

Abstract
Purpose
Biotinylated lipid prodrugs of acyclovir (ACV) were designed to target the sodium dependent multivitamin transporter (SMVT) on the cornea to facilitate enhanced cellular absorption of ACV.
Methods
All the prodrugs were screened for in vitro cellular uptake, interaction with SMVT, docking analysis, cytotoxicity, enzymatic stability and antiviral activity.
Results
Uptake of biotinylated lipid prodrugs of ACV (B-R-ACV and B-12HS-ACV) was significantly higher than biotinylated prodrug (B-ACV), lipid prodrugs (R-ACV and 12HS-ACV) and ACV in corneal cells. Transepithelial transport across rabbit corneas indicated the recognition of the prodrugs by SMVT. Average Vina scores obtained from docking studies further confirmed that biotinylated lipid prodrugs possess enhanced affinity towards SMVT. All the prodrugs studied did not cause any cytotoxicity and were found to be safe and non-toxic. B-R-ACV and B-12HS-ACV were found to be relatively more stable in ocular tissue homogenates and exhibited excellent antiviral activity.
Conclusions
Biotinylated lipid prodrugs demonstrated synergistic improvement in cellular uptake due to recognition of the prodrugs by SMVT on the cornea and lipid mediated transcellular diffusion. These biotinylated lipid prodrugs appear to be promising drug candidates for the treatment of herpetic keratitis (HK) and may lower ACV resistance in patients with poor clinical response.
Keywords: Acyclovir, herpetic keratitis, SMVT, cornea and antiviral activity
Introduction
Herpetic keratitis (HK) is a serious corneal complication which can lead to blindness, predominantly because of its recurrent nature (1-3). HK is caused by herpes simplex virus (HSV), a DNA virus that commonly affects humans. Herpes viruses are categorized into two types: HSV-1 and HSV-2. The former is mainly responsible for orofacial and ocular infections, while the latter is primarily responsible for genital infections. According to recent US statistics, the seroprevalence of HSV-1 and HSV-2 were reported to be approx. 50% and 20% respectively (4, 5). Following primary infection, HSV remains latent in the neuronal sensory ganglia, which then becomes an intermittent site for periodic reactivations (6-8). A recent report described that the cornea acts as a reservoir for HSV-1 and is a major cause of corneal latency (9, 10). Also, several studies reported the presence of HSV-1 DNA in the corneas of patients suffering from chronic HK (11-15), corneas deposited in the eye banks, (16-18) and even in the tears of some asymptomatic patients (19-21). Operational latency, persistent low-grade infection, and neuronal reactivation of latent HSV were hypothesized to be the major reasons for the expression of HSV-1 DNA in human corneas (9, 10). HSV infections have been found to be more severe in immunocompromised patients (4). Approximately 20,000 new cases of infections and more than 28,000 reactivations occur in the US annually. The recurrence rate of ocular HSV is approximately 20% within 2 years, 40% within 5 years, and 67% within 7 years (22, 23).
Since its introduction, acyclovir (ACV), a guanosine analogue has been the standard first line drug for prophylaxis and treatment of HSV infections, particularly HK. In HSV infected cells, ACV gets converted to ACV monophosphate by viral thymidine kinases. The monophosphate derivative is then subsequently phosphorylated by cellular kinases to ACV triphosphate which then serves as a substrate for viral DNA polymerase. It can also act as chain terminator. Though highly effective, ACV has poor permeation across the cornea due to its hydrophilic nature (24). Moreover, sufficient evidence demonstrates that long term prophylaxis with ACV could lead to the development of resistant viral isolates, and these resistance levels have been found to be increasing from 3.5-10% in immunocompromised patients (4, 25). Though the exact mechanism of resistance is not known, it is postulated to be mediated by either a mutation in thymidine kinase or viral DNA polymerase (26). High prevalence of ACV-resistant HSV-1 isolates was found in HK patients (11 of 173 tested), and nine out of the 11 patients with resistant isolates were refractory to ACV therapy. These remarkable results accentuate the need for physicians to consider ACV resistance in patients with poor clinical response to therapy (1, 27, 28).
Various strategies have been explored to improve the cellular absorption of ACV, including lipophilic prodrug derivatization and transporter targeted approach. Recently we have developed a novel targeted lipid prodrug strategy which combines both the lipid and transporter targeted delivery to generate synergistic effect (29). Results from these studies demonstrate that the targeted lipid prodrugs of ACV exhibited higher affinity for sodium dependent multivitamin transporter (SMVT). These encouraging results generated for us the idea of utilizing these novel conjugates in special drug delivery cases like topical ocular delivery of highly hydrophilic molecules where there is a rapid drug loss in tear fluids, and reduced uptake due to tight junctions. In such circumstances, enhanced lipophilicity to augment cellular uptake could be highly advantageous. This could result in achieving higher absorption of ACV and preventing ACV mediated resistance. Therefore, in this report we wanted to investigate the feasibility of utilizing targeted lipid prodrugs via topical application to the eye. In the present study, we have investigated the cellular uptake, cytotoxicity in human corneal epithelial cells (HCEC), prodrug interaction with SMVT on freshly excised rabbit corneas, and enzymatic stability of novel biotinylated prodrugs in tissue homogenates (cornea, iris-ciliary body and lens). Docking analysis was performed to examine the affinity of prodrugs towards hSMVT protein. Also, the antiviral activity of these biotinylated lipid prodrugs against herpes simplex viruses (HSV-1 and 2), human cytomegalovirus (HCMV) and epstein barr virus (EBV) was assessed.
Materials and Methods
Materials
[3H] Biotin (specific activity 60Ci/mMol) was procured from Perkin Elmer (Boston, MA). All the prodrugs of ACV were synthesized and purified in our laboratory as described earlier (29). Unlabeled biotin, HEPES, bovine insulin, human epidermal growth factor, penicillin, streptomycin, lactalbumin enzymatic dehydrolysate, sodium bicarbonate, and D-glucose were purchased from Sigma Chemical Co. Fetal Bovine Serum (FBS) was obtained from Atlanta biologicals (Lawrenceville, GA). Culture flasks (75cm2 growth area), 12-well plates (3.8cm2 growth area per well) and 96-well plates (0.32 cm2 growth area per well) were purchased from Corning Costar Corp. (Cambridge, MA, USA). Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) and minimum essential medium (MEM) were purchased from Invitrogen (Carlsbad, CA). All buffer components and organic solvents were purchased from Fisher Scientific Co. (Fair Lawn, NJ). New Zealand White (NZW) Rabbits weighing 6-7 lbs were purchased from Myrtle's Rabbitry (Thompson Station, TN). Sodium pentobarbital was obtained from the stock maintained by the UMKC School of Pharmacy and used under supervision.
Cell Culture
Human corneal epithelial cells (HCEC) were a generous gift from Dr. Araki-Sasaki (Kinki Central Hospital, Japan). HCEC cells were cultured at 37°C in an incubator with 95% air and 5% CO2. Culture medium consisted of DMEM/F-12 supplemented with 15 % fetal bovine serum (FBS) (heat inactivated), 15mM HEPES, 22mM NaHCO3, 100mg each of penicillin and streptomycin, 5μg/ml insulin and 10ng/ml of human epidermal growth factor (30, 31). Rabbit primary corneal epithelial cells (rPCEC) were cultured following previous reports from our laboratory (32, 33). Culture medium consisted of minimum essential medium (MEM), supplemented with 10% FBS, lactalbumin, HEPES, NaHCO3, 100mg each of penicillin and streptomycin. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 90% relative humidity. The medium was changed every alternate day. After reaching 80% confluency, the cells were passaged using TrypLE™ Express (superior replacement for trypsin). The cells were then plated in 12-well uptake plates at a density of 250,000 cells/well and in 96-well plates at a density of 10,000 cells/well. The cells were grown in a similar way in these plates and utilized for further studies.
Uptake Study
Cellular uptake studies were performed on HCEC cells after growing them for 7 days in 12-well plates. Prior to experimentation, the medium was removed and cells were rinsed 3 times with DPBS (130 mM NaCl, 0.03 mM KCl, 7.5 mM Na2HPO4, 1.5 mM KH2PO4, 1 mM CaCl2, 0.5 mM MgSO4, 20 mM HEPES and 5 mM glucose). Uptake was initiated by adding 1ml of either drug or prodrug solution into each well and incubated for a period of 30 min. After the incubation period, the drug solutions were removed and uptake was terminated using ice-cold stop solution (containing 200 mM KCl and 2 mM HEPES). Subsequently the cells were washed three times to remove the surface bound drug/prodrug and lysed overnight at -80°C with 500μL cremophore water (2 drops of cremophore gel in 50 ml of deionized water) in each well. The samples were then analyzed using LC/MS/MS and uptake was normalized to the protein content of each well. The amount of protein in the cell lysate was estimated using BioRad protein estimation kit (BioRad, Hercules, CA) using bovine serum albumin as an internal standard.
Following a similar method, the uptake of B-R-ACV, B-12HS-ACV and B-ACV was also carried out in the presence of a sodium free buffer to examine the involvement of sodium and delineate the sodium dependency of the transport system for translocation of biotin conjugated prodrugs via SMVT. Sodium free DPBS buffer was prepared by replacing sodium chloride (130 mM) and disodium phosphate (7.5 mM) with equimolar quantities of choline chloride and dipotassium phosphate.
Ex Vivo Rabbit Corneal Transport
Animal studies were conducted according to The Association for Research in Vision and Ophthalmology (ARVO) guidelines for the use of animals in ophthalmic and vision research. Animals were euthanized by administering sodium pentobarbital (50 mg/kg) through marginal ear vein. Eyes were immediately enucleated and rinsed with ice-cold DPBS of pH 7.4 to get rid of any traces of blood. Vitreous humor was aspirated with a 1 mL syringe following a small incision into the sclera. Cornea was carefully excised such that some scleral portion is adhered to the cornea. Sclera attached to cornea assists in securing the cornea between the half-cells during the course of transport experiment. Subsequently, the lens and the iris-ciliary body (ICB) were removed and separated from the cornea. Cornea was washed instantly with ice-cold DPBS and mounted on a side-by-side diffusion half-chamber (type-VSC-1, Crown Glass Company Inc, (Somerville, NJ) such that the epithelial side faces the donor compartment. Water was circulated through the jacketed chambers of the diffusion apparatus to maintain temperature at 34°C (corneal temperature in vivo). Drug solutions (3 mL) consisting of 0.5 μCi/mL of [3H] biotin in the presence or absence of unlabeled biotin and all biotinylated prodrugs were added on the epithelial side of the cornea (donor compartment) and DPBS (3.4 mL) was added to the other half-chamber (receiver compartment). This excess volume of DPBS in the receiver compartment maintains the curvature of the cornea due to elevated hydrostatic pressure. Magnetic stirrer bars were added into the chambers to ensure continuous stirring. Sink conditions were maintained throughout the experiment for 3 h. Aliquots (200μL) were removed from the receiver chamber at appropriate time intervals, and replaced with an equal volume of DPBS. Samples were immediately transferred to scintillation vials containing 3 mL of scintillation cocktail, and radioactivity was measured with a Beckman Scintillation Counter (Model LS-6500, Beckman Instruments, Inc.). Corneal integrity was also ensured by assessing the permeability of [14C] mannitol (0.25 μCi/mL), a paracellular marker.
Modeling of Human SMVT Protein and Docking of Biotinylated ACV Prodrugs
The entire SMVT protein sequence (Q9Y289) was retrieved from UNIPROT database (www.uniprot.org/uniprot/Q9Y289). The sequence was submitted to I-Tasser homology model-building server for model calculation (34-36). The selected model had an I-Tasser score of -1.453. Stereo chemical quality check was performed on the returned model. All biotin conjugated structures were sketched using GaussView (GaussView, Version 5, Dennington, R.; Keith, T.; Millam, J. Semichem Inc., Shawnee Mission KS, 2009). AutoDock Vina (Scripps Institute docking program) (37) was used to perform the docking studies using the default settings. The receptor grid was centered at the center of the protein (X: 71.8067; Y: 71.5789; Z: 72.2651). The grid was maximized with dimensions of X: 62.0908, Y: 71.4766, and Z: 73.3060 to encompass the entire protein. Each structure was independently docked to the targeted protein to produce nine different poses with the highest affinities. Vina scores were derived by utilizing a scoring function as described in Trott et. al (37).
Cell Proliferation Assay
Cytotoxicity assay was carried out with Cell Titer 96® Aqueous Non-Radioactive Cell Proliferation Assay Kit (Promega, Madison, WI) to test the toxicity of all biotinylated prodrugs including ACV. HCEC and rPCEC cells were grown on 96-well plates. Aliquots (100μL) of sterile prodrug solutions (100μM) in culture medium were added to each well and incubated for 48h. Cell proliferation in the presence of ACV and all biotinylated prodrugs was compared with a positive control (medium without drug) and a negative control (medium without cells). After 48 h of incubation, 20μL of freshly prepared dye solution was added to each well and incubated for two and half hours. The amount of farmazan formed was measured using a 96-well micro titer plate reader (SpectraFluor Plus, Tecan, Maennedorf, Switzerland) with absorbance set at 490 nm wavelength.
Ocular Tissue Homogenate Studies
Preparation of Ocular Tissues
NZW rabbits were euthanized and eyes were enucleated as described earlier. An incision was made on sclera and the lens was exposed after aspirating vitreous humor. The cornea was then carefully cut without any scleral tissue attached to it. The lens was separated followed by the iris-ciliary body. All the tissues were immediately washed with ice-cold DPBS and stored in -80°C freezer until further use. Prior to experimentation, each tissue was homogenized with a homogenizer (Tissue Tearer Model 985-370 Type 2, Dremel Multipro) in 3 mL ice-cold DPBS and the homogenate was centrifuged at 12,000 rpm for 30 min at 4°C using an ultracentrifuge (Beckman TL-100) to remove cellular debris. The obtained supernatant was utilized for hydrolysis studies. Protein content of each supernatant was estimated with BioRad assay with bovine serum albumin as the standard (38, 39).
Enzymatic Hydrolysis Procedure
The protein content of supernatant obtained from homogenates was adjusted to 0.5mg/mL and was equilibrated at 34°C for about 15 min prior to initiation of each experiment. Hydrolysis was initiated by the addition of a calculated amount of prodrug from respective stock to 1.25 mL of supernatant to adjust to 100 μM concentration in a microcentrifuge tube. Tubes were then placed in a shaking water bath set at 34°C. Aliquots (125 μL) were withdrawn at appropriate time intervals for up to 24 h. The samples were immediately diluted with equal amount of ice-cold organic mixture containing methanol and acetonitrile (4:5) to stop enzymatic hydrolysis and precipitate proteins. Samples were stored at −80°C until further analysis. Prior to analysis, samples were thawed and centrifuged at 5,000rpm for 10min and the supernatant was extracted by a well-established procedure and analyzed by LC/MS/MS (29). The apparent first order rate constants were calculated from the slope of log (prodrug concentration) vs. time plot and corrected for any chemical hydrolysis observed with the control.
LC/MS/MS Analysis
A fast and sensitive LC/MS/MS method has been developed in multiple reaction monitoring (MRM) with electrospray (ES) positive ionization mode for detection of ACV and novel biotinylated lipid prodrugs. QTrap® LC/MS/MS mass spectrometer (API 3200, Applied Biosystems/MDS Sciex, Foster City, CA, USA) was employed to analyze samples from non-radioactive cellular uptake studies. Chromatographic separation was achieved on XTerra® RP C8 MS column 50 × 4.6 mm I.D, 5μm particle size (Waters Corporation, Milford, MA) with isocratic mobile phase. The mobile phase consisted of 70 % acetonitrile, 30 % water and 0.1% formic acid which were pumped at a flow rate of 0.2 ml/min. Precursor ions of the analytes as well as internal standard were determined from spectra obtained during the infusion of standard drug/prodrug solutions using an infusion pump connected directly to the electrospray ionization source. Each of these precursor ions was subjected to collision-induced dissociation to determine their respective product ions. MRM transitions at m/z [M+H]+ generated were 226.4/152.2 for ACV, 452.3/301.3 for B-ACV, 506.3/488.6 for R-ACV, 732.3/257.4 for B-R-ACV, 507.34/488.5 for 12HS-ACV, 735.6/257.3 for B-12HS-ACV and 256/152 for GCV. Peak areas for all components were automatically integrated by using Analyst™ software and peak-area ratios (area of analytes to area of internal standard) were plotted vs concentration by weighted linear regression (1/concentration). The analytical data which resulted from prodrugs with MRM method showed a significant linearity. This method gave rapid and reproducible results.
Stocks and stock dilutions of ACV and respective prodrugs were prepared similarly following a previously published procedure (29, 31, 40). Samples were extracted by liquid-liquid extraction method. Ganciclovir (GCV) was used as an internal standard to ensure reproducibility and reliability of the method. Prior to analysis, samples were thawed at room temperature. Two hundred microliter sample along with 20μL of GCV (5 μg/mL) was extracted with 1mL of organic solvent containing 2:3 ratios of isopropanol and dichloromethane. The samples were vortexed for approximately 2 min and centrifuged at 12000 g for 15 min at 4°C. Organic layer (850 μL) was transferred into eppendorf tubes and evaporated to dryness under speed vacuum with a Speedvac (SAVANT Instruments, Inc., Holbrook, NY). The residue was then reconstituted in 100 μL of mobile phase, vortexed for 30 sec and transferred into pre-labeled vials with silanized inserts. Subsequently, 15 μL of the resulting solution was injected onto LC-MS/MS. Appropriate calibration standards of ACV and all prodrugs were prepared by spiking a known analyte concentration to blank tissue homogenate following similar procedures. A calibration curve was generated using calibration standards.
Antiviral Screening Procedures
Cell Culture and Virus Strains
Human foreskin fibroblast (HFF) cells prepared from human foreskin tissue were obtained from the University of Alabama at Birmingham tissue procurement facility, with approval from its IRB. The tissue was incubated at 4°C for 4 h in Clinical Medium consisting of minimum essential media (MEM) with Earl's salts supplemented with 10% FBS (Hyclone, Inc. Logan UT), L-glutamine, fungizone, and vancomycin. Tissue was then placed in phosphate buffered saline (PBS), minced, rinsed to remove the red blood cells, and resuspended in trypsin/EDTA solution. The tissue suspension is incubated at 37°C and gently agitated to disperse the cells, which are collected by centrifugation. Cells were resuspended in 4 ml Clinical Medium and placed in a 25 cm2 flask and incubated at 37°C in a humidified CO2 incubator for 24 h. The media was then replaced with fresh Clinical Medium, and the cell growth was monitored daily until a confluent monolayer was formed. HFF cells were then expanded through serial passages in standard growth medium of MEM with Earl's salts supplemented with 10% FBS, L-glutamine, penicillin, and gentamycin. The cells were passaged routinely and used for assays at or below passage 10 (41). Akata cells were kindly provided by John Sixbey (Louisiana State University, Baton Rouge, LA). Cells were maintained in RPMI 1640 (Mediatech, Inc., Herndon, VA) supplemented with 10% FBS, L-glutamine, penicillin and gentamicin at 37°C in a humidified 5% CO2 atmosphere. Cells were passaged 3 days prior to performing the assay, enumerated on a hemacytometer and diluted to a final concentration of 4×105 cells/ml. Latently infected cells were induced to undergo a lytic infection by adding a F(ab′)2 fragment of goat anti-human IgG antibody (MP Biomedicals, Aurora, OH) (42). Strains of HSV-1 (E-377 strain) and HSV-2 (MS strain) were a gift of Jack Hill (Burroughs Wellcome). The HCMV strain (AD169 strain) was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and the construction of RC314 with a K355M mutation in the UL97 kinase was reported previously (43). Akata cells latently infected with EBV were obtained from John Sixbey.
Antiviral Assays
Each experiment that evaluated the antiviral activity of the compounds included both positive and negative control compounds to ensure the performance of each assay. Concurrent assessment of cytotoxicity was also performed for each study (see below).
(i) Plaque Reduction Assays for HSV-1, HSV-2 and HCMV
Monolayers of HFF cells were prepared in 6-well plates and incubated at 37°C for 2 days to allow the cells to reach confluency. Media was then aspirated from the wells and 0.2 ml of virus was added to each of three wells to yield 20-30 plaques in each well. The virus was allowed to adsorb to the cells for 1 h and the plates were agitated every 15 minutes. Compounds were diluted in assay media consisting of MEM with Earl's salts supplemented with 2% FBS, L-glutamine, penicillin, and gentamicin. Solutions ranging from 300 μM to 0.1 μM were added to duplicate wells and the plates were incubated for various times, depending on the virus used. For HSV-1 and -2, the monolayers were then stained with 1% crystal violet in 20% methanol and the unbound dye was removed by washing with dH2O. For assays with HCMV, the cell monolayer was stained with 1% neutral red solution for 4 h, then the stain was aspirated and the cells were washed with PBS. For all assays, plaques were enumerated using a stereomicroscope and the concentration of compound that reduced plaque formation by 50% (EC50) and 90% (EC90) was interpolated from the experimental data.
(ii) DNA Hybridization Assay for EBV
Assay for EBV was performed in Akata cells that were induced to undergo a lytic infection with 50 μg/ml of a goat anti-human IgG antibody by methods reported previously (42). Experimental compounds were diluted in round bottom 96-well plates to yield concentrations ranging from 60 μM to 0.48 μM. Akata cells were added to the plates at a concentration of 4×104 cells per well and incubated for 72 h. Briefly, 100 μl of denaturation buffer (1.2M NaOH, 4.5M 80 NaCl) was added to each well to denature the DNA and a 50 μl aliquot was aspirated through an Immobilon nylon membrane (Millipore, Bedford, MA) using a Biodot apparatus (Bio-Rad, Hercules, CA). The membranes were then allowed to dry before equilibration in DIG Easy Hyb (Roche Diagnostics, Indianapolis, IN) at 56°C for 30min. A specific digoxigenin (DIG)-labeled probe was prepared following the manufacturer's protocol (Roche Diagnostics). The primers (forward) 5′-CCC AGG AGT CCC AGT AGT CA-3′ and (reverse) 5′-CAG TTC CTC GCCTTAGGTTG-3, amplified a fragment corresponding to coordinates 96802–97234 in EBV genome (AJ507799). Membranes with EBV DNA were hybridized overnight at 56°C followed by sequential washes 0.2× SSC with 0.1% SDS and 0.1× SSC with 0.1% SDS at the same temperature. Detection of specifically bound DIG probe was performed with anti-DIG antibody according to the manufacturer's protocol (Roche Diagnostics). An image of the photographic film was captured and quantified with QuantityOne software (Bio-Rad) and compound concentrations sufficient to reduce the accumulation of viral DNA by 50% (EC50) and 90% (EC90), were interpolated from the experimental data.
Cytotoxicity Assays on Viral Cells
Every antiviral assay included a parallel cytotoxicity assay with the same cells used for each virus, the same cell number, the same drug concentrations, and the same incubation times to provide the same drug exposure. To ensure that the cytotoxicity of all compounds could be compared directly, we also performed a standard neutral red uptake cytotoxicity assay for all compounds in confluent HFF cells with a 7 day incubation period.
(i) Neutral red uptake cytotoxicity assays
Each compound was evaluated in a standard cytotoxicity assay by standard methods (41). Briefly, HFF cells were seeded onto 96-well tissue culture plates at a density of 2.5 × 104 cells/well in standard growth medium. After 24 h of incubation, medium was replaced with MEM containing 2% FBS and compounds were added to the first row and then 5-fold serial dilutions were used to generate a series of compound concentrations with a maximum of 300 μM. Assay plates were then incubated for 7 days, and 100 μl of 0.66 mg/ml neutral red solution in PBS was added to each well and the plates were incubated for 1 h. The stain was then removed, the plates rinsed with PBS and the dye internalized by viable cells was solubilized in PBS supplemented with 50% ethanol and 1% glacial acetic acid. The optical density was then determined at 550 nm and CC50 values were interpolated from the experimental data. For all plaque reduction assays, neutral red cytotoxicity assays were performed on a parallel set of 6-well plates containing HFF cells that received the same compound concentrations as used for the antiviral assays, but remained uninfected. The cytotoxicity plates were removed from the incubator on the same day as each antiviral assay and the cell monolayer was stained for 6 h with 2 ml of a neutral red solution at a concentration of 0.165 mg/ml in PBS. The dye was then removed, residual dye rinsed from the cells with PBS, and cell monolayers were inspected visually for any signs of toxicity.
(ii) CellTiter Glo (Toxicity) assay
Viability of Akata cells were assessed with the CellTiter-Glo Luminescent Cell Viability Assay (Promega) following established protocols (42). Briefly, assay plates were incubated at ambient temperature for 30 min, 50 μl of CellTiter-Glo reagent was added to each well and the plates were mixed for 2 min on an orbital shaker to lyse the cells. The plates were then incubated for an additional 10 min at ambient temperature, and the luminescence was quantified on a luminometer. Standard methods were used to calculate drug concentrations that inhibited the proliferation of Akata cells by 50% (CC50).
Data Analysis
[3H] Biotin transport across freshly excised rabbit corneas in the presence of biotinylated prodrugs were calculated according to Eq. 1.
| Eq. 1 |
CPMsample and CPMdonor denote average values of Counts per Minute (CPM) of sample and donor (n=4) respectively; Cdonor represents the concentration of donor used and Csample represents the concentration of sample.
The flux and the permeability values were calculated according to Fick's 1st law of diffusion. Flux (Eq. 2) was calculated by dividing the slope (dM/dT) (obtained by plotting a cumulative amount of the [3H] Biotin permeated versus time) with the cross-sectional area (A) available for diffusion, i.e., 0.636 cm2.
| Eq. 2 |
Permeability values were calculated by dividing the flux values with the concentration of the respective donor. Permeability (P) value was then calculated according to Eq. 3.
| Eq. 3 |
Statistical Analysis
All the experiments were conducted at least in quadruplicate (n = 4), and results are expressed as mean ± standard deviation (SD). Statistical comparison of mean values was performed with a student t-test and/or one-way analysis of variance (ANOVA). A P-value of <0.05 was considered to be statistically significant.
Results
Uptake Study
Cellular accumulation of B-R-ACV, B-12HS-ACV, B-ACV, R-ACV, 12HS-ACV and ACV was examined on HCEC cells. Compared to ACV, the uptake of B-R-ACV and B-12HS-ACV increased by approx. 13.6 and 13.1 times respectively, whereas the uptake of B-ACV, R-ACV and 12HS-ACV increased only by 4.6, 1.8, 2.0 times respectively (Fig. 1). As depicted in Fig. 2, the uptake of B-R-ACV, B-12HS-ACV and B-ACV was significantly reduced to about 43%, 41% and 45% in the presence of a sodium free buffer.
Fig. 1.
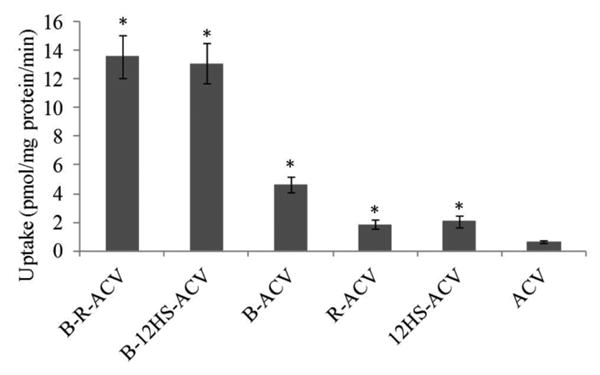
Cellular accumulation of B-R-ACV, B-12HS-ACV, B-ACV, R-ACV, 12HS-ACV and ACV on HCEC cells. Uptake was performed at 37°C with DPBS buffer for 30 min. Values represent mean ± standard deviation (n= 4). A P-value of less than 0.05 was considered to be statistically significant and denoted by asterisk (*).
Fig. 2.
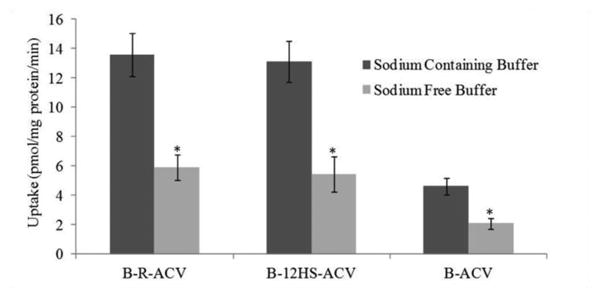
Cellular accumulation of B-R-ACV, B-12HS-ACV and B-ACV on HCEC cells in the presence and absence of sodium ions in DPBS buffer at 37°C. Data represents mean ± standard deviation (n= 4). A P-value of less than 0.05 was considered to be statistically significant and denoted by asterisk (*).
Rabbit Corneal Transport Study
Transport of [3H] biotin (0.5μCi/mL) across freshly excised rabbit corneas was assessed. Cumulative amount transported was calculated from the transport data and plotted against time. Unlabeled biotin and all biotinylated ACV prodrugs inhibited the transport of [3H] biotin (Fig. 3) demonstrating the ability of these compounds to interact with SMVT. B-R-ACV and B-12HS-ACV caused a greater inhibition in [3H] biotin transport than B-ACV suggesting the synergistic involvement of SMVT transporter and the lipid moiety in mediating cellular transport across the cornea. Permeabilities of [3H] biotin are 3.73 × 10−6 cm/sec in comparison to 1.40 × 10−6 cm/sec, 3.10 × 10−6 cm/sec, 2.35 × 10−6 cm/sec, 2.44 × 10−6 cm/sec in the presence of 50μM concentration of unlabeled biotin and B-ACV, B-R-ACV, and B-12HS-ACV, respectively (Fig. 4).
Fig. 3.
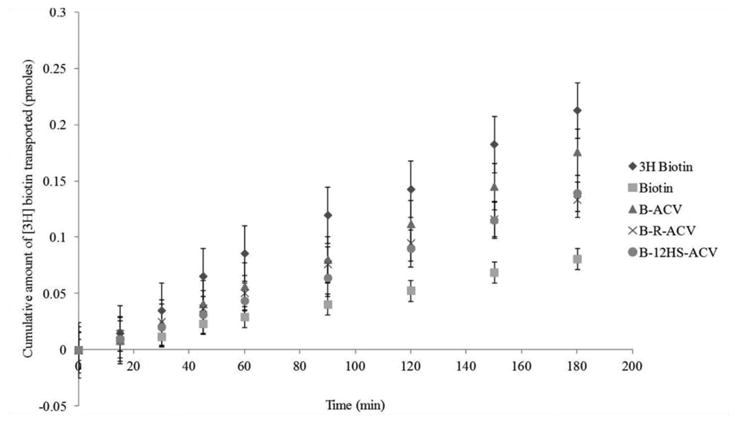
Transepithelial transport of [3H] biotin across freshly excised rabbit corneas in the absence and presence of 50μM unlabeled biotin, B-ACV, B-R-ACV, and B-12HS-ACV. Cumulative amount of [3H] biotin transported was measured in DPBS buffer at 34°C for 180 min. Data represents mean ± standard deviation (n= 4-6).
Fig. 4.
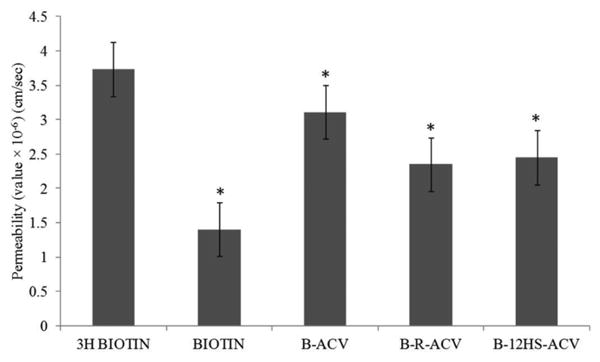
Comparison of permeabilities (cm/sec) of [3H] biotin alone, in the presence of 50μM unlabeled biotin, B-ACV, B-R-ACV, and B-12HS-ACV across freshly excised rabbit corneas. Data represents mean ± standard deviation (n= 4-6). A P-value of less than 0.05 was considered to be statistically significant and denoted by asterisk (*).
Docking Analysis
Docking analysis was carried out to examine the affinity of biotinylated prodrugs towards SMVT. The average Vina docking score for each predicted conformation is shown in Table I. The average Vina score was calculated using the nine highest Vina docking scores for each biotinylated prodrug of ACV. The highest scoring binding poses for each ligand, along with the neighboring amino acids of SMVT are shown in Fig. 5. Binding affinity varies inversely with docking score meaning; the lower the docking score, the stronger the binding affinity. Docking of biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) yielded lower docking scores relative to non-lipidated but biotinylated prodrug (B-ACV). These results signify that the lipid raft enhances the affinity of prodrugs towards SMVT protein.
Table 1.
Docking scores of biotinylated lipid prodrugs of acyclovir. Binding affinity varies inversely with docking score meaning; the lower the docking score, the stronger is the binding affinity.
| Structure of Prodrug | Average Docking Score |
|---|---|
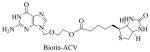
|
-6.61 |
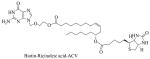
|
-7.49 |
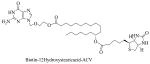
|
-7.90 |
Fig. 5.
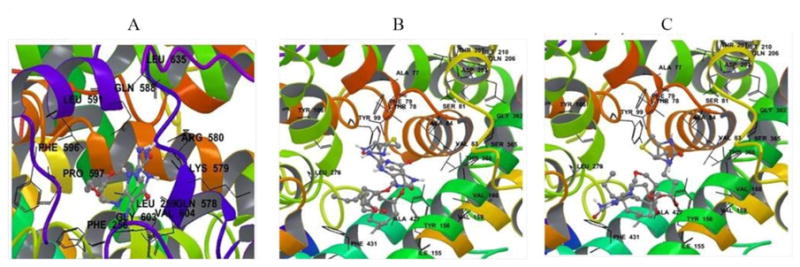
Biotinylated prodrugs of ACV are shown in their predicted binding site on SMVT with all amino acid residues within 4 Å of the ligand. A) B-ACV; B) B-R-ACV and C) B-12HS-ACV.
Cell Proliferation Assay
Results from cytotoxicity studies on HCEC and rPCEC cells following exposure for 48 h to B-R-ACV, B-12HS-ACV, B-ACV and ACV are shown in Fig. 6. DMSO (10% v/v) was used as a positive control and blank medium without any drug served as the negative control. All biotinylated prodrugs did not exhibit any significant cytotoxicity at the concentration tested, demonstrating that these compounds are safe and non-toxic. Simultaneous conjugation of biotin and lipid rafts to ACV did not show any sign of toxicity and were equivalent or even better than parent drug ACV.
Fig. 6.
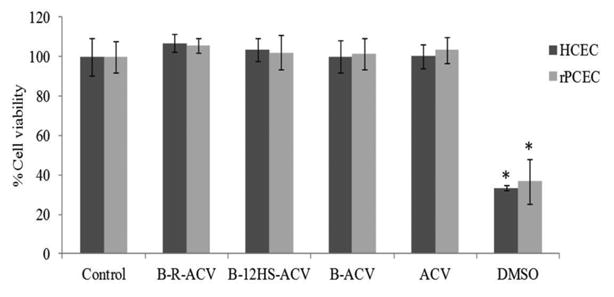
Cytotoxicity assay in the presence of B-R-ACV, B-12HS-ACV, B-ACV and ACV on HCEC and rPCEC cells for 48 h. DMSO (10% v/v) served as a positive control. Data represent mean percentage of viable cells ± standard deviation (n= 4). A P-value of less than 0.05 was considered to be statistically significant and denoted by asterisk (*).
Ocular Tissue Stability
Enzymatic degradation of all the prodrugs (B-R-ACV, B-12HS-ACV and B-ACV) was studied in ocular tissues such as cornea, ICB and lens. Biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) exhibited better stability and were less susceptible to enzymatic degradation compared to non-lipidated but biotinylated prodrug, B-ACV. Degradation rate constants of all prodrugs in rabbit ocular tissue homogenates are shown in Table II. All the prodrugs exhibited a first-order degradation rate in ocular tissue homogenates. Unlike B-ACV, biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) exhibited greater stability in ocular tissues (ICB and lens) indicating that these prodrugs were relatively stable, and less susceptible to enzymatic hydrolysis. Figures 7A and 7B highlight the varied susceptibility of B-R-ACV and B-12HS-ACV to ocular tissues such as cornea, ICB and lens. The half-lives of B-R-ACV and B-12HS-ACV were observed to be almost similar in each of the ocular tissue homogenates studied. On the contrary, complete degradation of B-ACV was observed in ICB and lens homogenates.
Table II.
First-order hydrolysis rate constants and half-lives of B-ACV, B-R-ACV and B-12HS-ACV in cornea, iris-ciliary body (ICB) and lens homogenates. Values are represented as mean ± standard deviation (n=4). Values reported are rounded off to significant digits at every step during calculations. ND = not determined due to rapid degradation.
| Prodrugs | Cornea | ICB | Lens | |||
|---|---|---|---|---|---|---|
| Rate constant (k×103 min−1 mg protein-1) | Half-life (hrs) | Rate constant (k×103 min−1 mg protein-1) | Half-life (hrs) | Rate constant (k×103 min−1 mg protein-1) | Half-life (hrs) | |
| B-R-ACV | 1.84 ± 0.30 | 6.37 ± 1.01 | 3.75 ± 0.28 | 3.09 ± 0.24 | 0.84 ± 0.02 | 13.70 ± 0.34 |
| B-12HS-ACV | 1.78 ± 0.29 | 6.62 ± 1.08 | 3.58 ±0.70 | 3.45 ± 1.06 | 0.85 ± 0.16 | 13.85 ± 2.32 |
| B-ACV | 1.99 ± 0.21 | 5.80 ± 0.49 | ND | - | ND | - |
Fig. 7.
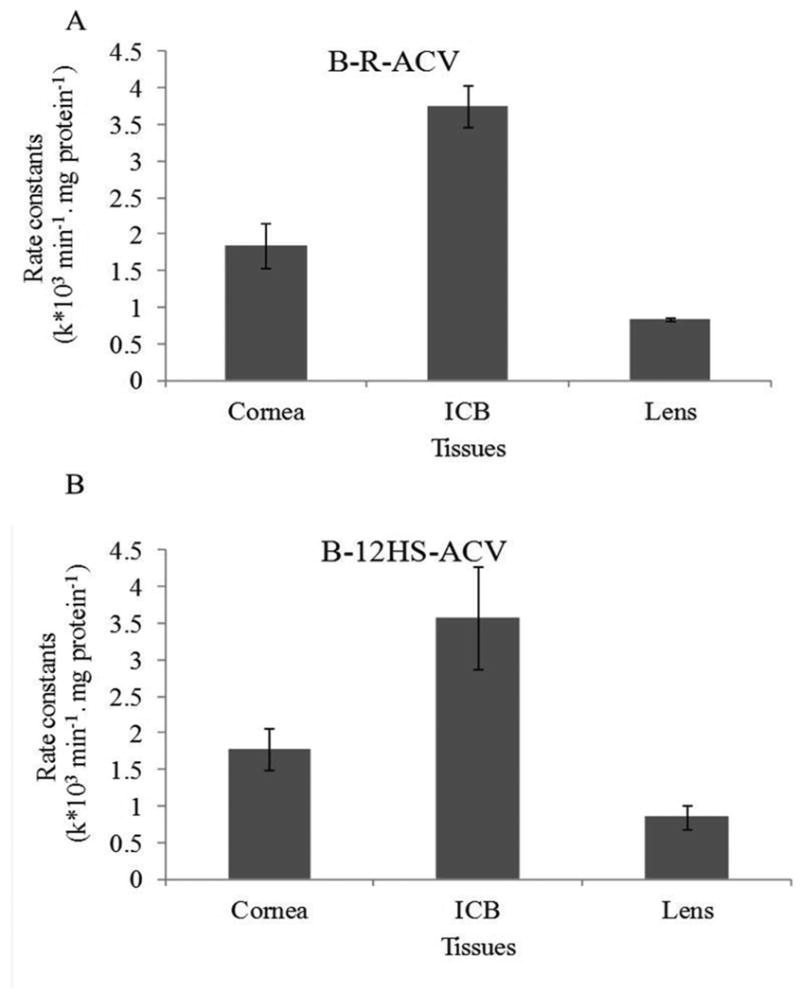
Comparison of hydrolytic rate constants for (A) B-R-ACV and (B) B-12HS-ACV in various ocular homogenates (cornea, ICB and lens). Data represents mean ± standard deviation (n= 4).
Antiviral Screening
To determine the in vitro antiviral activity of biotinylated ACV prodrugs against members of the α-herpesviruses, β-herpesvirus and γ-herpesviruses, all the compounds were tested against representative strains of HSV-1 and HSV-2, HCMV and EBV, respectively. Summary of viral strains, cell lines, and concentration ranges and assay methods employed for antiviral screening against HSV-1, HSV-2, HCMV and EBV are listed in Table III. The antiviral activities and cytotoxic potencies of ACV prodrugs have been summarized in Table IV. ACV served as a positive control for antiviral screening against HSV-1, HSV-2 and EBV, while GCV was used as a positive control for screening against HCMV. As evident by the EC50 values, 12HS-ACV demonstrated greater than 3 fold and 15 fold increase in antiviral activity against HSV-1 and HSV-2, respectively. B-12HS-ACV also displayed 34 fold and 60 fold increase in antiviral potency against HSV-1 and HSV-2, respectively. The EC50 value for B-R-ACV against HSV-2 was 0.27 μM, which is about 22 fold lower than that of ACV suggesting its greater antiviral potency. Both the biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) exhibited greater potency against HSV-1 and HSV-2. EC90 values clearly demonstrate that B-R-ACV is at least 4.5 and 8.7 times more potent against representative strains of HSV-1 and HSV-2, respectively relative to ACV. Surprisingly, B-12HS-ACV was approx. 200 and 21 times more potent against HSV-1 and HSV-2 relative to ACV. Both these prodrugs revealed very high selectivity indices (CC50/EC50) against HSV-1 and HSV-2 indicating that these compounds are highly selective and effective against virus infected cells.
Table III.
Summary of viral strains, cell lines, concentration ranges and assay methods employed for antiviral screening against HSV-1, HSV-2, HCMV and EBV.
| Virus Screened | Herpes Simplex Virus 1 (HSV-1) | Herpes Simplex Virus 2 (HSV-2) | Human Cytomegalovirus (HCMV) | Epstein-Barr Virus (EBV) |
|---|---|---|---|---|
| Virus Strain | E-377 | MS | AD169 | Akata |
| Cell Line | HFF | HFF | HFF | Akata |
| Vehicle | DMSO | DMSO | DMSO | DMSO |
| Prodrug Concentration Range | 0.1 - 300 μM | 0.1 - 300 μM | 0.1 - 300 μM | 0.48 - 60 μM |
| Control Concentration Range | 0.032 - 100 μM | 0.032 - 100 μM | 0.032 - 100 μM | 0.8 - 100 μM |
| Assay Method | Crystal Violet (Cytopathic effect/Toxicity) | Crystal Violet (Cytopathic effect/Toxicity) | Crystal Violet (Cytopathic effect/Toxicity) | DNA hybridization/CellTiter Glo (Toxicity) |
Table IV.
In vitro antiviral activity (EC50 and EC90), cytotoxicity (CC50) and their selectivity indices (SI) of prodrugs of ACV against HSV-1, HSV-2, HCMV and EBV.
EC50 = Compound concentration that reduces viral replication by 50%, in units of μM concentration.
EC90 = Compound concentration that reduces viral replication by 90%, in units of μM concentration.
CC50 = Compound concentration that reduces cell viability by 50%, in units of μM concentration.
SI50 = Selectivity index calculated as the CC50 divided by the EC50.
SI90 = Selectivity index calculated as the CC90 divided by the EC90.
| Drug/Prodrug | Herpes Simplex Virus-1 (HSV-1) | Herpes Simplex Virus-2 (HSV-2) | Human Cytomegalovirus (HC MV)* | Epstein-Barr Virus (EBV) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||||
| EC5o (μM) | EC90 (μM) | CC50 (μM) | SI50 (μM) | SI90 (μM) | EC50 (μM) | EC90 (μM) | CC50 (μM) | SI50 (μM) | SI90 (μM) | EC50 (μM) | EC90 (μM) | CC50 (μM) | SI50 (μM) | SI90 (μM) | EC50 (μM) | EC90 (μM) | CC50 (μM) | SI50 (μM) | SI90 (μM) | |
|
| ||||||||||||||||||||
| ACV/GCV* | 3.43 | >100 | >100 | >29.15 | 1 | 5.98 | >100 | >100 | >16.72 | 1 | 0.21 | 0.38 | >100 | >476.19 | >263.16 | 12.4 | 67.3 | >100 | >8.06 | >1.49 |
| B-ACV | 181.54 | >300 | >300 | >1.65 | 1 | >300 | >300 | >300 | 1 | 1 | >60 | >60 | 234.24 | <3.9 | <3.9 | 22.3 | 52.8 | >60 | >2.69 | >1.14 |
| R-ACV | 8.3 | 168.32 | >300 | >36.14 | >1.78 | 3.4 | 8.89 | >300 | >88.24 | >33.75 | 46.47 | >300 | >300 | >6.46 | 1 | 10.7 | 49.5 | >60 | >5.61 | >1.21 |
| B-R-ACV | 5.7 | 21.89 | >300 | >52.6 | >13.7 | 0.27 | 11.39 | >300 | >1111.11 | >26.34 | 70.07 | 99.96 | >300 | >4.28 | >3 | 2.1 | 41.5 | >60 | >28.57 | >1.45 |
| 12HS-ACV | 1.16 | 6.24 | 296.07 | 255.23 | 47.22 | 0.41 | >300 | 296.07 | 722.12 | 0.99 | 101.02 | 136.55 | >300 | >2.97 | >2.2 | 35.8 | 55 | 47.3 | 1.32 | 0.86 |
| B-12HS-ACV | <0.096 | 0.42 | >300 | >3125 | >714.29 | <0.096 | 4.65 | >300 | >3125 | >64.52 | >300 | >300 | >300 | 1 | 1 | 37.2 | >60 | >60 | >1.61 | 1 |
Since ACV is not indicated for the treatment of human CMV infections, all the prodrugs tested in this study were found to be inactive against HCMV strains (Table IV). An interesting observation was that B-R-ACV was highly active against representative strain of EBV. The EC50 and EC90 values were 2.1 and 41.5 μM indicating that this compound is at least 6 and 1.6 times, respectively more potent than ACV (EC50 = 12.4 μM and EC90 = 67.3 μM). Neutral red uptake cytotoxicity and CellTiter Glo (Toxicity) assays were performed to examine for any sign of toxicity. The obtained CC50 values from all the assays evaluated in this study were presumably very higher than EC50 values, confirming that these biotinylated lipid prodrugs are not cytotoxic to non-virus infected cells. All these prodrugs are currently under investigation by NIH/NIAID for screening the in vivo antiviral efficacy in virus infected animal models.
Discussion
Herpetic keratitis (HK) is a viral infection of the cornea and remains the leading cause of visual morbidity in developed nations like the United States. ACV has been the standard therapy for management of HK for many years. Prolonged treatment with ACV resulted in the incidence of ACV resistance in immunocompromised patients (4, 25). Recently we reported the development of novel conjugated compounds in which the hydrophilic drug (ACV) is linked to a substrate for membrane transporter (SMVT) via lipid rafts. This novel technology has enabled greater cellular uptake of therapeutic agents (29). The current study is focused on investigating the feasibility of utilizing biotinylated lipid prodrugs of ACV as potential drug candidates for the treatment of HSV infections, especially HK.
HCEC cells have been employed for performing cellular uptake studies because of the functional and molecular presence of SMVT. Also, it has been suggested as a good in vitro model for evaluating the cellular absorption of biotinylated prodrugs (31). Cellular uptake study was carried out in HCEC cells to determine the rate of cellular accumulation. Enhanced uptake of biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) was observed relative to biotinylated but non-lipidated prodrug (B-ACV). Such marked increase in intracellular drug accumulation might presumably be due to the synergistic effect of transcellular diffusion due to enhanced lipophilicity and significant carrier-mediated transport by SMVT (Fig. 1). Significant diminution in the uptake of B-R-ACV, B-12HS-ACV and B-ACV in the absence of sodium suggested that the prodrug uptake is mediated by SMVT, which is a highly sodium-dependent transporter (Fig. 2).
[3H] biotin, a well-established SMVT substrate, was employed to establish correlation with respect to interaction of biotinylated prodrugs with SMVT on the rabbit cornea (32). [3H] biotin permeability across freshly excised rabbit corneas was assessed in the presence of unlabeled biotin and biotinylated prodrugs (Fig. 3). Interaction of biotinylated prodrugs with SMVT is clearly indicated by inhibition of [3H] biotin transport. Biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) caused greater inhibition of [3H] biotin transport suggesting their enhanced affinity towards SMVT relative to B-ACV (Fig. 4). Such enhanced interaction could be attributed to the current approach which combines both lipid and transporter targeted delivery to generate synergistic effect.
Docking analysis was carried out to examine the affinity of all biotinylated prodrugs towards SMVT. Biotinylated prodrugs of ACV were shown in their predicted binding site on SMVT with all amino acid residues within 4 Å of the ligand (Fig. 5). Average Vina Scores obtained from docking studies suggest that the biotinylated lipid prodrugs of ACV possess enhanced affinity towards the SMVT protein than B-ACV (Table I). Moreover, from a drug delivery point of view, biotinylated lipid prodrugs of ACV may possess higher binding affinity for SMVT. Further, cell proliferation assay on HCEC and rPCEC cells suggest that all the prodrugs are safe and nontoxic to corneal epithelial cells (Fig. 6).
Prodrugs are designed to be therapeutically inactive until in vivo bioactivation to release the parent drug which then exerts its therapeutic effect. Therefore, ocular tissue homogenate studies were carried out to evaluate the enzymatic degradation of all biotinylated prodrugs. Targeted lipid prodrugs B-R-ACV and B-12HS-ACV exhibited better stability in cornea, ICB and lens homogenates relative to B-ACV (Table II). Such enhanced stability might be due to the presence of lipid rafts which may slow down the enzymatic cleavage by the various ocular tissues. Also, esterase activity has been reported to decrease due to the increased bulk of the lipid moiety leading to steric hindrance (44-46). B-R-ACV and B-12HS-ACV were relatively less stable in ICB compared to cornea and lens because of the abundant esterase activity in ICB followed by cornea and lens (Fig. 7) (44, 47, 48). Even though esterase activity is abundant in ICB, the activity in cornea is highly relevant since HSV establishes long term latency in the cornea, and the cornea acts as a major permeation pathway for topically administered drugs. Surprisingly, B-ACV was rapidly hydrolyzed in ICB and lens suggesting increased susceptibility of this compound to esterases.
Antiviral activity of the prodrugs was screened against HSV-1, HSV-2, HCMV and EBV. Biotinylated lipid prodrugs (B-R-ACV and B-12HS-ACV) were particularly highly effective against strains of HSV-1 and HSV-2 (Table IV). Greater antiviral activity of these two compounds may probably be due to enhanced cellular accumulation into the viral infected cells. On the other hand, B-R-ACV was found to be more potent against viral strains of EBV and could be a promising candidate for infections caused by EBV. As expected, all the compounds tested in this study were found to be inactive against viral strains of HCMV. Higher selectivity indexes of both the biotinylated lipid prodrugs with greater antiviral potency and lack of cytotoxicity to non-virus infected cells makes these compounds highly favorable for clinical development.
Conclusions
In summary, biotinylated lipid prodrugs of ACV were successfully designed and evaluated as potential drug candidates against various herpes viruses. The overall aim of this study was to improve the cellular absorption of ACV by employing targeted lipid prodrug strategy and investigating the feasibility of utilizing biotinylated lipid prodrugs for corneal drug delivery. Biotinylated lipid prodrugs of ACV have shown synergistic improvement in cellular uptake due to recognition of the prodrugs by SMVT on the cornea and lipid mediated transcellular diffusion. These compounds displayed better tissue stability and minimal/no cytotoxicity. Also, these novel compounds exhibited excellent antiviral activity against HSV-1, HSV-2 and EBV. These biotinylated lipid prodrugs appear to be promising drug candidates for the treatment of HK and thus may lower ACV resistance in patients with poor clinical response to therapy.
Acknowledgments
We would like to acknowledge Dr. Mark Prichard at The University of Alabama at Birmingham (UAB) for conducting the in vitro antiviral screening studies under NIH/NIAID contract. Also, we would like to thank Dr. Christopher Tseng and Miriam Perkins at National Institute of Allergy and Infectious Diseases (NIAID) for their support. This work has been supported by NIH grant R01EY009171. All these prodrugs are currently under investigation by NIH/NIAID for screening the in vivo antiviral efficacy in virus infected animal models.
Abbreviations
- ACV
Acyclovir
- SMVT
Sodium dependent multivitamin transporter
- B-R-ACV
Biotin-ricinoleicacid-acyclovir
- B-12HS-ACV
Biotin-12hydroxystearicacid-acyclovir
- B-ACV
Biotin-acyclovir
- R-ACV
Ricinoleicacid-acyclovir
- 12HS-ACV
12hydroxystearicacid-acyclovir
- HCEC
Human corneal epithelial cells
- rPCEC
Rabbit primary corneal epithelial cells
- HK
Herpetic keratitis
- HSV
Herpes simplex virus
- HCMV
Human cytomegalovirus
- EBV
Epstein - Barr virus
- LC/MS/MS
Liquid chromatography-tandem mass spectrometry
References
- 1.Duan R, de Vries RD, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir-resistant corneal HSV-1 isolates from patients with herpetic keratitis. J Infect Dis. 2008 Sep 1;198(5):659–63. doi: 10.1086/590668. [DOI] [PubMed] [Google Scholar]
- 2.Remeijer L, Osterhaus A, Verjans G. Human herpes simplex virus keratitis: the pathogenesis revisited. Ocul Immunol Inflamm. 2004 Dec;12(4):255–85. doi: 10.1080/092739490500363. [DOI] [PubMed] [Google Scholar]
- 3.Rowe AM, St Leger AJ, Jeon S, Dhaliwal DK, Knickelbein JE, Hendricks RL. Herpes keratitis. Progress in Retinal and Eye Research. 2013;32(0):88–101. doi: 10.1016/j.preteyeres.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011 Feb;55(2):459–72. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, et al. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA. 2006 Aug 23;296(8):964–73. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- 6.Al-Dujaili LJ, Clerkin PP, Clement C, McFerrin HE, Bhattacharjee PS, Varnell ED, et al. Ocular herpes simplex virus: how are latency, reactivation, recurrent disease and therapy interrelated? Future Microbiol. 2011 Aug;6(8):877–907. doi: 10.2217/fmb.11.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webre JM, Hill JM, Nolan NM, Clement C, McFerrin HE, Bhattacharjee PS, et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J Biomed Biotechnol. 2012;2012:612316. doi: 10.1155/2012/612316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vadlapudi AD, Vadlapatla RK, Mitra AK. Update On Emerging Antivirals For The Management Of Herpes Simplex Virus Infections: A Patenting Perspective. Recent Pat Antiinfect Drug Discov. 2013 Jan 15; doi: 10.2174/1574891x11308010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kennedy DP, Clement C, Arceneaux RL, Bhattacharjee PS, Huq TS, Hill JM. Ocular herpes simplex virus type 1: is the cornea a reservoir for viral latency or a fast pit stop? Cornea. 2011 Mar;30(3):251–9. doi: 10.1097/ICO.0b013e3181ef241d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kennedy DP, Clement C, Arceneaux RL, Bhattacharjee PS, Huq TS, Hill JM. Ocular Herpes Simplex Virus Type 1: Is the Cornea a Reservoir for Viral Latency or a Fast Pit Stop? Cornea. 2010 Nov 17; doi: 10.1097/ICO.0b013e3181ef241d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantin EM, Chen J, McNeill J, Willey DE, Openshaw H. Detection of herpes simplex virus DNA sequences in corneal transplant recipients by polymerase chain reaction assays. Curr Eye Res. 1991;10(Suppl):15–21. doi: 10.3109/02713689109020353. [DOI] [PubMed] [Google Scholar]
- 12.Easty DL, Shimeld C, Claoue CM, Menage M. Herpes simplex virus isolation in chronic stromal keratitis: human and laboratory studies. Curr Eye Res. 1987 Jan;6(1):69–74. doi: 10.3109/02713688709020071. [DOI] [PubMed] [Google Scholar]
- 13.Coupes D, Klapper PE, Cleator GM, Bailey AS, Tullo AB. Herpesvirus simplex in chronic human stromal keratitis. Curr Eye Res. 1986 Oct;5(10):735–8. doi: 10.3109/02713688609000013. [DOI] [PubMed] [Google Scholar]
- 14.Tullo AB, Easty DL, Shimeld C, Stirling PE, Darville JM. Isolation of herpes simplex virus from corneal discs of patients with chronic stromal keratitis. Trans Ophthalmol Soc U K. 1985;104(Pt 2):159–65. [PubMed] [Google Scholar]
- 15.Shimeld C, Tullo AB, Easty DL, Thomsitt J. Isolation of herpes simplex virus from the cornea in chronic stromal keratitis. Br J Ophthalmol. 1982 Oct;66(10):643–7. doi: 10.1136/bjo.66.10.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thuret G, Acquart S, Gain P, Dumollard JM, Manissolle C, Campos-Guyotat L, et al. Ultrastructural demonstration of replicative herpes simplex virus type 1 transmission through corneal graft. Transplantation. 2004 Jan 27;77(2):325–6. doi: 10.1097/01.TP.0000102458.30516.13. [DOI] [PubMed] [Google Scholar]
- 17.Remeijer L, Maertzdorf J, Doornenbal P, Verjans GM, Osterhaus AD. Herpes simplex virus 1 transmission through corneal transplantation. Lancet. 2001 Feb 10;357(9254):442. doi: 10.1016/S0140-6736(00)04011-3. [DOI] [PubMed] [Google Scholar]
- 18.Openshaw H, McNeill JI, Lin XH, Niland J, Cantin EM. Herpes simplex virus DNA in normal corneas: persistence without viral shedding from ganglia. J Med Virol. 1995 May;46(1):75–80. doi: 10.1002/jmv.1890460116. [DOI] [PubMed] [Google Scholar]
- 19.Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM. HSV-1 DNA in tears and saliva of normal adults. Invest Ophthalmol Vis Sci. 2005 Jan;46(1):241–7. doi: 10.1167/iovs.04-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abiko Y, Ikeda M, Hondo R. Secretion and dynamics of herpes simplex virus in tears and saliva of patients with Bell's palsy. Otol Neurotol. 2002 Sep;23(5):779–83. doi: 10.1097/00129492-200209000-00028. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto S, Shimomura Y, Kinoshita S, Nishida K, Yamamoto R, Tano Y. Detection of herpes simplex virus DNA in human tear film by the polymerase chain reaction. Am J Ophthalmol. 1994 Feb 15;117(2):160–3. doi: 10.1016/s0002-9394(14)73071-5. [DOI] [PubMed] [Google Scholar]
- 22.Uchoa UB, Rezende RA, Carrasco MA, Rapuano CJ, Laibson PR, Cohen EJ. Long-term acyclovir use to prevent recurrent ocular herpes simplex virus infection. Arch Ophthalmol. 2003 Dec;121(12):1702–4. doi: 10.1001/archopht.121.12.1702. [DOI] [PubMed] [Google Scholar]
- 23.Liesegang TJ. Herpes simplex virus epidemiology and ocular importance. Cornea. 2001 Jan;20(1):1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 24.Anand BS, Mitra AK. Mechanism of corneal permeation of L-valyl ester of acyclovir: targeting the oligopeptide transporter on the rabbit cornea. Pharm Res. 2002 Aug;19(8):1194–202. doi: 10.1023/a:1019806411610. [DOI] [PubMed] [Google Scholar]
- 25.Bacon TH, Levin MJ, Leary JJ, Sarisky RT, Sutton D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev. 2003 Jan;16(1):114–28. doi: 10.1128/CMR.16.1.114-128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morfin F, Thouvenot D. Herpes simplex virus resistance to antiviral drugs. J Clin Virol. 2003 Jan;26(1):29–37. doi: 10.1016/s1386-6532(02)00263-9. [DOI] [PubMed] [Google Scholar]
- 27.Choong K, Walker NJ, Apel AJ, Whitby M. Aciclovir-resistant herpes keratitis. Clin Experiment Ophthalmol. 2010 Apr;38(3):309–13. doi: 10.1111/j.1442-9071.2010.02209.x. [DOI] [PubMed] [Google Scholar]
- 28.Wilson SS, Fakioglu E, Herold BC. Novel approaches in fighting herpes simplex virus infections. Expert Rev Anti Infect Ther. 2009 Jun;7(5):559–68. doi: 10.1586/eri.09.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vadlapudi AD, Vadlapatla RK, Kwatra D, Earla R, Samanta SK, Pal D, et al. Targeted lipid based drug conjugates: A novel strategy for drug delivery. Int J Pharm. 2012 Sep 15;434(1-2):315–24. doi: 10.1016/j.ijpharm.2012.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karla PK, Quinn TL, Herndon BL, Thomas P, Pal D, Mitra A. Expression of multidrug resistance associated protein 5 (MRP5) on cornea and its role in drug efflux. J Ocul Pharmacol Ther. 2009 Apr;25(2):121–32. doi: 10.1089/jop.2008.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vadlapudi AD, Vadlapatla RK, Pal D, Mitra AK. Functional and Molecular Aspects of Biotin Uptake via SMVT in Human Corneal Epithelial (HCEC) and Retinal Pigment Epithelial (D407) Cells. Aaps J. 2012 Aug 18;14(4):832–42. doi: 10.1208/s12248-012-9399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janoria KG, Hariharan S, Paturi D, Pal D, Mitra AK. Biotin uptake by rabbit corneal epithelial cells: role of sodium-dependent multivitamin transporter (SMVT) Curr Eye Res. 2006 Oct;31(10):797–809. doi: 10.1080/02713680600900206. [DOI] [PubMed] [Google Scholar]
- 33.Dey S, Patel J, Anand BS, Jain-Vakkalagadda B, Kaliki P, Pal D, et al. Molecular evidence and functional expression of P-glycoprotein (MDR1) in human and rabbit cornea and corneal epithelial cell lines. Invest Ophthalmol Vis Sci. 2003 Jul;44(7):2909–18. doi: 10.1167/iovs.02-1142. [DOI] [PubMed] [Google Scholar]
- 34.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y. I-TASSER: fully automated protein structure prediction in CASP8. Proteins. 2009;77(Suppl 9):100–13. doi: 10.1002/prot.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010 Jan 30;31(2):455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katragadda S, Talluri RS, Mitra AK. Simultaneous modulation of transport and metabolism of acyclovir prodrugs across rabbit cornea: An approach involving enzyme inhibitors. Int J Pharm. 2006 Aug 31;320(1-2):104–13. doi: 10.1016/j.ijpharm.2006.04.010. Comparative Study Research Support, N.I.H., Extramural. [DOI] [PubMed] [Google Scholar]
- 39.Tak RV, Pal D, Gao H, Dey S, Mitra AK. Transport of acyclovir ester prodrugs through rabbit cornea and SIRC-rabbit corneal epithelial cell line. Journal of pharmaceutical sciences. 2001 Oct;90(10):1505–15. doi: 10.1002/jps.1101. Comparative Study Research Support, U.S. Gov't, P.H.S. [DOI] [PubMed] [Google Scholar]
- 40.Earla R, Boddu SH, Cholkar K, Hariharan S, Jwala J, Mitra AK. Development and validation of a fast and sensitive bioanalytical method for the quantitative determination of glucocorticoids--quantitative measurement of dexamethasone in rabbit ocular matrices by liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal. 2010 Aug 1;52(4):525–33. doi: 10.1016/j.jpba.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prichard MN, Keith KA, Quenelle DC, Kern ER. Activity and mechanism of action of N-methanocarbathymidine against herpesvirus and orthopoxvirus infections. Antimicrob Agents Chemother. 2006 Apr;50(4):1336–41. doi: 10.1128/AAC.50.4.1336-1341.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prichard MN, Daily SL, Jefferson GM, Perry AL, Kern ER. A rapid DNA hybridization assay for the evaluation of antiviral compounds against Epstein-Barr virus. J Virol Methods. 2007 Sep;144(1-2):86–90. doi: 10.1016/j.jviromet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gill RB, Frederick SL, Hartline CB, Chou S, Prichard MN. Conserved retinoblastoma protein-binding motif in human cytomegalovirus UL97 kinase minimally impacts viral replication but affects susceptibility to maribavir. Virol J. 2009;6:9. doi: 10.1186/1743-422X-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dias CS, Anand BS, Mitra AK. Effect of mono- and di-acylation on the ocular disposition of ganciclovir: physicochemical properties, ocular bioreversion, and antiviral activity of short chain ester prodrugs. J Pharm Sci. 2002 Mar;91(3):660–8. doi: 10.1002/jps.10072. [DOI] [PubMed] [Google Scholar]
- 45.Lambert DM. Rationale and applications of lipids as prodrug carriers. Eur J Pharm Sci. 2000 Oct;11(Suppl 2):S15–27. doi: 10.1016/s0928-0987(00)00161-5. [DOI] [PubMed] [Google Scholar]
- 46.Chang SC, Lee VH. Influence of chain length on the in vitro hydrolysis of model ester prodrugs by ocular esterases. Curr Eye Res. 1982;2(10):651–6. doi: 10.3109/02713688209019993. [DOI] [PubMed] [Google Scholar]
- 47.Talluri RSHS, Karla PK, Mitra AK. Drug delivery to cornea and conjunctiva–esterase and protease directed prodrug design, in Encyclopedia of the Eye. Elsevier Ltd; 2012. [Google Scholar]
- 48.Ashim M, Jignesh P, Clapton D, Harisha A, Giridhar T. Theory and Practice of Contemporary Pharmaceutics. CRC Press; 2004. Ocular Nasal Pulmonary, and Otic Routes of Drug Delivery; pp. 479–524. [Google Scholar]