

Abstract
We reported that tumor necrosis factor receptor I (TNFRI) is required for neuronal death induced by amyloid-β protein in the Alzheimer’s disease (AD) brain. However, whether TNF receptor subtypes are expressed and activated differentially in AD brains compared to non-demented brains remains unclear. Our studies on Western blot and ELISA measurements demonstrated that TNFRI levels are increased whereas TNFRII levels are decreased in AD brains compared to non-demented brains (p < 0.05). Immunohistochemical results demonstrated that both TNFRI and TNFRII are expressed in neurons in AD and non-demented brains. However, in situ hybridization studies showed little change in the mRNA levels of either type of TNF receptor in the neurons of AD brains compared to non-demented brains. To examine whether different levels of TNF receptors in AD brains are correlated with the alteration of functional binding of TNF receptors, by using 125I-TNF-α binding technique, we found that, in AD brains, 125I-TNF-α binding affinity to TNFRI is increased, whereas binding affinity to TNFRII is decreased (p < 0.01). These studies reveal a novel observation of abnormal TNF receptor activation in AD brains. Differential TNF receptor protein levels and binding affinities suggest distinct pathogenic mechanisms of neurodegeneration in the AD brain.
Keywords: Alzheimer’s disease, amyloid-β, neurodegeneration, receptor binding, TNF-α, TNFRI, TNFRII
INTRODUCTION
Produced mainly by cells of the immune system, tumor necrosis factor-α (TNF-α) operates in various parts of the body as a key cytokine in inflammation and immune processes [1]. In the peripheral immune system, activated macrophages and monocytes release TNF-α. In the brain, TNF-α is expressed by neurons and glia and promotes inflammatory responses by recruiting microglia or astrocytes to lesion sites, leading to glial cell activation. After its release, TNF-α binds to specific membrane glycoprotein receptors (TNFR I and II) to elicit biological effects by mechanisms not fully understood. The two receptor subtypes, TNFRI and TNFRII, differ considerably in amino acid sequence, with just 24% homology in the extracellular region and < 10% homology in the intracellular domain. The receptors also differ functionally. TNFRI, but not TNFRII, contains an intracellular “death domain” that activates NF-κB signaling pathways leading to apoptosis [2,3]. On the other hand, our previous gene knockout studies have shown that TNFRII plays a trophic or protective role in neuronal survival [4].
Once released by activated microglia or astrocytes, TNF-α can be trophic or toxic, depending on the stage of development, the target cell, and the receptor subtype. For example, TNF-α protects fetal and postnatal neurons after glucose deprivation [5], and it also protects adult cortical neurons after traumatic brain injury [6]. TNF-α has been shown to promote reparative remyelination in an experimental model of demyelination [7,8]. However, as a proinflammatory cytokine, TNF-α has been implicated in the neuronal damage caused by a variety of brain insults [9–15]. Additionally, the same doses of TNF-α can be protective to neurons from middle-age or embryonic rats, but toxic to old rat neurons [16]. Indeed, TNF-α can become destructive, and even toxic, during aging, injury, and in certain types of chronic neurodegenerative disease states such as Alzheimer’s disease (AD).
Using virally-infected primary neurons that overexpress TNFRI or neurons from TNFRI knockout mice, we have recently demonstrated that amyloid-β (Aβ) protein, a major component of plaques in the AD brain, induces neuronal apoptosis through TNFRI [17]. Our in vitro studies show that TNF-α has little effect on hippocampal neurons from TNFRI−/− knockout mice, whereas neurons from TNFRII−/− mice are typically vulnerable to TNF-α even at low doses [2]. Recently, we have reported that deletion of the TNFRI gene in APP23 transgenic mice (APP23/TNFRI−/−) inhibits Aβ generation, diminishes Aβ plaque formation, and prevents learning and memory deficits [18]. Together, these results suggest that TNFRI and TNFRII may play distinct roles in neurodegeneration as well as in Aβ plaque formation.
In the present study, we tested a hypothesis that distinct TNF receptor subtypes are expressed differently in AD brains, which is responsible for differential functional ligand binding of TNF receptor subtypes. We therefore studied the quantitative TNF receptor subtype protein expression, the receptor mRNA distribution, and the radioligand functional binding affinity of the TNF receptors in the cortices from AD and non-demented (ND) individuals postmortem.
PATIENTS AND METHODS
Brain samples for research
Human tissue used in this study was collected with informed consent of subjects or next of kin and with ethical approval from the institutional review boards of Sun Health Research Institute. All procedures involving experiments on human subjects are done in accord with the ethical standards of the Committee on Human Experimentation of Sun Health Research Institute. Frontal cortex samples from clinically diagnosed, neuropathologically confirmed AD and ND patients were frozen at autopsy and stored in vacuum-sealed plastic bags at −80°C until assayed. A board-certified neuropathologist using Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) [19] criteria performed neuropathology evaluations. Postmortem intervals averaged less than 3 hours. We randomly selected 24 cases, consisting of 12 AD cases and 12 aged-matched ND control cases. All of the cases we used died from lung or kidney failure, and they did not have any other central nervous system diseases, sudden death or infectious diseases (based on clinical records and pathological examination). However, it is possible that some particular infectious diseases might affect TNF receptor expression, and many respiratory and renal failures result from inflammatory, immunological, and collagen diseases occurring in lung and kidney. Therefore, we have already excluded patients with immunological disorders or severe infectious diseases. In AD group, 8 cases died from chronic renal failure due to hypertensive nephrosclerosis or urolithiasis, and 4 cases died from respiratory failure due to pulmonary embolism. In the ND group, 10 cases died from chronic renal failures due to hypertensive nephrosclerosis, renal vein thrombosis, or renal artery stenosis, and 2 cases died from respiratory failure due to pneumothorax. These two groups were well-matched regarding gender, age, postmortem interval, and sample preparations (Table 1). Analyses of TNFR mRNA and protein levels were carried out using the same tissue samples from cortical regions of AD patients and aged-matched ND controls.
Table 1.
Demographic data and 125 I-TNF-α binding parameter (Kd) values
| Group | Sample Size(n) | Gender (M/F) | Age (y) | History (y) | PMI (hrs) | TNFRI, Kd (nM) | TNFRII, Kd (nM) |
|---|---|---|---|---|---|---|---|
| AD | 12 | 6/6 | 82.57 ± 1.18 | 4.87 ± 2.18 | 2.15 ± 0.44 | 0.96* ± 0.21 | 12.85* ± 3.24 |
| ND | 12 | 6/6 | 84.41 ± 2.48 | 2.77 ± 0.64 | 1.67 ± 0.36 | 3.56 ± 0.97 |
Data are expressed as mean ± S.E.M. Each Kd is derived from 3–5 independent experiments.
p < 0.01, compared with ND. PMI: postmortem intervals.
Western blot analysis
For neuron specific enolase (NSE; Abcam, Cambridge, MA, 1:1000) and synaptophysin (Santa Cruz, CA, 1:1000) Western blots, AD and ND brain samples were individually homogenized in 5 volumes of homogenizing buffer containing 10 mM Tris-HCl, pH 7.4, 25 mM NaCl, 50 mM EDTA, 1 mM EGTA plus 0.5% Triton X-100, 10% SDS, and a protease inhibitor cocktail (Boehringer Mannheim, Indianapolis, IN). For TNFRI and TNFRII (R&D System, Minneapolis, MN, 1:1000) Western blots, tissue membrane samples were used. The brain samples were homogenized in 5 volumes of buffer containing 50 mM phosphate, pH 7.7, and a protease inhibitor cocktail. The tissue membrane samples (25000 × g, 30 min) were collected and dissolved in the homogenizing buffer. β-actin (Sigma, St. Louis, MO, 1:5000) was examined as a loading control.
ELISA assays of TNFRI and TNFRII
For TNFRI and TNFRII ELISAs, recombinant purified TNFRI standard, recombinant purified TNFRII standard, or brain cortex homogenates were added to polyclonal anti-TNFRI or anti-TNFRII (R&D System, Minneapolis, MN) antibody-coated plates and were incubated at 25°C for 2 hours to permit TNFRI or TNFRII be captured. To bind captured TNFRI or TNFRII, 100 μl of 0.3 μg/ml monoclonal anti-TNFRI antibody or anti-TNFRII antibody (R&D System, Minneapolis, MN) was added. Biotinylated anti-mouse IgG and avidin-conjugated HRP and OPD (o-phenylenediamine dihydrochloride) were employed to detect binding. The specificity of the ELISA protocols was verified by serial dilutions of purified TNFRI and TNFRII standards. Pilot studies with the TNFRI and TNFRII ELISA demonstrated that it could detect as little as 3–5 pg TNFRI or TNFRII/ml (data not shown). The unit for ELISA was pg/mg protein, meaning that every milligram total protein from wet brain tissue contains amount of TNFRI or TNFRII protein.
In situ hybridization
Riboprobes for both TNFRI and TNFRII were generated from full-length cDNAs of TNFRI and TNFRII in pBluescript II SK+ linearized with EcoRI and transcribed with T7 RNA polymerase. Briefly, cRNA probes were labeled with the random primer kit (Amersham Biosciences, Arlington Heights, IL) using [α-35S]dATP according to the manufacturer’s instructions. The labeling was carried out using 1 μg of linearized cRNA, incubated for two hours to a specific activity of approximately 1 × 109 cpm/μg. The probes were purified on Nuc Trap Column (Stratagene, La Jolla, CA) prior to use. Coronal sections (12 μM) of brain tissues were cut in a cryostat at −20°C. The sections were thawed onto slides precoated with poly-l-lysine (40 mg/ml) and were stored at −80°C until the slides were processed for in situ hybridization histochemistry. In each experiment, every three sections of human brain tissue were hybridized with either the antisense and sense TNFRI cRNA probes or the anti-sense and sense TNFRII cRNA probes. In brief, tissue sections were fixed in phosphate buffer saline (PBS) containing 5% paraformaldehyde for 30 min at room temperature and rinsed twice in PBS. The slides then were treated with 0.25% acetic anhydrite and 0.1 M triethanolamine for 10 min at room temperature. After dehydration in a graded ethanol solution (50%, 75%, 95%, and 100%), the slides were air-dried. Tissue sections were hybridized overnight (16 hours) at 42 °C in a mixture containing 50% deionized formamide, 10% dextran sulfate, 2X SSC, 300 μg/ml of yeast tRNA, 13 Denhardt’s solution 10% dextran sulfate, 0.5 mg/ml sheared salmon sperm DNA, 0.02 M Na3PO4, 0.05 M dithiothreitol, and 35S-labeled 107 cpm/ml probes of sense or antisense cRNA of TNFRI or TNFRII. After performing a final stringency wash at 63°C in 0.1X SSC and allowing the sections to air-dry, the sections were dipped in Kodak NTB-3 photo emulsion (diluted 1:1 in water), exposed for 6 weeks at 4°C, developed, and fixed. The tissues were then counterstained with crystal violet, and the cover slip was applied with Protex mounting medium.
Immunohistochemistry of TNF receptors in AD and ND brains
The superior frontal cortex tissues from AD and ND patients were fixed with 4% paraformaldehyde and sectioned to 30 μm. The sections were pre-treated with 0.3% triton x-100 and blocked with 10% donkey serum for 30 min. Primary antibodies were applied with goat anti-TNFRII (Santa Cruz Biotechnology, 1:200), rabbit anti-TNFRI (Abcam, 1:500), mouse anti-NeuN (Millipore, 1:100), and rabbit anti-GFAP (DAKO, 1:4000) and mouse anti-GFAP (Covance, 1:5000). The incubation was performed overnight at 4°C. Secondary antibodies of donkey against goat or mouse or rabbit with fluorescence 488 or 568 (Invitrogen, 1:1000) were incubated for 30 min. The images were taken with FluoView FV1000 confocal microscope (Olympus).
TNF receptor binding in brain tissue
Samples of the frontal cortex and hippocampus were dissected just prior to being assayed. The cortices were homogenized with a glass homogenizer in 50 volumes of ice-cold Tris buffer saline (TBS, 50 mM, pH 7.7) containing phenylmethyl-sulfonyl fluoride (PMSF, 0.1 mM), EDTA (1 mM) and NaCl (l00 mM). The homogenates were centrifuged (14,000 × g) for 30 min and resuspended in potassium phosphate buffer (50 mM), pH 7.2, bovine serum albumin (0.2%) and EDTA (10 mM). The tissue membrane suspensions were incubated with 5 concentrations of 125I-TNF-α (10–10,000 pM, Amersham Biosciences, Arlington Heights, IL) for 120 min at 4°C in the absence or presence of 100 μM cold TNF-α (R&D System, Minneapolis, MN) to measure total and non-specific binding. Specific binding (the difference between binding of 125I-TNF-α in the absence and presence of cold TNF-α) was usually >80% of the total 125I-TNF-α binding activity. At the end of the incubation, ice-cold TBS buffer was added to the tubes. After centrifugation, 3 ml of ice-cold buffer was added to the tubes, followed by filtration through a Brandell (Gaithersburg, MD) cell harvester, using GF/C glass fiber filters previously soaked in 0.05% polyethylenimine. The radioactivity on the filters was determined in a liquid scintillation counter with a counting efficiency of approximately 40% (Wallac, Gaithersburg, MD). The data preprocessing was performed using the Prism program, and the actual nonlinear curve-fitting was performed by LIGANDS software.
RESULTS
Different protein levels of TNF receptor subtypes in the AD brain
Since TNF receptor is required for amyloid protein induced neuron death [18], it is essential to understand whether protein levels of TNFR are changed in the AD brain. Densitometric analysis of TNFRI, TNFRII, NSE, and synaptophysin immunoreactive bands in Western blots from identical brain regions of the same patients showed a 24% reduction in synaptophysin and a 17% increase in TNFRI in AD frontal tissue compared to that found in ND (Fig. 1A and B) tissue. Interestingly, TNFRII has a significant decrease (35%) in AD brains compared to that in ND brains (Fig. 1A and B). We also note that the decreased level (43%) of TNFRII from ELISA data is lower than that from the Western blot study (35%) and the increased level (28%) of TNFRI from ELISA data is higher than that from Western blot study (17%), suggesting our TNFR ELISAs are more sensitive than regular Western analysis (Fig. 1C). Meanwhile, we have also used NSE, a neuronal cell body marker, to evaluate neurodegeneration in AD brains (Fig. 1A and B). Our results demonstrate that NSE has little change between AD and ND brains. These results suggest that TNFRI overexpression and TNFRII deficiency may cause neuronal vulnerability because high levels of “death-domain” containing neurons in the AD brain. Additionally, the TNFRII deficiency may not be entirely due to neuronal loss. The decrease of TNFRII levels may be due to the death of neurons that normally express the TNFRII protein, or to decreased expression of the protein in affected regions of the AD brain.
Fig. 1.

Different levels of TNFRI and TNFRII protein in AD brains. The frontal cortex region in AD and ND individuals were homogenated, and TNFRI and TNFRII protein levels were measured specifically by Western blot (A and B) and ELISA (C). Western blot studies on TNFRI and II, synaptophysin, and neuron specific enolase (NSE) in AD and ND brains are shown in (A). (B) Data represent the mean (± SD) quantification analysis using densitometry imaging (FluorChem 8900) showing that TNFRI immunoreactivity was increased while TNFRII was decreased in AD brains compared to ND, whereas synaptophysin was decreased about 24%. There is no significant change in NSE protein levels. β-actin was used as a loading control. (C) ELISA assay showed a 43% reduction in TNFRII and a 28% increase in TNFRI in AD frontal tissue compared to that found in ND. Data in bar graphs represent mean±SEM. All results were repeated at least three times from independent experiments. *p < 0.05; **p < 0.01.
Expression of TNFRI and TNFRII in the human brain at the mRNA and protein levels
To map the cellular distribution of TNFRI and TNFRII in the brain, sense and antisense 35S-labeled cRNA probes of these two genes were hybridized to coronal sections of AD and ND frontal cortex. Sense strand control probes showed little or no hybridization to frontal cortex sections from three different AD specimens (representative pictures are demonstrated in Fig. 2B, D, F, and H). In adjacent sections of the frontal cortex, specific antisense TNFRI hybridization was localized to the interneurons and pyramidal neurons in frontal cortex sections from all of the AD individuals and the enlargement of this region highlights the prominent laminar labeling consistent with localization to pyramidal and interneurons within this cortex area (Fig. 2A). These results suggest that interneurons and pyramidal neurons are the cell types that predominantly but not exclusively express TNFRI mRNA and there is no significant difference between AD and ND brains in our experimental condition (Fig. 2A and E). To confirm that neurons that synthesize TNFRI would be the same neuronal population expressing TNFRII, we used a cRNA probe for TNFRII to hybridize the same brain sections. Interestingly, we found that both TNF receptor transcripts were expressed in a similar pattern in the cortex (Fig. 2). Specifically, TNFRII transcripts were also detected in both pyramidal and interneurons of the cortex (Fig. 2C and G). However, not all cells were labeled; numerous small cells with darkly stained nuclei (possibly were microglia) were unlabeled in the area where there was specific labeling of adjacent small cells (non-pyramidal neurons and/or astrocytes). Similar to TNFRI, we did not observe any significant difference in TNFRII between AD and ND (Fig. 2C and G). Immunohistochemistry of TNFRI (Fig. 3A) and TNFRII (Fig. 3B) with neuron marker NeuN in neurons of the AD and ND superior cortex region further supported the in situ hybridization results that neurons do express TNFRI and TNFRII. Specifically, we found that TNFRI and TNFRII were immunostained on the cell membrane around neurons (verified by NeuN staining). Increased expression of TNFRI and the decreased expression of TNFRII were showed in AD patients compared to ND controls (Fig. 3 A and B). Astrocytes have little TNFRII expression with immunostaining even in the over-activated astrocytes of AD patients.
Fig. 2.

Neuronal expression of TNFRI and TNFRII mRNA in human brains using in situ hybridization. AD frontal cortex sections were hybridized to 35S-labeled sense and antisense cRNA probes for TNFRI and TNFRII. Resultant autoradiographs were used directly as negatives for photographic enlargement of frontal cortex. All of pictures in the left panel (A,C,E,G) are hybridization by antisense cRNA probes whereas the pictures in the right panel (B,D,F,H) are sense hybridization as controls. The frontal cortex sections from a 79-year-old male with AD hybridized with antisense cRNA probes for TNFRI (A) and TNFRII (C). Note the specific hybridization over the cortical strip of the frontal cortex. Furthermore, (A) and (C) also demonstrate that specific hybridization was noted in pyramidal neurons and interneurons in this area. Similarly, the frontal cortex sections from a non-demented aged matched control brain (82-year-old male) hybridized with antisense RNA probes for TNFRI (E) and TNFRII (G). We found similar neuronal distribution of both TNFRI and II transcripts, hybridized specifically in pyramidal neurons and interneurons in this brain region.
Fig. 3.
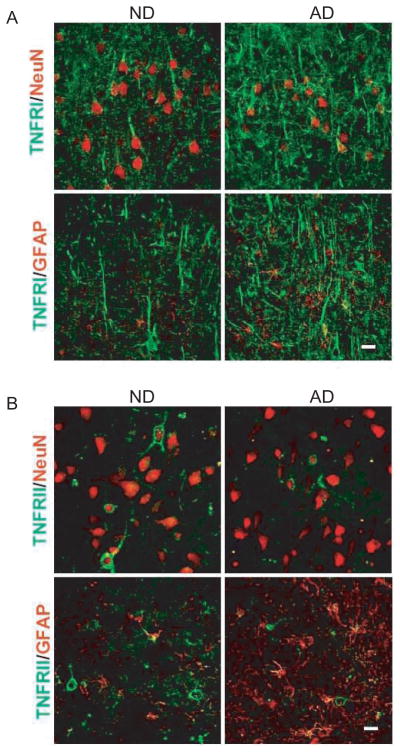
TNFRI and TNFRII protein expressions in AD and ND brains using immunohistochemistry. (A) TNFRI protein expression is increased in AD cortical tissues and much higher expression of TNFRI was observed on the dendrites. Compared to ND brains, there is an increased TNFR1 level in AD cortical tissues along with a decreased number of neurons (NeuN staining). There is little TNFR1 expression in activated astrocytes in ND and AD brains, even in the over-activated astrocytes in AD patients. Bar stands for 20 μm. (B) The TNFRII protein expression is decreased in AD cortical tissues compared to ND controls. A portion of neurons in the cortex are showed with strong TNFRII-positive immunostaining on the cell membrane around neuronal nuclei (verified by NeuN staining). Astrocytes have little TNFRII expression with immunostaining in both ND and AD brains. Bar stands for 20 μm.
Distinct TNF-α radioligand binding affinities to TNF receptor subtypes in AD Brains
To examine whether the binding activities of 125I-TNFα to these two receptors are altered in the AD brain, we compared radioligand binding in AD versus ND brain tissue membrane preparations. 125I-TNF-α binding is saturable with the first class of binding sites (average Kd = 1.67 nM) and the second class of binding sites (average Kd = 3.56 nM) in ND brains (Table 1). This suggests two classes of binding sites (high and low affinities) in the brain tissue, which may correspond to TNFRI and TNFRII, respectively. In AD brains, however, the binding affinity for the first class of binding sites is higher (Kd = 0.96 nM) whereas the second class binding affinity in AD brain tissue is significantly lower (Kd = 12.85 nM) compared to ND (Table 1). We also evaluated inhibition of 125I-TNF-α binding using unlabeled TNF-α in tissue from ND and AD brains. TNF-α is uniquely potent with IC50 values from 0.53 nM to 12.16 nM in ND brains for the two binding sites, respectively (Fig. 4). In AD brains, TNF-α displays an IC50 for the first class of binding sites of 0.36 nM (about 30% higher potency than in ND brains) and for the second class of binding sites of 21.43 nM (around 45% lower potency than in ND brains) as illustrated in Fig. 4. This is consistent with the binding results of higher affinity in the first class of binding sites and lower in the second class of binding sites in AD brains.
Fig. 4.

Scatchard analysis of 125I-TNF-α binding in human brain tissue from AD and ND patients (A) and representative competition curve for TNF-α inhibition of 100 pM 125I-TNF-α binding in human brain tissue from AD and ND patients (B). Incubation conditions were described in Methods. Computer analysis of the binding data revealed that competition curve is best described by two-components binding model, with different sets of IC50 values.
DISCUSSION
The results presented in this paper are the first to demonstrate that TNFRI and TNFRII are expressed and activated differentially in AD and ND brains. AD brains exhibit elevated TNFRI protein levels and reduced TNFRII levels compared to ND brains; however, the mRNA distribution of these two receptors does not differ significantly in these two groups. Moreover, TNFRI exhibits a higher affinity and TNFRII presents a lower affinity for 125I-TNF-α binding in AD brains.
Most chronic neurodegenerative diseases, including AD [21], are accompanied by a cytokine-mediated inflammatory response termed neuroinflammation. Though most cytokines, including TNF-α, are expressed at very low levels in the healthy brain [22], disease-related neuroinflammation can be detected years before neurons die in significant numbers. In AD patients, activated microglia cells, especially those associated with amyloid deposits, secrete high levels of TNF-α [23–25]. Elevated TNF levels also appear to correlate with disease progression, as high serum levels of TNF are present in patients with severe AD compared to individuals with mild-to-moderate disease [26]. The initial event in the action of TNF-α is its binding to specific receptors on cell surfaces. It has been recognized and established that TNF-α binds to responsive cells with both high and low affinities, suggesting two distinct classes of TNF receptors, i.e., TNFRI and TNFRII [2,27,28]. Our previous radiolabeled Aβ binding assay has shown that Aβ1–40 binds with high affinity and specificity to TNFRI [17]. Here, using radiolabeled TNF-α, we found that in the AD brain, which features plaques containing Aβ, TNFRI binding affinity is increased while TNFRII binding affinity is significantly decreased. One possible explanation is that Aβ might enhance TNF-α binding affinity to TNFRI but decrease binding affinity to TNFRII; therefore, these two types of TNF receptors are in fact activated differentially in the AD brain.
Depending on cellular context, TNFRI and TNFRII can activate many of the same downstream pathways and can either act synergistically or in opposing fashions. TNFR1 contains a cytoplasmic death domain that binds to adaptor TRADD (TNFR-associated death domain), which activates NF-κB or caspase cascades [29]. Although TNFRII lacks the death domain sequence, it recruits adaptor protein, TRAF2, allowing subsequent activation of NF-κB as well [30]. In general, TNFRI activates pro-apoptotic pathways [31–33], whereas TNFRII is associated with cell survival [2,3,34, 35]. Patients with AD-type dementia were reported to bear significantly enhanced numbers of both receptors on T lymphocytes [36]. TNFRI and TNFRII are mainly expressed in neurons although glial cells have also shown to express TNFRI and TNFRII [37,38]. In a study of postmortem AD and control brain tissue, the proportion of cortical neurons immunopositive for TNFRI, TRADD, activated caspase-3, or TNF-α was elevated 3- to 10-fold in AD over that in age-matched control brains, but the number of neurons that could be labeled for TNFRII was decreased, suggesting activation of the TNF death receptor pathway in AD [39]. However, in a more recent study, no variations in TNFRI and RII mRNA and protein expression were found to be associated with AD [40]. Our present study, using rapidly autopsied AD brain tissues, demonstrates that the two TNFR receptors, TNFRI and TNFRII, are mainly expressed in neurons from cortical regions (Fig. 3), suggesting that results from radioligand binding and biochemistry represent neuronal findings. While we observed that TNFRI expression is increased, expression of TNFRII is decreased in AD brains compared to ND brains. Furthermore, to support the results from immunohistochemistry, we found that the ratio of TNFRI/TMFRII is changed in AD patients such that TNFRI protein levels are upregulated, leading to increased neuron death, and TNFRII protein levels are downregulated, leading to decreased neuron survival. No significant differences in these two receptor mRNA expressions were observed between AD and ND, suggesting that alteration of TNF receptors in the AD brain might occur at the translational level.
Our results also demonstrate that synaptophysin was only reduced 24%, while TNFRI was increased 17–28% and TNFRII was significantly decreased 35–43% in AD brains. As our results demonstrate, the measurement of TNFRI and TNFRII by ELISA is much more sensitive than that by Western blot analysis. Surprisingly, we did not see a significant change in NSE protein levels in the AD brain compared to the ND brain, nor did we observe any significant changes in microtublin associated protein (MAP-2) or β III tublin (data not shown). One possibility is that these neuronal cell body markers are not sensitive enough to detect neuron loss in the AD brain compared to synaptophysin detection. The other possibility is that synaptic loss may be earlier than neuron loss in the AD brain. Although 24% reduction of synaptophysin and 35–43% decrease of TNFRII fall into the same range, TNFRII decrease may reflect the function of TNFRII is deficiency. This deficiency may not be parallel to the same degree of synaptophysin deficiency. Similar pathological findings we have observed in another immunological protein, CD59, in AD brains [41] and TNF receptors in vitro [2] and in vivo with Aβ interaction [17,18]. Taken together, these data further suggest that TNFRII deficiency may not, at least entirely, be due to synaptic or neuron cell body loss in the AD brain.
Overall, these results are the first to demonstrate that two types of TNF receptors in the AD brain are expressed and activated differentially compared to ND. TNFRI protein levels are increased, but TNFRII levels are decreased in the AD brain. TNFRI binding affinity is increased, while TNFRII binding affinity is significantly decreased. Thus, the ratio of the two TNF receptor subtypes might be involved in different signal transduction pathways and regulates neuronal death or survival. Our data give further support for the important roles of TNF-α and its receptors in the pathogenesis of AD. The results from these studies may lead to the exploration of TNF receptors as alternative therapeutic targets in the treatment of AD, providing the basis for developing agonist and antagonist systems for TNF receptor subtypes and also encouraging better strategies for treatments of AD as well as other brain disorders related to neuroinflammation.
Acknowledgments
We thank Mr. Spencer Guest, Mr. Mike Gurule, and Ms. Ronda Lee for technical assistance and Ms. Adrienne Bowers for editorial assistance during the manuscript preparation. This work was supported by grants from National Institute on Aging (NIH R01 AG025888), the Alzheimer’s Disease Association, and the Arizona Alzheimer’s Research Consortium.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=132).
References
- 1.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen Y, Li R, Shiosaki K. Inhibition of p75 tumor necrosis factor receptor by antisense oligonucleotides increases hypoxic injury and beta-amyloid toxicity in human neuronal cell line. J Biol Chem. 1997;272:3550–3553. [PubMed] [Google Scholar]
- 5.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 6.Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligo-dendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 8.Plant SR, Arnett HA, Ting JP. Astroglial-derived lymphotoxin-alpha exacerbates inflammation and demyelination, but not remyelination. Glia. 2005;49:1–14. doi: 10.1002/glia.20089. [DOI] [PubMed] [Google Scholar]
- 9.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 10.Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, LaFerla FM, Callahan LM, Federoff HJ, Bowers WJ. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol. 2008;173:1768–1782. doi: 10.2353/ajpath.2008.080528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 12.New DR, Maggirwar SB, Epstein LG, Dewhurst S, Gelbard HA. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem. 1998;273:17852–17858. doi: 10.1074/jbc.273.28.17852. [DOI] [PubMed] [Google Scholar]
- 13.Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. FASEB J. 2006;20:670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- 14.Stahel PF, Kariya K, Shohami E, Barnum SR, Eugster H, Trentz O, Kossmann T, Morganti-Kossmann MC. Intracerebral complement C5a receptor (CD88) expression is regulated by TNF and lymphotoxin-alpha following closed head injury in mice. J Neuroimmunol. 2000;109:164–172. doi: 10.1016/s0165-5728(00)00304-0. [DOI] [PubMed] [Google Scholar]
- 15.Villarroya H, Marie Y, Ouallet JC, Le Saux F, Tchelingerian JL, Baumann N. Expression of TNF alpha in central neurons of Lewis rat spinal cord after EAE induction. J Neurosci Res. 1997;49:592–599. doi: 10.1002/(SICI)1097-4547(19970901)49:5<592::AID-JNR9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Viel JJ, McManus DQ, Smith SS, Brewer GJ. Age-and concentration-dependent neuroprotection and toxicity by TNF in cortical neurons from beta-amyloid. J Neurosci Res. 2001;64:454–465. doi: 10.1002/jnr.1097. [DOI] [PubMed] [Google Scholar]
- 17.Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, Zhang D, Shen Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J Neurosci. 2004;24:1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 20.Konishi Y, Lindholm K, Yang LB, Li R, Shen Y. Isolation of living neurons from human elderly brains using the immunomagnetic sorting DNA-linker system. Am J Pathol. 2002;161:1567–1576. doi: 10.1016/S0002-9440(10)64435-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGeer PL, McGeer EG. Local neuroinflammation and the progression of Alzheimer’s disease. J Neurovirol. 2002;8:529–538. doi: 10.1080/13550280290100969. [DOI] [PubMed] [Google Scholar]
- 22.Vitkovic L, Bockaert J, Jacque C. “Inflammatory” cytokines: neuromodulators in normal brain? J Neurochem. 2000;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- 23.Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia. 1993;7:75–83. doi: 10.1002/glia.440070113. [DOI] [PubMed] [Google Scholar]
- 24.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Jr, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia. 2001;35:72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 26.Paganelli R, Di Iorio A, Patricelli L, Ripani F, Sparvieri E, Faricelli R, Iarlori C, Porreca E, Di Gioacchino M, Abate G. Proinflammatory cytokines in sera of elderly patients with dementia: levels in vascular injury are higher than those of mild-moderate Alzheimer’s disease patients. Exp Gerontol. 2002;37:257–263. doi: 10.1016/s0531-5565(01)00191-7. [DOI] [PubMed] [Google Scholar]
- 27.Hohmann HP, Brockhaus M, Baeuerle PA, Remy R, Kolbeck R, van Loon AP. Expression of the types A and B tumor necrosis factor (TNF) receptors is independently regulated, and both receptors mediate activation of the transcription factor NF-kappa B. TNF alpha is not needed for induction of a biological effect via TNF receptors. J Biol Chem. 1990;265:22409–22417. [PubMed] [Google Scholar]
- 28.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–497. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 30.Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 31.Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52:447–456. doi: 10.1016/j.neuint.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Q, Wu J, Rowan MJ, Anwyl R. Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci. 2005;22:2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x. [DOI] [PubMed] [Google Scholar]
- 33.Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- 35.Williams MA, Turchan J, Lu Y, Nath A, Drachman DB. Protection of human cerebral neurons from neurodegenerative insults by gene delivery of soluble tumor necrosis factor p75 receptor. Exp Brain Res. 2005;165:383–391. doi: 10.1007/s00221-005-2307-9. [DOI] [PubMed] [Google Scholar]
- 36.Bongioanni P, Romano MR, Sposito R, Castagna M, Boccardi B, Borgna M. T-cell tumour necrosis factor-alpha receptor binding in demented patients. J Neurol. 1997;244:418–425. doi: 10.1007/s004150050115. [DOI] [PubMed] [Google Scholar]
- 37.Kinouchi K, Brown G, Pasternak G, Donner DB. Identification and characterization of receptors for tumor necrosis factor-alpha in the brain. Biochem Biophys Res Commun. 1991;181:1532–1538. doi: 10.1016/0006-291x(91)92113-x. [DOI] [PubMed] [Google Scholar]
- 38.Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasserman AJ, Cotman CW. The induction of the TN-Falpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem Res. 2003;28:307–318. doi: 10.1023/a:1022337519035. [DOI] [PubMed] [Google Scholar]
- 40.Culpan D, Cornish A, Love S, Kehoe PG, Wilcock GK. Protein and gene expression of tumour necrosis factor receptors I and II and their promoter gene polymorphisms in Alzheimer’s disease. Exp Gerontol. 2007;42:538–544. doi: 10.1016/j.exger.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Yang LB, Li R, Meri S, Rogers J, Shen Y. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. J Neurosci. 2000;20:7505–7509. doi: 10.1523/JNEUROSCI.20-20-07505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]