

Abstract
A highly selective method for the synthesis of asymmetrically substituted carbocycles and heterocycles from unactivated aldehyde–olefin precursors has been achieved via enantioselective SOMO-catalysis. Addition of a catalytically generated enamine radical cation across a pendent olefin serves to establish a general asymmetric strategy towards the production of a wide range of formyl-substituted rings with alkene transposition. Conceptually, this novel mechanism allows direct access to “homo-ene” type products.
Carbocyclic and heterocyclic ring systems bearing asymmetric substitution patterns are widely distributed among medicinal agents and bioactive natural products.1 A preeminent goal of organic synthesis is the development of technologies to enable the rapid and enantioselective construction of these high–value cyclic substructures from simple starting materials.2 Along these lines, the powerful carbonyl-ene cyclization delivers stereochemically complex small ring systems from achiral aldehyde–olefin precursors in a routine and predictable fashion through a mechanism that does not require pre-functionalization of the olefin component.3 This fundamental transformation has been widely studied and a number of catalytic enantioselective carbonyl-ene protocols have been developed.4,5 Expanding upon this general concept, we sought to invent a one-carbon extended, “homo-ene” variant, wherein unactivated substrates undergo asymmetric α-carbonyl cyclization through a SOMO-activation mechanism, to stereoselectively generate cyclic adducts bearing synthetically useful aldehyde and olefin functional handles. We describe herein the development of the first asymmetric “homo-ene” cyclization, a transformation we anticipate will be of significant value to the chemical synthesis community.
Design Plan
In 2007, our laboratory introduced a mode of asymmetric activation termed SOMO (singly occupied molecular orbital) organocatalysis.6 Subsequent to our initial discovery, we have established SOMO organocatalysis as a remarkably robust and versatile activation platform, capable of facilitating a range of previously elusive transformations, including direct enantioselective α-allylic alkylation,7 α-enolation,8 α-vinylation,9 α-chlorination,10 and α-arylation,11 as well as polycyclization12 and cycloaddition13 to generate cyclohexyl rings and pyrrolidines. Recently, we questioned whether the SOMO platform might be leveraged for the development of an asymmetric α-carbonyl “homo-ene” cyclization of unactivated aldehyde-olefin substrates. While the traditional carbonyl-ene cyclization proceeds through a LUMO-lowered 2π-electron pathway, the SOMO activation mode is distinguished by an electrophilic 3π-electron species (1-he). Based on precedent from our lab,6–13 we anticipated that this enamine radical cation would add stereoselectively from the unshielded Re-face to the pendent olefin, thus generating a transient alkyl radical. Operation of a radical-polar crossover mechanism14 would serve to oxidize the radical to the corresponding carbocation (2). Finally, deprotonation and hydrolysis would regenerate the amine catalyst and deliver the enantioenriched cyclized product. Central to the proposed strategy was our expectation that cyclization would proceed via a highly ordered chair-E transition state15 (1-he) to deliver the product with trans diastereoselectivity in a fashion that is highly analogous to the venerable ene 2π-electron pathway (1-ene). Notably, this study would require only simple olefins as the tethered SOMO-phile component, a substantial expansion of the scope and utility of this enantioselective oxidation pathway.16
Results
The proposed transformation was first evaluated in the context of the amine-tethered prenyl aldehyde substrate 3. As presented in Table 1, treatment of this aldehyde with base and Fe(III)trisphenanthroline in the presence of an imidazolidinone catalyst (5, 6, or 7) led to the stereoselective formation of piperidine 4 with high levels of trans diastereoselectivity and enantiocontrol. Optimal selectivities were obtained with the naphthyl-bearing amine catalyst 7, presumably due to enhanced facial shielding of the SOMO intermediate (1-he). Variation of the counterion (X−) in the Fe(III)trisphenanthroline salt revealed the soluble trisbistriflamide salt to be most effective (entry 6).
Table 1.
Effect of catalyst structure and counterion on cyclization
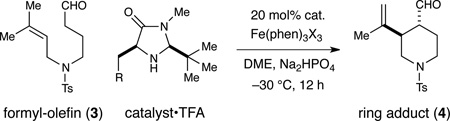 | ||||
|---|---|---|---|---|
| entry | catalyst (R) | X− | yield (%)a | ee (%)b |
| 1 | 5 (H) | SbF6− | 41% | 89% |
| 2 | 6 (Ph) | SbF6− | 41% | 97% |
| 3 | 7 (1-naphthyl) | SbF6− | 43% | 99% |
| 4 | 7 (1-naphthyl) | ClO4− | 12% | 99% |
| 5 | 7 (1-naphthyl) | PF6− | 73% | 99% |
| 6 | 7 (1-naphthyl) | Tf2N− | 95%c | 99% |
Determined by 1H NMR using internal standard. Diastereoselectivity >20:1 in all cases.
Determined by chiral SFC analysis of the corresponding alcohol, absolute configuration determined by chemical correlation.
Isolated in 93% yield.
Having identified optimal conditions for the organocatalytic “homo-ene” reaction, we next explored the scope of the reaction with respect to the tethering moiety. As shown in Table 2, the method was found to readily accommodate significant structural diversity in the linker, offering enantioselective access to a wide range of 5- and 6-membered carbocyclic and heterocyclic ring systems. Under our conditions, achiral substrates were cyclized to generate piperidine, pyrrolidine, tetrahydropyran, tetrahydrofuran, cyclopentane, and cyclohexane motifs with good to excellent diastereoselectivity (>20:1), enantioselectivity (85–99%) and yield (62–95%). Notably, the formation of tetrahydrofuran and pyrrolidine adducts was achieved without β-oxy or β-amino elimination from the corresponding aldehyde precursors (entries 2 and 3), demonstrating the mild conditions employed in generating the 3π-electron activated intermediate. The products depicted in Table 2 represent important structural motifs that are widely encountered throughout natural product synthesis and drug discovery.1
Table 2.
Scope of the enantioselective homo-ene ring products
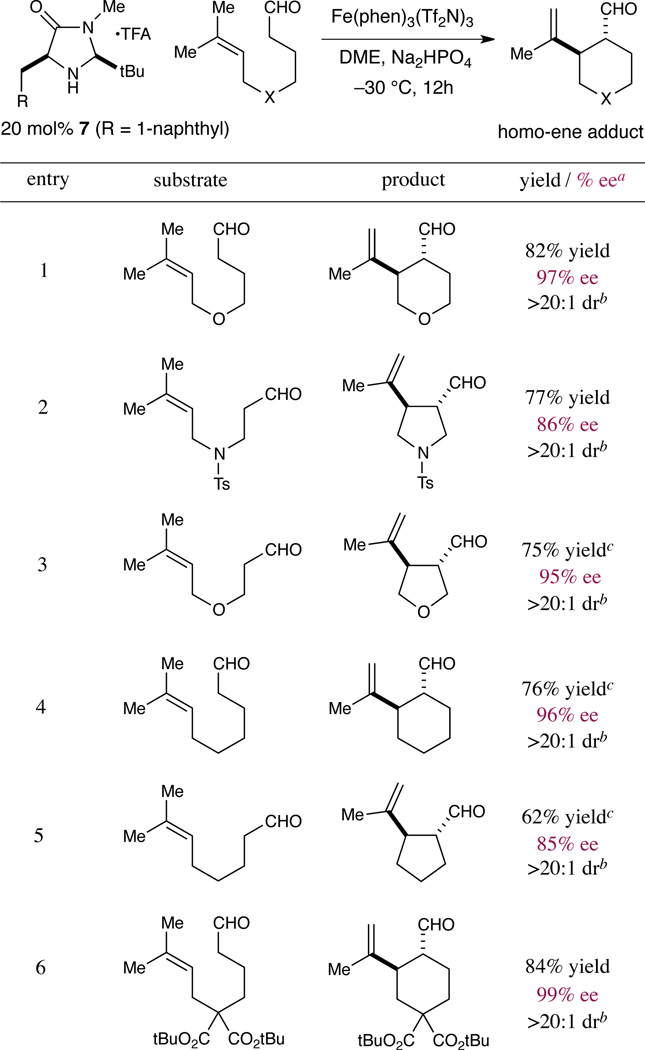 |
Determined by chiral HPLC analysis of the alcohol or aryl ester.
Determined by 1H-NMR analysis.
Determined by 1H-NMR analysis using an internal standard.
The reaction is also tolerant of a broad array of olefin systems as suitable π-nucleophiles for this homo-ene type cyclization. As highlighted in Table 3, alkylidene cycloalkanes of various ring sizes readily undergo enantioselective C–C bond formation to generate the corresponding bicyclic products in good yield and with excellent stereocontrol (entries 1–3). In addition, α-carbonyl cyclization using styrenyl olefins is readily accomplished (entry 4). Importantly, non-symmetrical 1,2,2-trisubstituted alkenes, e.g. bearing methyl and cyclohexyl groups, can generate the purported carbocation intermediate (2) before undergoing selective deprotonation to generate single olefin-transposition regioisomers (entry 6, >98:2 rr). We note that, at this stage, unfunctionalized 1,2-disubstituted olefins are not effective substrates for this transformation, presumably due to the higher oxidation potential for conversion of secondary radicals to secondary cations in comparison to their tertiary radical counterparts.17 Despite this current limitation in scope, cycloadducts bearing monosubstituted olefin substituents are nonetheless readily accessible from allyl silane precursors (entry 5).
Table 3.
Scope of π-nucleophile, selectivity of olefin transposition
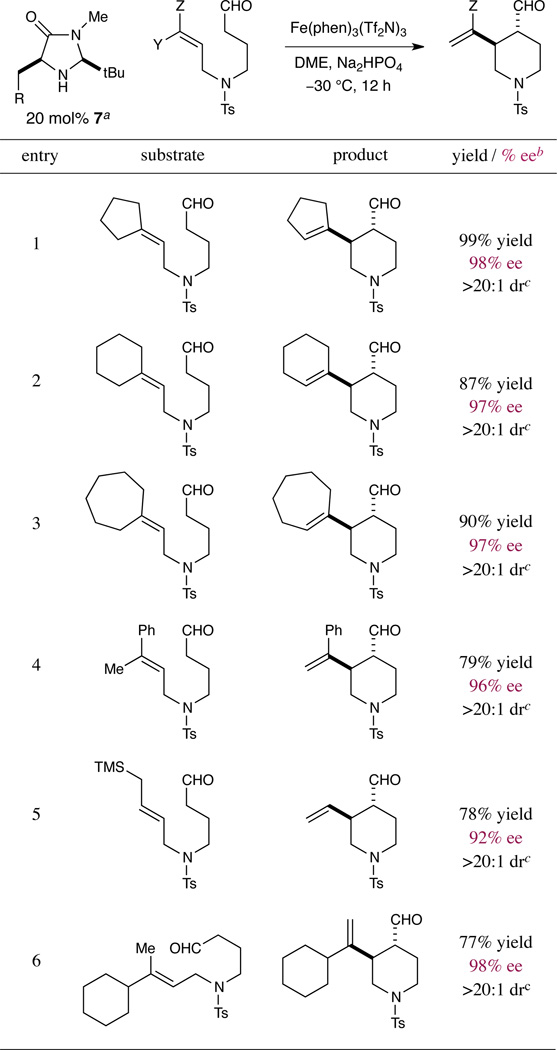 |
R = 1-naphthyl.
Determined by chiral HPLC analysis of the alcohol or aryl ester. Diastereoselectivities determined by 1H-NMR analysis.
Determined by 1H-NMR analysis using an internal standard.
We next sought to examine the ability of the amine catalyst to override the influence of stereochemical information already present on the intramolecular cyclization substrate. We recognized that if catalyst-controlled stereodifferentiation could be achieved, it should be possible to selectively generate a diverse array of highly complex cyclic systems bearing three or more contiguous stereocenters. As shown in Table 4, enantioenriched aldehyde substrates incorporating β-methyl substitution (99% ee) were subjected to our ring-closing conditions with either the S,S-7 or R,R-7 catalyst. Remarkably, catalyst-mediated cyclization in both enantiomeric series successfully delivered ring systems bearing three contiguous stereocenters, with excellent selectivity for the trans-stereochemical relationship across the newly forming bond. In each case, the amine catalyst strongly dictated the diastereochemical outcome, effectively overriding the influence of the substrate β-methyl stereocenter with either catalyst antipode. Presumably, the steric demand of the amine catalyst provides a major conformational lock, enforcing a chair-E transition state (see 8 and 9) and thereby matching or overriding the inherent substrate bias.
Table 4.
Production of rings with three contiguous stereocentersa
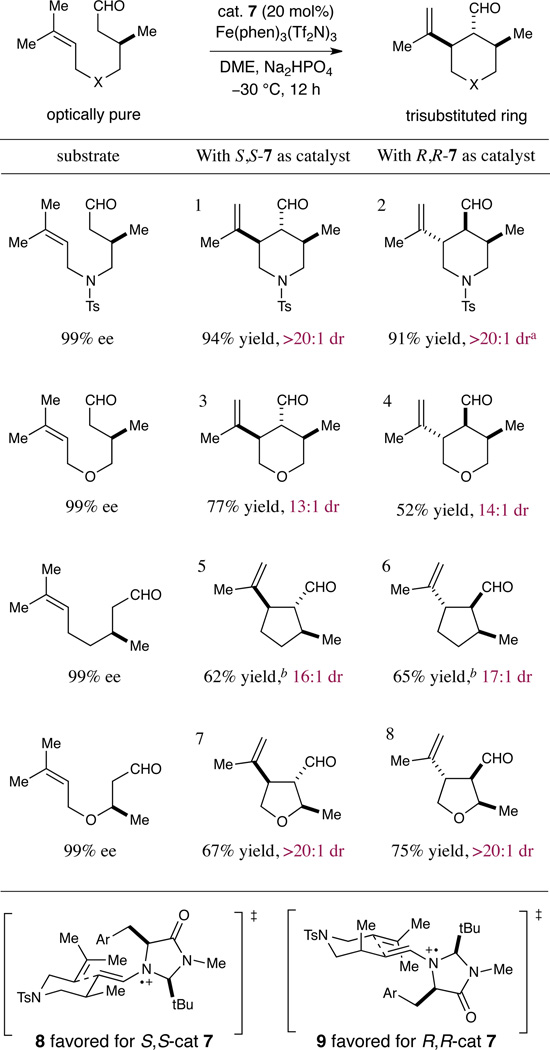 |
Diastereoselectivities determined by 1H-NMR analysis.
Determined by 1H-NMR analysis using an internal standard.
Finally, in an extension of the findings shown in Table 4, we have developed a simple two-step organocatalytic protocol by which to achieve the overall conversion of simple achiral aldehydes to stereochemically complex cyclic adducts. As shown in Scheme 1, substrates 10 and 11 were subjected to sequential enantioselective organocatalytic transfer hydrogenation (using catalyst 12) followed directly by asymmetric homo-ene cyclization (using either enantiomer of catalyst 7) to generate the observed products in good overall yield and with excellent selectivity. The transformations depicted in Scheme 1 serve to highlight the ability of the SOMO-mediated homo-ene technology to effect the rapid production of stereochemical complexity in cyclic architectures.
Scheme 1.

Sequential organocatalytic hydrogenation-cyclizationa
Conditions: Step 1) 2.5 equiv. 13, 20 mol% 12, CHCl3, −50 °C, 24 hours. Step 2) 2.5 equiv. Fe(phen)3(Tf2N)3, 2.5 equiv. Na2HPO4, 20 mol % 7, −30 °C, DME, 12 hours. aDiastereoselectivities determined by 1H-NMR analysis. Enantiomeric excess determined by chiral HPLC analysis of the corresponding alcohol or terephthalic acid monomethyl diester.
In summary, enantioselective SOMO-organocatalysis has been leveraged for the development of a potentially general approach toward the synthesis of stereochemically rich carbocycles and heterocycles from achiral precursors. This protocol bears analogy to the venerable carbonyl-ene cyclization, yet provides access to a differentiated array of complex cyclic scaffolds incorporating valuable aldehyde and olefin functional handles. We anticipate that this method will find broad application among practitioners of organic synthesis.
Supplementary Material
Figure 1.

Impetus and design elements of home-ene reaction
Acknowledgement
Financial support was provided by the NIHGMS (R01 GM103558-01) and kind gifts from Merck, Amgen and Abbvie.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Companyó X, Alba A-N, Rios R. In: Targets in Heterocyclic Systems. Attanasi OA, editor. Vol. 13. London: Royal Society of Chemistry; 2009. pp. 147–174. [Google Scholar]
- 2.a) Monfette S, Fogg D. Chem. Rev. 2009;109:3783. doi: 10.1021/cr800541y. [DOI] [PubMed] [Google Scholar]; b) Buffatt MGP. Tetrahedron. 2004;60:1701. [Google Scholar]; c) O’Hagan D. Nat. Prod. Rep. 2000;17:435. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]
- 3.a) Snider BB. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol 2. Oxford: Pergamon; 1991. pp. 527–561. [Google Scholar]; b) Oppolzer W. Angew. Chem. Int. Ed. 1984;23:876. [Google Scholar]; c) Snider BB. Acc. Chem. Res. 1980;13:426. [Google Scholar]
- 4.Some recent examples: a) Terida M, Soga K, Momiyama N. Angew. Chem. Int. Ed. 2008;47:4122. doi: 10.1002/anie.200800232. Kezuka S, Ikeno T, Yamada T. Org. Lett. 2001;3:1937. doi: 10.1021/ol015980m. Zheng K, Shi J, Liu X, Feng X. J. Am. Chem. Soc. 2008;130:15770. doi: 10.1021/ja808023y. Mikami K, Kawakami Y, Akiyama K, Aikawa K. J. Am. Chem. Soc. 2007;129:12950. doi: 10.1021/ja076539f. Koh JH, Larsen A, Gagné MR. Org. Lett. 2001;3:1233. doi: 10.1021/ol015702n. Evans DA, Wu J. J. Am. Chem. Soc. 2005;127:8006. doi: 10.1021/ja0522130. Rueping M, Thiessman T, Kuenkel A, Koenigs RM. Angew. Chem. Int. Ed. 2008;47:6798. doi: 10.1002/anie.200802139. Mikami K, Kakuno K, Aikawa K. Angew. Chem. Int. Ed. 2005;44:7257. doi: 10.1002/anie.200502682. Mikami K, Shimizu M. Chem. Rev. 1992;92:1021. Zhao J-F, Tjan T-BW, Loh T-P. Tetrahedron Lett. 2010:5649. Grachan ML, Tudge MT, Jacobsen EN. Angew. Chem. Int. Ed. 2008;47:1469. doi: 10.1002/anie.200704439. Bigot A, Breuninger D, Breit B. Org. Lett. 2008;10:5321. doi: 10.1021/ol8016148.
- 5.a) Mikami K, Sawa E, Terada M. Tetrahedron: Asymmetry. 1991;2:1403. [Google Scholar]; b) Andersen ED, Ernat JJ, Nguyen MP, Palma AC, Mohan RS. Tetrahedron Lett. 2005;46:7747. [Google Scholar]; c) Oppolzer W, Snieckus V. Angew. Chem. Int. Ed. 1978;17:476. [Google Scholar]; d) Cariou CAM, Kariuki BM, Snaith JS. Org. Biomol. Chem. 2008;6:3337. doi: 10.1039/b808644c. [DOI] [PubMed] [Google Scholar]; d) Williams JT, Bahia PS, Kariuki BM, Spencer N, Philp D, Snaith JS. J. Org. Chem. 2006;71:2460. doi: 10.1021/jo052532+. [DOI] [PubMed] [Google Scholar]; f) Suzuki T, Kobayashi S. Org. Lett. 2010;12:2920. doi: 10.1021/ol100935w. [DOI] [PubMed] [Google Scholar]; g) Nakagawa K, Okano T, Ozono K, Kato S, Kubodera N, Ohba S, Itoh Y, Mikami K. J. Fluor. Chem. 2007;128:654. [Google Scholar]
- 6.Jang HY, Hong JB, MacMillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]
- 7.a) Beeson TD, Mastracchio A, Hong J-B, Ashton K, MacMillan DWC. Science. 2007;316:582. [PubMed] [Google Scholar]; b) Mastracchio A, Warkentin AA, Walji AM, MacMillan DWC. Proc. Natl. Acad. Sci. 2010;107:20648. doi: 10.1073/pnas.1002845107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pham PV, Ashton K, MacMillan DWC. Chem. Sci. 2011;2:1470. doi: 10.1039/C1SC00176K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang H, Hong J, MacMillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]
- 9.Kim H, MacMillan DWC. J. Am. Chem. Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]
- 10.Amatore M, Beeson TD, Brown SP, MacMillan DWC. Angew. Chem. Int. Ed. 2009;48:5121. doi: 10.1002/anie.200901855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J. Am. Chem. Soc. 2009;131:11640. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rendler S, MacMillan DWC. J. Am. Chem. Soc. 2010;132:5027. doi: 10.1021/ja100185p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Jui NT, Lee ECY, MacMillan DWC. J. Am. Chem. Soc. 2010;132:10015. doi: 10.1021/ja104313x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jui NT, Garber JAO, Finelli FG, MacMillan DWC. J. Am. Chem. Soc. 2012;134:1140. doi: 10.1021/ja305076b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy JA. The Radical-Polar Crossover Reaction. In: Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Weinheim: Wiley-VCH; 2001. pp. 298–316. [Google Scholar]
- 15.a) Curran DP, Porter NA, Giese B. Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications. New York: VCH; 1995. [Google Scholar]; b) Renaud P. Stereoselectivity of Radical Reactions: Cyclic Systems. In: Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Weinheim: Wiley-VCH; 2001. pp. 298–316. [Google Scholar]
- 16.Intramolecular cyclizations using SOMO catalysis have previously been accomplished using π-rich activated olefins such as allylsilanes. This study expands the scope and utility of the enantioselective SOMO oxidation pathway via the use of simple olefins.
- 17.Unpublished results from our lab have indicated that the initial radical coupling step is reversible when oxidation of the resulting radical to a cation is relatively slow.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.