

Abstract
We report the identification of a new class of antimicrobial peptidomimetics-γ-AApeptides with potent and broad-spectrum activity, including clinically-relevant strains that are unresponsive to most antibiotics. They are also not prone to select for drug-resistance.
Antimicrobial peptides are small cationic amphiphilic peptides found in virtually all living organisms.1 They play an important role in innate immune defense against various infections.2 In the last decade, there has been significant interest in the development of antimicrobial peptides because of the emergence of antibiotic resistance.1,2 Compared with conventional antibiotics, which target specific metabolic processes in bacteria,3 antimicrobial peptides are able to form amphipathic structures, where cationic and hydrophobic groups are segregated into two regions, so as to facilitate interaction with the negatively charged bacterial cytoplasmic membrane.4 Such interaction is based on the global chemical properties of peptides, rather than their precise sequence.5 Therefore, antimicrobial peptides are unlikely to be hindered by the resistance mechanisms observed for current antibiotic treatments.1 Furthermore, unlike conventional antibiotics, antimicrobial peptides exhibit broad-spectrum activity against both Gram-positive and Gram-negative bacteria, and even fungi and viruses.1,2 They appear to be ideal antibiotic agents to supplement or replace existing treatments.4
However, despite significant enthusiasm, there are intrinsic drawbacks associated with the development of peptide antibiotics due to the peptidic nature of antimicrobial peptides. These include potential immunoreactivity and susceptibility to enzymatic degradation, etc.6 Non-natural peptidomimetic approaches that mimic antimicrobial peptides may circumvent these impediments by introducing amide bond isosteres, and modifying the peptide backbone so as to improve resistance to proteolytic hydrolysis.7 To this end, non-natural antimicrobial oligomers, such as β-peptides, peptoids, arylamides, and oilgourea have been developed.8 However, their rational design sometimes turns out not to be straightforward due to the difficulty of introducing a variety of functional groups to fine tune their activity and selectivity, and the inconsistency of their structure–activity-relationship.9 Furthermore, recent research findings from many groups suggest that helical conformations, in which lipophilic and cationic side chains are globally segregated, are not necessary for antimicrobial activity.10,11 Indeed, a pre-organized secondary structure seems not required for bacterial killing;5 instead, oligomers with a strong propensity for helical conformation or conformational rigidity may lead to high hemolytic activity.4,12 Potent antimicrobial activity may actually require the presence of flexible, or even random coiled backbones, where side groups are segregated into hydrophobic and cationic regions upon interaction with bacterial membranes,5,12 even if the amphiphilic conformation is irregular and non-helical.
Herein, we report a new class of antimicrobial peptide mimetics – γ-AApeptides developed by a simple design strategy. γ-AApeptides were recently developed by our group (Fig. S1†) in order to further facilitate drug discovery. Certain γ-AApeptides were able to disrupt protein-protein interactions13 and recognize nucleic acids with high affinity and specificity,14 and were highly resistant to protease degradation.13 Moreover, the synthesis and diversification of γ-AApeptides is efficient and straightforward,13 strengthening their potential to generate focused libraries for drug-lead screening.
To design antimicrobial peptidomimetics based on γ-AApeptides, we first followed the rationale: Global distribution of cationic and hydrophobic groups are most important for potent antimicrobial activity; and that secondary structure is irrelevant.5,10–12 We have also demonstrated this principle in the recent report of antimicrobial α-AApeptide design,15 in which amphiphilic α-AApeptide building blocks were linked together to display antimicrobial activity. We anticipated γ-AApeptides would also be perfect candidates as antimicrobial agents by joining amphiphilic γ-AApeptide building blocks together, due to their high stability, unlimited derivatizing potential, and capability to adopt globally amphipathic structures by easily adjusting their conformation upon interaction with bacterial cell membranes (Fig. S2†).
As such, we prepared some γ-AApeptide sequences (Fig. 1) and tested their antimicrobial activity against a range of clinically-relevant Gram-negative and Gram-positive bacteria, as well as fungus C. albicans. γ1, γ2, γ3 are γ-AApeptides of different length that are composed of amphiphilic γ-AApeptide building blocks. γ4 contains one hydrophobic building blocks while γ5 contains two hydrophobic building blocks, an attempt to tune overall hydrophobicity of γ-AApeptides. As controls, we included AMP magainin II (m1),15 a 14-mer conventional peptide p115 with alternative phenylalanine and lysine residues, and the most potent antimicrobial α-AApeptide α1 reported by us recently.15 The antimicrobial activity of these oligomers was tested and listed in Table 1. Their hemolytic activity was also tested to evaluate their selectivity.
Fig. 1.
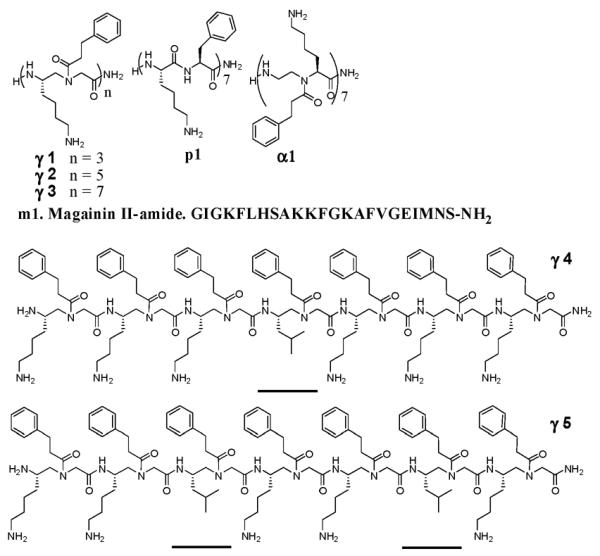
The structures of oligomers tested for their antimicrobial activity. Underlined building blocks are hydrophobic building blocks containing two hydrophobic side chains; the rest of the building blocks in the sequences are amphiphilic with one cationic and one hydro-phobic side chains.
Table 1.
The antimicrobial and hemolytic activities of oligomers. Magainin II is a natural amphipathic antimicrobial peptide used as the comparison here. The sequences showing broad-spectrum antimicrobial activity (γ3–γ5 and α1) are shaded in light grey
| MIC (μg ml−1) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Organism | γ1 | γ2 | γ3 | γ4 | γ5 | p1 | α1 | m1 |
| Gram-negative | ||||||||
| E. coli | >100 | 25 | 2.5–5 | 2.5–5 | 2.5–5 | >100 | 4.5 | 40 |
| K. pneumoniae | >100 | >100 | >100 | >100 | 5 | >100 | >100 | >100 |
| Gram-positive | ||||||||
| B. subtilis | >100 | 5 | 2.5 | 2.5 | 2 | >100 | 2 | 40 |
| Multi-drug resistant S. epidermidis | >100 | >100 | 6.3–12.5 | 3.1–6.3 | 3.1–6.3 | >100 | 10 | >100 |
| Vancomycin-resistant E. faecalis | >100 | >100 | 12.5–25 | 12.5–25 | 3.1–6.3 | >100 | 75 | 75 |
| Methicillin-resistant S. aureus | >100 | >100 | 12.5–25 | 3.1–6.3 | 5 | >100 | 75 | >100 |
| MRSA USA100 lineage | — | — | — | — | 5 | — | >100 | >100 |
| B. anthracis | — | — | 25–50 | 25–50 | 5 | — | >100 | >100 |
| Fungi | ||||||||
| C. albicans | >100 | >100 | 12.5–25 | 12.5–25 | 5–10 | >100 | 20–30 | 75 |
| Hemolysis (H10/H50) | >500/>500 | >500/>500 | >500/>500 | 400/>500 | 75/300 | 300/>500 | 400/>500 | >500/>500 |
As expected, a few sequences show very potent broad-spectrum activities against fungi, and a series of clinically-relevant Gram-positive and Gram-negative bacteria, including pathogens that are unresponsive to most antibiotics (Table 1). The results support our hypothesis very nicely, and display clear structure-function relationships. The results support our hypothesis very nicely, and display clear structure-function relationships. Active antimicrobial sequences can be generated by simply joining amphipathic building blocks together, as seen for γ3. It suggests that γ-AApeptides can adopt globally amphipathic structures upon binding onto bacterial membranes. Interestingly, the control sequence p1, which contains alternative Phe and Lys residues, with the functional groups identical to γ3, does not show any antimicrobial activity at all. Such observation can be explained by peptide's intrinsic folding propensity.15 The results demonstrate that the straightforward design of γ-AApeptide antimicrobial agents is not applicable to regular peptides.
Meanwhile, longer sequences are more potent and broad-spectrum active than shorter sequences with similar structures, as seen for the activities of γ1–γ3, which indicates that the number of cationic charge is important for interaction with bacterial membranes, consistent to what has been identified in the development of antimicrobial α-AApeptides.15 Notably, γ3 is more potent and broad-spectrum active than α3 of same functional groups, indicating γ-AApeptides might be more suitable than α-AApeptides for antimicrobial development.
The further development of antimicrobial γ-AApeptides based on the lead γ3 yields more potent sequences. As an initial attempt, we introduced a simple hydrophobic building block to tune the overall hydrophobicity and hydrophilicity of γ3, in order to evaluate the feasibility to tune antimicrobial activity of γ-AApeptides in the future. Such an effort led to the discovery of γ5, in which two amphiphilic building blocks are replaced with hydrophobic building blocks (containing two hydrophobic side chains) compared to γ3, and only one side chain difference from γ4. While γ4 are not active against Gram-negative K. pneumoniae, however, γ5 potently inhibited the growth of all tested Gram-negative and Gram-positive bacteria, and also the fungus C. albicans. In fact, γ5 is amongst the most potent and broad-spectrum antimicrobial peptidomimetics reported to date. Most significantly, it arrested the growth of the USA100 lineage MRSA strain with extreme potency. This observation is of particular importance as this strain displays resistance to a wealth of existing antimicrobial agents, and is broadly multi-drug resistant. As such, this may satisfy urgent needs in hospitals for new therapeutics, since this strain has been identified as the most prevalent cause of hospital-associated infections in the United States, and almost no current antibiotics can effectively target it. γ5 also potently inhibits B. anthracis, which is known to cause the highly lethal condition, anthrax. The findings may suggest that γ5 has the potential for use in bio-defence in the future. In the mean time, all the γ-AApeptides have excellent selectivity for bacteria than human blood cells, since the majority of them has H50 of more than 500 μg ml−1; even the most hemolytic sequence γ5 still have a selectivity of at least 60-fold. This is because mammalian cell membranes are almost neutrally charged, while bacterial membranes are negatively charged, which favors more for electrostatic interactions with cationic γ-AApeptide sequences.
One of the biggest challenges for conventional antibiotics is their susceptibility to the development of resistance, which quickly abolishes their efficacy. This situation has become more severe in recent years, and may lead to outbreaks of deadly infectious diseases that are untreatable. To investigate the potential for bacteria to develop resistance against the treatment of γ-AApeptides, methicillin-resistant S. aureus (ATCC 33592) was serially passaged in half-MIC concentrations of γ5, and new MIC values were determined every 24 h. As a positive control, parallel cultures were exposed to serial 2-fold dilutions of the antibiotic norfloxacin (Fig. S3†).16 After 17 days, virtually no change in the MIC occurred for γ5 over the 17 passages, whereas the MIC for norfloxacin started to increase after just three passages, and developed to profound resistance after 17 (MIC increased > 20-fold). These results demonstrate that MRSA strains do not readily develop resistance to γ-AApeptide γ5. Although additional studies to assess the ability of bacteria to evolve resistance to γ5 are required, we suggest that this preliminary data indicates that γ5 may actually mimic natural antimicrobial peptides which are notoriously difficult to develop resistance towards. This augments the potential of γ5 as a new generation of antibiotic lead compounds.
The antimicrobial mechanism of g-AApeptides in disrupting bacterial membranes was also assessed by fluorescence microscopy (Fig. S4†) using a double staining method with DAPI and PI. DAPI stains all bacterial cells irrespective of their viability, whereas PI only stains injured or dead cells with damaged membranes.17,18 After incubation with γ-AApeptide γ5 for 2 h, strongly PI-stained red fluorescent E. coli and B. subtilis were observed, demonstrating the membranes of these bacteria had been disrupted. The aggregation of dead cells is consistent to previously reported observation.18
In summary, we reported the identification of a new class of antimicrobial peptidomimetics-γ-AApeptides, with potency and broad-spectrum activity far superior to previous reported α-peptides.15 These γ-AApeptides likely inhibit bacterial growth by mimicking conventional antimicrobial peptides through membrane disruption. This is an important observation as it suggests the potential for virtually no development of resistance towards these agents. Additionally, although more extensive studies are needed, our initial attempt has demonstrated the ease and effectiveness of building block-joining strategy, which is unique in the development of antimicrobial AApeptides. Such approach avoids tedious and sometimes ineffective secondary structure development in the design of other classes of antimicrobial peptidomimetics. Coupled with virtually limitless side chain variation, bioavailability and enhanced stability, it is very straightforward to further tune the antimicrobial activity and selectivity of γ-AApeptides by changing hydrophobicity and hydrophilicity of building blocks and backbone lengths. This would be particularly effective if resistance to these compounds were ever observed, allowing rapid change and alteration to circumnavigate any such occurrence. These findings will also shed further light on the design and optimization of other non-natural antimicrobial oligomers in the future. For example, other class of peptidomimetics with alternative hydrophobic and hydrophilic residues could be potential antimicrobial agents since their backbones are different from natural α-peptides. Comprehensive studies of a wide variety of γ-AApeptides containing hydrophobic and amphiphilic building blocks of different functional groups, positions, and ratio are currently underway. Further investigation of bactericidal mechanisms is also ongoing.
Supplementary Material
Acknowledgments
This work is supported by USF start-up fund (JC) and NIH 1R01AI080626-01A2 (LNS).
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures and compound characterization data. Figures for fluorescence microscopy and drug-resistance study. See DOI: 10.1039/c1cc14476f
Notes and references
- 1.Marr AK, Gooderham WJ, Hancock RE. Curr. Opin. Pharmacol. 2006;6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Hancock RE, Sahl HG. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 3.Alekshun MN, Levy SB. Cell. 2007;128:1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott RW, DeGrado WF, Tew GN. Curr. Opin. Biotechnol. 2008;19:620–627. doi: 10.1016/j.copbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaiou M. J. Mol. Med. 2007;85:317–329. doi: 10.1007/s00109-006-0143-4. [DOI] [PubMed] [Google Scholar]
- 7.Violette A, Fournel S, Lamour K, Chaloin O, Frisch B, Briand JP, Monteil H, Guichard G. Chem. Biol. 2006;13:531–538. doi: 10.1016/j.chembiol.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Tew GN, Scott RW, Klein ML, Degrado WF. Acc. Chem. Res. 2009;43:30–39. doi: 10.1021/ar900036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fowler SA, Blackwell HE. Org. Biomol. Chem. 2009;7:1508–1524. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmitt MA, Weisblum B, Gellman SH. J. Am. Chem. Soc. 2007;129:417–428. doi: 10.1021/ja0666553. [DOI] [PubMed] [Google Scholar]
- 11.Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J. Am. Chem. Soc. 2007;129:15474–15476. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- 12.Ivankin A, Livne L, Mor A, Caputo GA, Degrado WF, Meron M, Lin B, Gidalevitz D. Angew. Chem., Int. Ed. 2010;49:8462–8465. doi: 10.1002/anie.201003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niu Y, Hu Y, Li X, Chen J, Cai J. New J. Chem. 2011;35:542–545. [Google Scholar]
- 14.Niu Y, Jones A, Wu H, Varani G, Cai J. Org. Biomol. Chem. 2011;9:6604–6609. doi: 10.1039/c1ob05738c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Padhee S, Hu Y, Niu Y, Bai G, Wu H, Costanza F, West L, Harrington L, Shaw LN, Cao C, Cai J. Chem. Commun. 2011;47:9729–9731. doi: 10.1039/c1cc13684d. [DOI] [PubMed] [Google Scholar]
- 16.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6968–6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsunaga T, Okochi M, Nakasono S. Anal. Chem. 1995;67:4487–4490. [Google Scholar]
- 18.Chen C, Pan F, Zhang S, Hu J, Cao M, Wang J, Xu H, Zhao X, Lu JR. Biomacromolecules. 2010;11:402–411. doi: 10.1021/bm901130u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.