

Traditional treatments for Plasmodium falciparum malaria such as chloroquine and pyrimethamine-sulfadoxine have in many areas succumbed to drug resistance, and evidence now suggests emerging resistance to the new first-line artemisinins in Western Cambodia.1 Infection is rampant, resulting in ~650,000 deaths per year.2 Clearly, a need exists for novel therapies that are orally bioavailable, economical, and safe and whose activities are not compromised by existing resistance.3-5
Our laboratories have been interested in fatty acid biosynthesis in both Plasmodium spp. and Mycobacterium tuberculosis. Initial investigations into the mycobacterial target of the front-line drug isoniazid (INH) led to the discovery that the INH-NAD adduct binds the enoyl acyl carrier protein reductase InhA (also known as FabI or ENR).6 This sparked efforts by us and others to study triclosan-inspired small molecule inhibitors of InhA that do not require KatG activation of INH for M. tuberculosis,7 and that inhibit the P. falciparum homolog (PfFabI, previously termed PfENR) in biochemical assays with purified enzyme.8-10 Interestingly, the plasmodial FabI is not essential for the intra-erythrocytic stage of the parasite and instead is required for normal progression of the liver-stage of infection.11, 12
Efforts to optimize the antimalarial efficacy of small molecule triclosan analogs began with the hypothesis that the phenol could be replaced by organic functionality capable of maintaining some of the hydrogen-bonding interactions that have been shown crystallographically to be important for enzyme inhibition.13 The key interactions in this case involve the PfFabI Tyr-277 phenol and the 2’-OH of the ribose unit of the bound NAD+ co-factor. The compounds (Table 1) were prepared via adaptation of standard synthetic methods common to diaryl ether assembly14 and feature a variety of 1-substituents, ranging from an ether and a nitrile to carboxylic acid derivatives, amines, sulfonamides, and ureas. Triclosan methyl ether was prepared by methylation of the parent with methyl iodide in the presence of potassium carbonate to afford 1. The synthesis of triclosan 1-substituted analogs relied significantly on the preparation of anilines 5a and 5b (Scheme 1). The anilines were synthesized beginning with the coupling of the respective o-chloronitrobenzene with 2,4-dichlorophenol in DMSO. The resulting diaryl ethers underwent reduction to afford anilines 5a and b. Aniline 5a was carried in three straightforward steps, involving diazotization, the Sandmeyer reaction, and the Rosenmund-von Braun reaction,15 to benzonitrile 2. The 1-cyano triclosan 2 was then hydrolyzed to afford amide 4 and carboxylic acid 3. Reduction of 2 with lithium aluminum hydride afforded aminomethyl 6. Utilization of Ciganek’s methylorganocerium reagent,16 generated in situ from cerium(III) chloride and one equivalent of methyllithium reduced 2 to α,α-dimethylamine 7. Aniline 5a provided a starting point for the synthesis of a limited set of sulfonamide and amide analogs 8 – 11 via reaction with a sulfonyl chloride, anhydride or acid chloride under traditional conditions (Scheme 2). Two routes to ureas were devised, involving either direct reaction of aniline 5a or 5b with a commercial isocyanate or activation with triphosgene followed by mixing with a commercial amine. Two simple urea analogs 12 and 13 were initially made.
Table 1.
In vitro activities of triclosan derivatives with a 1-substituent against cultured P. falciparum strains.
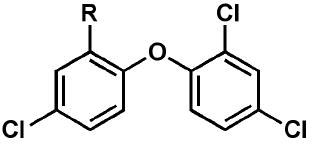
| |||
|---|---|---|---|
| Compound | R | EC50 3D7 (μM) | EC50 Dd2 (μM) |
| triclosan | OH | 2.9 | 3.8 |
| 1 | OCH3 | 100 | 76 |
| 2 | CN | 20 | 43 |
| 3 | COOH | >150 | >150 |
| 4 | CONH2 | 73 | 60 |
| 5a | NH2 | 52 | 55 |
| 6 | CH2NH2 | 2.3 | 4.3 |
| 7 | C(CH3)2NH2 | 7.2 | 1.8 |
| 8 | NHSO2Me | 38 | 60 |
| 9 | NHSO2Ph | 4.7 | 5.1 |
| 10 | NHAc | 33 | 48 |
| 11 | NHBz | 9.7 | 9.4 |
| 12 | NHC(O)NH2 | 18 | 18 |
| 13 | NHC(O)NHPh | 0.73 | 1.2 |
Scheme 1.
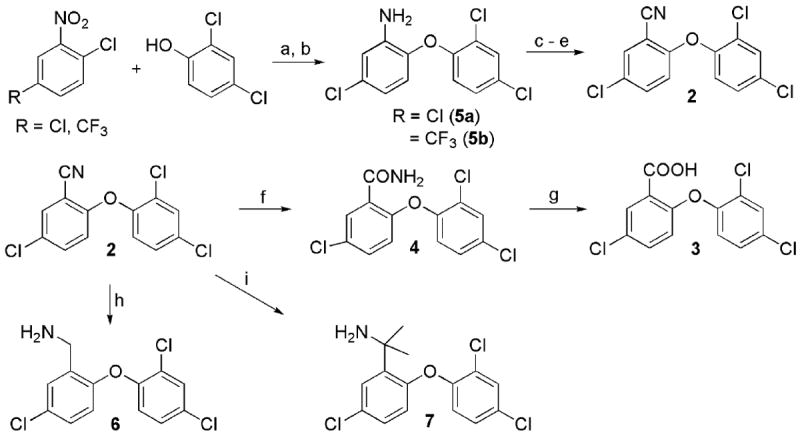
Synthesis of various 1-substituted triclosan derivatives. a) KOH, DMSO, 100 °C; b) H2, Ra-Ni, EtOH; c) t-BuONO, BF3 Et2O, THF, 0 °C ; d) NaI, acetone; e) CuCN, DMF, 150 °C; f) NaOH, H2O2, EtOH, 50 °C; g) 6 N HCl(aq), 2-methoxyethanol, 80 °C.
Scheme 2.
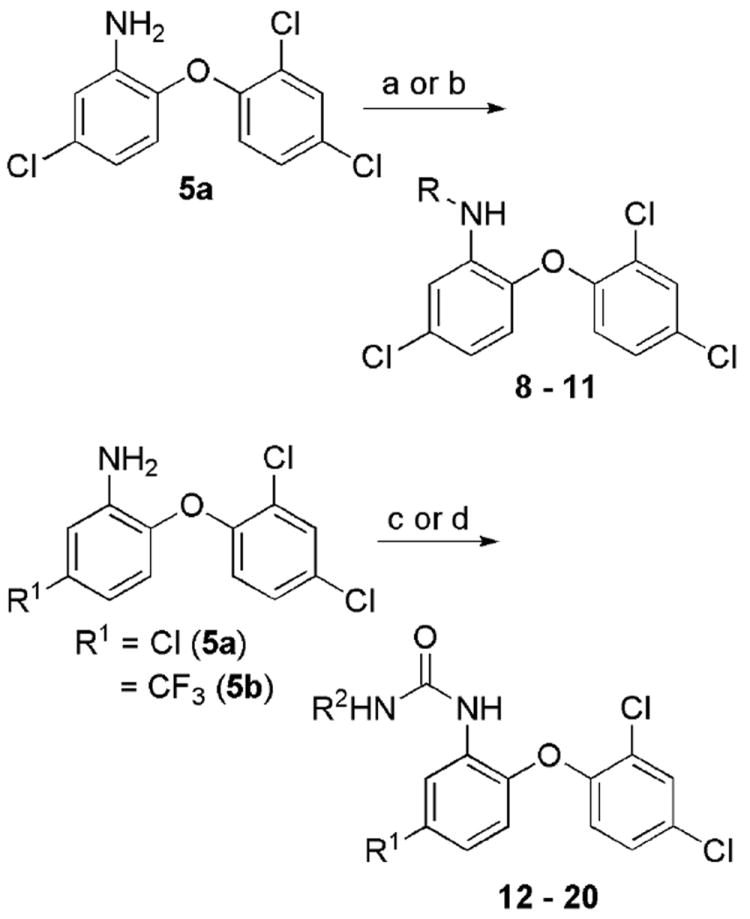
Synthesis of various 1-substituted triclosan amides, sulfonamides, and ureas. Reagents: (a) RSO2Cl, NEt3, DCM; (b) Ac2O or PhC(O)Cl and NEt3, DCM; (c) i. (Cl3CO)2CO, NEt3, DCM, -78 °C -> rt, ii. R2NH2; (d) R2NCO, tol.
While none of the phenol replacements displayed significant activity (IC50 > 10 μM) in a PfFabI inhibition assay,13 both aminomethyl 6 and diaryl urea 13 demonstrated efficacy in growth inhibition assays17 with cultured 3D7 (drug-sensitive) and Dd2 (resistant to chloroquine and pyrimethamine-sulfadoxine) P. falciparum strains. Molecular modeling studies rationalized how the loss of hydrogen-bonding (perhaps through both donating and accepting) upon replacement of the 1-OH may lead to abrogation of potent binding to, and hence reduced inhibition of PfFabI. Chemical inspection of 6 and 13 led to the prioritization of the diaryl urea for optimization based on its superior whole-cell efficacy and the potential to readily explore a range of aryl substituents, not belonging to the diaryl ether subunit.
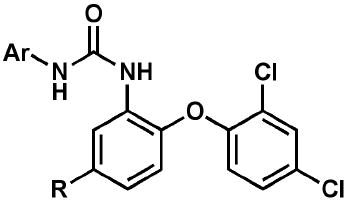
A set of follow-up diaryl ureas (Scheme 2; Table 2) was prepared to probe the structure-activity relationship pertinent to growth inhibition of in vitro-cultured P. falciparum, regardless of molecular target. It is clear from a select subset of analogs that the substitution pattern on the non-diaryl ether aryl moiety affected antimalarial activity against both strains of P. falciparum. In particular, the 3-position favored electron-withdrawing groups such as trifluoromethyl or cyano. The 4-position also preferred electron-withdrawing groups such as cyano, nitro, fluoro, and chloro. This was exemplified in the most potent compounds in this series: 18 (3-CF3-4-ClC6H3), 21 (3-CF3-4-NO2C6H3), and 24 (3,4-diCNC6H3).
Table 2.
Profile of diaryl ureas tested for in vitro activity against cultured P. falciparum strains and in vivo efficacy in a P. berghei mouse model.
| ||||||
|---|---|---|---|---|---|---|
| Compound | Ar | R | EC50 3D7 (μM) | EC50 Dd2 (μM) | SC Dose (mg/kg) | FSurvival @ 31da,b,c |
| 14 | 1-naphthyl | CF3 | 1.2 | 0.81 | 256 | NES |
| 15 | 2-naphthyl | CF3 | 0.61 | 0.41 | 256 | NES |
| 16 | 4-ClPh | Cl | 2.0 | 5.0 | 256 | NES |
| 17 | 3,4-Cl2Ph | Cl | 1.9 | 2.5 | 256 | NES |
| 18 | 3-CF3-4-ClPh | CF3 | 0.037 | 0.055 | 256 | 4/7 |
| 18 | 128 | EXT | ||||
| 19 | 3-CF3-4-FPh | CF3 | 0.13 | 0.15 | 256 | 1/7 |
| 19 | 128 | 2/7 | ||||
| 20 | 3-CF3-4-OMePh | CF3 | 0.18 | 0.18 | ndd | nd |
| nd | nd | |||||
| 21 | 3-CF3-4-NO2Ph | CF3 | 0.072 | 0.072 | nd | nd |
| 22 | 3-CF3-4-NH2Ph | CF3 | 1.5 | 3.6 | nd | nd |
| 23 | 3-CF3-4-CNPh | CF3 | 0.13 | 0.075 | nd | nd |
| 24 | 3,4-CN2Ph | CF3 | 0.081 | 0.14 | nd | nd |
| 25 | 3,4-Cl2Ph | CF3 | 0.20 | 0.20 | nd | nd |
FSurvival = proportion of animals living at day 31
NES = no extension of survival beyond infected control animals (8 days)
EXT = extension of survival beyond infected control animals (8 days), but no survival at day 31
nd = not determined
The in vivo activity of a subset of the urea derivatives was assessed utilizing the P. berghei rodent malaria model (Table 2).18 Briefly, ~5 weeks old CD-1 mice (Charles River Laboratories) were infected intraperitoneally with 106 P. berghei (KBG-173 line) parasitized red blood cells on day 0. Drug dosing (bid, divided dose) was initiated on day 3 and continued on days 4 and 5. Subcutaneous administration was achieved using a peanut oil suspension of the compound. Activity was defined by the fractional survival at day 31. The infected control mice survived an average of 8 days whereas non-infected control mice survived the entire 31 days of the study. Compounds with comparatively lower in vitro activity, such as 14 – 17, failed to exhibit an extension of survival beyond the control animals. 19, exhibiting better whole-cell efficacy, allowed survival of 2 of 7 and 1 of 7 animals at 31 days post-infection, at the 128 mg/kg and 256 mg/kg doses respectively. Diaryl urea 18, displaying the most potent whole-cell efficacy to date in this family, when dosed at 128 mg/kg, enabled extension of survival of the infected mice beyond the control. Promisingly, dosing at 256 mg/kg of 18 demonstrated 4 of 7 animals surviving 31 days post-infection. While many factors contribute to the in vivo efficacy of a small molecule antimalarial, it is clear that the diaryl ureas’ ability to inhibit the growth of P. falciparum in vitro was correlated with their efficacy in an in vivo P. berghei mouse model of infection.
It is interesting to note that phenoxy-substituted ureas have been previously reported as potent inhibitors of Plasmodium spp., based on solely in vitro data. These include the compound WR268961 (Supplementary Data Figure S1),19 where the urea linkage is para- with respect to the oxygen of the diaryl ether unit instead of ortho- as in 13 - 25. WR268961 abrogated parasite growth with an EC50 = 87 nM (W2 strain) and 460 nM (D6 strain), and modestly inhibited the P. falciparum cysteine protease plasmepsin 2 (PfPM2; IC50 = 17 μM). The diaryl ureas presented herein, however, do not appear to significantly target the plasmepsins as they equally inhibit the growth of both wild type and knockouts of PfPM1 through 4 (See Supplementary Data Table S1), attained via a genetic disruption methodology in the Dd2 background.20 GlaxoSmithKline disclosed the whole-cell efficacy of screening hit TCMDC-139010 (Supplementary Data Figure S1; XC50 = 930 nM vs. 3D7 strain), but without information concerning the biochemical target.4 We also reported in 2005 the preparation of triclosan-based 4’-ureas (Supplementary Data Figure S1) that were less potent against cultured parasite (EC50 values of ~100 μM) than 12 – 25, but exhibited IC50 values of ~100 nM against purified PfFabI assayed in vitro.14 More generally, the diaryl urea class of small molecules has been previously reported in the literature to exhibit potent efficacy against cultured parasites21-25 but without definitive biological target identification. This chemotype was also found amongst hits against P. falciparum dihydroorotate dehydrogenase26 that lacked whole-cell activity.
In order to more quantitatively compare the diaryl ureas reported herein with those disclosed in the literature as antimalarials, we leveraged a total of thirty-four diaryl ureas generated in our laboratories (Supplementary Data Table 1) to generate a common features pharmacophore (Accelrys Discovery Studio 2.5.5) with five hydrophobic features and two hydrogen bond acceptors. Figure 1 depicts the top 3 active compounds that mapped to it. This pharmacophore is also able to select 23 compounds out of the 451 with the diaryl urea substructure present in the GSK library of antimalarial hits4 (up to 100 conformers per molecule generated using the FAST algorithm in CAESAR). Slight variants on the diaryl urea scaffold, such as TCMDC-140251 (Supplementary Data Figure S1; XC50 = 190 nM against 3D7 strain) containing aryl and 1-indolinyl moieties, mapped well to the model. Interestingly, TCMDC-139010 exhibited a poor fit because it lacked three of the hydrophobic features. The pharmacophore differs from that constructed by Zhang et al. which contained 2-3 hydrophobic features and 2-3 hydrogen bond acceptors.25 Not surprisingly, the 3 most active diaryl ureas from the paper by Zhang et al. failed to map to our pharmacophore; most likely, the 4-aminoquinaldine-derived diaryl ureas present different features than the triclosan-derived diaryl ureas. Distinctions in the arrangement of hydrophobic and hydrogen bonding features in the diaryl ureas from Zhang et al. and in this study may enable these molecules to target different proteins in Plasmodium falciparum. The pharmacophore developed in this study may be leveraged to search other databases (e.g. compound vendor libraries and approved drugs) to identify novel compounds with antimalarial activity.
Figure 1.
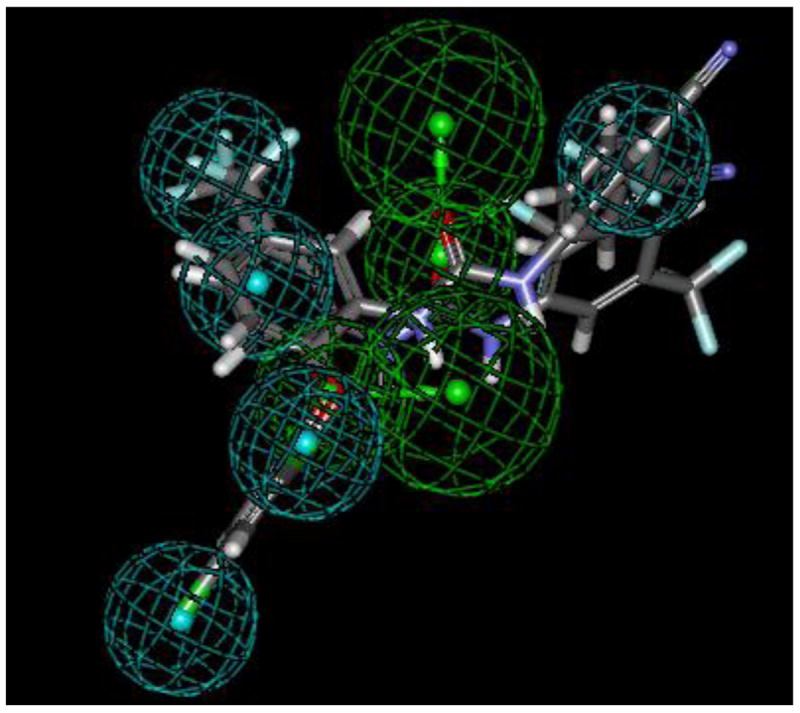
Common features pharmacophore for triclosan-inspired diaryl ureas showing the top 3 active molecules aligned to the 5 hydrophobic features (cyan) and 2 hydrogen bond acceptors (green).
A novel class of diaryl ureas derived initially from triclosan has been disclosed with regard to their potent in vitro efficacy against cultured drug-sensitive and drug-resistant strains of P. falciparum. Importantly, family members such as 18 demonstrate promising in vivo activity in a P. berghei mouse model of infection. Further investigation of the structure-activity relationship of these triclosan derivatives is necessary to further improve their antimalarial activity, in addition to their pharmacokinetic profiles. Biological studies are also important to determine the molecular target(s) of these potent compounds.
Supplementary Material
Acknowledgments
This work has been supported by funding from the Medicines for Malaria Venture. SE kindly acknowledges Accelrys Inc. for providing Discovery Studio.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. N Engl J Med. 2009;361:455. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. World Malaria Report 2011 [Google Scholar]
- 3.Fidock DA. Nature. 2010;465:297. doi: 10.1038/465297a. [DOI] [PubMed] [Google Scholar]
- 4.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. Nature. 2010;465:305. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 5.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jiménez-Díaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Jr, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery WM, Guy RK. Nature. 2010;465:311. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Jr, Sacchettini JC. Science. 1998;279:98. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 7.Pan P, Tonge PJ. Curr Top Med Chem. 2012;12:672. doi: 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freundlich JS, Wang F, Tsai H-C, Kuo M, Shieh H-M, Anderson JW, Nkrumah LJ, Valderramos J-C, Yu M, Kumar TRS, Valderramos SG, Jacobs WR, Jr, Schiehser GA, Jacobus DP, Fidock DA, Sacchettini JC. J Biol Chem. 2007;282:25436. doi: 10.1074/jbc.M701813200. [DOI] [PubMed] [Google Scholar]
- 9.Mishra S, Karmodiya K, Parasuraman P, Surolia A, Surolia N. Bioorg Med Chem. 2008;16:5536. doi: 10.1016/j.bmc.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Frecer V, Megnassan E, Miertus S. Eur J Med Chem. 2009;44:3009. doi: 10.1016/j.ejmech.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 11.Yu M, Kumar TR, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, Kelly BJ, Moura PA, Lakshmanan V, Freundlich JS, Valderramos JC, Vilcheze C, Siedner M, Tsai JH, Falkard B, Sidhu AB, Purcell LA, Gratraud P, Kremer L, Waters AP, Schiehser G, Jacobus DP, Janse CJ, Ager A, Jacobs WR, Jr, Sacchettini JC, Heussler V, Sinnis P, Fidock DA. Cell host & microbe. 2008;4:567. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaughan AM, O’Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, Cowman AF, Kappe SH. Cell Microbiol. 2009;11:506. doi: 10.1111/j.1462-5822.2008.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perozzo R, Kuo M, Sidhu AS, Valiyaveettil JT, Bittman R, Jacobs WR, Jr, Fidock DA, Sacchettini JC. J Biol Chem. 2002;277:13106. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 14.Freundlich JS, Anderson JW, Sarantakis D, Shieh H-M, Yu M, Valderramos J-C, Lucumi E, Kuo M, Jacobs WR, Jr, Schiehser GA, Fidock DA, Jacobus DP, Sacchettini JC. Bioorg and Med Chem Lett. 2005;15:5247. doi: 10.1016/j.bmcl.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 15.Lindley J. Tetrahedron. 1984;40:1433. [Google Scholar]
- 16.Freundlich JS, Yu M, Lucumi E, Kuo M, Tsai HC, Valderramos JC, Karagyozov L, Jacobs WR, Jr, Schiehser GA, Fidock DA, Jacobus DP, Sacchettini JC. Bioorg Med Chem Lett. 2006;16:2163. doi: 10.1016/j.bmcl.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 17.Fidock DA, Nomura T, Wellems TE. Mol Pharmacol. 1998;54:1140. doi: 10.1124/mol.54.6.1140. [DOI] [PubMed] [Google Scholar]
- 18.Ager AL. In: Handbook of experimental pharmacology: antimalarial drugs. Peters W, Richards WHG, editors. Vol. 68. Springer-Verlag; New York, NY: 1984. p. 225. [Google Scholar]
- 19.Jiang S, Prigge ST, Wei L, Gao Y-E, Hudson TH, Gerena L, Dame JB, Kyle DE. Antimicrob Agents Chemother. 2001:2577. doi: 10.1128/AAC.45.9.2577-2584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Omara-Opyene AL, Moura PA, Sulsona CR, Bonilla JA, Yowell CA, Fujioka H, Fidock DA, Dame JB. J Biol Chem. 2004;279:54088. doi: 10.1074/jbc.M409605200. [DOI] [PubMed] [Google Scholar]
- 21.Leban J, Pegoraro S, Dormeyer M, Lanzer M, Aschenbrenner A, Kramer B. Bioorg Med Chem Lett. 2004;14:1979. doi: 10.1016/j.bmcl.2004.01.083. [DOI] [PubMed] [Google Scholar]
- 22.Domínguez JN, León C, Rodrigues J, Gamboa de Domínguez N, Gut J, Rosenthal PJ. J Med Chem. 2005;48:3654. doi: 10.1021/jm058208o. [DOI] [PubMed] [Google Scholar]
- 23.Madapa S, Tusi Z, Mishra A, Srivastava K, Pandey SK, Tripathi R, Puri SK, Batra S. Bioorg Med Chem. 2009;17:222. doi: 10.1016/j.bmc.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Madapa S, Tusi Z, Sridhar D, Kumar A, Siddiqi MI, Srivastava K, Rizvi A, Tripathi R, Puri SK, Shiva Keshava GB, Shukla PK, Batra S. Bioorg Med Chem. 2009;17:203. doi: 10.1016/j.bmc.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Anderson M, Weisman JL, Lu M, Choy CJ, Boyd VA, Price J, Sigal M, Clark J, Connelly M, Zhu F, Guiguemde WA, Jeffries C, Yang L, Lemoff A, Liou AP, Webb TR, DeRisi JL, Guy RK. ACS Med Chem Lett. 2010;1:460. doi: 10.1021/ml100083c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. J Biol Chem. 2005;280:21847. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.