

Abstract
Occupational and/or environmental exposure to nickel has been implicated in various types of cancer, and in vitro exposure to nickel compounds results in accumulation of Ni(II) ions in cells. One of the major targets of Ni(II) ions inside the cell is Fe(II)- and αKG-dependent dioxygenases. Using JMJD2A and JMJD2C as examples, we show that JMJD2 family of histone demethylases, which are products of putative oncogenes as well as Fe(II)- and αKG-dependent dioxygenases, are highly sensitive to inhibition by Ni(II) ions. In this work, X-ray absorption spectroscopy (XAS) has been used to investigate the Fe(II) active site of truncated JMJD2A and JMJD2C (1 – 350 aa) in the presence and absence of αKG and/or substrate to obtain mechanistic details of the early steps in catalysis that precede O2 binding in histone demethylation by the JMJD2 family of histone demethylases. Zinc K-edge XAS has been performed on the resting JMJD2A (with iron in the active site) to confirm the presence of the expected structural zinc site. XAS of the Ni(II)-substituted enzymes has also been performed to investigate the inhibition of these enzymes by Ni(II) ions. Our XAS results indicate that the five-coordinate Fe(II) center in the resting enzyme is retained in the binary and ternary complexes. In contrast, the Ni(II) center is six-coordinate in the resting enzyme, binary and ternary complexes. XAS results indicate that both Fe(II) and Ni(II) bind αKG in the binary and ternary complexes. The electron density build-up that is observed at the Fe(II) center in the presence of αKG and substrate is not observed at the Ni(II) center. Thus, both electronic and steric factors are responsible for Ni-induced inhibition of the JMJD2 family of histone demethylases. Ni-induced inhibition of these enzymes may explain the alteration of the epigenetic mechanism of gene expression that is responsible for Ni-induced carcinogenesis.
Occupational and/or environmental exposure to nickel has been implicated in various types of cancer (1 – 6). However, the precise mechanism(s) of nickel-induced carcinogenesis are not fully understood (7, 8). Alterations of epigenetic mechanisms have been proposed to play an important role in nickel-induced carcinogenesis (9 – 11). In vitro exposure to nickel compounds results in accumulation of Ni(II) ions in cells, and recent studies have shown that Fe(II)- and αKG-dependent dioxygenases are one of the major targets of Ni(II) ions inside the cell (12). Further, increases in cellular nickel concentration correlate with increases in the global levels of mono-and di-methylated histone H3 Lys9 (H3K9Me1 and H3K9Me2) – not by affecting histone methyltransferases, but by inhibiting a group of Fe(II)- and αKG-dependent histone demethylases (13). It is known that Ni(II) ions inhibit both ABH2 (a DNA demethylase) and JMJD1A (a histone demethylase that demethylates H3K9Me2 and H3K9Me1) (14). It is possible that nickel causes alterations of epigenetic gene expression by inhibiting the JMJD2 family of histone demethylases, which are among the known Fe(II)- and αKG-dependent enzymes (12, 15).
The JMJD2 family of histone demethylases (a.k.a. KDM4, lysine specific demethylase 4 histone demethylases) catalyze the demethylation of di- and tri-methylated H3K9 and H3K36 (Figure 1) (16). JMJD2 proteins are candidate oncogenes, which contribute to tumor formation (17, 18). JMJD2A plays an important role in cell proliferation and oncogenesis (19). JMJD2C has been found to play some role in prostate and breast cancer progression, and has been implicated in the regulation of androgen receptor responsive genes (20). Inhibition of JMJD2A and JMJD2C is also known to affect cellular growth (21). However, there is limited data available on the enzymology of histone demethylases.
Figure 1.

Demethylation of methylated histone tails by JMJD2 proteins.
In this work, XAS has been used to investigate the Fe(II) active site structure of truncated forms of both JMJD2A and JMJD2C (1 – 350 aa) in the presence and absence of αKG and/or substrate to obtain mechanistic details of the early steps (prior to O2 binding) in histone demethylation by the JMJD2 family of histone demethylases. Full-length JMJD enzymes have high molecular weights (e.g., 150 kD) and cannot be expressed in large quantities in bacteria, but must be expressed in baculovirus and insect cells. The truncated forms of these enzymes, where large quantities of enzyme can be produced in bacteria, were therefore investigated. In addition, XAS has been performed on the Ni(II)-substituted enzymes to understand the nickel-induced inhibition of these enzymes. Crystal structures available for JMJD2 proteins suggest that these proteins have a zinc binding site involving three Cys and one His ligands, along with the Fe(II) site consisting of a facial triad of protein ligands (2-His-1-carboxylate) and water molecules (22 – 27). Zinc K-edge XAS has been performed on the resting JMJD2A (with iron in the active site) to confirm the presence of the expected structural zinc site. It should be noted that some of these crystal structures feature Ni(II) in the place of Fe(II), and also may substitute 2-hydroxyglutarate and N-oxalylglycine (N-OG) for αKG (Table 1). These are not competent enzymes, and therefore not directly relevant to the Fe(II)- and αKG-dependent demethylation reaction carried out by these enzymes. This study examines the structure of the competent forms of the enzymes in solution, and provides a comparison with two related enzymes, the available crystal structures, and with the Ni(II)-inhibited enzymes.
Table 1.
Crystallographically determined active site structures of the JMJD2 family of histone demethylases.
| PDB | Protein | Metal ions | Cofactor | Substrate | Ref |
|---|---|---|---|---|---|
| 3RVH | JMJD2A | Ni/Zn | 8-hydroxy-3-(piperazin-1- yl)quinoline-5-carboxylic acid |
none | * |
| 2Q8E | JMJD2A | Ni/Zn | N-OG | none | 24 |
| 2P5B | JMJD2A | Fe/Zn | N-OG | H3K36Me3 peptide | 23 |
| 2PXJ | JMJD2A | Fe/Zn | N-OG | H3K36Me1 peptide | 23 |
| 2Q8C | JMJD2A | Ni/Zn | αKG | H3K9Me3 peptide | 24 |
| 2OQ6 | JMJD2A | Ni/Zn | N-OG | H3K9Me3 peptide | 25 |
| 2OQ7 | JMJD2A | Ni/Zn | N-OG | none | 25 |
| 2OS2 | JMJD2A | Ni/Zn | N-OG | H3K36Me3 peptide | 25 |
| 2OT7 | JMJD2A | Ni/Zn | N-OG | H3K9Me1 peptide | 25 |
| 2OX0 | JMJD2A | Ni/Zn | N-OG | H3K9Me2 peptide | 25 |
| 2VD7 | JMJD2A | Ni/Zn | Pyridine-2,4-dicarboxylic acid | none | * |
| 4AI9 | JMJD2A | Ni/Zn | Daminozide | none | 28 |
| 2Q8D | JMJD2A | Ni/Zn | Succinic acid | H3K36Me2 peptide | 24 |
| 2YBK | JMJD2A | Ni/Zn | (2R)-2-hydroxypentanedioic acid | none | 26 |
| 2WWJ | JMJD2A | Ni/Zn | O-benzyl-N-(carboxycarbonyl)- D-tyrosine |
none | 29 |
| 4GD4 | JMJD2A | Ni/Zn | 2-(1H–pyrazol-3-yl)pyridine-4- caboxylic acid |
none | * |
| 3PDQ | JMJD2A | Ni/Zn | 4'-[(2-aminoethyl)carbamoyl]- 2,2'-bipyridine- 4-carboxylic acid |
none | 30 |
| 2GP3 | JMJD2A | Fe/Zn | none | none | 22 |
| 2YBP | JMJD2A | Ni/Zn | (2R)-2-hydroxypentanedioic acid | H3K36Me3 peptide | 26 |
| 2YBS | JMJD2A | Ni/Zn | (2S)-2-hydroxypentanedioic acid | H3K36Me3 peptide | 26 |
| 3NJY | JMJD2A | Ni/Zn | 5-carboxy-8-hydroxyquinoline | none | 31 |
| 2GP5 | JMJD2A | Fe/Zn | αKG | none | 22 |
| 3U4S | JMJD2A | Ni/Zn | N-(carboxylcabbonyl)-D-cysteine | H3K9Me3 peptide | 32 |
| 2XML | JMJD2C | Ni/Zn | N-OG | none | * |
| 3DXU | JMJD2D | Fe/Zn | N-OG | none | * |
| 4HOO | JMJD2D | Ni/Zn | none | none | 27 |
| 3DXT | JMJD2D | Zn | none | none | * |
| 4HON | JMJD2D | Ni/Zn | αKG | H3K9Me3 peptide | 27 |
these crystal structures have not been published but available in the PDB.
Experimental Section
Expression and purification of truncated histone demethylases
pGEX-4T-1-GST-JMJD2A and pET28a–His6-JMJD2C constructs were kindly provided by Dr. Yang Shi. The protein expression, purification, subsequent cleavage of the GST- or His6-tag, and characterization of the protein products have been reported previously from the Shi lab (16, 22). E. coli BL21(DE3)pLysS competent cells (Novagen) were transformed with pGEX-4T-1-GST-JMJD2A and pET28a–His6-JMJD2C and were plated and grown overnight at 37 °C on LB medium containing 34 µg/mL chloramphenicol and 100 µg/mL ampicillin (for JMJD2A) or 30 µg/mL kanamycin (for JMJD2C). Single colonies were grown overnight in 150 mL cultures containing the antibiotics mentioned above, and then diluted to 1:100 in 2 L of fresh LB medium. The cells were grown at 37 °C to an optical density of 0.8 at 600 nm and then induced with isopropyl β-D-1-thiogalactopyranoside (final conc. 0.2 mM). The cells were grown for an additional 18 h at 18 °C, and were harvested by centrifugation and resuspended in lysis buffer (50 mM HEPES, 300 mM NaCl, 20 mM imidazole, 0.5 mM TCEP and 5% glycerol at a pH of 7.5) and stored at −80 °C. Upon thawing, the cells were lysed in the presence of PMSF and DNAse and then centrifuged to collect the lysate.
Purification of JMJD2A
The supernatant was added to the MagneGST™ particles in a centrifuge tube (50 mL) and gently agitated for an hour using a shaker (Orbitron rotator II). Glutathione immobilized on MagneGST™ particles binds the GST-fusion protein. These particles were sequestered by a magnetic field, the liquid phase removed, and unbound proteins were washed away using wash buffer (50 mM HEPES, 300 mM NaCl, 0.5 mM TCEP and 5% glycerol at a pH of 7.5). Pure JMJD2A was released from the particles using the wash buffer containing 20 mM glutathione. The purity of JMJD2A was confirmed by a single band with MW ∼ 67 kD on an SDS-PAGE gel (see supporting information). Finally, the GST-tag was cleaved from purified protein using a Thrombin CleanCleave Kit (Sigma). Pure JMJD2A (yield ∼ 1.4 mg/l of cell culture) was separated from uncleaved JMJD2A and the GST-tag using a gel filtration column (23.5 mL, Superdex 75 10/300 GL, GE Healthcare Life Sciences) and buffer containing 10 mM HEPES, 300 mM NaCl, 0.5 mM TCEP and 5 % glycerol at a pH of 7.5. The cleavage of GST-tag was confirmed by SDS-PAGE (see supporting information). This cleavage leaves two extra amino acid residues (GS) on the N-terminus of JMJD2A (16, 22).
Purification of JMJD2C
The supernatant was loaded on to a Ni-NTA column (27 mL, Kontes FlexColumn, Kimble Chase Kontes) and gently mixed for an hour using a shaker (Orbitron rotator II). The column was washed with lysis buffer containing 40 mM imidazole. JMJD2C was eluted using lysis buffer with 300 mM imidazole. JMJD2C obtained from Ni-NTA column was buffer exchanged into a buffer containing 10 mM HEPES, 300 mM NaCl, 0.5 mM TCEP, 5 % glycerol at a pH of 7.5 and then loaded on to a gel filtration column (23.5 mL, Superdex 200 10/300 GL, GE Healthcare Life Sciences) that was previously equilibrated with the same buffer. JMJD2C was eluted using two column volumes (∼ 47 mL) of this buffer. The purity of JMJD2C obtained was confirmed by a single band with MW ∼ 44 kD on an SDS-PAGE gel (see supporting information). The His6-tag was subsequently cleaved from purified protein using a Thrombin CleanCleave Kit (Sigma) and removed by dialysis against a buffer containing 50 mM HEPES, 300 mM NaCl, 0.5 mM TCEP and 5 % glycerol at a pH of 7.5 (yield ∼ 1.1 mg/l of cell culture). The cleavage of the His6-tag was confirmed by SDS-PAGE (see supporting information). This cleavage will leave 17 extra amino acid residues (GSHMASMTGGQQMGRGS) on the N-terminus of JMJD2C (16, 22).
Activity assay of JMJD2 proteins
The demethylase activity of JMJD2A and JMJD2C was confirmed using an assay described previously (14), employing 5 µg of histone protein (Sigma) in 25 µL (for JMJD2A) or in 30 µL (for JMJD2C) of reaction buffer containing 50 µM HEPES (pH 7.8), 100 µM FeSO4, 100 µM ZnSO4, 1 mM αKG, 2 mM ascorbic acid. The demethylase activity assay of JMJD2A and JMJD2C (see supporting information) was used to determine the amount of enzyme used for the nickel-inhibition studies. In order to show the inhibition of JMJD2A and JMJD2C by Ni(II) ions, varying concentrations of NiCl2 were added to the reaction buffer prior to the addition of JMJD2 proteins. The reaction mixture was incubated at 37 °C for 1 hour. The reaction was terminated by the addition 6X SDS loading dye to a final concentration of 1X and heating at 100 °C for 5 minutes. The remaining amounts of modified H3K9Me3 in histones were determined by immunoblotting as previously described (Figure 2) (14). The IC50 value for the inhibition of JMJD2C by Ni(II) was determined from densitometry performed in the gel image using GraphPad Prisom 6 software (version 6.0a, July18, 2012) and a sigmoidal dose-response function (see supporting information).
Figure 2.

Demethylase activity assay of JMJD2A (top) and JMJD2C (bottom) in the presence of varying concentrations of Ni(II) ions done by immunoblotting. The lack of any band in the absence of Ni(II) ions indicates the demethylation of all the H3K9Me3. The appearance of bands in the presence of Ni(II) ions indicates that H3K9Me3 remains present due to the inhibition of the enzymes.
XAS Sample preparation
Apo-protein was prepared by adding a five-fold excess of EDTA to the protein solution and incubating it at 4 °C overnight. EDTA was then removed by dialysis against a buffer solution containing 50 mM HEPES, 300 mM NaCl, 2 mM ascorbic acid and 5 % glycerol at pH = 7.5. The protein concentration was determined by the Bradford assay using BSA as a standard (33). Metallated samples were prepared in a Coy™ chamber and frozen under N2 atmosphere. Both JMJD2A and JMJD2C were first metallated with zinc (both of these proteins have a zinc binding site along with the Fe(II) active site) by adding ZnSO4 (10 mM, 1.25 eq) to the corresponding apo-protein ( ∼ 45 µM). Unbound and non-specifically bound zinc was then removed by adding Chelex 100 beads to the sample, incubating 20 min., and then decanting the solution via pipette. This zinc containing protein solution was divided in to two portions – one portion was further metallated with iron and the other portion was metallated with nickel as described below. Fe(JMJD2A/JMJD2C) was prepared by adding a solution of (NH4)2Fe(SO4)2 (10 mM, 1.25 eq) in the buffer mentioned above to the zinc containing protein and incubating for 6 h. Unbound and non-specifically bound metal was then removed by adding Chelex 100 beads to the sample, incubating 20 min., and then decanting the solution via pipette. The iron and zinc content of the metallated proteins was determined by ICP-OES using the emissions at 238.208 and 206.2 nm, respectively. On the basis of a Bradford assay of protein concentration and the metal content found, the metallated JMJD2A and JMJD2C have Fe:Zn:protein ratios of 0.85:0.92:1 and 0.91:0.96:1, respectively.
The same procedure, only substituting NiCl2 (10 mM, 1.25 eq) for (NH4)2Fe(SO4)2, was used to prepare Ni(JMJD2A/JMJD2C) complexes. Analysis of the protein concentration and metal content using the same procedure as for the Fe(II) enzyme (nickel concentration was measured using the 231.604 nm emission) gave Ni:Zn:protein ratios of 0.90:0.94:1 for JMJD2A and 0.94:096:1 for JMJD2C.
From the Fe(JMJD2A/JMJD2C) preparation described above, a total of three samples of each protein were prepared for XAS experiments. The first sample contained only the metallated protein, hereafter referred to as Fe(JMJD2A/JMJD2C). The second sample contained Fe(JMJD2A/JMJD2C) plus 1.0 equiv. of αKG, hereafter referred to as Fe(JMJD2A/JMJD2C)-αKG. Finally, the third sample contained Fe(JMJD2A/JMJD2C), 1.0 equiv. of αKG, plus 2 equiv. of histone from calf thymus (Sigma), and is hereafter referred to as Fe(JMJD2A/JMJD2C)-αKG + substrate. The samples had an iron content of 0.64 mM (for 0.75 mM JMJD2A protein) and 0.62 mM (for 0.68 mM JMJD2Cprotein) for the Fe(JMJD2A) and Fe(JMJD2C) samples, respectively; 0.63 mM (for 0.74 mM JMJD2A protein) and 0.61 mM (for 0.67 mM JMJD2C protein) for the Fe(JMJD2A)-αKG and Fe(JMJD2C)-αKG samples, respectively; 0.61 mM (for 0.72 mM JMJD2A protein) and 0.59 mM (for 0.65 mM JMJD2C protein) for the Fe(JMJD2A)-αKG + substrate and Fe(JMJD2C)-αKG + substrate samples, respectively, as determined by ICP-OES. Three analogous samples were prepared from Ni(JMJD2A/JMJD2C). The samples had a nickel content of ∼ 0.60 mM (for 0.67 mM JMJD2A protein) and 0.63 mM (for 0.67 mM JMJD2C protein) for the Ni(JMJD2A) and Ni(JMJD2C) samples, respectively; 0.59 mM (for 0.66 mM JMJD2A protein) and 0.62 mM (for 0.66 mM JMJD2C protein) for the Ni(JMJD2A)-αKG and Ni(JMJD2C)-αKG samples, respectively; 0.57 mM (for 0.63 mM JMJD2A protein) and 0.60 mM (for 0.64 mM JMJD2C protein) for the Ni(JMJD2A)-αKG + substrate and Ni(JMJD2C)-αKG + substrate samples, respectively, as determined by ICP-OES. The protein examined by Zn K-edge XAS had a zinc content of 0.69 mM (for 0.75 mM JMJD2A protein). The samples (60 µL) were syringed into polycarbonate sample holders with kapton windows and frozen in liquid N2 for XAS data collection.
X-ray Absorption spectroscopy
XAS data collection and analysis were performed as reported previously (34, 35). Iron, nickel and zinc K-edge data were collected at 10 K using a liquid He cryostat (Oxford Instruments) on beam line 7–3 at the Stanford Synchrotron Radiation Lightsource (SSRL). Beamline optics consisted of a Si(220) double crystal monochromator and a single Rh-coated mirror for harmonic rejection. X-ray fluorescence was collected using a 30-element Ge detector (Canberra). Scattering was minimized by placing a set of Soller slits with a (Z-1) element (Mn, Co or Cu) filter between the sample chamber and the detector. The energy of each metal K-edge was calibrated to the first inflection point of the relevant metal foil (7112.5 eV for iron, 8331.6 eV for nickel and 9660.7 eV for zinc). Extended X-ray absorption fine structure (EXAFS) data were collected to k= 14 Å−1 above the edge energy for all the samples.
XAS data analysis was performed using EXAFS123 (36) for XANES analysis and SixPack (37) for EXAFS analysis. Scattering parameters for SixPack fitting were generated using the FEFF 8 (38). For the data presented here, 12 scans were averaged for iron containing samples, eight scans were averaged for nickel containing samples, and six scans were averaged for the zinc data. The averaged spectra were background corrected and normalized in SixPack, which uses AUTOBK routine of IFEFFIT (37, 39, 40). For XANES analysis, the edge energy reported was taken to be the maximum of first derivative in the edge region. For pre-edge XANES analysis, a Gaussian function was used to fit the baseline of the pre-edge region, with a 75% Gaussian and 25% Lorenzian function used to fit the rise in the fluorescence at the edge. Gaussian peaks were added to the baseline corrected spectrum to account for any peaks in the pre-edge region. The areas of these peaks were taken as a measure of their intensities. The intensity of the peak associated with a 1s → 3d electronic transition was then used to indicate the coordination number/geometry of the metal sites (44 – 46).
For EXAFS analysis, the averaged spectra were first converted to k-space. EXAFS data were fit over a k range of 2 – 12.5 Å−1 with the exception of Fe-JMJD2A-αKG and Ni-JMJD2C-αKG + substrate, where a k range of 2 – 12 Å−1 was used due to poorer signal:noise ratio at high values of k. For the zinc K-edge EXAFS analysis of the resting JMJD2A (with iron in the active site), a k range of 2 – 13.5 Å−1 was used. The k 3-weighted data were fit in r-space. Structural models of the metal sites involving coordination numbers from two to seven were systematically evaluated for all possible combinations of N/O- and S-donors by holding the number of scattering atoms in each shell to integer values (see supporting information). The number of histidine imidazole ligands involved in the coordination sphere was estimated by multiple-scattering analyses, as described previously (44 – 46). Amplitudes and phase shifts for multiple-scattering paths for Fe-Im ligands were generated using FEFF 8 with the coordinates of an Fe-Im ligand obtained from the crystal structure of human PHF8 (PDB: 3K3O). For Ni-Im and Zn-Im ligands, the crystal structures of PHF2 (PDB: 3PUA) and JMJD2A (PDB: 2GP3) were used, respectively. Scattering paths of similar lengths were combined in one shell as described by Tierney et al (45, 46) to decrease the number of variables in each fit. During the fitting process coordination numbers were constrained to be integer values and a scale factor of 0.9 was used. Bond lengths (r), σ2 and a single value of ΔE0 were allowed to vary in each fit. For αKG and ascorbate ligands, the best models including imidazole ligands were further screened using multiple-scattering analysis employing rigid O-C-C-O five membered chelate rings with parameters obtained from FEFF 8 and the structures mentioned above. The crystal structure of trimethylreductic acid (an ascorbic acid analogue) was used to generate the parameters for the chelate ring in the resting enzymes (47). The crystal structures of PHF2 (PDB: 3PUA) and PHF8 (PDB: 3K3O) were used to generate the parameters for the chelate ring in the αKG containing complexes. In this analysis, the distances in the chelate ring were constrained to vary with a single value of Δr and σ2. With the exception of resting enzymes with Fe(II) and Ni(II), the fits were improved by the substitution of two N/O donors in the first coordination sphere by the chelate ring, which adds two carbon atoms to the second coordination sphere in addition to replacing two N/O atoms in the first coordination sphere.
To compare different fits in the same data set IFEFFIT uses three goodness of fit parameters – χ2 (Equation 1), reduced χ2 and R (Equation 2) where Nidp is the number of independent data points, Nε2 = number of uncertainties to minimize, Re() = real part of the EXAFS fitting function, Im() = imaginary part of the EXAFS fitting function and χ͂(fi) is the Fourier-transformed data or model function. Reduced χ2 = χ2/(Nind – Nvarys) (where Nvarys = the number of refining parameters) and represents the degrees of freedom in the fit. IFEFFIT also calculates R for the fit, which is given by Equation 2 and is scaled to the magnitude of the data, making it proportional to χ2. To compare different models (fits) the R-factor and reduced χ2 parameters can be assessed, in which case both parameters should be minimized. Although R will always improve with an increasing number of shells (adjustable parameters), reduced χ2 will go through a minimum and then increase, indicating that the model is over fitting the data. Best fits were judged by using two goodness of fit parameters, reduced χ2 and R, and the deviation of σ2 from typical values.
| (1) |
| (2) |
Results
Activity Assay of JMJD2 proteins
The demethylase activity of JMJD2A shows that it took 9 µg of enzyme to demethylate all the H3K9Me3. JMJD2C was slightly more active; it took 6 µg of enzyme to demethylate all H3K9Me3 (see supporting information). These amounts of enzymes were subsequently used to show the inhibition of JMJD2A and JMJD2C by Ni(II) ions.
Inhibition of demethylase activity of JMJD2A and JMJD2C was observed in the presence of a varying concentration of Ni(II) ions (Figure 2). It should be noted that JMJD2C is inhibited at a much lower concentration of Ni(II) ions, 50 µM vs. 250 µM for JMJD2A. This is due in part to the fact that a smaller amount of enzyme was used for the JMJD2C assay than for the JMJD2A assay. The molar ratio of Ni(II):JMJD2 proteins for the inhibition of JMJD2A and JMJD2C are 28.4 and 10.6 respectively. This molar ratio indicates that JMJD2C is more sensitive to Ni(II) inhibition than is JMJD2A. We were able to calculate IC50 value for the inhibition of JMJD2C by Ni(II) ions using densitometry (see supporting information). The IC50 value for the inhibition of JMJD2C by Ni(II) ions is 20.5 µM, which is similar to that previously determined for JMJD1A (25 µM) (14).
X-ray absorption spectroscopy
XANES Analysis
The analysis of XANES data provides information about the coordination number and geometry of the metal site (44 – 46) and is summarized for the JMJD2A and JMJD2C proteins in Table 2 and in Figures 3 –5. Both Fe(II) and Ni(II) give rise to peaks associated with 1s → 3d electronic transitions (at ∼ 7113 eV and ∼8332 eV, respectively) that are observed in the pre-edge region of the K-edge XANES spectra in all the histone demethylase samples. The intensities (peak areas) of these peaks depend on the coordination number and geometry of the metal sites (44 – 46). Comparison of the peak areas measured for the histone demethylase samples with those obtained for coordination compounds with known coordination numbers and/or geometries provides a measure of coordination number and geometry for the enzyme metal sites. The peak areas obtained for all of the iron containing histone demethylase samples (10 – 13 × 10−2 eV) are indicative of five-coordinate sites (typical values are 8 – 13 ×10−2 eV) (42, 43). This is in agreement with the crystal structure of JMJD2A obtained in the presence or absence of αKG (22).
Table 2.
XAS analysis of JMJD2A and JMJD2C samples.
| Sample | XANES Analysis | EXAFS Analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Edge (ev) |
E | 1s→ peak (x10−2 |
3d area eV) |
Coord. No. |
Shell | r (Å)a | σ2 (x10−3 Å2)b |
ΔE0 (eV) |
Red χ 2 |
%Rc | |
| Fe(JMJD2A) | 7123.1(2) | 13(1) | 5 | 3 N/O | 2.00(2) | 2(2) | −9(2) | 9.27 | 2.3 | ||
| 2 N/O | 2.16(2) | 5(2) | |||||||||
| (2Im) | |||||||||||
| Fe(JMJD2A) | 9661.2(2) | NA | NA | 1 Im | 2.08(2) | 3(2) | −9(1) | 13.1 | 0.9 | ||
| Zn site | 3 S | 2.34(4) | 3(3) | ||||||||
| Fe(JMJD2A) | 7122.5(2) | 13(1) | 5 | 3 N/O | 2.09(2) | 2(2) | −12(4) | 7.88 | 7.6 | ||
| +αΚG | 2 N/O | 1.93(4) | 4(4) | ||||||||
| (2Im) | |||||||||||
| 3 N/O | 2.05(2) | 1(1) | 0(4) | 4.03 | 5.0 | ||||||
| (2Im) | |||||||||||
| 1 O | [1.90(3)]d | 2(2) | |||||||||
| 1 O | [2.11(3)] | ||||||||||
| 1 C | [2.62(3)] | ||||||||||
| 1 C | [2.72(3)] | ||||||||||
| Fe(JMJD2A) | 7121.9(2) | 12(1) | 5 | 3 N/O | 1.98(2) | 2(2) | −8(2) | 11.4 | 4.0 | ||
| +αΚG + | 2 N/O | 2.13(3) | 6(3) | ||||||||
| substrate | (2Im) | ||||||||||
| 3 N/O | 2.00(1) | 2(1) | 1(2) | 7.89 | 2.2 | ||||||
| (2Im) | |||||||||||
| 1 O | [1.96(5)] | 4(2) | |||||||||
| 1 O | [2.17(5)] | ||||||||||
| 1 C | [2.68(5)] | ||||||||||
| 1 C | [2.78(5)] | ||||||||||
| Ni(JMJD2A) | 8344.1(2) | 3.7(7) | 6 | 6 N/O | 2.08(1) | 4(1) | 5(1) | 6.26 | 2.7 | ||
| (2Im) | |||||||||||
| Ni(JMJD2A) | 8344.1(2) | 3.6(6) | 6 | 4 N/O | 2.14(1) | 1(1) | −2(2) | 5.77 | 3.3 | ||
| +αΚG | 2 N/O | 1.98(1) | 1(1) | ||||||||
| (2Im) | |||||||||||
| 4 N/O | 2.08(2) | 3(2) | 5(3) | 3.55 | 3.1 | ||||||
| (2Im) | |||||||||||
| 1 O | [2.00(6)] | 5(5) | |||||||||
| 1 O | [2.05(6)] | ||||||||||
| 1 C | [2.67(6)] | ||||||||||
| 1 C | [2.69(6)] | ||||||||||
| Ni(JMJD2A) | 8344.1 (2) | 2.9(8) | 6 | 4 N/O | 2.14(1) | 1(1) | −1(1) | 2.74 | 3.4 | ||
| +αΚG + | 2 N/O | 1.99(1) | 1(1) | ||||||||
| substrate | (2Im) | ||||||||||
| 4 N/O | 2.09(2) | 3(1) | 4(2) | 1.59 | 3.0 | ||||||
| (2Im) | |||||||||||
| 1 O | [1.98(4)] | 4(3) | |||||||||
| 1 O | [2.03(4)] | ||||||||||
| 1 C | [2.65(4)] | ||||||||||
| 1 C | [2.67(4)] | ||||||||||
| Fe(JMJD2C) | 7122.9(2) | 11(1) | 5 | 3 N/O | 1.99(1) | 1(1) | −11(1) | 1.37 | 2.8 | ||
| 2 N/O | 2.15(1) | 2(1) | |||||||||
| (2Im) | |||||||||||
| Fe(JMJD2C) | 7122.5(2) | 12(1) | 5 | 3 N/O | 2.01(2) | 3(2) | −9(2) | 2.08 | 2.5 | ||
| +αΚG | 2 N/O | 2.17(4) | 9(7) | ||||||||
| (2Im) | |||||||||||
| 3 N/O | 2.05(2) | 4(1) | −2(4) | 1.28 | 1.5 | ||||||
| (2Im) | |||||||||||
| 1 O | [1.92(7)] | 6(4) | |||||||||
| 1 O | [2.13(7)] | ||||||||||
| 1 C | [2.64(7)] | ||||||||||
| 1 C | [2.74(7)] | ||||||||||
| Fe(JMJD2C) | 7122.1(2) | 10(2) | 5 | 3 N/O | 2.19(2) | 2(1) | −10(2) | 32.4 | 4.3 | ||
| +αΚG + | 2 N/O | 2.04(2) | 3(2) | ||||||||
| substrate | (2Im) | ||||||||||
| 3 N/O | 2.16(1) | 2(1) | −1(2) | 24.7 | 3.3 | ||||||
| (2Im) | |||||||||||
| 1 O | [2.01(2)] | 2(2) | |||||||||
| 1 O | [2.22(2)] | ||||||||||
| 1 C | [2.73(2)] | ||||||||||
| 1 C | [2.83(2)] | ||||||||||
| Ni(JMJD2C) | 8344.3(2) | 2.9(1) | 6 | 6 N/O | 2.09(1) | 5(1) | 6(1) | 7.88 | 3.6 | ||
| (2Im) | |||||||||||
| Ni(JMJD2C) | 8344.5(2) | 2.8(7) | 6 | 4 N/O | 2.13(2) | 4(4) | −3(2) | 10.5 | 4.6 | ||
| +αΚG | 2 N/O | 1.98(4) | 3(2) | ||||||||
| (2Im) | |||||||||||
| 4 N/O | 2.04(2) | 4(3) | 8(2) | 7.89 | 3.5 | ||||||
| (2Im) | |||||||||||
| 1 O | [2.09(3)] | 3(3) | |||||||||
| 1 O | [2.14(3)] | ||||||||||
| 1 C | [2.76(3)] | ||||||||||
| 1 C | [2.78(3)] | ||||||||||
| Ni(JMJD2C) | 83441(2) | 2.7(9) | 6 | 6 N/O | 2.08(1) | 3(1) | 6(2) | 7.78 | 4.8 | ||
| +αΚG + | (2Im) | ||||||||||
| substrate | |||||||||||
| 4 N/O | 2.07(1) | 1(1) | 3(3) | 5.85 | 2.9 | ||||||
| (2Im) | |||||||||||
| 1 O | [1.96(8)] | 10(9) | |||||||||
| 1 O | [2.01(8)] | ||||||||||
| 1 C | [2.63(8)] | ||||||||||
| 1 C | [2.65(8)] | ||||||||||
r (Å) is the radial distance between metal and ligand.
σ2 is the root mean square disorder in the metal-ligand distance.
R is the goodness of fit. Numbers in parentheses represent standard deviation for least square fits.
Distances in [ ] correspond to atoms in a O-C-C-O chelate ring and were constrained to vary with a single value of Δr for the chelate ring.
Figure 3.

Top: Fe K-Edge XANES spectra; Fe(JMJD2A) (brown), Fe(JMJD2A)-αKG (red) and Fe(JMJD2A)-αKG + substrate (orange). Bottom: Ni K-Edge XANES spectra; Ni(JMJD2A) (blue), Ni(JMJD2A)-αKG (purple) and Ni(JMJD2A)-αKG + substrate (green). Inserts show the expansions of the pre-edge XANES region showing peaks associated with 1s → 3d transitions.
Figure 5.

Zn K-edge XAS of resting JMJD2A (with iron in the active site): XANES spectrum (top) and EXAFS analysis (bottom, Left: Unfiltered, k3-weighted EXAFS spectra in red and fit in black, from Table 2. Right: Fourier-transformed EXAFS data and fit.
XANES data (Table 2, Figures 3 and 4) for the iron containing samples show a decrease in iron K-edge energy from Fe(JMJD2A/JMJD2C) → Fe(JMJD2A/JMJD2C)-αKG → Fe(JMJD2A/JMJD2C)-αKG + substrate. This indicates an increase in the electron density at the iron center that will eventually bind O2 in the presence of both αKG cofactor and substrate. No changes in the coordination number of the iron site are observed in any of the iron containing samples. In contrast, XANES data (Table 2, Figures 3 and 4) for nickel containing JMJD2A and JMJD2C samples do not show any systematic lowering of edge energy in the presence of αKG and/or the substrate, indicating that very little change in the electron density occurs at the nickel site in the presence of αKG and/or the substrate. Unlike the analogous iron containing samples, the 1s → 3d peak areas (2.7 – 3.7 ×10−2 eV) are indicative of six-coordinate nickel sites (typical values are 0.6 – 4 ×10−2 eV) (41) in all the nickel containing samples. This result is in agreement with the crystal structures of nickel containing JMJD2A obtained in the presence of αKG and substrate (24, 25), and suggests that the electronic interaction of Ni(II) with αKG is quite different from Fe(II). The result is also in agreement with the crystal structure of JMJD2C complexed with Ni(II) and N-OG.
Figure 4.

Top: Fe K-Edge XANES spectra; Fe(JMJD2C) (brown), Fe(JMJD2C)-αKG (red) and Fe(JMJD2C)-αKG + substrate (orange). Bottom: Ni K-Edge XANES spectra; Ni(JMJD2C) (blue), Ni(JMJD2C)-αKG (purple) and Ni(JMJD2C)-αKG + substrate (green). Inserts show the expansions of the pre-edge XANES region showing peaks associated with 1s → 3d transitions.
Zn(II) does not give rise to peaks associated with 1s → 3d electronic transitions in the pre-edge XANES region of the K-edge spectra. However, the shape and intensity of the absorption edge can be used to obtain the structural information of the zinc site in resting JMJD2A (with iron in the active site, Figure 5). The intensity of the zinc K-edge XANES spectrum of the resting enzyme indicates the presence of a four-coordinate zinc site (48). The presence of two XANES peaks at 9664 eV and 9669 eV indicates the presence of a zinc site with a mixed environment of N/O and S ligands (48).
EXAFS analysis
EXAFS analysis provides information regarding the number (N) and the type of ligands bound to a metal and metric details of the metal site structure. The information obtained from the best fits of the data is summarized in Table 2, and Figures 5 – 7. For all the histone demethylase samples, the determined N values are in agreement with the XANES analysis (vide supra). The best fit for both Fe(JMJD2A) and Fe(JMJD2C) EXAFS data consist of five N/O donors, of which two are imidazole ligands. The remaining three ligands are consistent with coordination by the active site Glu ligand and two aqua ligands, consistent with the crystal structure of the catalytic domain of JMJD2A (22). Multiple-scattering analyses that include a five-membered O-C-C-O chelate ring do not support inclusion of ascorbate binding to iron center in any of the resting enzymes, as was seen for ABH2 (35) and 1-aminocyclopropane-1-carboxylate oxidase (ACCO) (49, 50). Upon reconstitution with Ni(II), the best fit for both JMJD2A and JMJD2C features six N/O-donors, of which two are His imidazole ligands. Multiple-scattering analyses involving O-C-C-O chelate rings do not show ascorbate binding to nickel center in either JMJD2A or JMJD2C. Thus, the remaining four ligands are consistent with the expected coordination of the active site Glu ligand and the three aqua ligands.
Figure 7.

EXAFS analysis for JMJD2C. Left: Unfiltered, k3-weighted EXAFS spectra (colored lines) and fits (black lines, from Table 2). Right: Fourier-transformed EXAFS data and fits.
The zinc K-edge EXAFS analysis of the resting JMJD2A (with iron in the active site) is shown in Table 2 and Figure 5. In agreement with expectations based on the XANES analysis (vide supra) and the amino acid sequence, the best fit for zinc K-edge EXAFS data of the resting JMJD2A consists of a four-coordinate tetrahedral zinc site with the ligation from one N/O-donor ligand (from imidazole) at 2.08 Å and three S-donor ligands (from cysteine) at 2.34 Å. The observed Zn-S distances are typical of tetrahedral zinc sites (2.33 Å) (51). The FT XAS spectrum for the resting JMJD2A shows outer peaks at 2.8 Å, 3.4 Å and 3.8 Å. These are similar to the peaks observed for a Zn(Cys)3His peptide complex (51), and are consistent with expectations from amino acid sequence homologies, which feature a C(X)5H(X)65CXC motif, and consistent with the crystal structure of the catalytic domain of JMJD2A (22) and JMJD2D (27).
Addition of αKG to either Fe(JMJD2A) or Fe(JMJD2C) does not cause any net change in the in the number of ligands in first-coordination sphere, as it still contains five N/O-donors including two imidazole ligands (Table 2, Figure 6 and Figure 7). This is in agreement with the crystal structure of Fe(JMJD2A) in the presence of αKG (22). However, changes in the second-coordination sphere are apparent (Figures 6 and 7). Multiple-scattering analyses involving O-C-C-O five-membered chelate rings were performed to determine αKG binding. These fits use a single distance parameter and a single value of σ2 for all the atoms in the chelate ring. Therefore, these fits do not contain more adjustable parameters than the corresponding fit without the chelate ring and are thus directly comparable. The results show that in contrast to the data for resting enzymes, both R and χ2 are improved by the replacement of two first coordination shell N/O donors by one O-C-C-O unit (Table 2). Using the chelate model, two second-coordination sphere carbon atoms at 2.62 Å and 2.72 Å for JMJD2A and 2.64 Å and 2.74 Å for JMJD2C are found. The presence of two second-coordination sphere carbon atoms is consistent with bidentate αKG binding to the iron center. While it is true that the EXAFS arising from second sphere carbon atoms is sometimes suppressed by destructive interference from other scattering atoms (52 – 54), this is not the case for the samples studied here. In the present case, the amplitude ratio for the αKG second sphere carbon atoms calculated by FEFF 8 (∼ 45, considering M-O to be ∼100) are comparable to that of second sphere carbon atoms in imidazole (∼ 40, considering M-N to be 100). This is consistent with the observation of second sphere carbon atoms using EXAFS in other samples (52 – 56). The decrease in the iron K-edge energy in the presence of αKG further supports the binding of αKG to the metal center. [It is important to note that the model obtained from the PHF8 crystal structure features two metal-carbon distances that are below the resolution of the EXAFS data (Table 2, Figure 6 and Figure 7)]. The similar metal-carbon distances, coupled with the errors (2.62(3) Å and 2.72(3) Å for JMJD2A and 2.64(7) Å and 2.74(7) Å for JMJD2C) indicate that the difference in the distances to the carbon atoms is not resolved in the EXAFS of JMJD2A or JMJD2C.
Figure 6.

EXAFS analysis for JMJD2A. Left: Unfiltered, k3-weighted EXAFS spectra (colored lines) and fits (black lines, from Table 2). Right: Fourier-transformed EXAFS data and fits.
Addition of αKG to either Ni(JMJD2A) and Ni(JMJD2C) does not change the first-coordination sphere, as the nickel site still consists of six N/O-donors, of which two are imidazole ligands. This is consistent with the crystal structure of Ni(JMJD2A) with N-OG (24) and Ni(JMJD2C) with N-OG, which shows the presence of a six-coordinate nickel site. Using the chelate model outlined above, two second-sphere carbon atoms at 2.67 Å and 2.69 Å for Ni(JMJD2A)-αKG and 2.76 Å and 2.78 Å for Ni(JMJD2C)-αKG are observed. The presence of the second coordination sphere carbon atoms indicates the bidentate binding of αKG to nickel center.
Addition of substrate to the αKG complexes of the iron- and nickel-containing histone demethylases does not change the structure of the metal sites significantly (Table 2, Figures 6 and 7). The iron site remains five-coordinate with five N/O-donor ligands, of which two are imidazoles. The O-C-C-O five membered chelate rings observed for the αKG complexes are retained in the presence of substrate. Using the chelate model for αKG binding, two second sphere carbon atoms at 2.68 Å and 2.78 Å for Fe(JMJD2A) and 2.73 Å and 2.83 Å for Fe(JMJD2C) are observed. The presence of the second sphere carbon atoms confirms that bidentate binding of αKG to the iron center is retained in the presence of substrate. The nickel site remains six-coordinate with six N/O donor ligands, of which two are imidazoles. This is in agreement with the crystal structure of JMJD2A in the ternary complex formed with a histone H3K9Me3 peptide and αKG (25). The O-C-C-O five membered chelate rings are also retained in the nickel containing samples in the presence of substrate as well. Using the chelate ring model, two second-sphere carbon atoms at 2.65 Å and 2.67 Å for Ni(JMJD2A) and 2.63Å and 2.65Å for Ni(JMJD2C) are observed. The presence of the second-coordination sphere carbon atoms indicates the bidentate binding of αKG to the nickel center is retained in the presence of substrate.
Discussion
Histone tails undergo a number of posttranslational modifications including acetylation, methylation, phosphorylation and ubiquitination (57). Histone methylation occurs on both lysine and arginine residues. Several lysine residues of histone H3 and H4 (H3K4, H3K9, H3K27, H3K36, H3K79 and H4K20) undergo methylation (58, 59). Histone methylation can contribute to several biological processes including heterochromatin formation, X-inactivation, genome imprinting and silencing of homeotic genes (60 – 62). Histone methylation was considered as a permanent modification for a long time (63, 64). However, the identification of the H3K4 specific lysine demethylase (LSD1) suggested that histone methylation is reversible (65). Histone demethylase JHDM1 (jumonji domain containing histone demethylase 1), which specifically demethylates H3K36Me2, also indicated that histone methylation is reversible (66). Both LSD1 and JHDM1 demethylate only mono- and di-methylated histones (65, 66). The inability of LSD1 and JHDM1 to demethylate trimethylated histone Lys residues raised the possibility of unknown histone demethylases able to demethylate trimethylated histones.
The JMJD2 family of histone demethylases consists of six members – the long group (JMJD2A, JMJD2B and JMJD2C) and the short group (JMJD2D, JMJD2E and JMJD2F) (67). Short group enzymes contain only JmjN and JmjC domains, whereas the long group enzymes contain two PHD domains and two Tudor domains in addition to the JmjN and JmjC domains (16). JMJD2A-C catalyze demethylation of di- and tri-methylated forms of H3K9 and di- and tri-methylated forms of H3K36 residues (Figure 1), but not mono-, di- or tri-methylated H3K4, H3K27 and H4K20 sites, while JMJD2D and JMJD2E demethylate di- and tri- methylated forms of only H3K9 (68). Sequence alignment of the JMJD2 family members shows a high degree of similarity (55.7 % identity and 81.71 % similarity) within the first 350 amino acids (22). JMJD2A and JMJD2C are the most studied members of this family (68). Studies on some of the truncated enzymes showed that they catalyze the demethylation reactions with the same site specificity as the full-length enzyme (16). Truncated enzymes were used in this study because of difficulties in the heterologous expression of the full-length enzymes in E. coli. Using JMJD2A and JMJD2C as examples, we show that the JMJD2 family of histone demethylases is sensitive to inhibition by Ni(II) ions (Figure 2).
A healthy unexposed adult has about 7.3 µg of Ni/kg body weight (69). The unexposed population in the United States has 2.6 µg/l (0.0443 µM) nickel in blood serum (70). Endocytosis of a 1.5 or 4.0 µm crystalline NiS particle can result in a intracellular Ni(II) concentration of 0.25 or 4.75 M respectively (69). Results obtained in this work indicate that JMJD2 proteins will not be inhibited by normal nickel exposures, but would be inhibited by nickel concentrations resulting from endocytosis of particulate nickel compounds. Inhibition of histone demethylases by Ni(II) ions changes global as well as gene specific levels of histone methylation (13). This would lead to alteration of gene expression via epigenetic mechanisms, which have been previously implicated in nickel-induced carcinogenesis. In this work, XAS was performed to examine the structures of Fe(II)- and Ni(II)-JMJD2A/2C complexes to obtain mechanistic details of histone demethylation by JMJD2 family of enzymes and their inhibition by Ni(II) ions.
Structures of the early intermediates in the JMJD2A and JMJD2C reaction cycles by XAS
The structural information obtained from XAS analysis (Table 3) is in general agreement with the expectations from the available crystal structures of the catalytic core domain of JMJD2 proteins (Table 1). Resting Fe(JMJD2A) and Fe(JMJD2C) show the presence of a five-coordinate iron site involving two imidazole ligands and three other N/O-donor ligands (Table 3), which are likely Glu (190 for JMJD2A and 192 for JMJD2C) and two water molecules observed in the crystal structures of JMJD2A (22). The two very long Fe-O distances involving two aqua ligands at 2.49 Å and 2.67 Å observed in the crystal structure of JMJD2A are not observed in XAS analysis (22). These results are consistent with the active site structures of other non-heme iron dioxygenases, which show ligation of the iron center by a 2-His-1- carboxylate facial triad and water molecules (71). XAS results obtained for resting JMJD2A (with iron in the active site) also show the presence of a four-coordinate zinc site involving one imidazole and three other sulfur donor ligands (Table 3). This is consistent with the Cys3HisZn site in the crystal structure of the catalytic domain of JMJD2A (22) and JMJD2D (27), and confirms the presence of the expected zinc site in the reconstituted samples used for XAS.
Table 3.
Results obtained from XAS analysis of JMJD2A and JMJD2C samples.
| Protein | Metal ion | Cofactor | Substrate | Active site |
|---|---|---|---|---|
| JMJD2A | Fe/Zn | No | No | 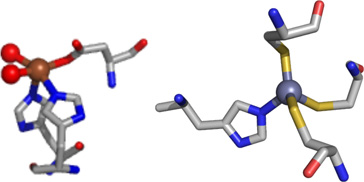 |
| JMJD2A | Fe/Zn | αKG | No | 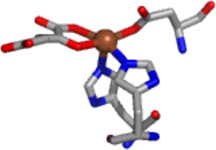 |
| JMJD2A | Fe/Zn | αKG | Yes | 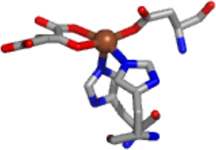 |
| JMJD2A | Ni/Zn | No | No | 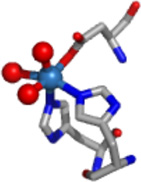 |
| JMJD2A | Ni/Zn | αKG | No |  |
| JMJD2A | Ni/Zn | αKG | Yes |  |
| JMJD2C | Fe/Zn | No | No |  |
| JMJD2C | Fe/Zn | αKG | No |  |
| JMJD2C | Fe/Zn | αKG | Yes | 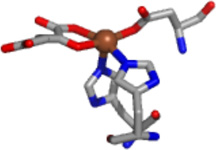 |
| JMJD2C | Ni/Zn | No | No |  |
| JMJD2C | Ni/Zn | αKG | No | 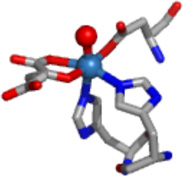 |
| JMJD2C | Ni/Zn | αKG | Yes | 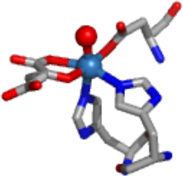 |
Crystal structures of the JMJD2 proteins in the presence of αKG or its analogue (Table 1) show that the cofactor binds to the metal center in a bidentate manner, forming a five-membered chelate ring. The EXAFS analysis of the samples of Fe(JMJD2A) and Fe(JMJD2C) prepared in the presence of αKG are also consistent with the bidentate coordination of αKG to the iron center. XAS results obtained for both Fe(JMJD2A) and Fe(JMJD2C) in the presence of αKG show the presence of a five-coordinate iron site involving two imidazole ligands, a bidentate αKG and one other N/O donor ligand, presumably the active site Glu residue (Table 3) observed in the crystal structure of JMJD2A in the presence of αKG (22). The distances obtained for the second sphere carbon atoms of αKG (2.62 Å and 2.72 Å for JMJD2A and 2.64 Å and 2.74 Å for JMJD2C) using XAS are much shorter than those observed in the crystal structure (3.26 Å and 3.29 Å) of Fe(JMJD2A) in the presence of αKG (22). However, similar M-C distances (obtained using XAS analysis) have been observed in human ABH2 complexed with αKG by XAS (35), the crystal structure of ABH3 complexed with αKG (2.82 – 2.84 Å) (72), the crystal structure of human FIH in complex with αKG (2.66 – 2.88 Å) (73), in the crystal structure of C. Elegans lysine specific demethylase 7 A (ceKDM7A) complexed with αKG (2.69 – 2.73 Å) (74), the crystal structure of human PHD2 complexed with αKG (2.77 – 2.87 Å) (75) and the crystal structure of human JMJD3 complexed with αKG (2.86 – 2.76 Å) (76). The shorter distances observed in XAS could be due to the fact that XAS data were collected at 4 K and therefore, one will expect slightly shorter distances. It is important to note that some of these crystal structures show considerable flexibility of the αKG chelate ring because of the shorter M-C distances than those required for symmetrical coordination of a bidentate αKG. In FIH and ceKDM7A, the five-membered O-C-C-O chelate ring undergoes a rotation with respect to the metal ion’s equatorial plane (M-O-C-C dihedral angle ∼ 19°). On the other hand, in ABH3, JMJD3 and PHD2 the rotation of the five-membered chelate ring with respect to the metal ion’s equatorial plane is much smaller (M-O-C-C dihedral angle ∼ 10°). Thus, the M-C distances obtained from XAS analysis for JMJD2A and JMJD2C reflect the range of these conformations. In the presence of both αKG and substrate, both JMJD2A and JMJD2C retain the five-coordinate iron site and the second sphere carbon atoms of αKG are observed at 2.68 Å and 2.78 Å for Fe(JMJD2A) and 2.73 Å and 2.83 Å for Fe(JMJD2C). These results are in agreement with the crystal structure of AlkB in complex with Fe(II) in the presence of both αKG and methylated trinucleotide (2.87 – 2.93 Å) (77), the crystal structure of E. coli. TauD complexed with Fe(II) in the presence of both αKG and substrate (2.57 – 2.85 Å) (78, 79) and in the crystal structure of FIH complexed with both αKG and substrate (2.64 – 2.70 Å) (80, 81).
The results obtained from XAS analysis of the Ni(II)-substituted JMJD2A and JMJD2C are in agreement with the crystal structure of JMJD2D (27). These structures show a six-coordinate nickel site involving two imidazole ligands and four other N/O donor ligands, which are identified in the crystal structure as Glu of the 2-His-1-carboxylate motif and water molecules. The EXAFS analysis of the samples of Ni(JMJD2A) and Ni(JMJD2C) prepared in the presence of αKG indicate the coordination of αKG to the nickel center in a bidentate manner (the second sphere carbon atoms of αKG are observed at 2.67 Å and 2.69 Å for JMJD2A and 2.76 Å and 2.78 Å for JMJD2C). In the presence of αKG, both Ni(JMJD2A) and Ni(JMJD2C) have six-coordinate nickel sites involving two imidazole ligands, a bidentate αKG and two other N/O donor ligands, which are likely active site Glu and a water molecule observed in the crystal structure of Ni(JMJD2C) in the presence of N-OG. These results are in agreement with the crystal structure of Ni(JMJD2A) with N-OG (2.85 – 2.90 Å) (24), the crystal structure of the KIAA1718 (a human jumonji demethylase) with N-OG (2.59 – 2.62 Å) (82), the crystal structure of human PHF2 with N-OG (2.82 – 2.86 Å) (83). In the presence of both αKG and substrate, both Ni(JMJD2A) and Ni(JMJD2C) retain the six-coordinate nickel site and the second coordination sphere carbon atoms of αKG are observed at 2.65 Å and 2.67 Å for Ni(JMJD2A) and 2.63Å and 2.65Å for Ni(JMJD2C). These results are in agreement with the crystal structure of human γ -butyrobetaine 2-oxoglutarate dioxygenase 1 (2.71 Å – 2.72 Å, PDB: 3MS5), the crystal structure of Ni(JMJD2A) with αKG and substrate (2.81 Å – 2.82 Å) (25) and crystal structure of a H3K9 jumonji demethylase (2.80 – 2.85 Å) (84).
Comparison between XAS results of DNA and histone demethylases
XAS was previously carried out for AlkB (85) (E. coli. DNA demethylase) and ABH2 (35) (human DNA demethylase). Both the DNA demethylases and the histone demethylases are Fe(II)- and αKG-dependent mononuclear non-heme iron enzymes. JMJD2A and JMJD2C have a five-coordinate iron site in the resting state, similar to the results obtained for Fe(AlkB) and Fe(ABH2). The maintenance of a five-coordinate iron site in the binary and ternary complex is observed for ABH2 and JMJD2 proteins. In both cases the iron K-edge energy decreases from the resting enzyme to the αKG complex, and again to the αKG-substrate complex. This is expected for an iron site that will eventually bind and reductively activate O2. Remarkably, the iron K-edge energy (7122.5 eV) of the αKG complex is same for ABH2 and JMJD2 proteins. This is due to the very similar active site (2-His-1-carboxylate and αKG) for ABH2 and JMJD2 proteins. However, the iron K-edge energy for the resting ABH2 and JMJD2 proteins are different (7125.9(2) eV for ABH2, vs. 7123.1(2) eV for JMJD2A and 7122.9(2) eV for JMJD2C). This could be attributed to the ascorbate binding shown for ABH2 (35). Also, the iron K-edge energies for the ABH2 and JMJD2 proteins are different (7121.5(2) eV for ABH2 vs. 7121.9(2) eV for JMJD2A and 7122.1(2) eV for JMJD2C) in the presence of the substrate. This could be due to differences in the nature of the substrate (DNA vs. histone protein) and also in active site environment for ABH2 and JMJD2 proteins. The Ni-complexes of ABH2 are five-(resting state and in the presence of αKG) and six-coordinate (in the presence of both αKG and substrate). All of the Ni-complexes are six-coordinate for JMJD2 proteins. The Ni K-edge energies for ABH2 do not show any systematic shift, and they are within experimental error for JMJD2 proteins. These results indicate very little change in electron density at the nickel center in the presence or absence of αKG or substrate for both ABH2 and JMJD2 proteins.
Mechanistic implications from the XAS results of iron-containing and nickel-substituted JMJD2 proteins
Characterization of a number non-heme Fe(II) oxygenases have revealed that they function by a sequential reaction mechanism (Scheme 1) (86 – 89). In the first step of the consensus mechanism, αKG displaces two aqua ligands (Scheme 1, B) from the six-coordinate metal site composed of the 2-His-1-carboxylate facial triad and three aqua ligands (Scheme 1, A). In the next step, an empty coordination site for O2 binding will open up due to the loss of the remaining aqua ligand in the presence of both αKG and substrate (Scheme 1, C). Subsequently, oxidative decarboxylation of αKG takes place, leading to the formation of an Fe(IV)=O species (90). In the case of the JMJD2 proteins, the Fe(IV)=O species will hydroxylate the methyl group on the N-atom of lysine in the histone tail, resulting into the formation of an alcohol, which then forms formaldehyde and a Lys with one less methyl group on the N-atom in the histone tail (Figure 1).
Scheme 1.

Consensus mechanism of non-heme Fe(II) dioxygenases (adapted from ref.
The XAS studies of the structures of early intermediates (prior to O2 binding) in the JMJD2A and JMJD2C reaction cycles show that they appear to operate by a variation of the consensus mechanism. XANES and EXAFS analysis are consistent with a five-coordinate Fe(II) site composed of N/O-donor ligands in both JMJD2A and JMJD2C in the resting state (Table 3). Analysis of features from scattering atoms in the second- and third-coordination spheres indicate that two of these ligands are histidine imidazoles, consistent with the available crystallographic data where the remaining ligands are presumably derived from Glu and water. The data from the complexes formed with αKG are consistent with the cofactor binding to Fe(II) in a bidentate manner in both JMJD2A and JMJD2C, while retaining the five-coordinate geometries of the complexes (Table 3). Thus, binding of αKG does not change the coordination number of the metal center, but perturbs the electronic environment of the metal center by replacing two aqua ligands with anionic O-donors. This is similar to the results obtained for Cu- or Fe-TfdA (44). Upon binding of the substrate, the iron site remains five-coordinate (Table 3) in both JMJD2A and JMJD2C. The formation of a five-coordinate iron site in the presence of both substrate and αKG has been observed in the consensus mechanism of other αKG-dependent enzymes (86 – 89). However, the iron K-edge energy decreases in the presence of substrate. This indicates a change in the electronic environment at the metal center in the presence of substrate. The presence of a five-coordinate iron site in the resting enzyme (Table 3) and its maintenance through ternary complex formation is not unprecedented in DNA or histone demethylases. The crystal structure of JMJD2A in the presence and absence of αKG reveals a five-coordinate iron site (22). The crystal structure of KIAA1718 in the presence of αKG also has a five-coordinate iron site (82).
JMJD2A and JMJD2C demethylate the same histone tail Lys residues (H3K9 and K3K36) (67). This similarity is consistent with their high sequence homology. They are also similar in their methylation state selectivity as they demethylate both dimethylated and trimethylated Lys residues (68). It has been suggested that methylation state selectivity will depend on the orientation of the substrate methyl groups towards a Fe(IV)=O intermediate in the enzyme-substrate complex (25). The XAS results presented here show that the resting enzyme, enzyme-αKG complex and enzyme-αKG + substrate complex are similar for both JMJD2A and JMJD2C (Table 3). Thus, these results are consistent with the similar activity of JMJD2A and JMJD2C. Substitution of iron by other metals in JMJD2 proteins would be expected to yield an enzyme with altered function because of the differences in the ability of the metal site to bind and decarboxylate αKG, and/or bind and activate O2. XANES and EXAFS analysis are consistent with a six-coordinate site composed of N/O-donor ligands for nickel in both JMJD2A and JMJD2C in the presence and absence of cofactor and/or substrate (Table 3). Analysis of features from scattering atoms in the second and third coordination shells indicate that two of these ligands are histidine imidazoles, consistent with the available crystallographic data and the remaining ligands are presumably derived from Glu and water (and/or αKG) (24, 25).
One possible mechanism of nickel induced inhibition of JMJD2 proteins would be to use protein conformation to block O2 access to the iron site as observed in AlkB, E. coli DNA demethylase that catalyzes demethylation of methylated DNA bases (91, 92). Like human JMJD2 proteins, AlkB is a mononuclear non-heme Fe(II) enzyme that utilizes αKG and O2 to carry out the oxidative demethylation of its substrate. Both of these enzymes also utilize the 2-His-1-carboxylate motif to bind Fe(II). There are several crystal structures of AlkB reported in the literature where the Fe(II) in the catalytic center has been replaced by Mn(II), Co(II) or Ni(II) in presence of both αKG and substrate (77, 93 – 95). All of the metal-substituted AlkB enzymes are catalytically inactive. Comparison of crystal structures of AlkB containing Fe(II), Co(II) or Mn(II) in presence of both αKG and substrate shows that the positioning of Trp178 and Leu184 is different in case of Mn(II) or Co(II) than that in case of Fe(II) (77, 95). In the case of Mn(II) and Co(II), Trp178 blocks the O2 binding site and Leu184 blocks the O2 diffusion tunnel. The positioning of these residues can also place O2 in a nonreactive mode (e.g., decreased O2 binding affinity) in a manner analogous to the distal H58Y mutation in hemoglobin (83).
There are several possible mechanisms of inhibition of JMJD2 proteins involving Ni(II). Among these possibilities are first, that Ni(II) in a ligand environment consisting of the 2-His-1-caboxylate motif and an αKG ligand would not be expected to possess a redox potential capable of reductively activating O2 (96 – 98). Consistent with this is the observation that the electron density build-up that occurs on the Fe(II) center upon binding αKG and then substrate is not observed for Ni(II). Second, the ternary complex formed between Ni(II)-JMJD2 proteins, αKG and substrate contains a six-coordinate nickel site (Table 3) that has no open coordination site for O2 binding. This six-coordinate Ni(II) complex is indistinguishable from the Ni(II) center in the crystal structure of JMJD2A in the presence of αKG and H3K9Me3 (24), JMJD2C in the presence of N-OG and Mn(II) center in the crystal structure of ABH2 in the presence of αKG and dsDNA (93).
In summary, using JMJD2A and JMJD2C as examples we show that JMJD2 family of histone demethylases are inhibited by Ni(II) ions. Using XAS, we show that the inhibition of these enzymes by nickel is due to the presence of nickel in the active site instead of iron. Also, the presence of nickel in the active site does not prevent the binding of αKG cofactor. This inhibition of JMJD2A and JMJD2C by Ni(II) appears to involve both electronic and steric factors.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Yang Shi for providing pGEX-4T-1-GST-JMJD2A and pET28a–His6-JMJD2C constructs. The Stanford Synchrotron Radiation Laboratory (SSRL), is a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural and Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program.
FUNDING
This work was supported by NIH grant ES005512-P30ES000260 (MC).
ABBREVIATIONS
- JMJD2
Jumonji domain containing histone demethylase 2
- αKG
2-Oxopentanedioic acid
- BSA
Bovine Serum Albumin
- DNAse
Deoxyribonuclease
- EDTA
Ethylenediaminetetraacetic acid
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- ICP-OES
Inductively Coupled Plasma Optical Emission Spectroscopy
- N-OG
N-oxalylglycine
- PMSF
Phenylmethylsulfonyl fluoride
- TCEP
Tris(2-carboxyethyl)phosphine
- EXAFS
Extended X-Ray Absorption Fine Structure
- XANES
X-Ray Absorption Near Edge Structure
- XAS
X-Ray Absorption Spectroscopy
Footnotes
Supporting information contains figures of enzyme activity assays of JMJD2A and JMJD2C and tables of EXAFS fits to iron, nickel and zinc K-edge data used to select the best-fit models. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Doll R, Morgan LG. Cancers of Lung and Nasal Sinuses in Nickel Workers. Brit. J. Cancer. 1970;24:623–632. doi: 10.1038/bjc.1970.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doll R, Mathews JD, Morgan LG. Cancers of Lung and Nasal Sinuses in Nickel Workers - Reassessment of Period of Risk. Brit. J. Ind. Med. 1977;34:102–105. doi: 10.1136/oem.34.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sunderman FW. Carcinogenicity of Nickel Compounds in Animals. IARC Sci. Publ. 1984;53:127–142. [PubMed] [Google Scholar]
- 4.Oller AR, Costa M, Oberdorster G. Carcinogenicity assessment of selected nickel compounds. Toxicol. Appl. Pharmacol. 1997;143:152–166. doi: 10.1006/taap.1996.8075. [DOI] [PubMed] [Google Scholar]
- 5.Grimsrud TK, Berge SR, Haldorsen T, Andersen A. Exposure to different forms of nickel and risk of lung cancer. Am. J. Epidemiol. 2002;156:1123–1132. doi: 10.1093/aje/kwf165. [DOI] [PubMed] [Google Scholar]
- 6.Rojas E, Herrera LA, Poirier LA, Ostrosky-Wegman P. Are metals dietary carcinogens? Mutat. Res.-Gen. Tox. En. 1999;443:157–181. doi: 10.1016/s1383-5742(99)00018-6. [DOI] [PubMed] [Google Scholar]
- 7.IARC. Chromium, Nickel and Welding. Lyon, France: World Health Organization; 1990. Monograph on the Evaluation of Carcinogenic Risks to Humans. [PMC free article] [PubMed] [Google Scholar]
- 8.IARC. Some Drinking-water Disinfectants and Contaminants, including Arsenic. Lyon, France: World Health Organization; 2004. Monographs on the Evaluation of Carcinogenic Risk to Humans. [PMC free article] [PubMed] [Google Scholar]
- 9.Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M. Carcinogenic Nickel Silences Gene-Expression by Chromatin Condensation and DNA Methylation - a New Model for Epigenetic Carcinogens. Mol. Cell. Biol. 1995;15:2547–2557. doi: 10.1128/mcb.15.5.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossman TG. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat. Res.-Fund. Mol. M. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Kargacin B, Klein CB, Costa M. Mutagenic Responses of Nickel Oxides and Nickel Sulfides in Chinese-Hamster V79 Cell-Lines at the Xanthine Guanine Phosphoribosyl Transferase Locus. Mutat. Res. 1993;300:63–72. doi: 10.1016/0165-1218(93)90141-y. [DOI] [PubMed] [Google Scholar]
- 12.Chen H, Costa M. Iron- and 2-oxoglutarate-dependent Dioxygenases: an emerging group of molecular targets for nickel toxicity and carcinogenicity. Biometals. 2009;22:191–196. doi: 10.1007/s10534-008-9190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen HB, Ke QD, Kluz T, Yan Y, Costa M. Nickel ions increase histone H3 lysine 9 dimethylation and induce transgene silencing. Mol. Cell. Biol. 2006;26:3728–3737. doi: 10.1128/MCB.26.10.3728-3737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen HB, Giri NC, Zhang RH, Yamane K, Zhang Y, Maroney M, Costa M. Nickel Ions Inhibit Histone Demethylase JMJD1A and DNA Repair Enzyme ABH2 by Replacing the Ferrous Iron in the Catalytic Centers. J. Biol. Chem. 2010;285:7374–7383. doi: 10.1074/jbc.M109.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chervona Y, Arita A, Costa M. Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics. 2012;4:619–627. doi: 10.1039/c2mt20033c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen ZZ, Spooner E, Li E, Zhang GY, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 17.Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S, Suzuki T. Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem. Biophys. Res. Commun. 2009;389:366–371. doi: 10.1016/j.bbrc.2009.08.155. [DOI] [PubMed] [Google Scholar]
- 18.Cloos PAC, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–1140. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gray SG, Iglesias AH, Lizcano F, Villanueva R, Camelo S, Jingu H, Teh BT, Koibuchi N, Chin WW, Kokkotou E, Dangond F. Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J. Biol. Chem. 2005;280:28507–28518. doi: 10.1074/jbc.M413687200. [DOI] [PubMed] [Google Scholar]
- 20.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- 21.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 22.Chen ZZ, Zang JY, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao QL, Pan CH, Dai SD, Hagman J, Hansen K, Shi Y, Zhang GY. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 23.Chen ZZ, Zang JY, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang CY, Whetstine J, Dai S, Hansen K, Shi Y, Zhang G. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat. Struct. Mol. Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 25.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BMR, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- 26.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IKH, Li XS, Woon ECY, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TDW, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. Embo Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krishnan S, Trievel RC. Structural and Functional Analysis of JMJD2D Reveals Molecular Basis for Site-Specific Demethylation among JMJD2 Demethylases. Structure. 2013;21:98–108. doi: 10.1016/j.str.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 28.Rose NR, Woon ECY, Tumber A, Walport LJ, Chowdhury R, Li XS, King ONF, Lejeune C, Ng SS, Krojer T, Chan MC, Rydzik AM, Hopkinson RJ, Che KH, Daniel M, Strain-Damerell C, Gileadi C, Kochan G, Leung IKH, Dunford J, Yeoh KK, Ratcliffe PJ, Burgess-Brown N, von Delft F, Muller S, Marsden B, Brennan PE, McDonough MA, Oppermann U, Klose RJ, Schofield CJ, Kawamura A. Plant Growth Regulator Daminozide Is a Selective Inhibitor of Human KDM2/7 Histone Demethylases. J. Med. Chem. 2012;55:6639–6643. doi: 10.1021/jm300677j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rose NR, Woon ECY, Kingham GL, King ONF, Mecinovic J, Clifton IJ, Ng SS, Talib-Hardy J, Oppermann U, McDonough MA, Schofield CJ. Selective Inhibitors of the JMJD2 Histone Demethylases: Combined Nondenaturing Mass Spectrometric Screening and Crystallographic Approaches. J. Med. Chem. 2010;53:1810–1818. doi: 10.1021/jm901680b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang K-H, King ONF, Tumber A, Woon ECY, Heightman TD, McDonough MA, Schofield CJ, Rose NR. Inhibition of Histone Demethylases by 4-Carboxy-2,2 ‘-Bipyridyl Compounds. ChemMedChem. 2011;6:759–764. doi: 10.1002/cmdc.201100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King ONF, Li XS, Sakurai M, Kawamura A, Rose NR, Ng SS, Quinn AM, Rai G, Mott BT, Beswick P, Klose RJ, Oppermann U, Jadhav A, Heightman TD, Maloney DJ, Schofield CJ, Simeonov A. Quantitative High-Throughput Screening Identifies 8-Hydroxyquinolines as Cell-Active Histone Demethylase Inhibitors. PLoS One. 2010;5:e15535. doi: 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woon ECY, Tumber A, Kawamura A, Hillringhaus L, Ge W, Rose NR, Ma JHY, Chan MC, Walport LJ, Che KH, Ng SS, Marsden BD, Oppermann U, McDonough MA, Schofield CJ. Linking of 2-Oxoglutarate and Substrate Binding Sites Enables Potent and Highly Selective Inhibition of JmjC Histone Demethylases. Angew. Chem. Int. Edit. 2012;51:1631–1634. doi: 10.1002/anie.201107833. [DOI] [PubMed] [Google Scholar]
- 33.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of Protein Using Bicinchoninic Acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 34.Higgins KA, Hu HDQ, Chivers PT, Maroney MJ. Effects of Select Histidine to Cysteine Mutations on Transcriptional Regulation by Escherichia coli RcnR. Biochemistry. 2013;52:84–97. doi: 10.1021/bi300886q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giri NC, Sun H, Chen HB, Costa M, Maroney MJ. X-ray Absorption Spectroscopy Structural investigation of Early Intermediates in the Mechanism of DNA Repair by Human ABH2. Biochemistry. 2011;50:5067–5076. doi: 10.1021/bi101668x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Padden KM, Krebs JF, MacBeth CE, Scarrow RC, Borovik AS. Immobilized metal complexes in porous organic hosts: Development of a material for the selective and reversible binding of nitric oxide. J. Am. Chem. Soc. 2001;123:1072–1079. doi: 10.1021/ja003282b. [DOI] [PubMed] [Google Scholar]
- 37.Webb SM. SIXpack: a graphical user interface for XAS analysis using IFEFFIT. Phys. Scr. T115. 2005:1011–1014. [Google Scholar]
- 38.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B. 1998;58:7565–7576. [Google Scholar]
- 39.Newville M. EXAFS analysis using FEFF and FEFFIT. J. Synchrotron Radiat. 2001;8:96–100. doi: 10.1107/s0909049500016290. [DOI] [PubMed] [Google Scholar]
- 40.Zabinsky SI, Rehr JJ, Ankudinov A, Albers RC, Eller MJ. Multiple-Scattering Calculations of X-Ray-Absorption Spectra. Phys. Rev. B. 1995;52:2995–3009. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- 41.Colpas GJ, Maroney MJ, Bagyinka C, Kumar M, Willis WS, Suib SL, Baidya N, Mascharak PK. X-Ray Spectroscopic Studies of Nickel-Complexes, with Application to the Structure of Nickel Sites in Hydrogenases. Inorg. Chem. 1991;30:920–928. [Google Scholar]
- 42.Randall CR, Shu LJ, Chiou YM, Hagen KS, Ito M, Kitajima N, Lachicotte RJ, Zang Y, Que L. X-Ray-Absorption Pre-Edge Studies of High-Spin Iron(II) Complexes. Inorg. Chem. 1995;34:1036–1039. [Google Scholar]
- 43.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. A multiplet analysis of Fe K-edge 1s->3d pre-edge features of iron complexes. J. Am. Chem. Soc. 1997;119:6297–6314. [Google Scholar]
- 44.Cosper NJ, Stalhandske CMV, Saari RE, Hausinger RP, Scott RA. X-ray absorption spectroscopic analysis of Fe(II) and Cu(II) forms of a herbicide-degrading alpha-ketoglutarate dioxygenase. J. Biol. Inorg. Chem. 1999;4:122–129. doi: 10.1007/s007750050295. [DOI] [PubMed] [Google Scholar]
- 45.Costello A, Periyannan G, Yang KW, Crowder MW, Tierney DL. Site-selective binding of Zn(II) to metallo-beta-lactamase L1 from Stenotrophomonas maltophilia. J. Biol. Inorg. Chem. 2006;11:351–358. doi: 10.1007/s00775-006-0083-z. [DOI] [PubMed] [Google Scholar]
- 46.Costello AL, Sharma NP, Yang KW, Crowder MW, Tierney DL. X-ray absorption spectroscopy of the zinc-binding sites in the class B2 metallo-beta-lactamase ImiS from Aeromonas veronii bv. sobria. Biochemistry. 2006;45:13650–13658. doi: 10.1021/bi061547e. [DOI] [PubMed] [Google Scholar]
- 47.Kim YJ, Feng XD, Lippard SJ. Synthesis, structure, and properties of a mixed-valent triiron complex of tetramethyl reductic acid, an ascorbic acid analogue, and its relationship to a functional non-heme iron oxidation catalyst system. Inorg. Chem. 2007;46:6099–6107. doi: 10.1021/ic700622a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacquamet L, Aberdam D, Adrait A, Hazemann JL, Latour JM, Michaud-Soret I. X-ray absorption spectroscopy of a new zinc site in the fur protein from Escherichia coli. Biochemistry. 1998;37:2564–2571. doi: 10.1021/bi9721344. [DOI] [PubMed] [Google Scholar]
- 49.Barlow JN, Zhang ZH, John P, Baldwin JE, Schofield CJ. Inactivation of 1-aminocyclopropane-1-carboxylate oxidase involves oxidative modifications. Biochemistry. 1997;36:3563–3569. doi: 10.1021/bi962521y. [DOI] [PubMed] [Google Scholar]
- 50.Zhang ZH, Barlow JN, Baldwin JE, Schofield CJ. Metal-catalyzed oxidation and mutagenesis studies on the iron(II) binding site of 1-aminocyclopropane-1-carboxylate oxidase. Biochemistry. 1997;36:15999–16007. doi: 10.1021/bi971823c. [DOI] [PubMed] [Google Scholar]
- 51.Garcia GA, Tierney DL, Chong SR, Clark K, PennerHahn JE. X-ray absorption spectroscopy of the zinc site in tRNA-guanine transglycosylase from Escherichia coli. Biochemistry. 1996;35:3133–3139. doi: 10.1021/bi952403v. [DOI] [PubMed] [Google Scholar]
- 52.Rohde JU, Torelli S, Shan XP, Lim MH, Klinker EJ, Kaizer J, Chen K, Nam WW, Que L. Structural insights into nonheme alkylperoxoiron(III) and oxoiron(IV) intermediates by x-ray absorption spectroscopy. J. Am. Chem. Soc. 2004;126:16750–16761. doi: 10.1021/ja047667w. [DOI] [PubMed] [Google Scholar]
- 53.Shearer J, Scarrow RC, Kovacs JA. Synthetic models for the cysteinate-ligated non-heme iron enzyme superoxide reductase: Observation and structural characterization by XAS of an Fe-III-OOH intermediate. J. Am. Chem. Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b. [DOI] [PubMed] [Google Scholar]
- 54.Padden KM, Krebs JF, Trafford KT, Yap GPA, Rheingold AH, Borovik AS, Scarrow RC. Probing the structure of immobilized metal sites in porous organic hosts by X-ray absorption spectroscopy. Chem. Mater. 2001;13:4305–4313. [Google Scholar]
- 55.Rohde J-U, Stubna A, Bominaar EL, Munck E, Nam W, Que L., Jr Nonheme oxoiron(IV) complexes of tris(2-pyridylmethyl) amine with cis-monoanionic ligands. Inorg. Chem. 2006;45:6435–6445. doi: 10.1021/ic060740u. [DOI] [PubMed] [Google Scholar]
- 56.Fujimori DG, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Jr, Krebs C, Riggs-Gelasco PJ. Spectroscopic evidence for a high-spin Br-Fe(IV)-Oxo intermediate in the alpha-ketoglutarate-dependent halogenase CytC3 from Streptomyces. J. Am. Chem. Soc. 2007;129:13408–13409. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 57.Peterson CL, Laniel MA. Histones and histone modifications. Curr. Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 58.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr. Opin. Genet. Dev. 2005;15:163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Gene Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 60.Kouzarides T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002;12:198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 61.Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 2002;14:286–298. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 62.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 63.Byvoet P, Shepherd GR, Noland BJ, Hardin JM. Distribution and Turnover of Labeled Methyl Groups in Histone Fractions of Cultured Mammalian-Cells. Arch. Biochem. Biophys. 1972;148:558–567. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- 64.Thomas G, Hempel K, Lange HW. Relative Stability of Lysine-Bound Methyl Groups in Arginine-Rich Histones and Their Subfractions of Ehrlich Ascites Tumor-Cells in-Vitro. H-S Zeitschrift Physiol. Chem. 1972;353:1423–1428. [PubMed] [Google Scholar]
- 65.Shi YJ, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear arnine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 66.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 67.Katoh M, Katoh M. Identification and characterization of JMJD2 family genes in silico. Int. J. Oncol. 2004;24:1623–1628. [PubMed] [Google Scholar]
- 68.Hillringhaus L, Yue WW, Rose NR, Ng SS, Gileadi C, Loenarz C, Bello SH, Bray JE, Schofield CJ, Oppermann U. Structural and Evolutionary Basis for the Dual Substrate Selectivity of Human KDM4 Histone Demethylase Family. J. Biol. Chem. 2011;286:41616–41625. doi: 10.1074/jbc.M111.283689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Munoz A, Costa M. Elucidating the mechanisms of nickel compound uptake: A review of particulate and nano-nickel endocytosis and toxicity. Toxicol. Appl. Pharm. 2012;260:1–16. doi: 10.1016/j.taap.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett BG. Environmental Nickel Pathways to Man. Iarc Sci. Publ. 1984:487–495. [PubMed] [Google Scholar]
- 71.Que L, Ho RYN. Dioxygen activation by enzymes with mononuclear non-heme iron active sites. Chem. Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 72.Sundheim O, Vagbo CB, Bjoras M, Sousa MML, Talstad V, Aas PA, Drablos F, Krokan HE, Tainer JA, Slupphaug G. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dann CE, Bruick RK, Deisenhofer J. Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15351–15356. doi: 10.1073/pnas.202614999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of alpha-Ketoglutarate-Dependent Dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosen MD, Venkatesan H, Peltier HM, Bembenek SD, Kanelakis KC, Zhao LX, Leonard BE, Hocutt FM, Wu XD, Palomino HL, Brondstetter TI, Haugh PV, Cagnon L, Yan W, Liotta LA, Young A, Mirzadegan T, Shankley NP, Barrett TD, Rabinowitz MH. Benzimidazole-2-pyrazole HIF Prolyl 4-Hydroxylase Inhibitors as Oral Erythropoietin Secretagogues. ACS Med. Chem. Lett. 2010;1:526–529. doi: 10.1021/ml100198y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kruidenier L, Chung CW, Cheng ZJ, Liddle J, Che KH, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu B, Hunt JF. Enzymological and structural studies of the mechanism of promiscuous substrate recognition by the oxidative DNA repair enzyme AlkB. Proc. Natl. Acad. Sci. U.S.A. 2009;106:14315–14320. doi: 10.1073/pnas.0812938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O’Brien JR, Schuller DJ, Yang VS, Dillard BD, Lanzilotta WN. Substrate-induced conformational changes in Escherichia coli taurine/alpha-ketoglutarate dioxygenase and insight into the oligomeric structure. Biochemistry. 2003;42:5547–5554. doi: 10.1021/bi0341096. [DOI] [PubMed] [Google Scholar]
- 79.Elkins JM, Ryle MJ, Clifton IJ, Hotopp JCD, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, Roach PL. X-ray crystal structure of Escherichia coli taurine/alpha-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry. 2002;41:5185–5192. doi: 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- 80.Yang M, Chowdhury R, Ge W, Hamed RB, McDonough MA, Claridge TDW, Kessler BM, Cockman ME, Ratcliffe PJ, Schofield CJ. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. Febs Journal. 2011;278:1086–1097. doi: 10.1111/j.1742-4658.2011.08022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ. Asparaginyl hydroxylation of the notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J. Biol. Chem. 2007;282:24027–24038. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 82.Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng XD. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010;17:38–43. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horton JR, Upadhyay AK, Hashimoto H, Zhang X, Cheng XD. Structural Basis for Human PHF2 Jumonji Domain Interaction with Metal Ions. J. Mol. Biol. 2011;406:1–8. doi: 10.1016/j.jmb.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Upadhyay AK, Rotili D, Han JW, Hu RG, Chang YQ, Labella D, Zhang X, Yoon YS, Mai A, Cheng XD. An Analog of BIX-01294 Selectively Inhibits a Family of Histone H3 Lysine 9 Jumonji Demethylases. J. Mol. Biol. 2012;416:319–327. doi: 10.1016/j.jmb.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mishina Y, Chen LX, He C. Preparation and characterization of the native iron(II)-containing DNA repair AlkB protein directly from Escherichia coli. J. Am. Chem. Soc. 2004;126:16930–16936. doi: 10.1021/ja045066z. [DOI] [PubMed] [Google Scholar]
- 86.Hausinger RP. Fe(II)/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 87.Costas M, Mehn MP, Jensen MP, Que L. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 88.Hanauskeabel HM, Gunzler VA. Stereochemical Concept for the Catalytic Mechanism of Prolylhydroxylase - Applicability to Classification and Design of Inhibitors. J. Theor. Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 89.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 90.Bollinger JM, Price JC, Hoffart LM, Barr EW, Krebs C. Mechanism of taurine: alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Eur. J. Inorg. Chem. 2005;21:4245–4254. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 91.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 92.Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 93.Yang CG, Yi CQ, Duguid EM, Sullivan CT, Jian X, Rice PA, He C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature. 2008;452:961–965. doi: 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu B, Edstrom WC, Benach J, Hamuro Y, Weber PC, Gibney BR, Hunt JF. Crystal structures of catalytic complexes of the oxidative DNA/RNA repair enzyme AlkB. Nature. 2006;439:879–884. doi: 10.1038/nature04561. [DOI] [PubMed] [Google Scholar]
- 95.Holland PJ, Hollis T. Structural and Mutational Analysis of Escherichia coli AlkB Provides Insight into Substrate Specificity and DNA Damage Searching. Plos One. 2010;5:e8680. doi: 10.1371/journal.pone.0008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mirza SA, Day RO, Maroney MJ. Oxidation of a dimeric nickel thiolate complex with O-2. Inorg. Chem. 1996;35:1992–1995. [Google Scholar]
- 97.Grapperhaus CA, Darensbourg MY. Oxygen capture by sulfur in nickel thiolates. Acc. Chem. Res. 1998;31:451–459. [Google Scholar]
- 98.Goldcamp MJ, Robison SE, Bauer JAK, Baldwin MJ. Oxygen reactivity of a nickel(II)-polyoximate complex. Inorg. Chem. 2002;41:2307–2309. doi: 10.1021/ic015590w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.