

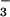
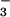
Abstract
Protein phosphoaspartate bonds play a variety of roles. In response regulator proteins of two-component signal transduction systems, phosphorylation of an aspartate residue is coupled to a change from an inactive to an active conformation. In phosphatases and mutases of the haloacid dehalogenase (HAD) superfamily, phosphoaspartate serves as an intermediate in phosphotransfer reactions, and in P-type ATPases, also members of the HAD family, it serves in the conversion of chemical energy to ion gradients. In each case, lability of the phosphoaspartate linkage has hampered a detailed study of the phosphorylated form. For response regulators, this difficulty was recently overcome with a phosphate analog, BeF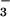 , which yields persistent complexes with the active site aspartate of their receiver domains. We now extend the application of this analog to a HAD superfamily member by solving at 1.5-Å resolution the x-ray crystal structure of the complex of BeF
, which yields persistent complexes with the active site aspartate of their receiver domains. We now extend the application of this analog to a HAD superfamily member by solving at 1.5-Å resolution the x-ray crystal structure of the complex of BeF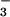 with phosphoserine phosphatase (PSP) from Methanococcus jannaschii. The structure is comparable to that of a phosphoenzyme intermediate: BeF
with phosphoserine phosphatase (PSP) from Methanococcus jannaschii. The structure is comparable to that of a phosphoenzyme intermediate: BeF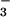 is bound to Asp-11 with the tetrahedral geometry of a phosphoryl group, is coordinated to Mg2+, and is bound to residues surrounding the active site that are conserved in the HAD superfamily. Comparison of the active sites of BeF
is bound to Asp-11 with the tetrahedral geometry of a phosphoryl group, is coordinated to Mg2+, and is bound to residues surrounding the active site that are conserved in the HAD superfamily. Comparison of the active sites of BeF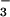 ⋅PSP and BeF
⋅PSP and BeF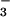 ⋅CeY, a receiver domain/response regulator, reveals striking similarities that provide insights into the function not only of PSP but also of P-type ATPases. Our results indicate that use of BeF
⋅CeY, a receiver domain/response regulator, reveals striking similarities that provide insights into the function not only of PSP but also of P-type ATPases. Our results indicate that use of BeF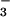 for structural studies of proteins that form phosphoaspartate linkages will extend well beyond response regulators.
for structural studies of proteins that form phosphoaspartate linkages will extend well beyond response regulators.
Protein phosphorylation plays critical roles in cellular control. Phosphorylation of aspartate residues is central to the function of two large protein families, response regulators and the haloacid dehalogenase (HAD) superfamily of hydrolases (1, 2).
Response regulators and their cognate histidine kinases constitute two-component systems (3), which dominate signal transduction in bacteria and are also present in archaea, microbial eukarya, and higher plants. Response regulators have in common a homologous regulatory module of ≈120-aa residues called a receiver domain. Receiver domains have an (α/β)5 fold and an active site defined by a quintet of highly conserved residues composed of three aspartates, a lysine, and either a threonine or a serine. Phosphorylation of the active site aspartate residue in a receiver domain is coupled to a conformational change that regulates the function of its output domain or downstream target protein. CheY, the central regulator of bacterial chemotaxis, is a well-studied response regulator. When phosphorylated, this single domain protein, which can also be described as a receiver domain, binds to the FliM protein in the basal body of the flagellum to reverse the direction of flagellar rotation (4, 5).
For many years, the lability of phosphoaspartate linkages in receiver domains limited structural studies to their unphosphorylated inactive forms. Recently, several structures of activated receiver domains were obtained by using a variety of means to overcome the problem of lability (6–10). Among these, formation of a complex with BeF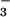 , which yields an excellent aspartyl phosphate mimic, appears to be a simple general solution (11). BeF
, which yields an excellent aspartyl phosphate mimic, appears to be a simple general solution (11). BeF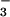 readily forms persistent activated complexes with many response regulators, regardless of the half-lives of their phosphorylated states.
readily forms persistent activated complexes with many response regulators, regardless of the half-lives of their phosphorylated states.
Members of the HAD superfamily of hydrolases have diverse functions (12–14). Although 2-haloacid dehalogenase, the founding member of this large protein family, does not catalyze hydrolysis of phosphorylated substrates or depend on a divalent cation, most other members do. For example, the phosphotransferase subgroup includes several phosphatases, among them phosphoserine phosphatase (PSP), and some phosphomutases. The subgroup of P-type ATPases serves in transport of cations across biological membranes (15–17). Phosphoenzyme intermediates have been detected among the phosphotransferases of the HAD superfamily (14, 18–20) and are known to form during the catalytic cycle of P-type ATPases (1, 15–17). In both cases, phosphorylation requires a divalent cation, normally Mg2+, and occurs at a conserved aspartate residue. In addition, there is biochemical evidence that vanadate binds to phosphatases and phosphomutases in the HAD superfamily (19, 21) and that vanadate, beryllofluoride, and aluminum fluoride, all of which can act as phosphate analogs, bind to P-type ATPases (22–28).
The core catalytic domains of HAD family phosphotransferases and P-type ATPases share the same quintet of highly conserved residues found in receiver domains (12, 29), although a circular permutation is required to align the primary sequences of members of these two large families (29). High-resolution crystal structures of HAD superfamily members [haloacid dehalogenases from Pseudomonas sp. YL and Xanthobacter autotrophicus GJ10 (30, 31), the Ca2+-ATPase from rabbit sarcoplasmic reticulum (32), and PSP from Methanococcus jannaschii (33)] indicate that the catalytic domains of these proteins form a typical α/β Rossmann fold that is similar to that in CheY (29). The quintet of conserved residues noted above surrounds the active site, as it does in receiver domains. These commonalities led to proposals of a common reaction mechanism for the formation of the phosphoaspartate bond (12, 29). They also raise the question of whether BeF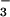 might yield a persistent aspartyl phosphate analog in members of the HAD superfamily and thus facilitate structural studies of their conformations during catalysis.
might yield a persistent aspartyl phosphate analog in members of the HAD superfamily and thus facilitate structural studies of their conformations during catalysis.
We now report the structure of BeF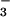 complexed with PSP from the hyperthermophile M. jannaschii. NMR spectroscopy and biochemical studies indicated that BeF
complexed with PSP from the hyperthermophile M. jannaschii. NMR spectroscopy and biochemical studies indicated that BeF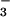 binds to the active site of PSP in solution and forms a tight persistent complex. Crystals of the complex, grown under conditions optimized by NMR spectroscopy, yielded a structure of BeF
binds to the active site of PSP in solution and forms a tight persistent complex. Crystals of the complex, grown under conditions optimized by NMR spectroscopy, yielded a structure of BeF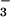 ⋅PSP at 1.5-Å resolution. This allowed detailed definition of the active site and comparison of the overall structure and arrangement of active site residues to those in BeF
⋅PSP at 1.5-Å resolution. This allowed detailed definition of the active site and comparison of the overall structure and arrangement of active site residues to those in BeF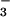 ⋅CheY.
⋅CheY.
Materials and Methods
Protein Samples and NMR.
PSP from M. jannaschii was expressed and purified by using methods previously described (33). Uniformly 15N-labeled samples of protein for NMR spectroscopy were prepared from cells grown in M9 minimal medium with [15N]ammonium chloride as the sole nitrogen source. 1H-15N fast heteronuclear single quantum correlation (FHSQC) spectra were collected on an AMX 600 NMR spectrometer (Bruker Instruments, Billerica, MA).
Crystallization.
PSP was concentrated to 54 mg/ml in a buffer containing 20 mM Tris⋅HCl, pH 7.5, 0.3 M NaCl, 1 mM EDTA, and 10 mM DTT. Crystals were grown by using the hanging drop vapor diffusion method with seeding. Concentrated NaF, BeCl2, and MgCl2 were added to the protein sample to final concentrations of 54 mM, 10.8 mM, and 90 mM, respectively. (CAUTION: Beryllium is toxic and immunogenic. Exercise care in its handling.) One microliter of this sample was then mixed with 1 μl of the reservoir well solution containing 0.1 M sodium acetate buffer at pH 4.5, 0.2 M sodium phosphate dihydrate, and 22% polyethylene glycol 2000 monomethylether (PEG2K MME). Microseeding was performed 1 h after the drop was set up. Crystals appeared within 12 h and reached a maximum size of 0.3 × 0.5 × 0.5 mm. The concentration of PEG2K MME was then raised to 30% to stabilize the crystals.
Data Collection and Structure Refinement.
A crystal from the crystallization drop was used directly for cryocrystallography data collection. X-ray diffraction data were collected at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA) beam line 5.0.2 by using an Area Detector System Quantum 4 charge-coupled device detector placed 130 mm from the crystal. The data were processed by using the programs denzo and scalepack (34). X-ray data statistics are shown in Table 1.
Table 1.
Summary of crystallographic analysis
| Measurement | Value |
|---|---|
| Data collection | |
| Resolution limit, Å | 1.5 |
| Total reflections | 288,900 |
| Unique reflections | 67,186 |
| Completeness, % | 93.8 |
| Rsym,* (%) | 6.7 |
| Refinement | |
| Resolution, Å | 20–1.5 |
| Number of protein atoms | 3709 |
| Number of water molecules | 402 |
| Rcryst,† % | 19.0 |
| Rfree,‡ % | 21.1 |
| Average B, Å2 | 14.77 |
| rmsd bond length, Å | 0.014 |
| rmsd bond angles, degrees | 1.80 |
rmsd, rms deviation.
Rsym = ΣhΣi|Ih,i − 〈I〉h|/Σ|〈I〉h|, where I is the scaled intensity of the given reflection h.
Rcryst = Σh|Foh − Fch|/ΣhFoh, where Foh, and Fch are the observed and calculated structure amplitudes for reflection h.
Value of Rfree for 10% of randomly selected reflections excluded from the refinement.
The protein part of a previously determined PSP structure was used for initial rigid-body refinement to obtain a preliminary model (33). An electron density map calculated from this model clearly showed densities consistent with the fluorine atoms of a BeF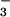 that is bound to Asp-11 with a beryllium–oxygen bond distance of 1.55 Å. This density was used to build the beryllofluoride aspartate residue into the model that was further refined against data up to 1.5 Å by using the programs cns (35) and o (36). The noncrystallographic symmetry constraints and restraints were completely released during the refinement. Ten percent of the data were randomly picked for free R-factor crossvalidation. The refinement statistics are shown in Table 1.
that is bound to Asp-11 with a beryllium–oxygen bond distance of 1.55 Å. This density was used to build the beryllofluoride aspartate residue into the model that was further refined against data up to 1.5 Å by using the programs cns (35) and o (36). The noncrystallographic symmetry constraints and restraints were completely released during the refinement. Ten percent of the data were randomly picked for free R-factor crossvalidation. The refinement statistics are shown in Table 1.
Phosphoserine Phosphatase Assay.
The hydrolytic activity of PSP was measured at the desired temperature by the release of Pi from l-phosphoserine and is expressed as pmol Pi/min/pmol PSP. Assays were performed in triplicate in mixtures (50 μl) containing 20 mM Tris⋅Cl (pH 7.5), 1 mM MgCl2, 5 mM l-phosphoserine, and different amounts of PSP. NaF (5 mM), BeCl2 (100 μM), AlCl3 (100 μM), or combinations were included in the mixture for inhibition assays. The reaction was stopped after 10 min by adding 5 μl of 0.2 M EDTA and placed on ice. The amount of inorganic phosphate released was determined with an EnzChek Phosphate Assay Kit (Molecular Probes).
Results
Inhibition of PSP Activity by BeF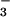 and Other Phosphate Analogues.
and Other Phosphate Analogues.
Like phosphorylation, BeF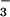 activates various response regulators (10, 11). If BeF
activates various response regulators (10, 11). If BeF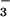 yielded an aspartyl phosphate analog in PSP, it would inhibit the activity of the enzyme by preventing substrate binding and/or formation of the phospho-PSP intermediate. At 50°C, the hydrolytic activity of PSP (100 nM) from M. jannaschii (840 ± 20 units) was markedly inhibited (>95%) in the presence of both BeCl2 and NaF. The latter was provided at concentrations that yield predominantly BeF3− in situ. Either BeCl2 or NaF alone had a much smaller inhibitory effect (35 or 20%, respectively). Similar inhibition by BeF
yielded an aspartyl phosphate analog in PSP, it would inhibit the activity of the enzyme by preventing substrate binding and/or formation of the phospho-PSP intermediate. At 50°C, the hydrolytic activity of PSP (100 nM) from M. jannaschii (840 ± 20 units) was markedly inhibited (>95%) in the presence of both BeCl2 and NaF. The latter was provided at concentrations that yield predominantly BeF3− in situ. Either BeCl2 or NaF alone had a much smaller inhibitory effect (35 or 20%, respectively). Similar inhibition by BeF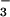 was observed at room temperature or at 70°C (data not shown), the temperature at which the thermostable PSP from M. jannaschii showed maximum activity (33).
was observed at room temperature or at 70°C (data not shown), the temperature at which the thermostable PSP from M. jannaschii showed maximum activity (33).
At 50°C, PSP activity (840 units, as above) was also inhibited in the presence of AlCl3 and NaF (>85 vs. 30% in the presence of AlCl3 alone), which should generate predominantly AlF3 and AlF4− in situ (37). Likewise, AlFx yields an aspartyl phosphate analogue in response regulator CheY, activating it in a manner similar to phosphorylation or formation of a BeF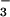 complex (D.Y. and H.C., unpublished results). Orthovanadate, another phosphate analogue, inhibited the activity of M. jannaschii PSP (data not shown), as it did the activity of the human enzyme (21). The major difference among these phosphate analogs is the difference in their coordination geometries (37–39). Beryllofluoride yields a tetrahedral ground state analog of phosphate, whereas aluminum fluoride and vanadate often yield planar complexes that are analogues of phosphate during the transition state for transfer or hydrolysis.
complex (D.Y. and H.C., unpublished results). Orthovanadate, another phosphate analogue, inhibited the activity of M. jannaschii PSP (data not shown), as it did the activity of the human enzyme (21). The major difference among these phosphate analogs is the difference in their coordination geometries (37–39). Beryllofluoride yields a tetrahedral ground state analog of phosphate, whereas aluminum fluoride and vanadate often yield planar complexes that are analogues of phosphate during the transition state for transfer or hydrolysis.
Monitoring Formation of BeF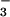 ⋅PSP Complexes by NMR Spectroscopy.
⋅PSP Complexes by NMR Spectroscopy.
Using 1H-15N FHSQC spectra, we could readily determine that BeF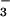 formed a persistent complex with PSP. As BeF
formed a persistent complex with PSP. As BeF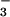 was added, many resonances doubled (Fig. 1). The lack of broadening of crosspeaks indicated that the dissociation rate of BeF
was added, many resonances doubled (Fig. 1). The lack of broadening of crosspeaks indicated that the dissociation rate of BeF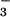 ⋅PSP complexes was slow (≤10 sec−1). Surprisingly, addition of BeF
⋅PSP complexes was slow (≤10 sec−1). Surprisingly, addition of BeF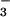 to PSP changed the chemical shifts (crosspeak positions) of most of the backbone amides. By contrast, formation of BeF
to PSP changed the chemical shifts (crosspeak positions) of most of the backbone amides. By contrast, formation of BeF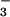 complexes of receiver domains changed the chemical shifts of the amides near the active site but of fewer farther away (10, 11); the latter effects occurred as a consequence of propagated conformational changes.
complexes of receiver domains changed the chemical shifts of the amides near the active site but of fewer farther away (10, 11); the latter effects occurred as a consequence of propagated conformational changes.
Figure 1.

1H-15N FHSQC spectrum of BeF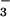 ⋅PSP. PSP (0.5 mM) was incubated with 5 mM MgCl2, 3 mM NaF, and 0.6 mM BeCl2. Red crosses represent the crosspeak positions in the absence of NaF and BeCl2. Spectra were recorded at 308 K and pH 6.5.
⋅PSP. PSP (0.5 mM) was incubated with 5 mM MgCl2, 3 mM NaF, and 0.6 mM BeCl2. Red crosses represent the crosspeak positions in the absence of NaF and BeCl2. Spectra were recorded at 308 K and pH 6.5.
The FHSQC spectra provided a convenient means of optimizing conditions for quantitative conversion of PSP to BeF ⋅PSP complexes. Unfortunately, PSP could not be maintained in a phosphorylated state long enough to collect data for the phosphoenzyme intermediate (data not shown).
⋅PSP complexes. Unfortunately, PSP could not be maintained in a phosphorylated state long enough to collect data for the phosphoenzyme intermediate (data not shown).
Crystal Structure of BeF ⋅PSP.
⋅PSP.
In the crystal structure of BeF ⋅PSP (Fig. 2A), the active site in the core α/β domain is covered by the four-helix bundle domain. BeF
⋅PSP (Fig. 2A), the active site in the core α/β domain is covered by the four-helix bundle domain. BeF is bound to Asp-11 in the α/β domain, which is the site of phosphorylation (14) and is surrounded by the sidechains of residues that are highly conserved in the HAD superfamily of proteins. The overall protein structure is very similar to a recently solved structure of PSP containing a phosphate (PO
is bound to Asp-11 in the α/β domain, which is the site of phosphorylation (14) and is surrounded by the sidechains of residues that are highly conserved in the HAD superfamily of proteins. The overall protein structure is very similar to a recently solved structure of PSP containing a phosphate (PO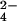 ) in the active site (33).
) in the active site (33).
Figure 2.

Ribbon diagram of BeF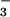 ⋅PSP (A) and stereoview of the active site (B). Ball-and-stick representations of the sidechain carbon, nitrogen, and oxygen atoms are colored green, blue, and red, respectively. In A, the sidechains of highly conserved active site residues, Asp-11, Ser-99, Lys-144, Asp-167, and Asp-171, are shown in ball-and-stick form. BeF
⋅PSP (A) and stereoview of the active site (B). Ball-and-stick representations of the sidechain carbon, nitrogen, and oxygen atoms are colored green, blue, and red, respectively. In A, the sidechains of highly conserved active site residues, Asp-11, Ser-99, Lys-144, Asp-167, and Asp-171, are shown in ball-and-stick form. BeF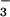 (red balls and orange bonds) is bonded to Asp-11 Oδ in the center of the active site (33). In B, the 2Fo − Fc electron density map covering the magnesium (cyan) and fluorine (red) atoms was calculated in the absence of these atoms and is shown contoured at 1σ. The dashed lines represent hydrogen bonds, salt bridges, and metal–ligand interactions. Three of the Mg2+ ligands (two water molecules and Asp-13-C = O) are not shown.
(red balls and orange bonds) is bonded to Asp-11 Oδ in the center of the active site (33). In B, the 2Fo − Fc electron density map covering the magnesium (cyan) and fluorine (red) atoms was calculated in the absence of these atoms and is shown contoured at 1σ. The dashed lines represent hydrogen bonds, salt bridges, and metal–ligand interactions. Three of the Mg2+ ligands (two water molecules and Asp-13-C = O) are not shown.
Active Site of BeF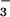 ⋅PSP.
⋅PSP.
In M. jannaschii PSP, the quintet of conserved residues found in HAD family members that form a phosphoaspartate intermediate are: Asp-11, the site of phosphorylation, Ser-99, Lys-144, Asp-167, and Asp-171. With the exception of Ser-99 to Thr, mutational change of any of the corresponding residues in human PSP dramatically decreased catalytic efficiency (40). These conserved residues come together in the active site to bind BeF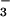 and the essential divalent cation Mg2+ (Fig. 2B). The Asp 11-Oδ1 is bound to beryllium with an O-Be distance of 1.5 Å. Asp-11-Oδ2, along with a fluorine, Asp-13-C = O, Asp-167-Oδ, and two water molecules occupy the six coordination sites of Mg2+. Ser-99-Oγ forms a hydrogen bond with BeF
and the essential divalent cation Mg2+ (Fig. 2B). The Asp 11-Oδ1 is bound to beryllium with an O-Be distance of 1.5 Å. Asp-11-Oδ2, along with a fluorine, Asp-13-C = O, Asp-167-Oδ, and two water molecules occupy the six coordination sites of Mg2+. Ser-99-Oγ forms a hydrogen bond with BeF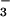 . Lys-144-Nζ forms salt bridges with BeF
. Lys-144-Nζ forms salt bridges with BeF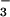 and Asp-171-Oδ1. Asp-171-Oδ2 forms hydrogen bonds with the backbone amides of Asp-167 and Gly-168. These two hydrogen bonds appear to stabilize the conformation of the backbone around Asp-167 such that the Asp-167 sidechain is restrained to a position that allows it to coordinate Mg2+ (see Discussion).
and Asp-171-Oδ1. Asp-171-Oδ2 forms hydrogen bonds with the backbone amides of Asp-167 and Gly-168. These two hydrogen bonds appear to stabilize the conformation of the backbone around Asp-167 such that the Asp-167 sidechain is restrained to a position that allows it to coordinate Mg2+ (see Discussion).
Discussion
Comparison of BeF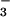 ⋅PSP and BeF
⋅PSP and BeF ⋅CheY: The “Stabilized Mg2+ Chelation Loop.”
⋅CheY: The “Stabilized Mg2+ Chelation Loop.”
Despite the circular permutation required to align their primary sequences (29) (Fig. 3A), superposition of the structurally homologous β-strand residues of BeF ⋅PSP and BeF
⋅PSP and BeF ⋅CheY (41) results in an rms deviation of only 0.6 Å and shows that the core β-sheets have a similar curvature (Fig. 3B). By contrast, the α-helices in the α/β domains of the two proteins show large differences in position. Comparison of their active sites reveals that the conserved quintet of residues in PSP and CheY chelate BeF
⋅CheY (41) results in an rms deviation of only 0.6 Å and shows that the core β-sheets have a similar curvature (Fig. 3B). By contrast, the α-helices in the α/β domains of the two proteins show large differences in position. Comparison of their active sites reveals that the conserved quintet of residues in PSP and CheY chelate BeF and Mg2+ similarly and form very similar hydrogen bonds and salt bridges (Fig. 3C). PSP residues Asp-11, Ser-99, and Asp-167, whose roles are discussed in Results, are functionally and structurally equivalent to CheY residues Asp-57, Thr-87, and Asp-13, respectively. PSP residues Lys-144 and Asp-171 appear to be functionally equivalent to CheY residues Lys-109 and Asp-12, respectively, although these residues originate from different secondary structural elements. Lys-144 (PSP) originates from an α-helix, whereas Lys-109 (CheY) originates from a loop; likewise, Asp-171 (PSP) originates from an α-helix, whereas Asp-12 (CheY) originates from the C terminus of a β-strand. Despite the differences in their position of origin, the amino groups of Lys-144 (PSP) and Lys-109 (CheY) reach comparable positions in the active site and form salt bridges with BeF
and Mg2+ similarly and form very similar hydrogen bonds and salt bridges (Fig. 3C). PSP residues Asp-11, Ser-99, and Asp-167, whose roles are discussed in Results, are functionally and structurally equivalent to CheY residues Asp-57, Thr-87, and Asp-13, respectively. PSP residues Lys-144 and Asp-171 appear to be functionally equivalent to CheY residues Lys-109 and Asp-12, respectively, although these residues originate from different secondary structural elements. Lys-144 (PSP) originates from an α-helix, whereas Lys-109 (CheY) originates from a loop; likewise, Asp-171 (PSP) originates from an α-helix, whereas Asp-12 (CheY) originates from the C terminus of a β-strand. Despite the differences in their position of origin, the amino groups of Lys-144 (PSP) and Lys-109 (CheY) reach comparable positions in the active site and form salt bridges with BeF and an aspartate residue, Asp-171 (PSP) and Asp-12 (CheY).
and an aspartate residue, Asp-171 (PSP) and Asp-12 (CheY).
Figure 3.

Alignment and stereoviews showing the superposition of BeF ⋅PSP and BeF
⋅PSP and BeF ⋅CheY. Structurally homologous β-strand residues (PSP: 7–11, 94–98, 116–120, 162–167, 180–182, and CheY: 53–57, 82–86, 104–108, 8–13, 34–36) were used for the superposition. PSP and CheY are colored purple and yellow, respectively. (A) Diagram of the circular permutation required to align the conserved active site residues of PSP and CheY (29). (B) Ribbon structures of PSP and CheY. The molecules are oriented with the H4-β5-H5 face of CheY directed toward the reader. The N and C termini of CheY are denoted. Some PSP residues that have no structural counterparts in CheY are not shown. (C) A top-down view of the superimposed active sites. This orientation was obtained by applying a −90° rotation around the x-axis starting from the orientation in B. The active site residues of PSP and CheY are labeled in purple and black, respectively. The sidechains are shown in ball-and-stick form with carbon, nitrogen, and oxygen atoms colored green, blue, and red, respectively. BeF
⋅CheY. Structurally homologous β-strand residues (PSP: 7–11, 94–98, 116–120, 162–167, 180–182, and CheY: 53–57, 82–86, 104–108, 8–13, 34–36) were used for the superposition. PSP and CheY are colored purple and yellow, respectively. (A) Diagram of the circular permutation required to align the conserved active site residues of PSP and CheY (29). (B) Ribbon structures of PSP and CheY. The molecules are oriented with the H4-β5-H5 face of CheY directed toward the reader. The N and C termini of CheY are denoted. Some PSP residues that have no structural counterparts in CheY are not shown. (C) A top-down view of the superimposed active sites. This orientation was obtained by applying a −90° rotation around the x-axis starting from the orientation in B. The active site residues of PSP and CheY are labeled in purple and black, respectively. The sidechains are shown in ball-and-stick form with carbon, nitrogen, and oxygen atoms colored green, blue, and red, respectively. BeF is represented by red balls with orange bonds. The cyan- and blue-green-colored balls, lower and upper, respectively, adjacent to BeF
is represented by red balls with orange bonds. The cyan- and blue-green-colored balls, lower and upper, respectively, adjacent to BeF represent the Mg2+ ions in the PSP and CheY structures, respectively. The purple arrow points to D167 of PSP.
represent the Mg2+ ions in the PSP and CheY structures, respectively. The purple arrow points to D167 of PSP.
The pairs of functionally analogous aspartate residues Asp-167 and Asp-171 of PSP and Asp-13 and Asp-12 of CheY play surprisingly similar roles in Mg2+ coordination (Figs. 2B and 3C), despite the fact that they are inverted with respect to one another and separated by different numbers of residues. In PSP, the first residue of the pair in the primary sequence, Asp-167, acts as a direct ligand to Mg2+, whereas in CheY it is the second, Asp-13. Asp-167 (PSP) and Asp-13 (CheY) are located in similar loops that are stabilized by main chain hydrogen bonds to the other aspartates of the respective pairs, Asp-171 (PSP) and Asp-12 (CheY) (“stabilized Mg2+ chelation loops”). The stabilizing aspartates also form salt bridges to the highly conserved lysine residues, Lys-144 (PSP) and Lys-109 (CheY) and hence are precisely positioned. Remarkably, the aspartate that functions as a direct Mg2+ ligand is not conserved in HAD family proteins that do not require a divalent cation for activity (12, 13).
Hypotheses Regarding the Function of PSP and the Ca2+-ATPase.
The two domains of PSP appear to be in a “closed configuration” in the BeF ⋅PSP complex (Fig. 2A) (33): the helical domain, which presumably participates in binding the substrate phosphoserine, completely shields the active site of the α/β domain from solvent. In this closed configuration of PSP, it would be impossible for substrate to enter or product to be released. It is likely that the widespread changes in the 1H-15N FHSQC spectrum of PSP on addition of BeF
⋅PSP complex (Fig. 2A) (33): the helical domain, which presumably participates in binding the substrate phosphoserine, completely shields the active site of the α/β domain from solvent. In this closed configuration of PSP, it would be impossible for substrate to enter or product to be released. It is likely that the widespread changes in the 1H-15N FHSQC spectrum of PSP on addition of BeF (Fig. 1) are at least partially caused by a transition of its two domains from a more open to this closed configuration. Such a transition might contribute to a high degree of specificity of the enzyme for phosphoserine (21), a necessity for the function of an intracytoplasmic phosphatase, and/or to the poor reversibility of phosphoserine hydrolysis. The closed configuration of BeF
(Fig. 1) are at least partially caused by a transition of its two domains from a more open to this closed configuration. Such a transition might contribute to a high degree of specificity of the enzyme for phosphoserine (21), a necessity for the function of an intracytoplasmic phosphatase, and/or to the poor reversibility of phosphoserine hydrolysis. The closed configuration of BeF ⋅PSP presumably mimics one from which phosphate would be hydrolyzed (33).
⋅PSP presumably mimics one from which phosphate would be hydrolyzed (33).
Although the Ca2+-ATPase of rabbit sarcoplasmic reticulum has a much more complicated domain structure than PSP or CheY (32) (Fig. 4A), there are nonetheless striking parallels among these three proteins. The phosphorylated (P) domain of the Ca2+-ATPase is structurally homologous to the core α/β domain of PSP and to CheY. Asp-703 in the P domain lies in a stabilized Mg2+ chelation loop similar to those of PSP and CheY. Asp-703 is correctly positioned to coordinate Mg2+ by means of hydrogen bonds between this loop and the stabilizing aspartate Asp-707. Conceptually, the nucleotide-binding (N) domain of the Ca2+-ATPase, which binds ATP and delivers its γ phosphate to the active-site aspartate in the P domain, has a role related to that of the helical domain of PSP or that of CheA, the cognate histidine autokinase for CheY. The small actuator domain of the ATPase, its third cytoplasmic domain, is unique.
Figure 4.

Ribbon diagrams of the Ca2+-ATPase from rabbit sarcoplasmic reticulum (A) and its catalytic P domain (B) [drawn from coordinates of Toyoshima et al. (32)]. (A) The N (nucleotide-binding), A (actuator), and M (Ca2+-binding) domains are shown in pink, yellow, and gray, respectively. The latter traverses the membrane of the sarcoplasmic reticulum. The two Ca2+ ions are shown in red. The catalytic P domain is shown in blue (helices), green (β-strands), and orange (loops). (B) This top-down view was obtained by applying a −90° rotation around the x-axis starting from the orientation in A. The quintet of conserved residues in the P domain is shown in ball-and-stick form in red. Mg2+ was apparently not present in this domain.
As is the case for CheY and other receiver domains, the P domain of the intact Ca2+-ATPase can be phosphorylated by acetylphosphate rather than ATP (16, 17, 42), an activity that presumably does not depend on its N domain. Phosphorylation by either donor acts as a key switch during Ca2+ transport (16, 17, 42). Formation of the first phosphoenzyme intermediate, E1MgP⋅(Ca2+)2, results in occlusion of calcium within the membrane-spanning portion of the protein. In a conformational change(s) that is a rate-limiting step in the catalytic cycle, this intermediate then yields E2MgP as a consequence of diffusion of the occluded calcium to lower affinity sites and its release into the lumen of the sarcoplasmic reticulum. Finally, the enzyme is regenerated by dephosphorylation. Because the site of phosphorylation in the Ca2+-ATPase is more than 50 Å away from the Ca2+-binding sites (32), formation of the phosphoaspartate bond must be coupled to a conformational change(s) within the P domain that is propagated to the transmembrane, Ca2+-binding domain. Given the comparable interactions in the active sites of BeF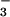 ⋅PSP and BeF
⋅PSP and BeF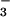 ⋅CheY (Fig. 3C), the phosphorylation-induced conformational change within the P domain of the Ca2+-ATPase is likely to be driven by a mechanism similar to that used in receiver domains. In CheY, this mechanism appears to entail the formation of a hydrogen bond between the hydroxyl group of Thr-87 and aspartyl phosphate (or BeF
⋅CheY (Fig. 3C), the phosphorylation-induced conformational change within the P domain of the Ca2+-ATPase is likely to be driven by a mechanism similar to that used in receiver domains. In CheY, this mechanism appears to entail the formation of a hydrogen bond between the hydroxyl group of Thr-87 and aspartyl phosphate (or BeF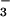 ) and the replacement of a salt bridge between the amino group of Lys-109 and the carboxyl group of the active site aspartate by one to the modified aspartate (6, 7, 10, 43). Analogies between interactions in the active sites of BeF
) and the replacement of a salt bridge between the amino group of Lys-109 and the carboxyl group of the active site aspartate by one to the modified aspartate (6, 7, 10, 43). Analogies between interactions in the active sites of BeF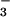 ⋅PSP and activated CheY (Fig. 3C) provide evidence that Thr-625 and Lys-684, respectively, of the Ca2+-ATPase form the corresponding hydrogen bond and salt bridge to the phosphoaspartate, as previously postulated (29). In the structure of the unphosphorylated calcium-bound form of the ATPase that has been determined (32), Lys-684 appears to form a salt bridge to Asp-351 that is similar to the salt bridge between Lys-109 and Asp-57 in inactive CheY (44). However, the sidechains of Thr-625 and Asp-351 are separated by 7.4 Å, a distance that cannot be bridged by a hydrogen bond and a phosphoryl group without a conformational change. As in receiver domains, the movement of the sidechains of Thr-625 and Lys-684 on phosphorylation of Asp-351 might stabilize a conformational change within the P domain of the Ca2+-ATPase that is propagated into the downstream transmembrane domain. Given that E1MgP⋅(Ca2+)2 and E2MgP are likely to be extremely difficult to trap for structural work, these hypothetical sidechain movements and the accompanying more extensive conformational changes might best be evaluated by crystallization of the corresponding BeF
⋅PSP and activated CheY (Fig. 3C) provide evidence that Thr-625 and Lys-684, respectively, of the Ca2+-ATPase form the corresponding hydrogen bond and salt bridge to the phosphoaspartate, as previously postulated (29). In the structure of the unphosphorylated calcium-bound form of the ATPase that has been determined (32), Lys-684 appears to form a salt bridge to Asp-351 that is similar to the salt bridge between Lys-109 and Asp-57 in inactive CheY (44). However, the sidechains of Thr-625 and Asp-351 are separated by 7.4 Å, a distance that cannot be bridged by a hydrogen bond and a phosphoryl group without a conformational change. As in receiver domains, the movement of the sidechains of Thr-625 and Lys-684 on phosphorylation of Asp-351 might stabilize a conformational change within the P domain of the Ca2+-ATPase that is propagated into the downstream transmembrane domain. Given that E1MgP⋅(Ca2+)2 and E2MgP are likely to be extremely difficult to trap for structural work, these hypothetical sidechain movements and the accompanying more extensive conformational changes might best be evaluated by crystallization of the corresponding BeF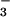 complexes. These would be structurally equivalent to the phosphorylated complexes but persistent.
complexes. These would be structurally equivalent to the phosphorylated complexes but persistent.
Use of BeF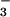 as a Phosphate Analogue.
as a Phosphate Analogue.
Originally, BeF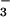 was used in biochemical and structural studies of G proteins and later of myosin, the F1-ATPase, nitrogenase, and some kinases (38, 39, 45). In these earlier cases, high-affinity binding of BeF
was used in biochemical and structural studies of G proteins and later of myosin, the F1-ATPase, nitrogenase, and some kinases (38, 39, 45). In these earlier cases, high-affinity binding of BeF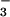 required the presence of a purine nucleoside diphosphate (GDP or ADP) in the nucleotide-binding site of the enzyme, and BeF
required the presence of a purine nucleoside diphosphate (GDP or ADP) in the nucleotide-binding site of the enzyme, and BeF served as an analogue of the γ-phosphate in its ground state. Recently, BeF
served as an analogue of the γ-phosphate in its ground state. Recently, BeF has been used to generate an analogue of aspartyl phosphate in response regulators, providing a convenient and apparently general means of mimicking their active phosphorylated forms (11). As discussed in this work, we have now extended the use of BeF
has been used to generate an analogue of aspartyl phosphate in response regulators, providing a convenient and apparently general means of mimicking their active phosphorylated forms (11). As discussed in this work, we have now extended the use of BeF to structural studies of members of the HAD superfamily. The BeF
to structural studies of members of the HAD superfamily. The BeF ⋅PSP complex mimics a phosphoenzyme intermediate for this enzyme (half-life ≤ 0.1 sec at 50°C), which was so labile that the phosphorylation of aspartate could not be confirmed chemically (14). Biochemical studies indicate that BeF
⋅PSP complex mimics a phosphoenzyme intermediate for this enzyme (half-life ≤ 0.1 sec at 50°C), which was so labile that the phosphorylation of aspartate could not be confirmed chemically (14). Biochemical studies indicate that BeF forms an aspartyl phosphate mimic in the Ca2+-ATPase from rabbit sarcoplasmic reticulum (24). The stable BeF
forms an aspartyl phosphate mimic in the Ca2+-ATPase from rabbit sarcoplasmic reticulum (24). The stable BeF -ATPase complex is rapidly disrupted by luminal Ca2+ (24), and hence it appears to resemble most closely E2MgP. Structural analogies between PSP, CheY, and the Ca2+-ATPase make it likely that BeF
-ATPase complex is rapidly disrupted by luminal Ca2+ (24), and hence it appears to resemble most closely E2MgP. Structural analogies between PSP, CheY, and the Ca2+-ATPase make it likely that BeF complexes will be useful in defining the conformational changes that allow P-type ATPases to couple the energy available from ATP hydrolysis to transport of cations (15–17, 32).
complexes will be useful in defining the conformational changes that allow P-type ATPases to couple the energy available from ATP hydrolysis to transport of cations (15–17, 32).
Acknowledgments
We thank our colleagues B. T. Nixon for critical review of the manuscript and Ore Carmi for help in its preparation. This work was supported by U.S. Department of Energy Contract No. DE-AC03–76SF0098 to D.W., S.-H.K., and R.K., National Institutes of Health (NIH) Grant P50 GM62412 to S.-H.K. and R.K., NIH Grant GM62163 to D.W., and NIH Grant GM38361 to S.K.
Abbreviations
- HAD
haloacid dehalogenase
- PSP
phosphoserine phosphatase
- FHSQC
fast heteronuclear single quantum correlation
- P domain
phosphorylated domain
Footnotes
Data deposition: The atomic coordinates reported in this paper have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1J97).
See commentary on page 8170.
References
- 1.Post R L, Kume S. J Biol Chem. 1973;248:6993–7000. [PubMed] [Google Scholar]
- 2.Weiss V, Magasanik B. Proc Natl Acad Sci USA. 1988;85:8919–8923. doi: 10.1073/pnas.85.23.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoch J A, Silhavy T J. Two-Component Signal Transduction. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 4.Djordjevic S, Stock A M. J Struct Biol. 1998;124:189–200. doi: 10.1006/jsbi.1998.4034. [DOI] [PubMed] [Google Scholar]
- 5.Falke J J, Bass R B, Butler S L, Chervitz S A, Danielson M A. Annu Rev Cell Dev Biol. 1997;13:457–512. doi: 10.1146/annurev.cellbio.13.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama J P. Struct Folding Des. 1999;7:1505–1515. doi: 10.1016/s0969-2126(00)88341-0. [DOI] [PubMed] [Google Scholar]
- 7.Lewis R J, Brannigan J A, Muchová K, Barák I, Wilkinson A J. J Mol Biol. 1999;294:9–15. doi: 10.1006/jmbi.1999.3261. [DOI] [PubMed] [Google Scholar]
- 8.Kern D, Volkman B F, Luginbühl P, Nohaile M J, Kustu S, Wemmer D E. Nature (London) 1999;402:894–898. doi: 10.1038/47273. [DOI] [PubMed] [Google Scholar]
- 9.Halkides C J, McEvoy M M, Casper E, Matsumura P, Volz K, Dahlquist F W. Biochemistry. 2000;39:5280–5286. doi: 10.1021/bi9925524. [DOI] [PubMed] [Google Scholar]
- 10.Cho H S, Lee S Y, Yan D, Pan X, Parkinson J S, Kustu S, Wemmer D E, Pelton J G. J Mol Biol. 2000;297:543–551. doi: 10.1006/jmbi.2000.3595. [DOI] [PubMed] [Google Scholar]
- 11.Yan D, Cho H S, Hastings C A, Igo M M, Lee S Y, Pelton J G, Stewart V, Wemmer D E, Kustu S. Proc Natl Acad Sci USA. 1999;96:14789–14794. doi: 10.1073/pnas.96.26.14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aravind L, Galperin M Y, Koonin E V. Trends Biochem Sci. 1998;23:127–129. doi: 10.1016/s0968-0004(98)01189-x. [DOI] [PubMed] [Google Scholar]
- 13.Koonin E V, Tatusov R L. J Mol Biol. 1994;244:125–132. doi: 10.1006/jmbi.1994.1711. [DOI] [PubMed] [Google Scholar]
- 14.Collet J F, Stroobant V, Pirard M, Delpierre G, Van Schaftingen E. J Biol Chem. 1998;273:14107–14112. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- 15.Jencks W P. Biosci Rep. 1995;15:283–287. doi: 10.1007/BF01788360. [DOI] [PubMed] [Google Scholar]
- 16.McIntosh D B. Nat Struct Biol. 2000;7:532–535. doi: 10.1038/76735. [DOI] [PubMed] [Google Scholar]
- 17.MacLennan D H, Green N M. Nature (London) 2000;405:633–634. doi: 10.1038/35015206. [DOI] [PubMed] [Google Scholar]
- 18.Seal S N, Rose Z B. J Biol Chem. 1987;262:13496–13500. [PubMed] [Google Scholar]
- 19.Pirard M, Collet J F, Matthijs G, Van Schaftingen E. FEBS Lett. 1997;411:251–254. doi: 10.1016/s0014-5793(97)00704-7. [DOI] [PubMed] [Google Scholar]
- 20.Collet J F, Gerin I, Rider M H, Veiga-da-Cunha M, Van Schaftingen E. FEBS Lett. 1997;408:281–284. doi: 10.1016/s0014-5793(97)00438-9. [DOI] [PubMed] [Google Scholar]
- 21.Veeranna, Shetty K T. Neurochem Res. 1990;15:1203–1210. doi: 10.1007/BF01208581. [DOI] [PubMed] [Google Scholar]
- 22.Cantley L C, Jr, Cantley L G, Josephson L. J Biol Chem. 1978;253:7361–7368. [PubMed] [Google Scholar]
- 23.Missiaen L, Wuytack F, De Smedt H, Vrolix M, Casteels R. Biochem J. 1988;253:827–833. doi: 10.1042/bj2530827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy A J, Coll R J. J Biol Chem. 1993;268:23307–23310. [PubMed] [Google Scholar]
- 25.Pick U. J Biol Chem. 1982;257:6111–6119. [PubMed] [Google Scholar]
- 26.Rapin-Legroux C, Troullier A, Dufour J-P, Dupont Y. Biochim Biophys Acta. 1994;1184:127–133. [Google Scholar]
- 27.Robinson J D, Davis R L, Steinberg M. J Bioenerg Biomembr. 1986;18:521–531. doi: 10.1007/BF00743148. [DOI] [PubMed] [Google Scholar]
- 28.Troullier A, Girardet J L, Dupont Y. J Biol Chem. 1992;267:22821–22829. [PubMed] [Google Scholar]
- 29.Ridder I S, Dijkstra B W. Biochem J. 1999;339:223–226. [PMC free article] [PubMed] [Google Scholar]
- 30.Hisano T, Hata Y, Fujii T, Liu J Q, Kurihara T, Esaki N, Soda K. J Biol Chem. 1996;271:20322–20330. doi: 10.1074/jbc.271.34.20322. [DOI] [PubMed] [Google Scholar]
- 31.Ridder I S, Rozeboom H J, Kalk K H, Janssen D B, Dijkstra B W. J Biol Chem. 1997;272:33015–33022. doi: 10.1074/jbc.272.52.33015. [DOI] [PubMed] [Google Scholar]
- 32.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Nature (London) 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Kim R, Jancarik J, Yokota H, Kim S H. Struct Folding Des. 2001;9:65–72. doi: 10.1016/s0969-2126(00)00558-x. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 36.Jones T A, Zou J Y, Cowan S W, Kjeldgaard Acta Crystallogr. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 37.Martin R B. Biochem Biophys Res Commun. 1988;155:1194–1200. doi: 10.1016/s0006-291x(88)81266-x. [DOI] [PubMed] [Google Scholar]
- 38.Chabre M. Trends Biochem Sci. 1990;15:6–10. doi: 10.1016/0968-0004(90)90117-t. [DOI] [PubMed] [Google Scholar]
- 39.Petsko G A. Proc Natl Acad Sci USA. 2000;97:538–540. doi: 10.1073/pnas.97.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collet J F, Stroobant V, Van Schaftingen E. J Biol Chem. 1999;274:33985–33990. doi: 10.1074/jbc.274.48.33985. [DOI] [PubMed] [Google Scholar]
- 41.Lee S Y, Cho H S, Pelton J G, Yan D, Henderson R K, King D S, Huang L S, Kustu S, Berry E A, Wemmer D E. Nat Struct Biol. 2001;8:52–56. doi: 10.1038/83053. [DOI] [PubMed] [Google Scholar]
- 42.Bodley A L, Jencks W P. J Biol Chem. 1987;262:13997–14004. [PubMed] [Google Scholar]
- 43.Lee S-Y, Cho H S, Pelton J G, Yan D, Berry E, Wemmer D E. J Biol Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 44.Stock A M, Mottonen J M, Stock J B, Schutt C E. Nature (London) 1989;337:745–749. doi: 10.1038/337745a0. [DOI] [PubMed] [Google Scholar]
- 45.Xu Y W, Moréra S, Janin J, Cherfils J. Proc Natl Acad Sci USA. 1997;94:3579–3583. doi: 10.1073/pnas.94.8.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]