

Abstract
2-Amino-1,4-dihydropyrimidines were reacted with bis-electrophiles to produce novel fused bi-pyrimidine, pyrimido-aminotriazine, and pyrimido-sulfonamide scaffolds. In addition, a quinazoline library was constructed using a guanidine Atwal-Biginelli reaction with 1-(quinazolin-2-yl)guanidines. The product heterocycles have novel constitutions with high nitrogen atom counts and represent valuable additions to screening libraries for the discovery of new modulators of biological targets.
Keywords: Guanidines, Pyrimidines, Quinazolines, Atwal-Biginelli reaction, Screening library
1. Introduction
Diversity Oriented Synthesis (DOS) inspires chemists to design or apply chemical methodologies that result in the synthesis of structurally diverse, functionally rich and often architecturally complex small molecule scaffolds.1,2 In many cases, new biological targets can be explored more effectively with DOS-derived probes and natural products than traditional aromatic “drug-like” heterocycles.3 Novel molecular scaffolds are therefore valuable additions to high throughput screening libraries and improve the odds of identifying lead compounds in drug discovery initiatives.4,5
As a continuation of our efforts in the preparation of rare heterocyclic scaffolds,6 we became interested in developing a convergent synthetic route to 4H-pyrimido[1,2-a]pyrimidines. While there is evidence in the literature demonstrating melanin-concentrating hormone receptor (MCH1R) antagonistic effects,7 protein kinase inhibition,8 treatment of atherosclerosis and restenosis,9 antiviral activities,10 inhibition of platelet aggregation,11 and anti-MRSA dihydrofolate reductase activity12 of 4H-pyrimido[1,2-a]pyrimidines 1, their synthesis and application is strikingly rare compared to the structurally and pharmacologically related ring-contracted imidazo[1,2-a]pyridines 2 (Figure 1). The latter heterocycle is an abundant pharmacophore in medicinal chemistry and has been used in antiviral, antibacterial, anti-inflammatory, analgesic, antipyretic, and anxioselective indications. Derivatives of 2 are β-amyloid formation inhibitors and constitute a novel class of orally active nonpeptide bradykinin B2 receptor antagonists.13 Several imidazo[1,2-a]pyridines are already clinically used, including zolimidine (an anti-ulcer drug), zolpidem (a hypnotic drug), and alpidem (a nonsedative anxiolytic). Imidazo[1,2-a]pyrimidine moieties are also important as benzodiazepine receptor agonists, antiviral agents, antibacterials, antifungal agents, and calcium channel blockers.13
Fig. 1.
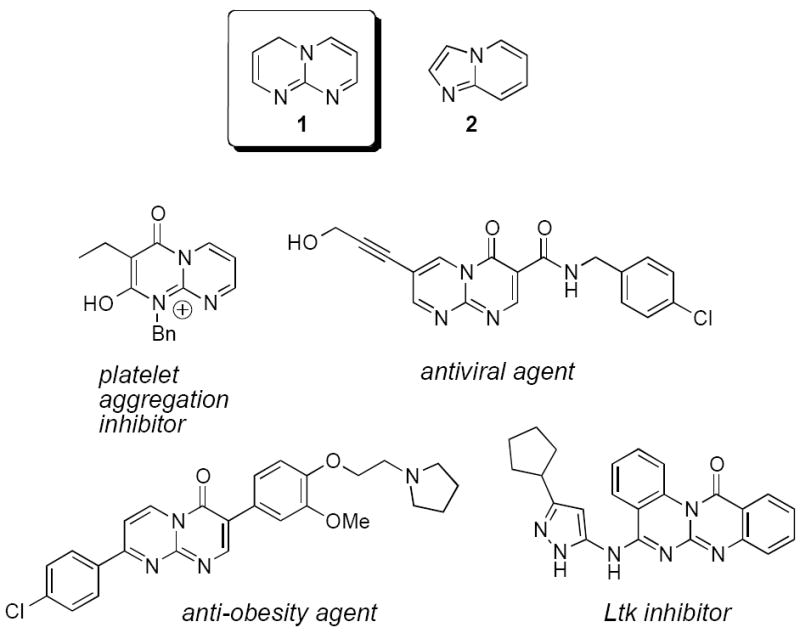
The 4H-pyrimido[1,2-a]pyrimidine and imidazo[1,2-a]pyridine scaffolds 1 and 2, and selected biologically active derivatives.
Using a variant of the Biginelli 3-component reaction for the preparation of cyclic guanidines,14,15 we were able to develop efficient strategies for a tandem condensation with bis-electrophiles to give bipyrimidines 3, pyrimido-aminotriazines 4, cyclic pyrimido-sulfonamides 5 and pyrimido-pyrimidines 6 (Scheme 1). In addition, a small sub-library of quinazoline-containing heterocycles 7 was assembled using Atwal-Biginelli conditions.16,17
Scheme 1.
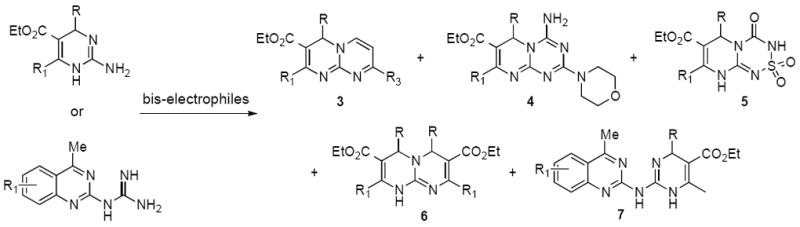
Overview of heterocycles obtained by condensation of guanidines with bis-electrophiles.
New strategies for accessing structurally diverse guanidine-based Biginelli products have previously been reported by the Overman group.18 Specifically, triazone-protected guanidines were found to endure the harsh acidic conditions frequently used for the Biginelli cyclization.18 In order to access C-4 alkyl-substituted cyclic guanidines 10, we first envisioned using thioureas 8 as starting materials followed by an aminolysis of the corresponding methylthioureas 9, in a manner similar to that reported by Kappe (Scheme 2).19
Scheme 2.
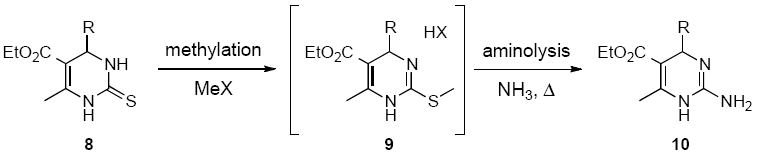
Thiourea methylation followed by aminolysis of the thioimidate provides access to 2-aminodihydropyrimidines.
2. Results and Discussion
Thioureas 8 with R = methyl, phenethyl, and iso-propyl were prepared through Biginelli multicomponent condensations from β-ketoesters, aldehydes, and thiourea under standard acid-catalyzed conditions.20 The reaction parameters for the one-pot methylation/aminolysis were optimized on substrate 8a (R = Me) (Table 1, Entries 1-5). Initial investigations using methyl iodide resulted in dealkylation of the newly formed methylthiourea 9a, most likely due to the presence of nucleophilic iodide ions in the vigorously heated aminolysis reaction mixture.21 Accordingly, methyl triflate was used as an alternative electrophile for the S-alkylation and proved effective for the formation of the desired guanidine 10a in good isolated yields. The use of dimethyl sulfate also provided 10a in 57% yield but required prolonged reaction times (Entry 5). High yields of product 10a were achieved when THF was used as the reaction solvent. However, the methylation step was problematic due to the propensity of methyl triflate to polymerize THF.22 The optimal solvent for this two-step reaction sequence was found to be chlorobenzene (PhCl), which was both inert to the reaction conditions and could safely be heated to 95-100 °C under sealed tube conditions (Table 1, Entry 3). The aminolysis step was found to proceed smoothly on multigram scale (2.5 g) with ammonium acetate or ammonium chloride as additives in the presence of ammonia. Following this one-pot reaction, the hydrochloride salts of 10a-c were prepared and subsequently used as substrates for further cyclization reactions (vide infra).
Table 1.
Conversion of thioureas (8a, R = CH3; 8b, R = (CH2)2Ph; 8c, R = CH(CH3)2) to thioimidates 9a-c and 2-aminopyrimidines 10a-c according to Scheme 2.
| Entry | 8/Conditions | 9/Additive/Time | 10/Yield %a |
|---|---|---|---|
| 1 | 8a/MeOTf, THF, 1.5 h | 9a/NH4OAc/20 h | 10a/77 |
| 2 | 8a/MeOTf, ClCH2CH2Cl, 0.75 h | 9a/NH4OAc/15 h | 10a/30 |
| 3 | 8a/MeOTf, PhCl, 3.5 h | 9a/NH4OAc/17 h | 10a/61 |
| 4 | 8a/MeOTf, PhCl, 15 h | 9a/NH4Cl/21 h | 10a/51 |
| 5 | 8a/(MeO)2SO2, THF, 60 h | 9a/NH4OAc/17 h | 10a/57 |
| 6 | 8b/MeOTf, PhCl, 3.5 h | 9b/NH4OAc/17 h | 10b/39 |
| 7 | 8c/MeOTf, PhCl, 3.5 h | 9c/NH4OAc/20 h | 10c/33 |
Isolated yield of purified product.
The 4-arylated 2-aminopyrimidine 13•TFA was obtained in a 2-step sequence starting from PMB-guanidine 11 (Scheme 3).23 Treatment of a solution of 11 in hexafluoroisopropanol with enone 12 and sodium bicarbonate at 90 °C provided the PMB-protected 13 in 42% yield. Deprotection of the PMB group was accomplished in 70% yield in TFA buffered with water, triethylsilane, and thioanisole.24 A 3-component Biginelli reaction with guanidine hydrochloride 14•HCl, ethyl 3-oxo-3-phenylpropanoate 15 and aldehydes 16 and 17 was performed to access heterocycles 18, 19 and the hydrochloride salt 18•HCl (Scheme 4).
Scheme 3.
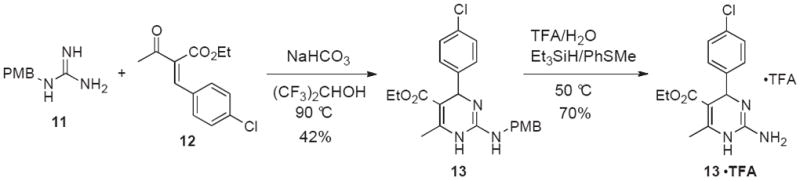
2-Step preparation of 2-aminopyrimidine 13•HCl from PMB-protected guanidine 11.
Scheme 4.
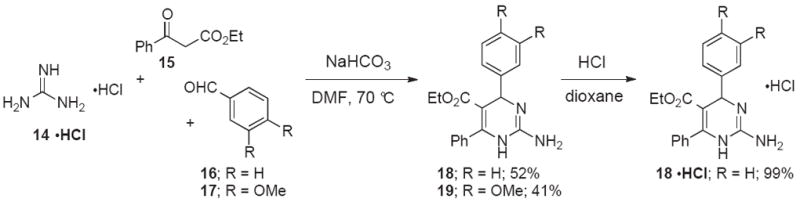
Preparation of 2-aminopyrimidines from guanidine hydrochloride.
Cyclization reactions of guanidines with the bis-electrophile 1,1,3,3-tetramethoxypropane (20) were explored under microwave conditions (Scheme 5 and Table 2).25,26 We found that these reactions proceeded smoothly in 42-76% yield when the hydrochloride salts of 10a-c or 18 were combined with 20 in a microwave vial and heated to 160 °C in trifluoroethanol (21) (Entries 1-4). The use of the hydrochloride salts was essential since the corresponding reactions with free amines did not afford the cyclocondensation products. We also briefly explored the reaction of 18 with 4-methoxybut-3-enone 26 to give 8-methyl 4H-pyrimido[1,2-a]pyrimidine 27 in 64% yield (Table 2, Entry 5).
Scheme 5.
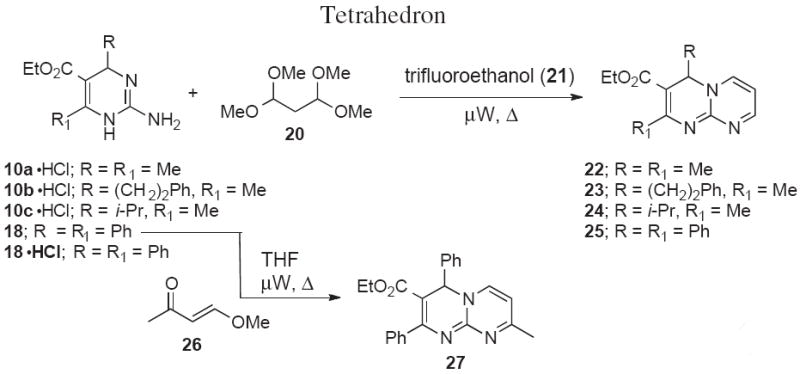
Synthesis of 4H-pyrimido[1,2-a]pyrimidine-3-carboxylates
Table 2.
Cyclocondensation reactions of guanidines with bis-electrophiles 20 and 26 according to Scheme 5.
| Entry | Reactants | Solvent/Temperature/Time | Product/Yield %a |
|---|---|---|---|
| 1 | 10a•HCl and 20 | 21/160 °C/1 h | 22/76 |
| 2 | 10b•HCl and 20 | 21/160 °C/2 h | 23/71 |
| 3 | 10c•HCl and 20 | 21/160 °C/2 h | 24/71 |
| 4 | 18•HCl and 20 | 21/160 °C/1.5 h | 25/42 |
| 5 | 18 and 26 | THF/150 °C/0.25 h | 27/64 |
Isolated yield of purified product.
Additional substituents on pyrimido[1,2-a]pyrimidines could be introduced by cyclocondensations of 18•HCl with heterocyclic TMS-alkynones (Scheme 6).27 In the presence of sodium carbonate and catalytic amounts of DMAP, microwave heating at 170 °C for 30 min provided the 8-heteroaryl substituted pyrimidopyridines 30 and 31 in modest yields. In order to further improve the synthesis of these interesting derivatives, we developed a one-pot Sonogashira coupling/cyclocondensation approach (Scheme 7). Room temperature conversion of acid chlorides 32-34 and trimethylsilyl acetylene in the presence of Pd(0)-catalysts led to ynones 35-37, which were condensed with pyrimidines 18 and 19 under microwave irradiation to give pyrimidopyridines 38a-d in improved overall yields of 28-60% (Table 3). A modest preference for NCy2Me over Et3N in the Sonogashira coupling was noted for acid chloride 32 (Entries 1, 2, 5, and 6), whereas the yield of the electron-rich acyl chloride 33 was almost unchanged (Entries 3 and 7). The efficiency of the conversion of the electron-deficient acyl chloride 34 decreased in the presence of NCy2Me (Entries 4 and 8). Copper- and solvent-free reaction conditions28 also lowered yields (Entries 9 and 10).
Scheme 6.
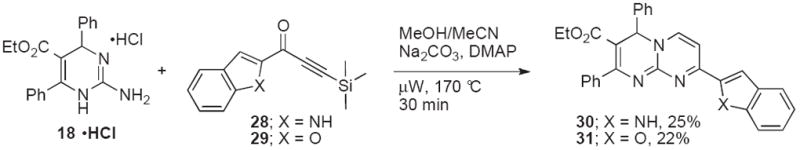
Synthesis of 8-heteroaryl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylates.
Scheme 7.
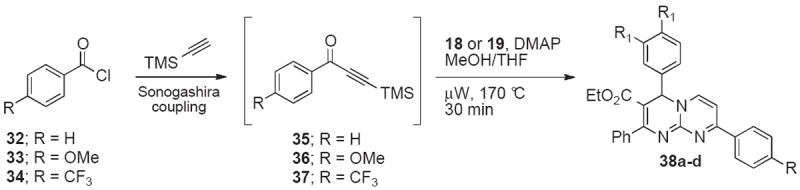
One-pot Sonogashira coupling - cyclocondensation for the synthesis of 8-aryl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylates.
Table 3.
Sonogashira couplings - cyclocondensations according to Scheme 7.
| Entry | Reactants | Sonogashira Coupling Conditions | Product/R/R1/Yield %a |
|---|---|---|---|
| 1 | 32 and 18 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), Net3, THF, rt | 38a/H/H/54 |
| 2 | 32 and 19 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), Net3, THF, rt | 38b/H/OMe/39 |
| 3 | 33 and 18 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), NEt3, THF, rt | 38c/OMe/H/32 |
| 4 | 34 and 18 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), Net3, THF, rt | 38d/CF3/H/55 |
| 5 | 32 and 18 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), NCy2Me, THF, rt | 38a/H/H/60 |
| 6 | 32 and 19 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), NCy2Me, THF, rt | 38b/H/OMe/51 |
| 7 | 33 and 18 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), NCy2Me, THF, rt | 38c/OMe/H/28 |
| 8 | 34 and 18 | Pd(PPh3)2Cl2 (2 mol %); CuI (4 mol %), NCy2Me, THF, rt | 38d/CF3/H/29 |
| 9 | 32 and 18 | Pd(OAc)2 (1 mol %); NCy2Me, 40 °C | 38a/H/H/30 |
| 10 | 34 and 18 | Pd(OAc)2 (1 mol %), NCy2Me, 40 °C | 38d/CF3/H/44 |
Isolated yield of purified product.
Pyrimidopyridines 27, 30, 31 and 38a-d were isolated as single regioisomers, and their assignment was supported by an x-ray analysis of 38d (Figure 2). The regiochemical preference for substitution at C-8 can be explained by a destabilizing steric hindrance exerted between the pseudoaxial 4-aryl group and a peri-substituent at the 6-position.
Fig. 2.
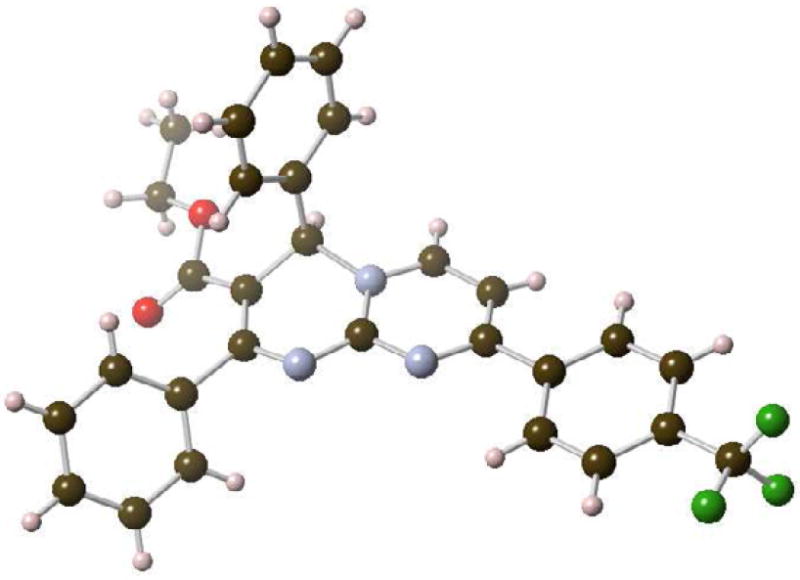
X-ray structure of 38d (CCDC 932463).
Starting with 2-aminopyrimidines 10a-c and 18, we were able to access two additional novel heterocyclic scaffolds, the 2-morpholino-6H-pyrimido[1,2-a][1,3,5]triazine-7-carboxylates 40-43, and the 4-oxo-3,4,6,9-tetrahydropyrimido[2,1-c][1,2,4,6]thiatriazine-7-carboxylate 2,2-dioxides 44 and 45 (Scheme 8). Condensation of pyrimidines 10a-c and 18 with in situ generated cyanoimine 39 furnished the pyrimidotriazines in 25-53% yield under microwave irradiation conditions. A similar transformation has recently been reported for the synthesis of aminotriazine-based anti-HIV agents.29 Alternatively, treatment of 10a and 18 with chlorosulfonylisocyanate afforded the cyclic sulfamides 44 and 45 in 15% and 40% yield, respectively. We were able to obtain an x-ray structure of 44 that confirmed the assignment (Figure 3).
Scheme 8.
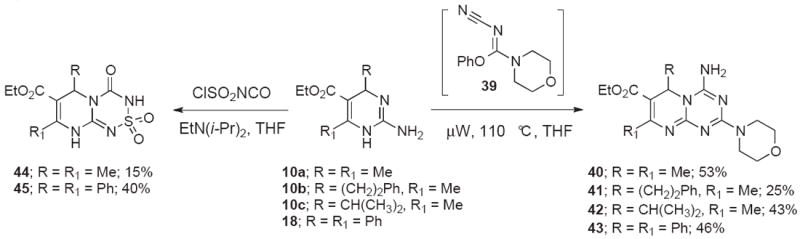
Synthesis of 2-morpholino-6H-pyrimido[1,2-a][1,3,5]triazine-7-carboxylates 40-43 and 4-oxo-3,4,6,9-tetrahydropyrimido[2,1-c][1,2,4,6]thiatriazine-7-carboxylate 2,2-dioxides 44-45.
Fig. 3.
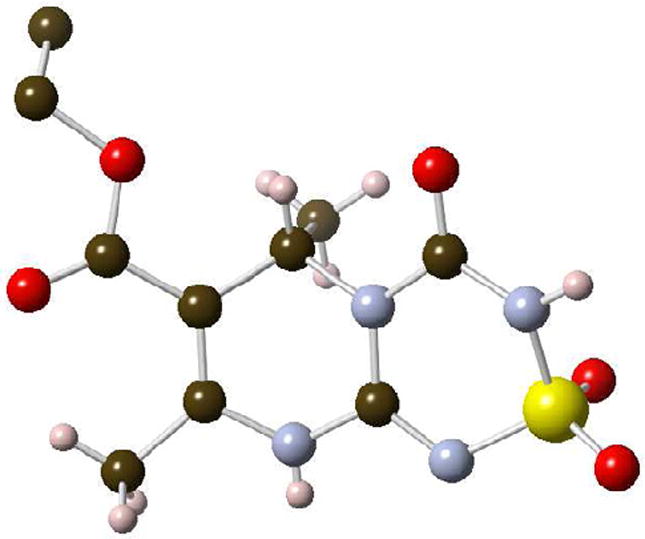
X-ray structure of 44 (CCDC 935121). Due to static disorder in the ethyl ester group, the five ethyl hydrogen positions are uncertain and are therefore not shown.
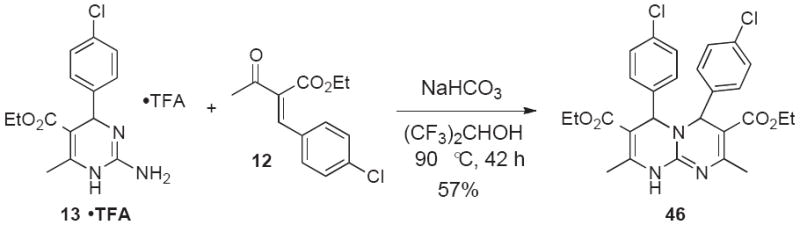
Heterocycles 40-43 and 44-45 are representative examples for target scaffolds 4 and 5 shown in Scheme 1. We were also interested in further extending our conversions of 2-aminopyrimidines and guanidinoquinazolines to the structural variants 6 and 7, which are only featured in very few reported examples in the literature.30,31 In particular, we wanted to determine if aminopyrimidine 13•TFA could be used as a substrate for a second Atwal-Biginelli condensation reaction. Although prolonged reaction times were required, we were pleased to find that the expected fused bicycle 46 was indeed formed as a 4:1 mixture of anti-syn-isomers in 57% yield (Scheme 9). The major anti-isomer was assigned based on literature precedence30 as well as ab initio HF-6-311G* calculations (Figure 4).32 The latter provided a 5.8 kcal/mol gas-phase energy difference in favor of anti-46, suggesting that the reaction was not under complete thermodynamic control. Indeed, upon heating a sample of the product mixture for 26 h in the presence of deuterated acetic acid at 90 °C in DMSO-d6, the signals characteristic for syn-46 at 7.00-6.92 and 5.35 ppm disappeared in the 1H NMR.
Scheme 9.
A second Atwal-Biginelli cyclocondensation reaction converts 2-aminopyrimidine 13•TFA into pyrimidopyrimidine 46.
Fig. 4.
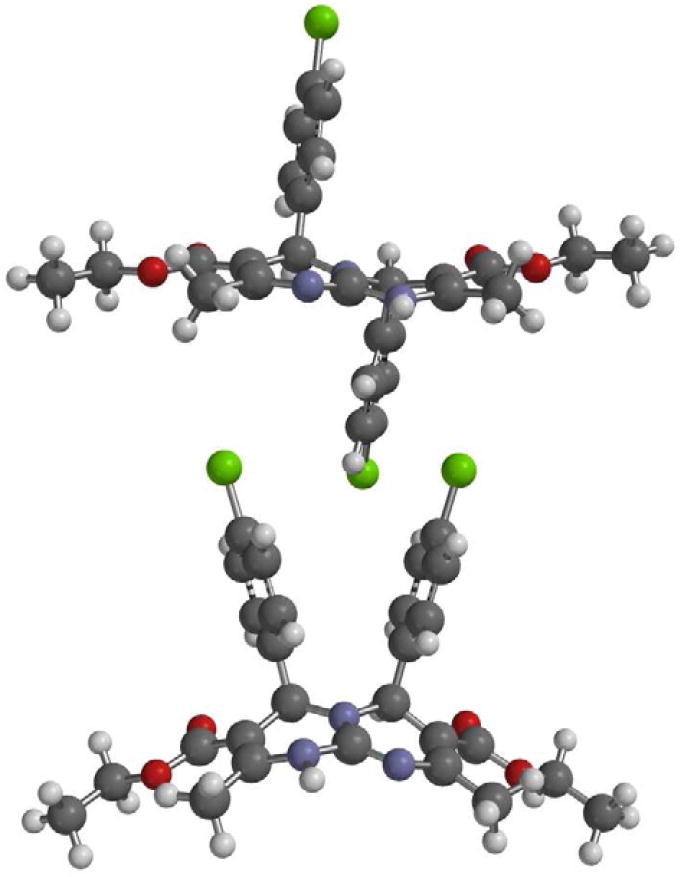
HF-6-311G* structures of anti- (top) and syn- (bottom) 46.
In an analogous synthetic sequence, guanidinoquinazolines 47a-d were converted in moderate to high yields to the amine-linked pyrimidyl-quinazolines 49a-i upon heating with 12 and 48a-d in hexafluoroisopropanol (Scheme 10 and Table 4). The cyclocondensation worked particularly well for the morpholine-substituted enone 48d (Entry 8), whereas the yield for the dimethylamino-substituted enone 48a was consistently low (Entries 4 and 9). Quinazolines 47a-d were prepared in a three-step sequence from the corresponding anilines.33
Scheme 10.
Atwal-Biginelli cyclocondensation reactions with guanidine-quinazolines 47a-d.
Table 4.
Atwal-Biginelli reactions according to Scheme 10.
| Entry | Reactants | Substituents R/R1/R2/R3 | Product/Yield %a |
|---|---|---|---|
| 1 | 47a and 12 | Cl/H/Cl/H | 49a/56 |
| 2 | 47b and 12 | Me/H/Cl/H | 49b/62 |
| 3 | 47c and 12 | F/H/Cl/H | 49c/52 |
| 4 | 47c and 48a | F/H/NMe2/H | 49d/40 |
| 5 | 47b and 48b | Me/H/H/NO2 | 49e/67 |
| 6 | 47d and 12 | -OCH2O-/Cl/H | 49f/41 |
| 7 | 47d and 48c | -OCH2O-/Br/H | 49g/57 |
| 8 | 47d and 48d | -OCH2O-/N((CH2CH2)2O)/H | 49h/81 |
| 9 | 47d and 48a | -OCH2O-/NMe2/H | 49i/45 |
Isolated yield of purified product.
3. Conclusion
Cyclocondensations of heterocyclic guanidines with bis-electrophiles provide a rapid access to previously unexplored or sparsely documented heterocyclic scaffolds. Specifically, we were able to devise concise routes to new bipyrimidines, pyrimidoaminotriazines, pyrimidosulfonamides, pyrimidopyrimidines, and amine-linked pyrimidoquinazolines. Several versatile methodologies were explored in this work: a) a one-pot methylation/aminolysis sequence for the preparation of 2-aminodihydropyrimidine intermediates (Scheme 2); b) an alternative Atwal-Biginelli cyclization of a PMB-protected guanidine for the construction of 2-aminodihydropyrimidines (Scheme 3); c) a one-pot tandem Sonogashira coupling–alkynone cyclization to produce fused bipyrimidines (Scheme 7); d) diverse cyclocondensation strategies, including consecutive Atwal-Biginelli reactions, to afford pyrimidopyrimidines, pyrimidotriazines and pyrimidothiatriazines (Schemes 5, 8 and 9). Finally, quinazoline-dihydropyrimidine amines were prepared by extending Atwal-Biginelli conditions to quinazoline guanidines (Scheme 10). We are currently exploring the activity profile of these new heterocycles in a variety of biological screens.
4. Experimental Section
4.1. General Experimental Procedures
All moisture-sensitive reactions were performed under an atmosphere of dry nitrogen and all glassware was either dried in an oven at 140 °C or flame dried under high vacuum prior to use. THF and Et2O were dried by distillation over Na/benzophenone, and CH2Cl2 and toluene were purified using an alumina filtration system. Reactions were monitored by either 1H NMR at 300 MHz in DMSO-d6, high resolution LC/MS (Thermo Scientific Exactive spectrometer), or TLC analysis (EM Science pre-coated silica gel 60 F254 plates). Visualization of TLCs was accomplished with a 254 nm UV light and by staining with a p-anisaldehyde solution (2.5 mL of p-anisaldehyde, 2 mL of AcOH, and 3.5 mL of conc. H2SO4 in 100 mL of 95% EtOH) or a KMnO4 solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of a 0.1% NaOH solution). Flash chromatography was performed on 40-63 μm silica gel (Silicycle) or on a Teledyne ISCO CombiFlash Rf. Melting points were determined using a Laboratory Devices Mel-Temp II and are not corrected. Infrared spectra were obtained on either a Nicolet Avatar 360, a Smiths Detection IdentifyIR FT-IR spectrometer, or Perkin Elmer ATR IR. Mass spectra were obtained on either a Micromass Autospec double focusing instrument or High Res LC/MS instrument (Thermo Scientific Exactive spectrometer). Purities were determined using an Agilent Technologies 385-ELSD. Microwave reactions were performed in a Biotage Initiator 2.0 Microwave reactor. 1H and 13C NMR spectra were obtained on a Bruker Avance 300, 400, 500, or 700 MHz instrument at room temperature unless otherwise noted. Chemical shifts (δ) are reported in parts per million (ppm) with the residual solvent peak used as an internal standard (CHCl3 δ 7.26 ppm for 1H and 77.00 ppm for 13C, DMSO δ 2.50 ppm for 1H and 40.45 ppm for 13C). 1H NMR spectra are tabulated as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, br=broad), integration, and coupling constant(s) (J) in Hertz (Hz). 13C NMR spectra were recorded using a proton-decoupled pulse sequence with a d1 of 5 sec, and are tabulated by the chemical shifts of the observed peaks. X-ray crystallographic data for 38d (deposition number CCDC 932463) and 44 (CCDC 935121) were deposited with the Cambridge Crystallographic Data Center.
4.2. Ethyl 4,6-dimethyl-2-thioxo-1,3,6-trihydropyrimidine-5-carboxylate (8a).20 General Procedure A
A suspension of ethyl 3-oxobutanoate (2.50 mL, 19.7 mmol), thiourea (2.20 g, 28.9 mmol), ethanal (1.10 mL, 19.7 mmol) and benzyltriethylammonium chloride (0.386 g, 1.97 mmol) was heated in a sealed tube at 75° C for 15 min. The reaction mixture turned light yellow in color and was heated at 105-100 °C for 20 h, allowed to cool to room temperature, diluted with water (20 mL), and stirred for 1 h. The resulting solid residue was isolated by vacuum filtration, washed with water (70 mL), recrystallized from boiling EtOH:H2O (3:1, 50 mL), washed with EtOH:H2O (1:1), followed by water, and dried in vacuo to afford 8a (1.81 g, 43%) as pale yellow crystals: 1H NMR (300 MHz, DMSO-d6) δ 10.12 (s, 1H), 9.19 (s, 1H), 4.15-4.03 (m, 3H), 2.19 (s, 3H), 1.19 (t, 3H, J=6.9 Hz), 1.09 (d, 3H, J=6.3 Hz). The spectral data are consistent with the literature values.20
4.3. Ethyl 4-methyl-6-(2-phenylethyl)-2-thioxo-1,3,6-trihydropyrimidine-5-carboxylate (8b).34
According to general procedure A, a suspension of thiourea (1.72 g, 22.4 mmol), benzyltriethylammonium chloride (0.29 g, 1.50 mmol), hydrocinnamaldehyde (2.08 mL, 15.0 mmol) and ethyl 3-oxobutanoate (2.50 mL, 15.0 mmol) was heated at 120 °C for 3 h. The viscous orange reaction mixture was allowed to cool to room temperature, diluted with EtOAc (150 mL), and washed with water (3 × 100 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude light orange residue was recrystallized from hexanes:EtOAc (2:1), isolated by vacuum filtration, washed with hexanes and dried in vacuo to afford 8b as a pale yellow solid (2.24 g, 49%, ~90% pure): 1H NMR (300 MHz, DMSO-d6) 10.19 (s, 1H), 9.45 (s, 1H), 7.30-7.18 (m, 5H), 4.18-4.16 (m, 1H), 4.07-4.02 (m, 2H), 2.63-2.51 (m, 2H), 2.22 (s, 3H), 1.78-1.66 (m, 2H), 1.13 (t, 3H, J=7.2 Hz). The spectral data are consistent with the literature values.34
4.4. Ethyl 4-methyl-6-(methylethyl)-2-thioxo-1,3,6-trihydropyrimidine-5-carboxylate (8c).20
According to general procedure A, a suspension of thiourea (2.75 g, 35.9 mmol), citric acid (2.30 g, 12.0 mmol), isobutyraldehyde (2.22 mL, 23.9 mmol) and ethyl 3-oxobutanoate (4.0 mL, 23.2 mmol) was heated at 120 °C for 15 h. The viscous yellow/orange reaction mixture was allowed to cool to room temperature, diluted with EtOAc (300 mL), and washed with water (3 × 200 mL). The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. The resulting light orange residue was slowly triturated with EtOAc (30 mL) and hexanes (60 mL) at room temperature. The suspension was cooled to -20 °C for 24 h and the resulting off-white precipitate was isolated by vacuum filtration, washed with hexanes:EtOAc (3:1, 100 mL), followed by hexanes (30 mL), and dried in vacuo to afford 8c as an off-white solid (2.13 g, 33%, ~90% pure): 1H NMR (300 MHz, DMSO-d6) 10.09 (s, 1H), 9.23 (s, 1H), 4.15-4.02 (m, 2H), 4.00-3.97 (m, 1H), 2.23 (s, 3H), 1.71-1.65 (m, 1H), 1.20 (t, 3H, J=7.2 Hz), 0.82 (d, 3H, J=6.0 Hz), 0.76 (d, 3H, J=6.0 Hz). The spectral data are consistent with the literature values.20
4.5. Ethyl 2-amino-4,6-dimethyl-1,4-dihydropyrimidine-5-carboxylate (10a). General Procedure B
To a light yellow suspension of 8a (1.50 g, 7.00 mmol) in chlorobenzene (40 mL) at 0 °C was added methyl trifluoromethanesulfonate (0.91 mL, 7.7 mmol) in one portion. The reaction mixture was allowed to warm to room temperature and stirred for 3.5 h. Ammonium acetate (5.4 g, 70 mmol) was added and ammonia gas was bubbled through the cold (0 °C) reaction mixture for 10 min. The reaction tube was sealed and heated at 95-100 °C for 17 h. The reaction mixture was cooled to room temperature, diluted with saturated Na2CO3 (250 mL) and water (250 mL), extracted with EtOAc (3 × 400 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude residue was recrystallized from hexanes:EtOAc (1:1, 150 mL), isolated by vacuum filtration, washed with hexanes, and dried in vacuo to afford 10a as a colorless solid (0.84 g, 61%, ELSD purity 97.3%): Mp 196-198 °C; IR (ATR) 3390, 3303, 3088, 2975, 1661, 1584, 1493, 1370, 1329, 1303, 1226, 1154, 1092, 1066 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 6.81 (br s, 1H), 6.12 (br s, 2H), 4.18 (q, J=6.3 Hz, 1H), 4.05-3.95 (m, 2H), 2.10 (s, 3H), 1.16 (t, 3H, J=7.2 Hz), 0.97 (d, 3H, J=6.3 Hz); 13C NMR (75 MHz, DMSO-d6,) δ 166.5, 160.8, 156.0, 98.6, 58.4, 45.5, 23.9, 15.0; HRMS (TOF ES+) m/z calcd for C9H16N3O2 198.1243, found 198.1242.
4.6. Ethyl 2-amino-6-methyl-4-(2-phenethyl)-1,4-dihydropyrimidine-5-carboxylate (10b)
According to general procedure B, a mixture of 8b (2.50 g, 8.21 mmol) and methyl trifluoromethanesulfonate (1.1 mL, 9.0 mmol) in chlorobenzene (80 mL) was treated with ammonium acetate (6.3 g, 82 mmol) and ammonia gas to give the crude product which was dissolved in CH2Cl2 (20 mL) and triturated with hexanes to yield a tan oil. This oil was isolated by decantation and again dissolved in CH2Cl2 (20 mL), triturated with hexanes (30 mL) and cooled to -20 °C to afford 10b as a pale yellow solid (0.92 g, 39%, ELSD purity 97.3%): Mp 129-134 °C; IR (ATR) 3390, 3348, 2975, 1650, 1584, 1465, 1374, 1238, 1172, 1206, 1146, 1077, 1029 cm-1; 1H-NMR (300 MHz; DMSO-d6) δ 7.29-7.15 (m, 6H), 6.87 (br s, 2H), 4.14 (m, 1H), 3.98-3.88 (m, 2H), 2.71-2.59 (m, 1H), 2.54-2.44 (m, 1H), 2.13 (s, 3H), 1.66-1.56 (m, 2H), 1.10 (t, 3H, J=6.9 Hz); 13C NMR (75 MHz, DMSO-d6) δ 165.8, 158.9, 155.1, 141.7, 128.3 (2 C), 128.2 (2 C), 125.6, 97.8, 58.2, 48.5, 38.2, 29.7, 22.6, 14.4; HRMS (TOF ES+) m/z calcd for C16H22N3O2 288.1712, found 288.1720.
4.7. Ethyl 2-amino-4-(methylethyl)-6-methyl-1,4-dihydropyrimidine-5-carboxylate (10c)
According to general procedure B, a mixture of 8c (1.80 g, 7.42 mmol) and methyl trifluoromethanesulfonate (0.96 mL, 8.2 mmol) in chlorobenzene (70 mL) was treated with ammonium acetate (6.0 g, 78 mmol) and ammonia gas to give the crude product which was purified by chromatography through a plug of SiO2 (EtOAc to EtOH). The product slowly eluted with EtOH and was recrystallized from boiling CH2Cl2:hexanes (1:1, 40 mL) to afford 10c as a white crystalline solid (0.581 g, 33%, ELSD purity 100.0%): Mp 171-173 °C; IR (ATR) 3497, 3353, 2951, 2869, 1653, 1596, 1489, 1377, 1225, 1154, 1068, 995, 816 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 6.87 (br s, 1H), 6.07 (br s, 2H), 4.04-3.92 (m, 3H), 2.12 (s, 3H), 1.61-1.54 (m, 1H), 1.15 (t, 3H, J=7.2 Hz), 0.78 (d, 3H, J=6.9 Hz), 0.71 (d, 3H, J=6.9 Hz); 13C NMR (75 MHz; DMSO-d6) δ 166.5, 161.1, 156.2, 95.7, 57.9, 54.4, 34.6, 23.5, 18.1, 15.9, 14.4; HRMS (TOF ES+) m/z calcd for C11H20N3O2 226.1556, found 226.1569.
4.8. Ethyl 2-imino-4,6-dimethyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate hydrochloride (10a•HCl). General Procedure C
To a suspension of 10a (0.500 g, 2.54 mmol) in MeOH (5.0 mL) was added a solution of HCl in dioxane (1.5 mL, 3.0 mmol, 2.0 M) dropwise at room temperature. After 10 min, the solution was clear and the solvents were removed under reduced pressure. The resulting oil was triturated with ether (2 × 10 mL), isolated by vacuum filtration, and dried in vacuo to afford 10a•HCl as a colorless foam (0.54 g, 91%): IR (ATR) 3361, 3236, 3055, 2975, 2869, 1681, 1551, 1241, 1096, 1070 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 10.90 (s, 1H), 9.06 (s, 1H), 7.89 (br s, 2H), 4.38-4.36 (m, 1H), 4.19-4.08 (m, 2H), 2.26 (s, 3H), 1.22 (t, 3H, J=7.2 Hz), 1.16 (d, 3H, J=6.3 Hz); 13C NMR (75 MHz, DMSO-d6) δ 164.9, 151.8, 144.1, 104.5, 60.5, 45.4, 22.9, 17.9, 14.6. For additional characterization data, see 10a.
4.9. Ethyl 2-imino-6-methyl-4-(2-phenethyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylate hydrochloride (10b•HCl)
According to general procedure C, 10b (0.150 g, 0.522 mmol) was combined with a solution of HCl in ether (0.31 mL, 0.63 mmol, 2.0 M) and MeOH (1.0 mL) at room temperature to afford 10b•HCl as a pale yellow solid (0.143 g, 85%): 1H NMR (300 MHz, DMSO-d6) δ 10.59 (br s, 1H), 9.18 (br s, 1H), 7.76 (br s, 2H), 7.31-7.16 (m, 5H), 4.33-4.30 (m, 1H), 4.14-4.02 (m, 2H), 2.73-2.49 (m, 2H), 2.26 (s, 3H), 1.78-1.09 (m, 2H), 1.15 (t, 3H, J=6.9 Hz). For additional characterization data, see 10b.
4.10. Ethyl 2-imino-4-(methylethyl)-6-methyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate hydrochloride (10c•HCl)
According to general procedure C, 10c (0.150 g, 0.666 mmol) was combined with a solution of HCl in ether (0.40 mL, 0.80 mmol, 2.0 M) in MeOH (1.5 mL) at room temperature to afford 10c•HCl as a pale yellow solid (0.121 g, 69%): 1H NMR (300 MHz, DMSO-d6) δ 10.77 (br s, 1H), 9.14 (br s, 1H), 7.84 (br s, 2H), 4.19-4.07 (m, 3H), 2.28 (s, 3H), 1.78-1.72 (m, 1H), 1.21 (t, 3H, J=7.2 Hz), 0.87 (d, 3H, J=6.9 Hz), 0.77 (d, 3H, J=6.9 Hz). For additional characterization data, see 10c.
4.11. 1-(4-(4-Chlorophenyl)-2-((4-methoxybenzylamino)-6-methyl-1,4-dihydropyrimidin-5yl)butan-1-one (13). General Procedure D
A microwave vial was charged with 1123 (0.63 g, 2.8 mmol) and (CF3)2CHOH (5.0 mL). To this homogeneous solution was added 1236,37 (1.4 g, 5.5 mmol) and NaHCO3 (0.26 g, 3.0 mmol). The vial was sealed and immersed into a pre-heated oil bath at 90 °C and heated for 16 h. The solution was allowed to cool to room temperature, filtered through a small plug of Celite, and rinsed with CH2Cl2. The filtrate was concentrated under reduced pressure and purified by chromatography on SiO2 (ISCO-Rf, 0-10%MeOH/CH2Cl2) to afford 13 as an orange crystalline solid (0.485 g, 42%, ELSD purity 99.7%): Mp 60-64 °C; IR (neat) 3311, 2929, 2830, 1661, 1574, 1512, 1212, 1088, 818 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.22-7.11 (m, 6H), 6.78 (d, 2H, J=8.4 Hz), 5.29 (s, 1H), 4.33 (d, 1H, J=15.3 Hz), 4.25 (d, 1H, J= 15.0 Hz), 4.03 (q, 2H, J=7.2 Hz), 3.78 (s, 3H), 2.38 (s, 3H), 1.14 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 166.6, 159.2, 153.0, 143.2, 133.2 (2 C), 128.9, 128.6 (2 C), 128.3 (2 C), 127.9 (2 C), 114.2 (2 C), 100.0, 59.5, 55.2, 53.7, 44.6, 22.4, 14.2; HRMS (ESI+) m/z calcd for C22H25O3N3Cl (M+H) 414.1579, found 414.1562.
4.12. Ethyl 2-amino-4-(4-chlorophenyl)-6-methyl-1,4-dihydroprimidine-5-carboxylate 2,2,2-trifluoroacetate (13•TFA)
A solution of 13 (0.420 g, 1.01 mmol) in a mixture of TFA/Et3SiH/thioanisole/H2O (92.5:2.5:2.5:2.5, 23 mL) was stirred at 50 °C for 22 h, allowed to cool to room temperature and concentrated under reduced pressure. The crude orange residue was purified by trituration with tert-butyl methyl ether, isolated by vacuum filtration, and dried in vacuo to give trifluoroacetate salt 13•TFA (0.29 g, 70%, ELSD purity 99.9%) as a white solid: Mp 207-218 °C (dec.); IR (neat) 3241, 3205, 3084, 2963, 2901, 1709, 1666, 1239, 1200, 1186, 1139, 1096, 848, 828, 800, 771, 726 cm-1; 1H NMR (300 MHz, CDCl3:MeOD, 1:1) δ 7.03 (d, 2H, J=8.4 Hz), 6.94 (d, 2H, J=8.4 Hz), 5.14 (s, 1H), 3.80 (q, 2H, J=7.2 Hz), 2.13 (s, 3H), 0.87 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, CDCl3:MeOD, 1:1) δ 164.0, 150.3, 143.1, 139.7, 133.7, 128.3(2 C), 127.5 (2 C), 103.1, 60.1, 52.1, 16.4, 12.8; HRMS (ESI+) m/z calcd for C14H17O2N3Cl (M+H) 294.1004, found 294.0999.
4.13. Ethyl 2-amino-4,6-diphenyl-1,4-dihydropyrimidine-5-carboxylate (18).30 General Procedure E
To a solution of benzaldehyde (16, 2.0 mL, 20 mmol), urea 14•HCl (3.32 g, 35.0 mmol) and NaHCO3 (6.72 g, 80.0 mmol) in DMF (40 mL) was added ethyl 3-oxo-3-phenylpropanoate (15, 3.80 mL, 22.5 mmol) at room temperature. The reaction mixture was heated to 70 °C for 17 h, cooled to room temperature, and slowly diluted with water (100 mL). The aqueous mixture was stirred at room temperature for 1 h and the resulting solid was separated by decanting and vacuum filtration. The crude product was purified by heating in hexanes:EtOAc (1:1, 100 mL) for 30 min, separated by vacuum filtration, and dried in vacuo to afford 18 as a white solid (3.35 g, 52%): 1H NMR (300 MHz, DMSO-d6,) δ 7.48 (s, 1H), 7.39-7.31 (m, 4H), 7.27-7.21 (m, 6H), 6.29 (s, 2H), 5.30 (s, 1H), 3.67 (q, 2H, J=7.2 Hz), 0.73 (t, 3H, J=7.2 Hz); MS (EI+) m/z 321 (M+, 43), 292 (38), 274 (32), 248 (52), 244 (100), 104 (37), 76 (42); HRMS (EI+) m/z calcd for C19H19N3O4 321.1477, found 321.1476. The spectral data are consistent with the literature values.18
4.14. Ethyl 2-imino-4,6-diphenyl-1,2,3,4-tetrahydropyrimidine-5-carboxylate hydrochloride (18•HCl)
According to general procedure C, 18 (1.28 g, 3.97 mmol) and a solution of HCl in dioxane (1.25 mL, 5.00 mmol, 4.0 M) and methanol (5.0 mL) afforded 18•HCl as a white crystalline solid (1.42 g, 99%): 1H NMR (300 MHz, DMSO-d6) δ 10.86 (s, 1H), 9.75 (s, 1H), 8.09 (br s, 2H), 7.51-7.33 (m, 10H), 5.55 (d, 1H, J=3.6 Hz), 3.78 (q, 2H, J=6.9 Hz), 0.74 (t, 3H, J=6.9 Hz); 13C NMR (75 MHz, DMSO-d6) δ 164.6, 151.7, 144.6, 142.2, 133.7, 130.3, 129.4 (2 C), 129.1 (2 C), 128.8, 128.6 (2 C), 127.1 (2 C), 104.2, 60.4, 52.8, 13.7. For additional characterization data, see 18.
4.15. Ethyl 2-amino-4-(3,4-dimethoxyphenyl)-6-phenyl-1,4-dihydropyrimidine-5-carboxylate (19)
According to general procedure E, ethyl 3-oxo-3-phenylpropanoate (15, 3.80 mL, 22.5 mmol), 3,4-dimethoxybenzaldehyde (17, 3.32 g, 20.0 mmol), urea 14•HCl (2.31 g, 24.0 mmol) and NaHCO3 (6.33 g, 75.4 mmol) in DMF (40 mL) provided the crude product which was recrystallized from EtOAc:hexanes (2:1, 150 mL), isolated by vacuum filtration, washed with hexanes (50 mL) and dried in vacuo to afford 19 as a white crystalline solid (3.11 g, 41%, ELSD purity 100.0%): Mp 261-263 °C (dec.); IR (ATR) 3403, 3340, 2974, 1655, 1627, 1565, 1372, 1232, 1135, 1100, 1027 cm-1; 1H NMR (300 MHz, DMSO-d6,) δ 7.44 (s, 1H), 7.25-7.21 (m, 5H), 6.98 (s, 1H), 6.93-6.86 (m, 2H), 6.28 (br s, 2H), 5.23 (s, 1H), 3.72 (s, 6H), 3.67, (q, 2H, J=7.2 Hz), 0.75 (t, 3H, J=7.2 Hz); 13C NMR (75 MHz, DMSO-d6,) δ 166.7, 161.6, 155.9, 149.0, 148.4, 143.2, 139.3, 128.5 (2 C), 127.4, 127.3 (2 C), 118.7, 112.2, 110.8, 98.0, 58.5, 56.0, 55.8, 52.9, 14.2; MS (EI+) m/z 381 (M+, 36), 352 (31), 308 (86), 244 (100), 198 (35), 77 (56); HRMS (EI+) m/z calcd for C21H23N3O4 381.1688, found 381.1687.
4.16. Ethyl 2,4-dimethyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (22). General Procedure F
A suspension of 10a•HCl (0.100 g, 0.428 mmol) and 1,1,3,3-tetramethoxypropane (20, 0.09 mL, 0.5 mmol) in trifluoroethanol (21, 1.5 mL) was heated in a microwave reactor at 160 °C for 1 h. The resulting mixture was dissolved in EtOAc (50 mL), washed with saturated sodium carbonate (50 mL), and extracted with EtOAc (2 × 50 mL). The organic layers were combined, dried (Na2SO4), filtered, and concentrated under reduced pressure. The resulting red/orange oil was purified via chromatography on neutral Al2O3 (0-2% MeOH/CH2Cl2) to afford 22 as a red/orange solid (0.076 g, 76%): Mp 151-152 °C; IR (ATR) 3076, 2970, 1657, 1622, 1585, 1491, 1437, 1396, 1273, 1245, 1206, 1109, 1070, 990 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.51 (dd, 1H, J=2.4, 1.8 Hz), 8.24 (dd, 1H, J=2.1, 6.3 Hz), 6.70 (dd, 1H, J=3.9, 6.3 Hz), 5.26 (q, 1H, J=6.6 Hz), 4.18-4.03 (m, 2H), 2.25 (s, 3H), 1.23 (t, 3H, J=7.2 Hz), 1.16 (d, 3H, J=6.6 Hz); 13C NMR (75 MHz, DMSO-d6) δ 165.1, 163.4, 157.6, 151.5, 147.2, 110.3, 100.3, 59.5, 56.8, 23.4, 22.2, 14.8; HRMS (ESI+) m/z calcd for C12H16N3O2 234.1243, found 234.1243.
4.17. Ethyl 2-methyl-4-phenethyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (23)
According to general procedure F, a mixture of 10b•HCl (0.138 g, 0.426 mmol) and 1,1,3,3-tetramethoxypropane (20, 0.09 mL, 0.5 mmol) in trifluoroethanol (21, 2.0 mL) was heated in a microwave reactor at 160 °C for 2 h. The crude red/orange oil was purified by chromatography on neutral Al2O3 (EtOAc to 5% MeOH/CH2Cl2) to afford 23 as a yellow/brown oil (0.098 g, 71%, ELSD purity 99.5%): IR (ATR) 2981, 2925, 1665, 1618, 1579, 1491, 1430, 1393, 1275, 1238, 1202, 1096, 1063, 774, 697 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.52 (dd, 1H, J=3.9, 2.1 Hz), 8.18 (dd, 1H, J=6.6, 2.1 Hz), 7.26-7.08 (m, 5H), 6.70 (dd, 1H, J=3.9, 6.3 Hz), 5.21 (t, 1H, J=5.1 Hz), 4.20-4.07 (m, 2H), 2.46-2.37 (m, 2H), 2.28 (s, 3H), 1.86-1.76 (m, 2H), 1.19 (t, 3H, J=7.2 Hz); 13C NMR (75 MHz, DMSO-d6) δ 164.7, 162.6, 158.0, 151.3, 147.0, 140.5, 128.4 (2 C), 128.0 (2 C), 125.9, 109.2, 99.0, 59.8, 59.1, 36.3, 30.3, 23.1, 14.4; HRMS (TOF ESI+) m/z calcd for C19H22N3O2 324.1712, found 324.1707.
4.18. Ethyl 4-isopropyl-2-methyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (24)
According to general procedure F, a mixture of 10c•HCl (0.113 g, 0.432 mmol) and 1,1,3,3-tetramethoxypropane (20, 0.076 mL, 0.45 mmol) in trifluoroethanol (21, 1.5 mL) was heated in a microwave reactor at 160 °C for 2 h. The crude red/orange oil was purified by chromatography on neutral Al2O3 (0-2% MeOH/CH2Cl2) to afford 24 as a dark red oil (0.080 g, 71%, ELSD purity 98.4%): IR (ATR) 3064, 2964, 1670, 1620, 1579, 1491, 1432, 1378, 1294, 1234, 1204, 1094, 1063, 999, 774 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 8.52 (dd, 1H, J=4.0, 2.4 Hz), 8.21 (dd, 1H, J=6.4, 2.0 Hz), 6.70 (dd, 1H, J=6.4, 4.0 Hz), 4.94 (d, 1H, J=6.4 Hz), 4.18-4.04 (m, 2H), 2.28 (s, 3H), 1.79-1.74 (m, 1H), 1.22 (t, 3H, J=7.2 Hz), 0.77 (d, 3H, J=6.8 Hz), 0.63 (d, 3H, J=6.8 Hz); 13C NMR (100 MHz, DMSO-d6) δ 165.4, 162.3, 157.6, 151.5, 147.4, 108.7, 98.3, 64.6, 59.0, 34.1, 23.1, 18.0, 17.4, 14.4; HRMS (TOF ESI+) m/z calcd for C14H20N3O2 262.1556, found 262.1558.
4.19. Ethyl 2,4-diphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (25)
According to general procedure F, a solution of 18•HCl (0.100 g, 0.311 mmol) and 1,1,3,3-tetramethoxypropane (20, 0.055 mL, 0.33 mmol) in trifluoroethanol (21, 1.5 mL) was heated in a microwave reactor at 150 °C for 1.5 h. The crude red/orange oil was purified by chromatography on SiO2 (EtOAc) to afford 25 as a red/orange solid containing ~5-6% EtOAc (0.047 g, 42%, ELSD purity 100.0%): Mp 185-186.5 °C; IR (ATR) 3070, 2982, 2869, 1694, 1612, 1689, 1558, 1493, 1476, 1441, 1277, 1232, 1189, 1074 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 8.55-8.54 (m, 2H), 7.42-7.30 (m, 10H), 6.78 (dd, 1H, J=4.8. 5.6 Hz), 6.45 (s, 1H), 3.80 (q, 2H, J=7.2 Hz), 0.80 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, DMSO-d6) δ 165.6, 163.5, 158.5, 151.7, 147.5, 141.5, 141.2, 129.5 (2 C), 129.1 (2 C), 128.5, 127.7 (2 C), 126.7 (2 C), 110.7, 98.7, 63.1, 59.6, 14.0; HRMS (TOF ESI+) m/z calcd for C22H20N3O2 358.1556, found 358.1556.
4.20. Ethyl 8-methyl-2,4-diphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (27)
A suspension of 18 (0.161 g, 0.500 mmol) and 26 (0.051 mL, 0.500 mmol) in distilled THF (1.0 mL) was heated in a microwave reactor for 15 min at 150 °C. The crude reaction mixture was recrystallized from hexanes:EtOAc (4:1, 20 mL), isolated by vacuum filtration, washed with hexanes, and dried in vacuo to afford 27 as a yellow-orange solid (0.12 g, 64%, ELSD purity 99.5%): Mp 185-187 °C; IR (ATR) 3068, 2969, 1705, 1625, 1511, 1448, 1241, 1196, 1085 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.38 (d, 1H, J=6.9 Hz), 7.42-7.32 (m, 10H), 6.72 (d, 1H, J=6.6 Hz), 6.38 (s, 1H), 3.79 (q, 2H, J=7.2 Hz), 2.32 (s, 3H), 0.79 (t, 3H, J=7.2 Hz); 13C NMR (75 MHz, DMSO-d6) δ 173.3, 165.7, 159.1, 151.4, 146.2, 141.8, 141.4, 129.4 (2 C), 129.1, 129.0 (2 C), 128.5, 127.6 (2 C), 126.7 (2 C), 111.2, 98.1, 62.8, 59.5, 25.1, 14.0; MS (EI+) m/z 371 (M+, 43), 342 (51), 298 (100), 278 (63), 93 (69), 66 (52); HRMS (EI+) m/z calcd for C23H21N3O2 371.1634, found 371.1618.
4.21. Ethyl 8-(1H-indol-2-yl)-2,4-diphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (30). General Procedure G
A suspension of 18•HCl (0.150 g, 0.419 mmol), 28 (0.111 g, 0.461 mmol), sodium carbonate (0.067 g, 0.63 mmol), and DMAP (0.013 g, 0.10 mmol) in MeOH (0.34 mL, 8.4 mmol) and acetonitrile (2.5 mL) was heated in a microwave reactor at 170 °C for 30 min. The dark red/orange reaction mixture was concentrated under reduced pressure and purified by chromatography on SiO2 (1:1, EtOAc:hexanes). The resulting product was recrystallized from boiling hexanes:EtOAc (6:1), isolated by vacuum filtration, washed with hexanes, and dried under high vacuum to afford 30 as a red solid (0.050 g, 25%, ELSD purity 99.6%): Mp 170-172 °C; IR (ATR) 3202, 3057, 2975, 1666, 1610, 1472, 1435, 1277, 1251, 1221, 1195, 1092, 753 cm-1; 1H-NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 8.49 (d, 1H, J=6.8 Hz), 7.63 (d, 1H, J=8.0 Hz), 7.50-7.32 (m, 13H), 7.22 (t, 1H, J=7.2 Hz), 7.05 (t, 1H, J=7.2 Hz), 6.42 (s, 1H), 3.83 (q, 2H, J=7.2 Hz), 0.83 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, DMSO-d6) δ 165.3, 160.2, 158.7, 151.3, 146.2, 141.5, 140.8, 138.6, 133.7, 129.0 (2 C), 128.8 (2 C), 128.5, 128.1, 127.7, 127.1 (2 C), 126.2 (2 C), 124.9, 121.7, 120.2, 112.7, 108.1, 106.6, 97.8, 62.2, 59.0, 13.6; HRMS (TOF ES+) m/z calcd for C30H25N4O2 473.1978, found 473.1993.
4.22. Ethyl 8-(benzofuran-2-yl)-2,4-diphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (31)
According to general procedure G, a suspension of 18•HCl (0.150 g, 0.419 mmol), 29 (0.112 g, 0.461 mmol), sodium carbonate (0.067 g, 0.63 mmol), and DMAP (0.013 g, 0.10 mmol) in MeOH (0.34 mL, 8.4 mmol) and acetonitrile (2.5 mL) provided a crude product which was purified by chromatography on SiO2 (1:1, EtOAc:hexanes). The resulting product was recrystallized (1:15, EtOAc:hexanes, 16 mL), isolated by vacuum filtration, washed with hexanes (5 mL) and dried in vacuo to afford 31 as a red/orange solid containing ~5% EtOAc (0.044 g, 22%, ELSD purity 99.1%): Mp 195-196 °C; IR (ATR) 3064, 2975, 1614, 1586, 1504, 1474, 1446, 1430, 1374, 1305, 1281, 1247, 1224, 1206, 1165, 1146, 1092, 761 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.68 (d, 1H, J=6.6 Hz), 7.98 (s, 1H), 7.82 (d, 1H, J=7.8 Hz), 7.71 (d, 1H, J=8.4 Hz), 7.53-7.33 (m, 13H), 6.51 (s, 1H), 3.83 (q, 2H, J=6.9 Hz), 0.82 (t, 3H, J=6.9 Hz); 13C NMR (125 MHz, DMSO-d6) δ 165.5, 159.0, 157.6, 156.1, 151.7, 151.2, 148.3, 141.3, 140.3, 129.6 (2 C), 129.2 (3 C), 128.8, 128.4, 128.1, 127.8 (2 C), 126.9 (2 C), 124.6, 123.6, 113.0, 112.4, 107.2, 99.3, 63.0, 59.8, 14.0; HRMS (TOF ES+) m/z calcd for C30H24N3O3 474.1818, found 474.1802.
4.23. 1-Phenyl-3-(trimethylsilyl)prop-2-yn-1-one (35).27,35
To a suspension of bis(triphenylphosphine)palladium(II) chloride (0.035 g, 0.050 mmol) and copper iodide (0.019 g, 0.10 mmol) in freshly distilled THF (10 mL) were added sequentially Et3N (0.35 mL, 2.5 mmol), benzoyl chloride (0.29 mL, 2.5 mmol) and TMS-acetylene (0.35 mL, 2.5 mmol). The reaction mixture turned from dark orange to pale yellow forming the Et3N•HCl precipitate over a 1 h period. TLC analysis (10:1, hexanes:EtOAc, product Rf = 0.59) showed full conversion after 1 h. The Et3N•HCl salt was filtered off and the filtrate was concentrated under reduced pressure. The crude product was purified by chromatography on SiO2 (10:1:0.1, hexanes:EtOAc:Et3N) to afford 35 as a pale orange oil (0.48 g, 95%): Rf 0.59 (10:1, hexanes:EtOAc); 1H-NMR (300 MHz, CDCl3) δ 8.18-8.14 (m, 2H), 7.63-7.60 (m, 1H), 7.53-7.48 (m, 2H), 0.33 (s, 9H); MS (EI+) m/z 202 (M+, 43), 187 (100), 159 (38), 105 (24), 76 (31); HRMS (EI) m/z calcd for C12H14OSi 202.0814, found 202.0814. The spectral data are consistent with the literature values.27,35
4.24. Ethyl 2,4,8-triphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (38a). General Procedure H
To a suspension of bis(triphenylphosphine)palladium(II) chloride (0.007 g, 0.01 mmol) and copper iodide (0.004 g, 0.02 mmol) in freshly distilled THF (0.75 mL) was added sequentially N,N-dicyclohexylmethyl amine (0.11 mL, 0.50 mmol), benzoyl chloride (0.058 mL, 0.50 mmol) and TMS-acetylene (0.071 mL, 0.50 mmol). The reaction mixture turned from dark orange to pale yellow forming the Cy2MeN•HCl precipitate over a 1.5 h period. TLC analysis (10:1 hexanes:EtOAc; Rf = 0.62) showed a quantitative formation of 35 after 1.5 h. The mixture was diluted to 0.15 M with THF (2.3 mL), and 18 (0.13 g, 0.40 mmol), DMAP (0.012 g, 0.10 mmol), and MeOH (0.40 mL) were added. The solution was heated in a microwave reactor at 170 °C for 30 min. The dark red-black reaction mixture was diluted with CH2Cl2 (30 mL) and washed with saturated NaHCO3 (1 × 30 mL). The organic layer was dried (Na2SO4) and concentrated under reduced pressure. The resulting dark red oil was purified by chromatography on SiO2 (1:1, hexanes:EtOAc) to afford 38a as a red solid (0.11 g, 60%, ELSD purity 100%): Mp 210-211 °C; IR (ATR) 3068, 2995, 1705, 1616, 1502, 1446, 1210, 1075 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.63 (d, 1H, J=6.9 Hz), 8.16 (d, 2H, J=6.9 Hz), 7.60-7.32 (m, 14H), 6.49 (s, 1H), 3.81 (q, 2H, J=6.9 Hz), 0.80 (t, 3H, J=6.9 Hz); 13C NMR (100 MHz, DMSO-d6) δ 166.9, 165.2, 158.5, 151.1, 147.4, 141.3, 140.9, 134.9, 132.4, 129.0 (4 C), 128.6 (3 C), 128.0, 127.8 (2 C), 127.2 (2 C), 126.3 (2 C), 106.7, 98.2, 62.3, 59.1, 13.6; HRMS (TOF MS ES) m/z calcd for C28H24N3O2 (M+H) 434.1869, found 434.1882.
4.25. Synthesis of 38a under copper- and solvent-free conditions
A mixture of palladium(II) acetate (0.001 g, 0.005 mmol), benzoyl chloride (0.058 mL, 0.50 mmol), TMS-acetylene (0.078 mL, 0.55 mmol) and N,N-dicyclohexylmethylamine (0.11 mL, 0.50 mmol) was heated in a sealed tube at 40 °C for 2 h. The reaction mixture turned heterogeneous and dark brown. After 2 h, TLC analysis (10:1, hexanes:EtOAc; Rf = 0.63) showed a nearly quantitative formation of 35. The reaction mixture was diluted to a concentration of 0.15 M with THF (3.0 mL) and 18 (0.129 g, 0.400 mmol), DMAP (0.012 g, 0.10 mmol), and MeOH (0.40 mL) were added. The solution was heated in a microwave reactor at 170 °C for 30 min. The resulting dark red reaction mixture was diluted with CH2Cl2 (30 mL) and washed with saturated NaHCO3 solution (1 × 30 mL). The organic layer was dried (Na2SO4), concentrated under reduced pressure, and the crude residue was purified by chromatography on SiO2 (2:1 to 1:1, hexanes:EtOAc). The resulting red solid was dissolved in EtOAc (1.0 mL) and crystallized by slowly adding hexanes (20 mL) at 0 °C. The solid was isolated by vacuum filtration, washed with hexanes, and dried in vacuo to afford 38a (0.053 g, 30%). The spectral data are consistent with the values obtained in 4.24.
4.26. Ethyl 4-(3,4-dimethoxyphenyl)-2,8-diphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (38b)
According to general procedure H, a solution of bis(triphenylphosphine)palladium(II) chloride (0.007 g, 0.01 mmol), copper iodide (0.004 g, 0.02 mmol), benzoyl chloride (0.058 mL, 0.50 mmol), TMS-acetylene (0.071 mL, 0.50 mmol) and N,N-dicyclohexylmethylamine (0.11 mL, 0.50 mmol) in freshly distilled THF (0.75 mL) provided 35. After addition of THF (2.3 mL), treatment with 19 (0.15 g, 0.40 mmol), DMAP (0.012 g, 0.10 mmol) and MeOH (0.40 mL) provided the crude product which was purified by chromatography on SiO2 (1:1 to 2:1, EtOAc:hexanes). The resulting solid was dissolved in EtOAc (1.0 mL) and crystallized by slowly adding hexanes (20 mL) at 0 °C. The precipitated solid was isolated by vacuum filtration, washed with hexanes, and dried in vacuo to afford 38b as an orange solid (0.10 g, 51%, ELSD purity 99.9%): Rf 0.21 (2:1, EtOAc:hexanes); Mp 95-96 °C; IR (ATR) 3086, 2987, 2932, 1662, 1618, 1517, 1435, 1215, 1075, 1027 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.66 (d, 1H, J=6.9 Hz), 8.14 (d, 2H, J=7.2 Hz), 7.60-7.51 (m, 3H), 7.46-7.36 (m, 6H), 7.14 (s, 1H), 7.00-6.94 (m, 2H), 6.39 (s, 1H), 3.79 (q, 2H, J=7.2 Hz), 3.73 (s, 3H), 3.71 (s, 3H), 0.81 (t, 3H, J=7.2 Hz); 13C NMR (75 MHz, DMSO-d6) δ 166.8, 165.2, 158.3, 150.9, 149.0, 148.8, 147.3, 141.1, 134.9, 133.7, 132.3, 129.0 (2 C), 128.5 (2 C), 127.9, 127.8 (2 C), 127.2 (2 C), 118.9, 112.0, 110.2, 106.6, 98.3, 62.3, 59.0, 55.5 (2 C), 13.6; MS (EI+) m/z 493 (M+, 29), 420 (100), 338 (34), 155 (21); HRMS (EI+) m/z calcd for C30H27N3O4 493.2001, found 493.1977.
4.27. Ethyl 8-(4-methoxyphenyl)-2,4-diphenyl-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (38c)
According to general procedure H, a solution of bis(triphenylphosphine)palladium(II) chloride (0.007 g, 0.009 mmol), copper iodide (0.004 g, 0.02 mmol), p-anisoyl chloride (0.099 g, 0.58 mmol), TMS-acetylene (0.083 mL, 0.58 mmol) and N,N-dicyclohexylmethyl amine (0.12 mL, 0.58 mmol) in freshly distilled THF (0.75 mL) led to 36 (Rf = 0.17; 20:1, hexanes:EtOAc) which was dissolved in THF (2.3 mL) and treated with 18 (0.150 g, 0.467 mmol), DMAP (0.012 g, 0.093 mmol) and MeOH (0.4 mL) to provide a crude product which was purified by chromatography on SiO2 (1:1, hexanes:EtOAc). The resulting red/orange solid was dissolved in CH2Cl2 (1.0 mL) and crystallized by slowly adding hexanes (5.0 mL) and cooling to -20 °C. The crystalline solid was isolated by vacuum filtration, washed with hexanes, and dried in vacuo to afford 38c as a red/orange solid (0.060 g, 28%, ELSD purity 99.8%): Rf 0.31(1:1, hexanes:EtOAc); Mp 210-212 °C; IR (ATR) 2969, 1700, 1594, 1493, 1439, 1297, 1241, 1212, 1169, 1075, 1023, 697 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.54 (d, 1H, J=6.9 Hz), 8.15 (d, 2H, J=9.0 Hz), 7.48-7.29 (m, 11H), 7.08 (d, 2H, J=8.7 Hz), 6.44 (s, 1H), 3.84 (s, 3H), 3.77 (q, 2H, J=7.2 Hz), 0.80 (t, 3H, J=7.2 Hz); 13C NMR (125 MHz, DMSO-d6) δ 166.2, 165.2, 162.8, 158.8, 151.2, 146.8, 141.4, 141.0, 129.8 (2 C), 129.0 (2 C), 128.6 (3 C), 127.9, 127.1 (3 C), 126.3 (2 C), 114.4 (2 C), 106.2, 97.9, 62.2, 59.0, 55.5, 13.5; HRMS (TOF ES+) m/z calcd for C29H26N3O3 464.1974, found 464.1975.
4.28. Ethyl 2,4-diphenyl-8-(4-(trifluoromethyl)phenyl)-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate (38d)
According to general procedure H, a solution of bis(triphenylphosphine)palladium(II) chloride (0.007 g, 0.01 mmol), copper iodide (0.004 g, 0.02 mmol), 4-(trifluoromethyl)benzoyl chloride (0.074 mL, 0.50 mmol), TMS-acetylene (0.071 mL, 0.50 mmol) and Et3N (0.071 mL, 0.50 mmol) in freshly distilled THF (3.0 mL) led to 37 (Rf = 0.64; 10:1, hexanes:EtOAc) which was treated with 18 (0.129 g, 0.400 mmol), DMAP (0.012 g, 0.10 mmol) and MeOH (0.40 mL) to give a crude product which was purified by chromatography on SiO2 (2:1 to 1:1, EtOAc:hexanes) to afford 38d as a red crystalline solid (0.11 g, 55%, ELSD purity 100.0%): Rf 0.31 (1:1, EtOAc:hexanes); Mp 206-208 °C; IR (ATR) 3068, 2974, 1705, 1618, 1506, 1456, 1323, 1114, 1066 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.70 (d, 1H, J=6.9 Hz), 8.35 (d, 2H, J=8.1 Hz), 7.91 (d, 2H, J=8.4 Hz), 7.54-7.30 (m, 11H), 6.52 (s, 1H), 3.81 (q, 2H, J=6.9 Hz), 0.80 (t, 3H, J=7.2 Hz); 13C NMR (150 MHz, DMSO-d6) δ 165.6, 165.2, 158.2, 150.9, 148.0, 141.1, 140.7, 138.7, 131.7 (q, JCF = 33.0 Hz), 129.1 (2 C), 128.7, 128.6 (4 C), 128.1, 127.2 (2 C), 126.4 (2 C), 125.8 (q, 2 C, JCF = 4.5 Hz), 123.9 (q, JCF = 271.5), 107.0, 98.5, 62.3, 59.1, 13.5; MS (EI) m/z 501 (M+, 11), 428 (100), 278 (14), 223 (17); HRMS (EI) m/z calcd for C29H22N3O2F3 501.1664, found 501.1658. X-ray crystallographic data for 38d were deposited with the Cambridge Crystallographic Data Center (deposition number CCDC 932463).
4.29. Ethyl 4-amino-6,8-dimethyl-2-morpholino-6H-pyrimido[1,2-a][1,3,5]triazine-7-carboxylate (40). General Procedure I
To a solution of 3,3-diphenoxy-2-azaprop-2-enenitrile (0.133 g, 0.558 mmol) in THF (1.0 mL) was added morpholine (0.049 mL, 0.56 mmol) in one portion at room temperature. The homogeneous colorless reaction mixture was stirred at room temperature for 2 h to form 39 (Rf = 0.33; 1:1, hexanes:EtOAc). To the reaction mixture was added 10a (0.100 g, 0.507 mmol) and the resulting suspension was heated in a microwave reactor at 110 °C for 45 min. The mixture was purified by chromatography on SiO2 (EtOH) to afford 40 as a white foam (0.090 g, 53%, ELSD purity 99.8%): Rf 0.17 (EtOH); IR (ATR) 3327, 2969, 2906, 2855, 1651, 1584, 1551, 1482, 1420, 1264, 1232, 1107, 1059, 1023, 885, 775 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 7.89 (br s, 2H), 5.15 (q, 1H, J=6.0 Hz), 4.14-4.01 (m, 2H), 3.65-3.58 (m, 8H), 2.16 (s, 3H), 1.22 (t, 3H, J=7.2 Hz), 1.08 (d, 3H, J=6.0 Hz); 13C NMR (75 MHz, DMSO-d6) δ 165.2, 160.8, 158.4, 156.2, 152.1, 100.3, 65.9 (2 C), 58.8, 47.7, 43.5 (2 C), 23.2, 19.4, 14.4; HRMS (TOF ESI+) m/z calcd for C15H23N6O3 335.1832, found 335.1825.
4.30. Ethyl 4-amino-8-methyl-2-morpholino-6-phenethyl-6H-pyrimido[1,2-a][1,3,5]triazine-7-carboxylate (41)
According to general procedure I, a solution of 3,3-diphenoxy-2-azaprop-2-enenitrile (0.091 g, 0.38 mmol) and morpholine (0.034 mL, 0.38 mmol) in THF (1.0 mL) led to 39 which was treated with 10b (0.100 g, 0.348 mmol). The reaction mixture was purified by chromatography on SiO2 (EtOH) to afford 41 as a pale yellow foam (0.037 g, 25%): Rf 0.82 (EtOH); IR (ATR) 3308, 2913, 2850, 1638, 1581, 1551, 1482, 1424, 1266, 1232, 1096, 1059, 1023, 889, 781 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 7.90 (br s, 2H), 7.25-7.06 (m, 5H), 5.31 (t, 1H, J=5.4 Hz), 4.17-4.02 (m, 2H), 3.65-3.59 (m, 8H), 2.58-2.40 (m, 2H), 2.20 (s, 3H), 1.86-1.73 (m, 2H), 1.23 (t, 3H, J=7.2 Hz); 13C NMR (125 MHz, DMSO-d6) δ 165.4, 160.6, 159.4, 156.3, 152.6, 141.3, 128.3 (2 C), 127.9 (2 C), 125.7, 98.6, 65.9 (2 C), 58.8, 51.3, 43.6 (2 C), 35.6, 29.6, 23.1, 14.4; HRMS (TOF ESI+) m/z calcd for C22H29N6O3 425.2301, found 425.2301.
4.31. Ethyl 4-amino-6-isopropyl-8-methyl-2-morpholino-6H-pyrimido[1,2-a][1,3,5]triazine-7-carboxylate (42)
According to general procedure I, a solution of 3,3-diphenoxy-2-azaprop-2-enenitrile (0.116 g, 0.488 mmol) and morpholine (0.043 mL, 0.49 mmol) in THF (1.0 mL) led to 39 which was treated with 10c (0.100 g, 0.444 mmol). The reaction mixture was purified by chromatography on SiO2 (EtOH) to afford 42 as a pale yellow foam (0.070 g, 43%, ELSD purity 99.7%): Rf 0.80 (EtOH); IR (ATR) 3341, 3152, 2964, 1655, 1577, 1547, 1480, 1424, 1275, 1227, 1111, 1079, 1022, 893, 787 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 7.86 (br s, 2H), 5.09 (d, 1H, J=5.4 Hz), 4.16-4.00 (m, 2H), 3.65-3.50 (m, 8H), 2.19 (s, 3H), 1.86-1.77 (m, 1H), 1.23 (t, 3H, J=6.9 Hz), 0.75 (d, 3H, J=6.9 Hz), 0.72 (d, 3H, J=6.9 Hz); 13C NMR (75 MHz, DMSO-d6) δ 166.0, 160.5, 158.6, 156.7, 152.7, 98.0, 65.9 (2 C), 58.7, 55.4, 43.6 (2 C), 34.1, 23.0, 17.8, 17.6, 14.4; HRMS (TOF ESI+) m/z calcd for C17H27N6O3 363.2145, found 363.2136.
4.32. Ethyl 4-amino-2-morpholino-6,8-diphenyl-6H-pyrimido[1,2-a][1,3,5]triazine-7-carboxylate (43)
According to general procedure I, a solution of 3,3-diphenoxy-2-azaprop-2-enenitrile (0.163 g, 0.685 mmol) and morpholine (0.060 mL, 0.68 mmol) in THF (1.0 mL) led to 39 which was treated with 18 (0.200 g, 0.622 mmol). The reaction mixture was cooled to room temperature, diluted with ether (1.5 mL) and cooled to -20 °C for 1 h. The resulting precipitate was isolated by vacuum filtration and washed with ether (15 mL). The crude precipitate was recrystallized from boiling hexanes:EtOAc (4:1, 35 mL), isolated by vacuum filtration, washed with hexanes, and dried in vacuo to afford 43 as a yellow/green foam (0.130 g, 46%, ELSD purity 99.9%): IR (ATR) 3334, 3165, 2975, 2855, 1646, 1549, 1476, 1418, 1271, 1230, 1105, 1087, 1021, 995, 764, 695 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 8.04 (s, 2H), 7.47-7.44 (m, 2H), 7.37-7.27 (m, 8H), 6.30 (s, 1H), 3.79-3.69 (m, 4H), 3.58 (s, 6H), 0.77 (t, 3H, J=7.2 Hz); 13C NMR (75 MHz, DMSO-d6) δ 165.5, 160.6, 159.0, 156.3, 152.5, 141.7, 141.5, 128.6 (2 C), 128.2 (2 C), 128.1, 127.5, 127.0 (2 C), 126.5 (2 C), 100.4, 65.9 (2 C), 58.7, 54.6, 43.6, 43.4, 13.5; HRMS (TOF ESI+) m/z calcd for C25H27N6O3 459.2145, found 459.2135.
4.33. Ethyl 6,8-dimethyl-4-oxo-3,4,6,9-tetrahydropyrimido[2,1-c][1,2,4,6]thiatriazine-7-carboxylate 2,2-dioxide (44). General Procedure J
To a suspension of 10a (0.100 g, 0.507 mmol) and DIPEA (0.13 mL, 0.76 mmol) in THF (2.5 mL) at 0 °C was added chlorosulfonyl isocyanate (0.068 mL, 0.76 mmol) in one portion. The reaction mixture became homogeneous within 5 min after the addition of the isocyanate. The solution was stirred for 3 h at 0 °C and warmed to room temperature for 14 h. 1H NMR analysis indicated a 47% conversion, and therefore the mixture was treated at 0 °C with additional DIPEA (0.084 mL, 0.51 mmol) and chlorosulfonyl isocyanate (0.045 mL, 0.51 mmol). The reaction mixture was stirred at 0 °C for 2 h, allowed to warm to room temperature, and stirred for an additional 4 h. The resulting clear pale yellow solution was diluted with EtOAc (30 mL) and concentrated under reduced pressure after addition of SiO2 (1.0 g). The resulting mixture was added directly to the top of a pre-flushed column and purified by chromatography on SiO2 (1:1, hexanes:EtOAc to EtOAc). The resulting product was recrystallized from boiling CH2Cl2:hexanes (1:1.5, 2.5 mL), isolated by vacuum filtration, washed with hexanes and dried in vacuo to afford 44 as a white solid (0.024 g, 15%): Mp 219-220 °C; IR (ATR) 3308, 3239, 3184, 2988, 1707, 1609, 1471, 1329, 1294, 1253, 1189, 1161, 1083, 902, 753, 738, 662 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 10.89 (s, 1H), 5.45 (q, 1H, J=6.3 Hz), 4.25-4.08 (m, 2H), 2.27 (s, 3H), 1.24 (t, 3H, J=6.9 Hz), 1.17 (d, 3H, J=6.3 Hz); 13C NMR (75 MHz, DMSO-d6) δ 168.0, 151.9, 151.1, 149.2, 108.5, 64.1, 49.9, 23.8, 21.0, 18.1; HRMS (TOF ESI+) m/z calcd for C10H15N4O5S 303.0763, found 303.0755. X-ray crystallographic data for 44 were deposited with the Cambridge Crystallographic Data Center (deposition number CCDC 935121).
4.34. Ethyl 4-oxo-6,8-diphenyl-3,4,6,9-tetrahydropyrimido[2,1-c][1,2,4,6]thiatriazine-7-carboxylate 2,2-dioxide (45)
According to general procedure J, a solution of 18 (0.250 g, 0.778 mmol), DIPEA (0.19 mL, 1.2 mmol), and chlorosulfonyl isocyanate (0.10 mL, 1.2 mmol) in THF (2.5 mL) led to crude product which was diluted with EtOAc (30 mL), concentrated under reduced pressure with SiO2 (1.0 g), and purified by chromatography on SiO2 (1:1, hexanes:EtOAc to EtOAc) to afford 45 as a white foam (0.132 g, 40%, ELSD purity 99.7%): IR (ATR) 3202, 3057, 2982, 1700, 1594, 1444, 1331, 1249, 1163, 1100, 979, 749, 693 cm-1; 1H NMR (300 MHz, DMSO-d6) δ 11.18 (s, 1H), 7.47-7.37 (m, 11H), 6.66 (s, 1H), 3.84 (q, 2H, J=6.9 Hz), 0.78 (t, 3H, J=6.9 Hz); 13C NMR (75 MHz, DMSO-d6) δ 164.1, 148.5, 147.5, 146.4, 139.1, 132.9, 129.8, 129.1 (2 C), 129.0 (2 C), 128.7, 128.0 (2 C), 126.2 (2 C), 104.5, 60.2, 51.8, 13.4; HRMS (TOF ESI+) m/z calcd for C20H19N4O5S 427.1076, found 427.1077.
4.35. Diethyl 4,6-bis(4-chlorophenyl)-2,8-dimethyl-4,6-dihydro-1H-pyrimido[1,2-a]pyrimidine-3,7-dicarboxylate (46)
According to general procedure D, a solution of 13•TFA (0.030 g, 0.074 mmol), 12 (0.074 g, 0.29 mmol) and NaHCO3 (0.014 g, 0.016 mmol) in (CF3)2CHOH (0.25 mL) was heated for 24 h. LC/MS analysis demonstrated an approximately 50% conversion of 13, and 2 more equivalents of 12 (0.037 g, 0.15 mmol) were added. The reaction mixture was stirred at 90 °C for an additional 18 h after which complete consumption of the starting material was observed via LC/MS. Product 46 (0.022 g, 57%) was obtained as a yellow crystalline 4:1 mixture of anti-syn-isomers that could not be further separated: Rf 0.42 (MeOH:CH2Cl2, 0.2:10); Mp 70-75 °C; IR (CH2Cl2) 2979, 2922, 1677, 1622, 1544, 1490, 1446, 1369, 1274, 1228, 1199, 1088, 1014, 830, 736 cm-1; 1H NMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 11.95 (s, 0.2H), 7.43-7.41 (d, 2H, J=8.4 Hz), 7.31-7.29 (d, 2 × 2H, J=8.4 Hz), 6.97-6.94 (d, 2 × 0.5H, J=8.8 Hz), 5.38 (s, 2 × 0.25H), 5.18 (s, 2 × 1H), 4.10-4.01 (m, 2 × 4H), 2.49 (s, 2 × 3H), 1.17-1.13 (t, 2 × 3H, J=7.2 Hz); 13C NMR (100 MHz, CDCl3) δ 164.3, 164.0 (2C), 148.2, 145.6, 144.7, 138.6, 137.7, 135.3, 134.4, 129.5 (2C), 129.1, 128.9 (2C), 128.3, 105.1, 103.4, 64.8, 62.3, 60.8, 56.6, 19.3, 14.0; HRMS (ESI+) m/z calcd for C27H28O4N3Cl2 (M+H) 528.1451, found 528.1441.
4.36. Ethyl 2-(4-morpholinobenzylidene)-3-oxobutanoate (48d).36
A solution of L-proline (1.21 g, 10.5 mmol) in DMSO (10 mL) was treated with 4-morpholinebenzaldehyde (1.12 g, 5.86 mmol) at room temperature. After 10 min, ethyl acetoacetate (0.80 mL, 6.3 mmol) was added and the mixture was stirred for 17 h. The yellow solution was treated with EtOAc, followed by water, and the organic layer was washed with brine, dried (Na2SO4), filtered, and concentrated. The resulting oil was purified by chromatography on SiO2 (ISCO-Rf, 20-100% EtOAc-hexanes, 15 min gradient) to give 48d as a sticky yellow 4.4:1 mixture of inseparable Z/E isomers (0.713 g, 40%): Mp 94-96 °C; IR (neat) 2974, 1713, 1588, 1514, 1260, 1163, 982 cm-1; 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 0.2 H), 7.43 (s, 1H), 7.35 (d, 2H, J=7.8Hz), 7.28 (d, 0.4H, J=8.8 Hz), 6.82-6.78 (overlap d, 2.4H), 4.33 (q, 2H, J=7.2 Hz), 4.23 (q, 0.4H, J=7.2 Hz), 3.80-3.78 (m, 5H), 3.24-3.21 (m, 5H), 2.37 (s, 0.6H), 2.34 (s, 3H), 1.31-1.28 (2 overlapping t, 3.7H); 13C NMR (100 MHz, CDCl3) δ 204.0, 194.5, 168.4, 164.6, 152.4, 152.1, 141.1, 140.3, 131.5, 131.4, 130.6, 129.7, 122.9, 122.6, 114.0, 113.9, 66.2, 61.2, 60.9, 47.3, 47.2, 31.0, 26.0, 14.0, 13.8; HRMS (ESI+) m/z calcd for C17H22NO4 (M+H) 304.1543, found 304.1535.
4.37. Ethyl 2-(6-chloro-4-methylquinazolin-2-ylamino)-4-(4-chlorophenyl)-6-methyl-1,4-dihydropyrimidine-5-carboxylate (49a)
According to general procedure D, a solution of 47a (0.029 g, 0.12 mmol), 1236,37 (0.23 g, 0.90 mmol) and NaHCO3 (0.026 g, 0.31 mmol) in (CF3)2CHOH (0.40 mL) afforded 49a as a light yellow solid (0.053 g, 56%, ELSD purity 95.4%): Mp >200 °C; IR (neat) 3059, 2975, 1650, 1575, 1461, 1420, 1305, 1215, 1094, 838 cm -1; 1H NMR (400 MHz, DMSO-d6) δ 10.87 (br s, 1H), 10.43 (br s, 1H), 8.11 (d, 1H, J=1.6 Hz), 7.85-7.79 (m, 2H), 7.39 (d, 2H, J=8.8 Hz), 7.36 (d, 2H, J=8.8 Hz), 5.70 (s, 1H), 4.07 (q, 2H, J=7.2 Hz), 2.77 (s, 3H), 2.39 (s, 3H), 1.17 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, DMSO-d6) δ 169.3, 165.6, 161.8, 151.8, 149.0, 148.8, 144.0, 134.3, 132.5, 128.9, 128.7, 128.5, 124.9, 120.8, 100.4, 59.8, 52.7, 21.9, 18.9, 14.6; HRMS (ESI+) m/z calcd for C23H22N5O2Cl2 (M+H) 470.1145, found 470.1146.
4.38. Ethyl 4-(4-chlorophenyl)-2-(4,6-dimethylquinazolin-2-ylamino)-6-methyl-1,4-dihydropyrimidine-5-carboxylate (49b)
According to general procedure D, a solution of 47b (0.055 g, 0.26 mmol), 1236,37 (0.28 g, 1.09 mmol) and NaHCO3 (0.049 g, 0.58 mmol) in (CF3)2CHOH (0.40 mL) afforded 49b as a light yellow solid (0.073 g, 62%, ELSD purity 98.4%): Mp >200 °C; IR (neat) 3058, 2975, 1655, 1535, 1450, 1420, 1316, 1215, 1077, 839 cm -1; 1H NMR (400 MHz, DMSO-d6) δ 10.72 (br s, 2H), 7.83 (s, 1H), 7.70 (d, 1H, J=8.4 Hz), 7.63 (d, 1H, J=8.4 Hz), 7.38 (app s, 4H), 5.71 (s, 1H), 4.06 (q, 2H, J=6.4 Hz), 2.75 (s, 3H), 2.45 (s, 3H), 2.39 (s, 3H), 1.16 (t, 3H, J=6.8 Hz); 13C NMR (100 MHz, DMSO-d6, 100 °C) δ 168.1, 164.8, 159.3, 150.4, 149.1, 147.4, 143.3, 135.0, 133.2, 131.4, 127.8, 127.5, 125.4, 123.6, 119.3, 99.3, 58.6, 51.9, 20.7, 20.3, 18.4, 13.5; HRMS (ESI+) m/z calcd for C24H25N5O2Cl (M+H) 450.1691, found 450.1680.
4.39. Ethyl 4-(4-chlorophenyl)-2-(6-fluoro-4-methylquinazolin-2-ylamino)-6-methyl-1,4-dihydropyrimidine-5-carboxylate (49c)
According to general procedure D, a solution of 47c (0.048 g, 0.22 mmol), 12 (0.22 g, 0.86 mmol) and NaHCO3 (0.043 g, 0.51 mmol) in (CF3)2CHOH (0.40 mL) afforded 49c as a yellow solid (0.051 g, 52%, ELSD purity 90%): Mp >200 °C; IR (neat) 3059, 2975, 1663, 1536, 1450, 1420, 1310, 1215, 1077, 826 cm -1; 1H NMR (400 MHz, DMSO-d6) δ 10.71 (br s, 2H), 7.93-7.83 (m, 2H), 7.72 (ddd, 1H, J=2.4, 8.8, 8.8 Hz), 7.39-7.35 (m, 4H), 5.70 (s, 1H), 4.07 (q, 2H, J=7.2 Hz), 2.75 (s, 3H), 2.39 (s, 3H), 1.16 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, DMSO-d6, 100 °C) δ 168.5, 164.7, 159.9, 157.6 (d, JCF = 243.5 Hz), 150.5, 148.2, 146.3, 143.0, 131.5, 128.2 (d, JCF = 8.1 Hz), 127.9, 127.7, 127.5, 122.7 (d, JCF = 25.2 Hz), 119.3 (d, JCF = 8.1 Hz), 108.6 (d, JCF = 22.1 Hz), 99.5, 58.7, 51.9, 20.9, 18.1, 13.5; HRMS (ESI+) m/z calcd for C23H22N5O2ClF (M+H) 454.1441, found 454.1435.
4.40. Ethyl 4-(4-dimethylaminophenyl)-2-(6-fluoro-4-methylquinazolin-2-ylamino)-6-methyl-1,4-dihydropyrimidine-5-carboxylate (49d)
According to general procedure D, a solution of 47c (0.032 g, 0.15 mmol), 48a36,37 (0.15 g, 0.59 mmol) and NaHCO3 (0.027 g, 0.32 mmol) in (CF3)2CHOH (0.60 mL) gave 49d as an orange solid (0.027 g, 40%, ELSD purity 99.4%): Mp >220 °C; IR (neat) 3059, 2974, 1655, 1543, 1420, 1307, 1215, 1077, 838 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 10.57 (br s, 2H), 7.84-7.81 (m, 2H), 7.71-7.67 (m, 1H), 7.15 (d, 2H, J=8.4 Hz), 6.63 (d, 2H, J=8.4 Hz), 5.57 (s, 1H), 4.06 (q, 2H, J=6.8 Hz), 2.82 (s, 6H), 2.74 (s, 3H), 2.38 (s, 3H); 1.19 (t, 3H, J= 6.8 Hz); 13C NMR (100 MHz, DMSO-d6) δ 168.9 (d, JCF = 4.0 Hz), 165.3, 161.3, 157.8 (d, JCF = 242.5 Hz), 151.6, 149.7, 147.4, 146.5, 132.2, 128.6 (d, JCF = 8.1 Hz), 126.7, 123.1 (d, JCF = 25.2 Hz), 119.5 (d, JCF = 9.1 Hz), 112.3, 109.3 (d, JCF = 22.1 Hz), 100.2, 59.2, 51.8, 40.1, 21.8, 18.2, 14.2; HRMS (ESI+) m/z calcd for C25H28N6O2F (M+H) 463.2252, found 463.2248.
4.41. Ethyl 2-(4.6-dimethylquinazolin-2-ylamino)-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyrimidine-5-carboxylate (49e)
According to general procedure D, a solution of 47b (0.043 g, 0.20 mmol), 48b36,38 (0.16 g, 0.60 mmol) and NaHCO3 (0.057 g, 0.68 mmol) in (CF3)2CHOH (0.60 mL) gave 49e as a light yellow solid (0.062 g, 67%, ELSD purity 90.4%): Mp >200 °C; IR (neat) 3059, 2974, 1668, 1525, 1450, 1420, 1340, 1215, 1077, 734, 692 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 10.94 (br s, 1H), 10.53 (br s, 1H), 8.23 (app t, 1H, J=1.6 Hz), 8.12-8.09 (m, 1H), 7.85 (s, 1H), 7.80 (d, 1H, J=7.6 Hz), 7.73 (d, 1H, J=8.4 Hz), 7.67-7.63 (m, 2H), 5.85 (s, 1H), 4.08 (q, 2H, J=7.2 Hz), 2.77 (s, 3H), 2.47 (s, 3H), 2.40 (s, 3H), 1.17 (t, 3H, J=7.2 Hz); 13C NMR (75 MHz, DMSO-d6) δ 169.0, 165.1, 159.8, 151.1, 147.8, 147.5, 147.0, 135.6, 133.8, 132.8, 130.3, 126.0, 124.3, 122.3, 121.0, 119.6, 98.7, 59.4, 51.9, 21.6, 21.0, 19.0, 14.1; HRMS (ESI+) m/z calcd for C24H25N6O4 (M+H) 461.1932, found 461.1913.
4.42. Ethyl 4-(4-chlorophenyl)-6-methyl-2-(8-methyl-[1,3]dioxolo[4,5-g]quinazolin-6-ylamino)-1,4-dihydropyrimidine-5-carboxylate (49f)
According to general procedure D, a solution of 47d (0.029 g, 0.12 mmol), 1236,37 (0.035 g, 0.14 mmol) and NaHCO3 (0.026 g, 0.31 mmol) in (CF3)2CHOH (0.40 mL) afforded 49f as a light yellow solid (0.024 g, 41%, ELSD purity 98.2%): Mp >200 °C; IR (neat) 3059, 2974, 1649, 1450, 1420, 1215, 1094, 1077, 768 cm -1; 1H NMR (300 MHz, DMSO-d6) δ 10.76 (br s, 2H), 7.43 (s, 1H), 7.38 (d, 2H, J=8.8 Hz), 7.34 (d, 2H, J=8.7 Hz), 7.26 (s, 1H), 6.19 (br s, 2H), 5.65 (s, 1H), 4.06 (q, 2H, J=7.2 Hz), 2.65 (s, 3H), 2.37 (s, 3H), 1.16 (t, 3H, J=7.2 Hz); 13C NMR (125 MHz, DMSO-d6) δ 166.7, 165.2, 159.6, 153.3, 150.9, 148.5, 145.8, 143.7, 131.8, 128.5, 127.9, 115.6, 102.9, 102.1, 101.0, 99.0, 59.2, 51.6, 21.8, 18.9, 14.2; HRMS (ESI+) m/z calcd for C24H23N5O4Cl (M+H) 480.1433, found 480.1431.
4.43. Ethyl 4-(4-bromophenyl)-6-methyl-2-(8-methyl-[1,3]dioxolo[4,5-g]quinazolin-6-ylamino)-1,4-dihydropyrimidine-5-carboxylate (49g)
According to general procedure D, a solution of 47d (0.041 g, 0.17 mmol), 48c36,37 and NaHCO3 (0.031 g, 0.37 mmol) in (CF3)2CHOH (0.40 mL) afforded 49g as a yellow solid (0.039 g, 57%, ELSD purity 98.2%): Mp >200 °C; IR (neat) 3059, 2974, 1700, 1629, 1536, 1420, 1192, 1077, 1028, 839 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 10.61 (br s, 2H), 7.51 (d, 2H, J=8.4 Hz), 7.41 (s, 1H), 7.29 (d, 2H, J=8.4 Hz), 7.25 (s, 1H), 6.20 (br s, 2H), 5.66 (s, 1H), 4.06 (q, 2H, J=7.2 Hz), 2.66 (s, 3H), 2.38 (s, 3H), 1,16 (t, 3H, J=7.2 Hz); 13C NMR (100 MHz, DMSO-d6) δ 166.6, 165.2, 159.7, 153.2, 151.0, 148.5, 145.8, 144.1, 131.4, 128.3, 120.3, 115.5, 102.9, 102.1, 101.0, 98.9, 59.2, 51.7, 21.8, 19.0, 14.2; HRMS (ESI+) m/z calcd for C24H23N5O4Br (M+H) 524.0928, found 524.0915.
4.44. Ethyl 6-methyl-2-(8-methyl-[1,3]dioxolo[4,5-g]quinazolin-6-ylamino)-4-(4 morpholinophenyl)-1,4-dihydropyrimidine-5-carboxylate (49h)
According to general procedure D, a solution of 47d (0.046 g, 0.17 mmol), 48d (0.21 g, 0.70 mmol) and NaHCO3 (0.032 g, 0.38 mmol) in CF3CHOH (0.40 mL) gave 49h as a yellow solid (0.073 g, 81%, ELSD purity 99.7%): Mp >220 °C; IR (neat) 3058, 2816, 1655, 1543, 1450, 1420, 1316, 1215, 1077, 1028, 956 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 10.56 (br s, 2H), 7.41 (s, 1H), 7.24 (s, 1H), 7.19 (d, 2H, J=8.0 Hz), 6.86 (d, 2H, J=8.0 Hz), 6.19 (br s, 2H), 5.58 (s, 1H), 4.06 (app t, 2H, J=6.0 Hz), 3.68 (br s, 4H), 3.03 (br s, 4H), 2.65 (s, 3H), 2.37 (s, 3H), 1.17 (t, 3H, J=6.8 Hz); 13C NMR (100 MHz, DMSO-d6) δ 166.5, 165.4, 160.1, 153.2, 151.2, 150.2, 148.6, 145.7, 135.4, 126.7, 115.4, 115.0, 102.9, 102.1, 101.0, 99.8, 66.0, 59.1, 51.6, 48.3, 21.9, 18.7, 14.2; HRMS (ESI+) m/z calcd for C28H31N6O2 (M+H) 531.2350, found 531.2338.
4.45. Ethyl 4-(4-(dimethylamino)phenyl)-6-methyl-2-(8-methyl-[1,3]dioxolo[4,5-g]quinazolin-6-ylamino)-1,4-dihydropyrimidine-5-carboxylate (49i)
According to general procedure D, a solution of 47d (0.043 g, 0.17 mmol), 48a36,37 (0.18 g, 0.69 mmol) and NaHCO3 (0.031 g, 0.37 mmol) in (CF3)2CHOH (0.60 mL) gave 49i as an off-white solid (0.038 g, 45%, ELSD purity 100%): Mp >220 °C; IR (neat) 3059, 2934, 1663, 1549, 1450, 1420, 1215, 1077, 1029, 839 cm-1; 1H NMR (400 MHz, DMSO-d6) δ 10.61 (br s, 2H), 7.42 (s, 1H), 7.24 (s, 1H), 7.14 (d, 2H, J=8.4 Hz), 6.64 (d, 2H, J= 8.8 Hz), 6.19 (br s, 2H), 5.54 (s, 1H), 4.06 (q, 2H, J=6.8 Hz), 2.82 (s, 6H), 2.66 (s, 3H), 2.37 (s, 3H), 1.18 (t, 3H, J=6.8 Hz); 13C NMR (100 MHz, DMSO-d6) δ 166.4, 165.4, 160.1, 153.1, 151.1, 149.7, 148.6, 145.6, 132.4, 126.7, 115.4, 112.3, 102.9, 102.1, 101.0, 100.0, 59.1, 51.6, 40.1, 21.8, 18.7, 14.2; HRMS (ESI+) m/z calcd for C26H29N6O4 (M+H) 489.2245, found 489.2237.
Supplementary Material
Acknowledgments
We thank Dr. Steven Geib for x-ray analyses, Dr. Dimas J. da Paz Lima for the preparation of intermediates 47a-d, Dr. Ki Bum Hong for the preparation of ynones 28 and 29, and the National Institutes of Health (GM067082; P50 Centers for Chemical Methodologies and Library Development, as well as P41GM081275) for financial support.
Footnotes
Submitted in honor of Prof. Paul Wender, the recipient of the 2012 Tetrahedron Prize for Creativity in Organic Chemistry
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Nielsen TE, Schreiber SL. Angew Chem Int Ed. 2008;47:48–56. doi: 10.1002/anie.200703073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Arya P, Joseph R, Gan Z, Rakic B. Chem Biol. 2005;12:163–180. doi: 10.1016/j.chembiol.2005.01.011. [DOI] [PubMed] [Google Scholar]; (c) Tan DS. Nat Chem Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wipf P. DOS. 2012:6–10. [Google Scholar]; (b) Huryn DM, Brodsky JL, Brummond KM, Chambers PG, Eyer B, Ireland AW, Kawasumi M, LaPorte MG, Lloyd K, Manteau B, Nghiem P, Quade B, Seguin SP, Wipf P. Proc Nat Acad Sci USA. 2011;108:6757–6762. doi: 10.1073/pnas.1015251108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zaware N, LaPorte MG, Farid R, Liu L, Wipf P, Floreancig PE. Molecules. 2011;16:3648–3662. doi: 10.3390/molecules16053648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hammill JT, Contreras-Garcia J, Virshup AM, Beratan DN, Yang W, Wipf P. Tetrahedron. 2010;66:5852–5862. doi: 10.1016/j.tet.2010.04.112. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wipf P, Stephenson CRJ, Walczak MAA. Org Lett. 2004;6:3009–3012. doi: 10.1021/ol0487783. [DOI] [PubMed] [Google Scholar]
- 3.Kim Y-K, Arai MA, Arai T, Lamenzo JO, Dean EF, Patterson N, Clemons PA, Schreiber SL. J Am Chem Soc. 2004;126:14740–14745. doi: 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]
- 4.Wipf P, Fang Z, Ferrié L, Ueda M, Walczak MAA, Yan Y, Yang M. Pure Appl Chem. 2013;85 in press. http://dx.doi.org/10.1351/PAC-CON-12-09-03. [Google Scholar]
- 5.(a) Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DVS, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. Nat Rev Drug Discov. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]; (b) Haggarty SJ. Curr Opin Chem Biol. 2005;9:296–303. doi: 10.1016/j.cbpa.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 6.(a) Walczak MAA, Wipf P. J Am Chem Soc. 2008;130:6924–6925. doi: 10.1021/ja802906k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wipf P, Walczak MAA. Angew Chem Int Ed. 2006;45:4172–4175. doi: 10.1002/anie.200600723. [DOI] [PubMed] [Google Scholar]
- 7.Guo T, Hunter RC, Zhang R, Greenlee WJ. Tetrahedron Lett. 2007;48:613–615. [Google Scholar]
- 8.Jimenez J-M, Patel S, Kay D, Knegtel R, Philps O. 7,855,214 B2. U S Patent. 2010
- 9.Wathen MW, Wathen LK. 019940 A1. WO. 2004
- 10.Bundy GL, Ciske FL, Genin MJ, Heasley SE, Larsen SD, Lee BH, May PD, Palmer JR, Schnute ME, Vaillancourt VA, Thorarensen A, Wolf AJ, Wicnienski NA, Wilhite D. 04444 A2. WO. 2002
- 11.Hellberg M, Stubbins JF, Glennon RA. Bioorg Med Chem. 2000;8:1917–1923. doi: 10.1016/s0968-0896(00)00123-1. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wyatt EE, Fergus S, Galloway WRJD, Bender A, Fox DJ, Plowright AT, Jessiman AS, Welch M, Spring DR. Chem Commun. 2006:3296–3298. doi: 10.1039/b607710b. [DOI] [PubMed] [Google Scholar]; (b) Wyatt EE, Galloway WRJD, Thomas GL, Welch M, Loiseleur O, Plowright AT, Spring DR. Chem Commun. 2008:4962–4964. doi: 10.1039/b812901k. [DOI] [PubMed] [Google Scholar]
- 13.See: Katritzky AR, Xu Y-J, Tu H. J Org Chem. 2003;68:4935–4937. doi: 10.1021/jo026797p., and references cited therein.
- 14.(a) Biginelli P. Gazz Chim Ital. 1893;23:360–416. [Google Scholar]; (b) Kappe CO, Stadler A. Org React. 2004;63:1–116. [Google Scholar]
- 15.(a) Mishra JK, Wipf P, Sinha SC. J Comb Chem. 2010;12:609–612. doi: 10.1021/cc1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Werner S, Turner DM, Lyon MA, Huryn DM, Wipf P. Synlett. 2006:2334–2338. [Google Scholar]; (c) Studer A, Jeger P, Wipf P, Curran DP. J Org Chem. 1997;62:2917–2924. doi: 10.1021/jo970095w. [DOI] [PubMed] [Google Scholar]; (d) Wipf P, Cunningham A. Tetrahedron Lett. 1995;36:7819–7822. [Google Scholar]
- 16.Atwal KS, Rovnyak GC, O’Reilly BC, Schwartz J. J Org Chem. 1989;54:5898–5907. [Google Scholar]
- 17.Tabouazat M, El Louzi A, Ahmar M, Cazes B. Synlett. 2008:2495–2499. [Google Scholar]
- 18.Nilsson BL, Overman LE. J Org Chem. 2006;71:7706–7714. doi: 10.1021/jo061199m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kappe CO. Bioorg Med Chem Lett. 2000;10:49–52. doi: 10.1016/s0960-894x(99)00572-7. [DOI] [PubMed] [Google Scholar]
- 20.(a) Mobinikhaledi A, Forughifar N, Safari JA, Amini E. J Heterocyclic Chem. 2007;44:697–699. [Google Scholar]; (b) Ramu E, Kotra V, Bansal N, Varala R, Adapa SR. Rasayan J Chem. 2008;1:188–194. [Google Scholar]
- 21.(a) Elliott MC, Long MS. Org Biomol Chem. 2004;2:2003–2011. doi: 10.1039/b404679j. [DOI] [PubMed] [Google Scholar]; (b) Butters M, Davies CD, Elliott MC, Cousins JH, Kariuki BM, Ooi L-L, Wood JL, Wordingham SV. Org Biomol Chem. 2009;7:5001–5009. doi: 10.1039/b914744f. [DOI] [PubMed] [Google Scholar]
- 22.(a) Oike H, Yoshioka Y, Kobayashi S, Nakashima M, Tezuka Y, Goethals EJ. Macromol Rapid Commun. 2000;21:1185–1190. [Google Scholar]; (b) Hoogenboom R, Andres PR, Kickelbick G, Schubert US. Synlett. 2004;10:1779–1783. [Google Scholar]
- 23.Hammoud H, Schmitt M, Bihel F, Antheaume C, Bourguignon J-J. J Org Chem. 2012;77:417–423. doi: 10.1021/jo202018w. [DOI] [PubMed] [Google Scholar]
- 24.Wipf P, Hopkins CR. J Org Chem. 1999;64:6881–6887. doi: 10.1021/jo990089v. [DOI] [PubMed] [Google Scholar]
- 25.Künstlinger M, Breitmaier E. Synthesis. 1983;2:161–162. [Google Scholar]
- 26.Seto H, Takesue T, Ikemura T. Bull Chem Soc Jpn. 1985;58:3431–3435. [Google Scholar]
- 27.Karpov AS, Müller TJJ. Org Lett. 2003;5:3451–3454. doi: 10.1021/ol035212q. [DOI] [PubMed] [Google Scholar]
- 28.Palimkar SS, Kumar PHK, Jogdand NR, Daniel T, Lahoti RJ, Srinivasan KV. Tetrahedron Lett. 2006;47:5527–5530. [Google Scholar]
- 29.Ludovici DW, Kavash RW, Kukla MJ, Ho CY, Ye H, De Corte BL, Andries K, de Bethune M-P, Azijn H, Pauwels R, Moereels HEL, Heeres J, Koymans LMH, de Jonge MR, Van Aken KJA, Daeyaert FFD, Lewi PJ, Das K, Arnold E, Janssen PAJ. Bioorg Med Chem Lett. 2001;11:2229–2234. doi: 10.1016/s0960-894x(01)00411-5. [DOI] [PubMed] [Google Scholar]
- 30.Milcent R, Malanda J-C, Barbier G, Vaissermann J. J Heterocyclic Chem. 1997;34:329–336. [Google Scholar]
- 31.Pryadeina MV, Burgart YV, Kodess MI, Saloutin VI, Chupakhin ON. Russ Chem Bull Int Ed. 2004;53:1261–1266. [Google Scholar]
- 32.Spartan, Wavefunction, Inc., Irvine, CA.
- 33.Webb TR, Lvovskiy D, Kim S-A, Ji X-D, Melman N, Linden J, Jacobson KA. Bioorg Med Chem. 2003;11:77–85. doi: 10.1016/s0968-0896(02)00323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Qiam C, Tiam He, Ma Y. Synth Commun. 2003;33:1459–1468. [Google Scholar]
- 35.Jackson MM, Leverett C, Toczko JF, Roberts JC. J Org Chem. 2002;67:5032–5035. doi: 10.1021/jo025682i. [DOI] [PubMed] [Google Scholar]
- 36.Ghorai MK, Halder S, Das RK. J Org Chem. 2010;75:7061–7072. doi: 10.1021/jo101680f. [DOI] [PubMed] [Google Scholar]
- 37.Feng C, Lu C, Chen Z, Dong N, Shi J, Yang G. J Org Chem. 2010;47:671–676. [Google Scholar]
- 38.Joslyn AF, Luchowski E, Triggle DJ. J Med Chem. 1988;31:1489–1492. doi: 10.1021/jm00403a002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.