FIV. Treatment with 10 nM LM11A-31 prevented the loss of dendrites (p<0.001). The decrease in the neuronal perikarya is illustrated in Figure 4C. FIV induced a 40% reduction in the cell body area. This loss was partially reversed by LM11A-31 but did not reach significance. The distribution of neuron perikarya areas is illustrated in Figure 4D. Control neurons have a broad distribution of sizes from about 150 to 350 μm2 likely reflecting different types. In the presence of FIV neurons tend to shift to a single distribution similar to the smaller control neurons. Treatment with LM11A-31 pushes the distribution significantly to the right again reflecting a broader distribution (X2 = 27.49, 6 df, p <0.01) that begins to resemble the control cell distribution. The neuron density in these same cultures did not change significantly between conditions: 150.6 ± 20.2, 143.8 ± 18.5 and 155.3 ± 16.6 neurons/mm2 for untreated controls, FIV and FIV+LM11A-31, respectively. LM11A-31-associated recovery was significant at FIV titers of 104–106 TCID50/ml but decreased in magnitude to 56% at the highest viral load (Figure 5).
Figure 5.
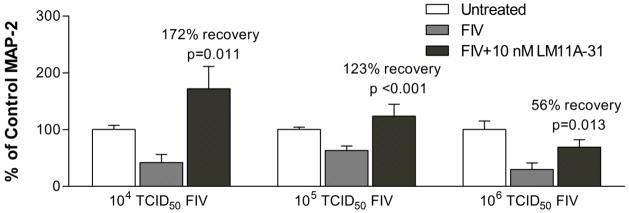
LM11A-31 efficacy varies with FIV load. FIV titers of 104, 105 and 106 TCID50/ml produced similar decreases in MAP-2 staining intensity. Addition of 10 nM LM11A-31 resulted in a significant recovery of MAP-2 in each case but the extent of recovery decreased from 172% (n=6) to 123% (n=7) and 56% (n=10) as the amount of FIV increased. Values are normalized to controls (100%).
FIV associated changes in astrocytes and microglia
After inoculation of the neural cultures notable changes were seen in both astrocytes and microglia. The morphology of the astrocytes often became less flattened with a more fibrous appearance as illustrated in Figure 3. In cultures showing the greatest damage, the retraction of astrocyte processes was often more severe. An analysis of the density of GFAP+ astrocytes in the cultures, however, showed only a small (12% p=0.0631), non-significant decrease in the mean area occupied by the astrocytes.
Increased numbers of microglia were observed in the cultures treated with FIV particularly at the longer durations. In neural cultures inoculated at day 6–8 with FIV microglial density is initially very low (1.8 ± 1.2 microglia/mm2; 0.8% of the neuron density). Seven days after inoculation when damage is evident, the density of microglia increased to 12.4 ± 0.7/mm2, a small (~5% of the neuron density) but significant increase (p=0.0170). In contrast, between seven and 16 days the density of microglia increased dramatically to 918 ± 83/mm2. To determine if treatment with LM11A-31 influenced the accumulation of microglia we labeled the microglia with 2 μg/ml DiI acetylated LDL and counted the number in five fields/culture. We started with matched cultures with visible microglia (day 12 in vitro) and measured the microglial density/mm2 at 7 days post-inoculation (day 19 in vitro). As illustrated in Figure 6A, FIV induced a significant increase in microglia which was reversed in the presence of LM11A-31. To determine if the increase was a direct effect of FIV on the microglia in the relative absence of neurons, enriched microglial cultures were prepared and challenged with FIV or FIV+LM11A-31. As illustrated in Figure 6C, inoculation of isolated microglia with FIV or FIV+LM11A-31 failed to show any changes in microglial density. Similar effects were seen in cultures with a lower initial density of purified microglia (not shown). Quantification of microglial death (Figure 6D) also showed little death (0.2–0.6%) and failed to show any significant changes due to the treatments.
Figure 6.

Microglial changes in response to FIV and LM11A-31. A. Neural cultures inoculated with 105 TCID50 FIV showed a significant increase in microglia after 7 days (p=0.0148, FIV vs. Control, n=15 and 11, respectively) which was largely reversed in the presence of 10 nM LM11A-31 (p=0.0297, FIV vs. FIV+LM11A-31, n=15 and 14, respectively). B. Large increases in microglia typically appeared after prolonged exposure to FIV. A significant increase in microglia was seen seven days post inoculation (Day 7 p.i., n=23) but numbers were still very low relative to neurons in the culture (~5% of the neuron density). By day 16 post-inoculation (n=10), microglia were abundant. C. Inoculation of microglia enriched cultures with FIV or treatment with LM11A-31 in the relative absence of neurons failed to affect density (n=4–6 cultures each). D. Inoculation of enriched microglia enriched cultures with FIV or treatment with LM11A-31 - did not produce significant microglial death (0.2–0.6%, n=4–6 cultures each).
LM11A-31 suppresses the neuronal calcium destabilization and damage in response to macrophage conditioned medium
To recapitulate FIV neuropathogenesis in vitro we used medium collected from feline choroid plexus macrophages 3 days after infection with 106 TCID50/ml FIV. The FIV dose was chosen to provide a robust response with no observable damage to the cells. When applied to feline neurons, the FIV-challenged feline macrophage conditioned medium (fMCM) produces a rapid increase in intracellular calcium followed by a late destabilization of calcium. Dendritic beading and pruning appear during the late destabilization (Figure 7A) but show early signs of destabilization and swelling as early as 5.8 minutes after stimulation. Neurons challenged with the same conditioned medium in the presence of 10 nM LM11A-31 (Figure 7B) failed to show the delayed destabilization of calcium even when the initial calcium rise was matched to the FIV cultures (Figure 7, 0.5 Min). In the absence of the delayed calcium rise, dendritic swelling or other signs of damage at 66 min were minimal or absent. The mean calcium response of feline neurons exposed to fMCM is illustrated in Figure 8A. After a slight delay, the macrophage conditioned medium induced a rapid rise in calcium which did not recover. After 6–10 min, a progressive increase in calcium was seen. In the presence of LM11A-31 the acute rise was similar but there was no late destabilization of calcium. Values were corrected for the effects of medium alone which induced a small acute rise in calcium (+65 fluorescence units) and a gradual upward drift in the delayed phase (+96 fluorescence units). Calcium levels remained stable throughout the experiment in cells challenged with aCSF.
Figure 7.
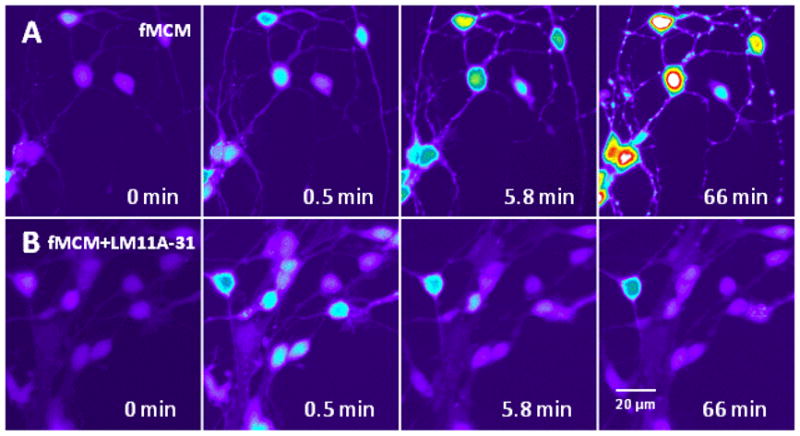
Feline macrophage conditioned medium (fMCM) from FIV-inoculated macrophages (106 TCID50/ml) induced a destabilization of intracellular calcium that was inhibited in the presence of LM11A-31. Conditioned medium from feline choroid plexus macrophages collected 3 days after inoculation with FIV was applied to primary feline neurons at a dilution of 1:5 in aCSF. Fluorescence intensity (calcium concentration) is pseudocolored to highlight changes (white>red>yellow>green>blue>dark blue). A. The conditioned medium caused a modest acute rise in intracellular calcium (0.5 min) which failed to recover leading to the appearance of focal calcium increases and swelling in dendrites by 5.8 min followed by a large calcium destabilization and dendritic beading after 66 min. B. Cultures pre-treated with 10 nM LM11A-31 for 10 min and challenged with the same feline macrophage-conditioned medium as above showed a similar acute rise but minimal delayed destabilization.
Figure 8.

Summary of the mean ± sem calcium changes (Fluorescence Increase) in response to fMCM or fMCM+LM11A-31. A. After a brief delay, fMCM (solid circles, n=119 neurons) induced a rise in calcium with no recovery over the first 6 min. A gradual destabilization of calcium developed over the next 60 min giving rise to high intracellular calcium. Co-stimulation with LM11A-31 did not change the acute calcium rise but largely prevented the destabilization (n=114 neurons). Neurons treated with aCSF (n=101 neurons) showed stable calcium levels throughout illustrating the integrity of the neurons in the absence of stimulation. B. Astrocytes (n=60) in the same cultures as A showed small increases in intracellular calcium (note change in scale versus A) but minimal long-term destabilization. The addition of LM11A-31 (n=58) suppressed the calcium changes to near background (aCSF) levels. C. Example of the typical calcium profile of most astrocytes showing negligible acute and small long-term changes relative to aCSF controls. D. Example of calcium responses seen in a subset of astrocytes which showed pulses of calcium during the delayed phase. This activity was not synchronized between astrocytes and occurred after the appearance of calcium elevations in the neurons.
Although these cultures are highly enriched in neurons, a small number of astrocytes were present which allowed a parallel evaluation of the astrocyte calcium responses. The average astrocyte response to the conditioned medium, illustrated in Figure 8B, was about 25% of the neuronal response in the acute phase and 9% in the delayed phase. However, the astrocytes showed varied response patterns. Most exhibited only a very small gradual increase in intracellular calcium above baseline as illustrated for a typical cell in Figure 8C. A subpopulation of astrocytes exhibited multiple brief spikes of calcium in the delayed phase in response to the conditioned medium. The typical pattern is illustrated in Figure 8D where repetitive spikes are seen after about 20 min. There was little synchrony of the patterns between cells and the onset was almost always after destabilization was seen in the neurons. Untreated astrocytes would also occasionally show an isolated spike of activity as illustrated in a cell exposed to aCSF (arrow), but this was rare relative to the challenged cells.
FIV production in the presence of LM11A-31
Peripheral blood mononuclear cell (PBMC), macrophage and microglial cultures were infected with an FIV mix at an MOI of 1 in the presence or absence of 10 nM LM11A-31. The cultures were washed at four hours and FIV was measured in the medium every 3 days over a period of 21 days. Cumulative FIV production from 6 to 21 days was calculated to compensate for any variation in the timing of FIV expression and is illustrated as mean log cumulative FIV ± sem (n=3) in Figure 9. No difference was seen in FIV content in the PBMC cultures. LM11A-31 was associated with a slight increase in FIV in the macrophage cultures and a very slight decrease in the microglial cultures, neither of which was significant.
Figure 9.
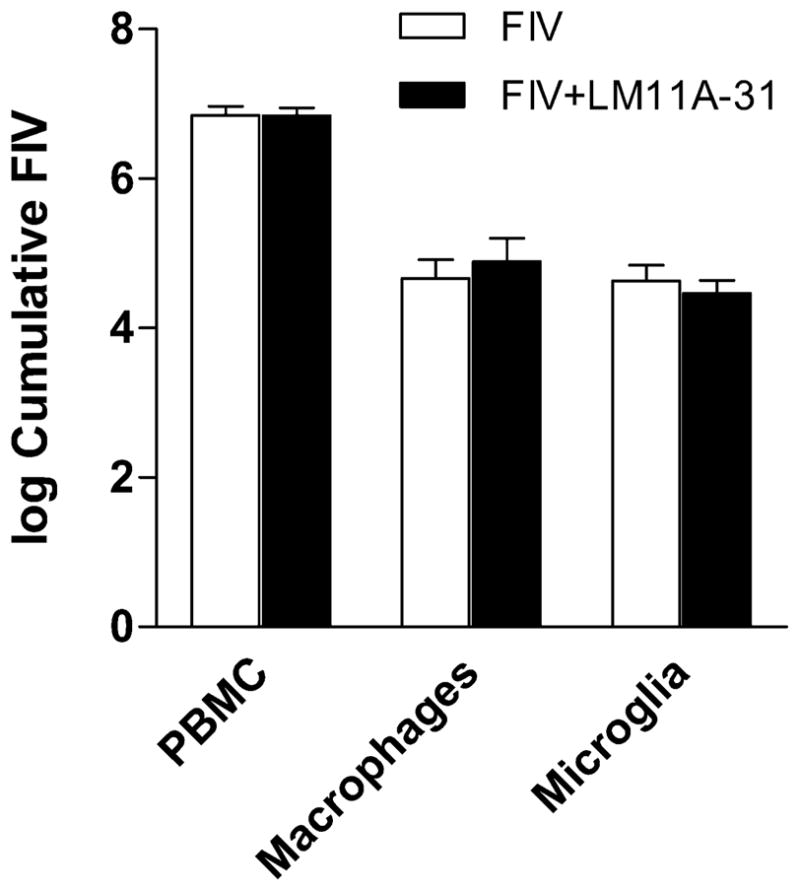
Recovery of FIV from PBMCs, choroid plexus macrophages or microglia inoculated with FIV at an MOI of 1. Log cumulative FIV recovery from day 6 to day 21 in culture was calculated and the mean ± sem values (n=3 cultures each) plotted. FIV replication in PBMCs was the same in the presence or absence of LM11A-31. A small but non-significant increase was seen in macrophages treated with LM11A-31. A very small non-significant decrease was seen in microglia.
Discussion
Feline neural cultures inoculated with FIV or treated with medium from FIV-infected macrophages develop pathological changes such as dendritic beading and pruning that are thought to reflect the underpinnings of HIV-associated neural dysfunction. These changes are preceded by the destabilization of neuronal intracellular calcium. Since this pathological sequence is likely to reflect changes that initiate HIV neuropathogenesis in vivo, it provides an opportunity to explore the actions of putative therapeutic compounds in a model that should translate to natural infection. The current studies provide the first assessment of a novel neuroprotective neurotrophin ligand with this model. Deficits in neurotrophins have been reported in HIV-infected patients suggesting a loss of protective neurotrophin signaling. LM11A-31 is a novel ligand that penetrates the blood-brain barrier and mimics the protective actions of NGF at the p75NTR. It competes with NGF for high affinity binding and triggers the activation of Akt and neurite outgrowth (Massa, 2006). Thus, it has the potential to improve neurotrophin signaling and protect against virus-associated damage. Neurotrophic activity of LM11A-31 was demonstrated in these studies by showing enhanced feline neuron survival and outgrowth at low seeding densities. Enhanced neuronal outgrowth was substantial after 2 weeks of treatment illustrating both the long-term beneficial effects of the compound as well as the absence of toxicity.
The suppression of damage from infectious FIV in these cultures at nanomolar concentrations further illustrated the potential utility of this compound for prevention of FIV-associated neural damage. No significant changes were seen in FIV replication although a very small effect was seen on macrophage FIV synthesis in agreement with Souza, et al. (Souza et al., 2011). Although this will have to be verified in vivo, these observations indicate that LM11A-31 can be used in infected cats with minimal risk of accelerating viral replication. The neuroprotective effect of LM11A-31 was most prominent on the elaboration of dendrites, consistent with neurotrophic actions. Treatment of FIV infected cultures with 10 nM LM11A-31 completely restored the average neuronal dendritic tree with no significant changes in the density of neurons under any condition. The putative shrinkage of neurons was partially reversed by LM11A-31 with a significant change in the size distribution toward larger sizes similar to controls. The protective effect was almost complete for the primary endpoint of MAP-2 stained area except at the highest viral burdens and with severe challenges such as combinations of concentrated macrophage toxins and glutamate (not shown). Thus, LM11A-31 offered protection over a wide range of toxic activity and had diminished efficacy only under highly toxic conditions.
It is noteworthy that the inoculation of the cultures with FIV also triggered an accumulation of microglia and changes in astrocyte morphology. The accumulation of microglia could certainly contribute to pathogenesis by increasing the number ot toxin-producing cells. The suppression of microglial accumulation by LM11A-31 could contribute to neuroprotection but several observations indicate that this effect was not the primary action responsible for the neuroprotection. The high accumulation of microglia was apparent only in the cultures with longer exposures to FIV. The accumulation was still relatively low after 7 days when damage was readily apparent suggesting that the increase may be secondary to neural damage. In addition, the effects of LM11A-31 in low density cultures and in the calcium assays were seen in the almost complete absence (<1%) of microglia. The potential impact of LM11A-31 interactions with the microglia remains an open question. Neurotrophin receptors have been identified on monocytes and macrophages raising the possibility of direct actions of LM11A-31 on cells of monocyte lineage(Dowling et al., 1999; Barouch et al., 2001; Samah et al., 2008). The role of neurotrophins in the regulation of microglia is poorly understood and this is an important area for future studies. In contrast to the microglia, the change in astrocyte morphology was not paralleled by a significant decrease in average GFAP+ astrocyte density. A very small increase in intracellular calcium was seen in the astrocytes in response to macrophage conditioned medium which was largely due to small bursts of activity in a subpopulation of cells. The increase was suppressed by pretreatment with LM11A-31. The bursts of activity typically appeared late in the run, after the onset of calcium accumulation and dendritic swelling in the neurons. Thus, like the microglia we believe the response of the astrocytes may be secondary to changes in the neurons. Each of these cells probably play a role at various stages of FIV pathogenesis and more work is needed to better understand the interactions that either drive pathogenesis or support recovery.
An important observation was the demonstration of the ability of LM11A-31 to inhibit the acute calcium rise and delayed calcium destabilization in neurons in response to conditioned medium from FIV-treated feline macrophages. Exposure to macrophage toxins(Giulian et al., 1990; Pulliam et al., 1991; Kaul et al., 2001) and the disruption of calcium homeostasis(Dreyer et al., 1990; Haughey et al., 1999; Holden et al., 1999; Meeker et al., 1999; Bragg et al., 2002; Meeker, 2007) are considered important early events in the development of neural damage. Indeed the calcium rise typically preceded the development of visible pathology. FIV alone added to the neural cultures did not produce a similar effect. This reinforced the idea that the interaction of FIV with the macrophages is instrumental in the development of the toxic response. The mechanisms responsible for the calcium rise and the protective effects of LM11A-31 are not completely understood. The relationship between p75NTR signaling and calcium homeostasis is also unknown but is likely to be indirect. The destabilization of calcium in this model and the development of damage resemble excitotoxicity (Tymianski et al., 1993) but are also different in important ways. First, unlike excitotoxic challenges where the strong initial activity is thought to drive the delayed calcium deregulation, the deregulation in our experiments was unrelated to the acute rise in calcium. Our previous studies suggested a dysfunction of the sodium-calcium exchanger (NCX) may contribute to the calcium dysregulation(Bragg et al., 2002; Meeker et al., 2005). The role of forward and reverse calcium exchange in different settings is still controversial (Bano et al., 2005; Brustovetsky et al., 2010) and the relationship between the calcium dysregulation and structural pathology is unclear. Various hypotheses have implicated dysfunctional mitochondria (Greenwood et al., 2007), loss of actin structure(Zeng et al., 2007) or disruption of neurofilaments (Kim et al., 2010). By activating Akt and targeting early events in the pathogenesis, LM11A-31 may suppress destructive events at many levels. More studies are needed to precisely identify the molecular pathways that lead to neuronal dysfunction and the role of neurotrophin receptors in the control of these pathways.
The use of the FIV model for the development of a new therapeutic strategy has set the stage for efficient translation to an in vivo model. The FIV model will continue to provide important resources for the evaluation of interactions between LM11A-31 and FIV under both in vitro and in vivo conditions. Exploration of these interactions with various cell types will help to define the therapeutic and non-therapeutic actions of neurotrophin ligands.
Acknowledgments
Source of funding
This work was supported by NIH grants MH079726, MH079626 and MH063646 to R. Meeker
Footnotes
conflict of interest
Dr. Longo is founder of PharmatrophiX Inc., a company focused on the development of neurotrophin ligands including LM11A-31. Other authors have no affiliation with PharmatrophiX Inc.
References
- Bachis A, Mocchetti I. Brain-Derived Neurotrophic Factor Is Neuroprotective against Human Immunodeficiency Virus-1 Envelope Proteins. Ann NY Acad Sci. 2005;1053:247–257. doi: 10.1196/annals.1344.022. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Barouch R, Appel E, Kazimirsky G, Brodie C. Macrophages express neurotrophins and neurotrophin receptors. Regulation of nitric oxide production by NT-3. J Neuroimmunol. 2001;112:72–77. doi: 10.1016/s0165-5728(00)00408-2. [DOI] [PubMed] [Google Scholar]
- Bragg DC, Boles JC, Meeker RB. Destabilization of neuronal calcium homeostasis by factors secreted from choroid plexus macrophage cultures in response to feline immunodeficiency virus. Neurobiol Dis. 2002;9:173–186. doi: 10.1006/nbdi.2001.0459. [DOI] [PubMed] [Google Scholar]
- Brown WC, Bissey L, Logan KS, Pedersen NC, Elder JH, Collisson EW. Feline immunodeficiency virus infects both CD4+ and CD8+ T lymphocytes. J Virol. 1991;65:3359–3364. doi: 10.1128/jvi.65.6.3359-3364.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner D, Pedersen NC. Infection of peritoneal macrophages in vitro and in vivo with feline immunodeficiency virus. J Virol. 1989;63:5483–5488. doi: 10.1128/jvi.63.12.5483-5488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky T, Bolshakov A, Brustovetsky N. Calpain activation and Na+/Ca2+ exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. Journal of Neuroscience Research. 2010;88:1317–1328. doi: 10.1002/jnr.22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow SW, Poss ML, Hoover EA. Feline immunodeficiency virus: a neurotropic lentivirus. J Acquir Immune Defic Syndr. 1990;3:658–668. [PubMed] [Google Scholar]
- Dow SW, Mathiason CK, Hoover EA. In vivo monocyte tropism of pathogenic feline immunodeficiency viruses. J Virol. 1999;73:6852–6861. doi: 10.1128/jvi.73.8.6852-6861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling P, Ming X, Raval S, Husar W, Casaccia-Bonnefil P, Chao M, Cook S, Blumberg B. Up-regulated p75NTR neurotrophin receptor on glial cells in MS plaques. Neurology. 1999;53:1676–1682. doi: 10.1212/wnl.53.8.1676. [DOI] [PubMed] [Google Scholar]
- Dreyer E, Kaiser P, Offerman J, Lipton S. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–367. doi: 10.1126/science.2326646. [DOI] [PubMed] [Google Scholar]
- English RV, Nelson P, Johnson CM, Nasisse M, Tompkins WA, Tompkins MB. Development of clinical disease in cats experimentally infected with feline immunodeficiency virus. J Infect Dis. 1994;170:543–552. doi: 10.1093/infdis/170.3.543. [DOI] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- Greenwood SM, Mizielinska SM, Frenguelli BG, Harvey J, Connolly CN. Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J Biol Chem. 2007;282:26235–26244. doi: 10.1074/jbc.M704488200. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem. 1999;73:1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x. [DOI] [PubMed] [Google Scholar]
- Holden CP, Haughey NJ, Nath A, Geiger JD. Role of Na+/H+ exchangers, excitatory amino acid receptors and voltage- operated Ca2+ channels in human immunodeficiency virus type 1 gp120- mediated increases in intracellular Ca2+ in human neurons and astrocytes. Neuroscience. 1999;91:1369–1378. doi: 10.1016/s0306-4522(98)00714-3. [DOI] [PubMed] [Google Scholar]
- Hurtrel M, Ganiere J, Guelifi J, Chakrabarti L, Maire M, Gray F, Montagnier L, Hurtrel B. Comparison of early and late feline immunodeficiency virus encephalopathies. AIDS. 1992;6:399–406. doi: 10.1097/00002030-199204000-00007. [DOI] [PubMed] [Google Scholar]
- Johnston JB, Silva C, Hiebert T, Buist R, Dawood MR, Peeling J, Power C. Neurovirulence depends on virus input titer in brain in feline immunodeficiency virus infection: evidence for activation of innate immunity and neuronal injury. J Neurovirol. 2002;8:420–431. doi: 10.1080/13550280260422721. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, Casaccia P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci. 2010;13:180–189. doi: 10.1038/nn.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Liu P, Hudson LC, Tompkins MB, Vahlenkamp TW, Colby B, Rundle C, Meeker RB. Cerebrospinal fluid is an efficient route for establishing brain infection with feline immunodeficiency virus and transfering infectious virus to the periphery. J Neurovirol. 2006;12:294–306. doi: 10.1080/13550280600889567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo FM, Massa SM. Small molecule modulation of p75 neurotrophin receptor functions. CNS Neurol Disord Drug Targets. 2008;7:63–70. doi: 10.2174/187152708783885093. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- Meeker R, English R, Tompkins M. Enhanced excitotoxicity in primary feline neural cultures exposed to feline immunodeficiency virus (FIV) Journal of Neuro-AIDS. 1996;1:1–27. doi: 10.1300/j128v01n03_01. [DOI] [PubMed] [Google Scholar]
- Meeker RB. Feline immunodeficiency virus neuropathogenesis: from cats to calcium. J Neuroimmune Pharmacol. 2007;2:154–170. doi: 10.1007/s11481-006-9045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker RB, Robertson K, Barry T, Hall C. Neurotoxicity of CSF from HIV-infected humans. J Neurovirology. 1999;5:507–518. doi: 10.3109/13550289909045380. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Boles JC, Robertson KR, Hall CD. Cerebrospinal fluid from human immunodeficiency virus--infected individuals facilitates neurotoxicity by suppressing intracellular calcium recovery. J Neurovirol. 2005;11:144–156. doi: 10.1080/13550280590922757. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Thiede BA, Hall C, English R, Tompkins M. Cortical cell loss in asymptomatic cats experimentally infected with feline immunodeficiency virus. AIDS Research and Human Retroviruses. 1997;13:1131–1140. doi: 10.1089/aid.1997.13.1131. [DOI] [PubMed] [Google Scholar]
- Mocchetti I, Bachis A. Brain-derived neurotrophic factor activation of TrkB protects neurons from HIV-1/gp120-induced cell death. Crit Rev Neurobiol. 2004;16:51–57. doi: 10.1615/critrevneurobiol.v16.i12.50. [DOI] [PubMed] [Google Scholar]
- Nosheny RL, Mocchetti I, Bachis A. Brain-derived neurotrophic factor as a prototype neuroprotective factor against HIV-1-associated neuronal degeneration. Neurotox Res. 2005;8:187–198. doi: 10.1007/BF03033829. [DOI] [PubMed] [Google Scholar]
- Pedersen NC, Ho EW, Brown ML, et al. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science. 1987;235:790–793. doi: 10.1126/science.3643650. [DOI] [PubMed] [Google Scholar]
- Peruzzi F, Gordon J, Darbinian N, Amini S. Tat-induced deregulation of neuronal differentiation and survival by nerve growth factor pathway. J Neurovirol. 2002;8(Suppl 2):91–96. doi: 10.1080/13550280290167885. [DOI] [PubMed] [Google Scholar]
- Phillips T, Prospero-Garcia O, Puaoi D, Lerner D, Fox H, Olmsted R, Bloom F, Heriksen S, Elder J. Neurological abnormalities associated with feline immunodeficiency virus infection. Jof General Virology. 1994;75:979–987. doi: 10.1099/0022-1317-75-5-979. [DOI] [PubMed] [Google Scholar]
- Podell M, Oglesbee M, Mathes L, Krakowka S, Olmstead R, Lafrado L. AIDS-Associated Encephalopathy with Experimental Feline Immunodeficiency Virus Infection. Journal of AIDS. 1993;6:758–771. [PubMed] [Google Scholar]
- Power C, McArthur JC, Nath A, Wehrly K, Mayne M, Nishio J, Langelier T, Johnson RT, Chesebro B. Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998;72:9045–9053. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam L, Herndier B, Tang N, McGrath M. Human Immunodeficiency Virus-infected Macrophages Produce Soluable Factors that cause Histological and Neurochemical Alterations in Cultured Human Brains. Journal of Clinical Investigation. 1991;87:503–512. doi: 10.1172/JCI115024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB. Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem. 2001;78:874–889. doi: 10.1046/j.1471-4159.2001.00467.x. [DOI] [PubMed] [Google Scholar]
- Ryan G, Klein D, Knapp E, Hosie MJ, Grimes T, Mabruk MJ, Jarrett O, Callanan JJ. Dynamics of viral and proviral loads of feline immunodeficiency virus within the feline central nervous system during the acute phase following intravenous infection. J Virol. 2003;77:7477–7485. doi: 10.1128/JVI.77.13.7477-7485.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samah B, Porcheray F, Gras G. Neurotrophins modulate monocyte chemotaxis without affecting macrophage function. Clin Exp Immunol. 2008;151:476–486. doi: 10.1111/j.1365-2249.2007.03578.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soontornniyomkij V, Wang G, Pittman CA, Wiley CA, Achim CL. Expression of brain-derived neurotrophic factor protein in activated microglia of human immunodeficiency virus type 1 encephalitis. Neuropathol Appl Neurobiol. 1998;24:453–460. doi: 10.1046/j.1365-2990.1998.00134.x. [DOI] [PubMed] [Google Scholar]
- Souza TM, Rodrigues DQ, Passaes CP, Barreto-de-Souza V, Aguiar RS, Temerozo JR, Morgado MG, Fontes CF, Araujo EG, Bou-Habib DC. The nerve growth factor reduces APOBEC3G synthesis and enhances HIV-1 transcription and replication in human primary macrophages. Blood. 2011;117:2944–2952. doi: 10.1182/blood-2010-05-287193. [DOI] [PubMed] [Google Scholar]
- Sparger EE, Luciw PA, Elder JH, et al. Feline immunodeficiency virus is a lentivirus associated with an AIDS-like disease in cats. AIDS. 1989;3:S43–S49. doi: 10.1097/00002030-198901001-00006. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. JNeurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Rensing NR, Sinatra PM, Rothman SM, Wong M. Kainate seizures cause acute dendritic injury and actin depolymerization in vivo. J Neurosci. 2007;27:11604–11613. doi: 10.1523/JNEUROSCI.0983-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]