

Abstract
Antibiotic use in humans has been associated with outgrowth of fungi. Here we used a murine model to investigate the gut microbiome over 76 days of treatment with vancomycin, ampicillin, neomycin, and metronidazole and subsequent recovery. Mouse stool was studied as a surrogate for the microbiota of the lower gastrointestinal tract. The abundance of fungi and bacteria was measured using quantitative PCR, and the proportional composition of the communities quantified using 454/Roche pyrosequencing of rRNA gene tags. Prior to treatment, bacteria outnumbered fungi by >3 orders of magnitude. Upon antibiotic treatment, bacteria dropped in abundance >3 orders of magnitude, so that the predominant 16S sequences detected became transients derived from food. Upon cessation of treatment, bacterial communities mostly returned to their previous numbers and types after 8 weeks, though communities remained detectably different from untreated controls. Fungal communities varied substantially over time, even in the untreated controls. Separate cages within the same treatment group showed radical differences, but mice within a cage generally behaved similarly. Fungi increased ∼40-fold in abundance upon antibiotic treatment but declined back to their original abundance after cessation of treatment. At the last time point, Candida remained more abundant than prior to treatment. These data show that 1) gut fungal populations change radically during normal mouse husbandry, 2) fungi grow out in the gut upon suppression of bacterial communities with antibiotics, and 3) perturbations due to antibiotics persist long term in both the fungal and bacterial microbiota.
Introduction
The effects of antibiotic use on the human microbiome can be challenging to study–confounding factors include complications of the underlying diseases states and concomitant use of additional forms of therapy [1]. Despite these difficulties, outgrowth of fungi has been repeatedly linked to antibiotic treatment at multiple body sites [2]–[8]. Fungal infection associated with antibiotic use is of particular concern in immunocompromised states such as HIV/AIDS [9]–[11], some cancers [8], [12], [13], and transplantation [14]–[18]. Many of these conditions necessitate the use of corticosteroids, which further predisposes the host to fungal infection [19]. Invasive fungal infections have been increasing in recent decades [1], [12], and the rise of azole-resistant species of Candida [7], [20], Aspergillus [21], [22], and Cryptococcus [9], [23] brings further urgency to understanding the interaction between commensal fungi and bacteria during antibiotic treatment.
Rodent models have been used to study the effects of antibiotics on the mammalian gut, using culture based [24], [25], metagenomic [26], and immunologic [26], [27] methods. Antibiotic treatment can predispose the host to infection by pathogens [28], [29] and alter microbial communities long term [29]. Induced exposure to Candida albicans shapes the bacterial composition of the murine gut during antibiotic recovery [25] and can cause gastritis [30], while Candida tropicalis has been associated with increased severity in ulcerative colitis [31]. Phenotypic effects have been found even after treatment with subclinical doses of antibiotics [32]. In studies of the role of the vertebrate microbiome in mice, antibiotic treatment is often used to suppress the host bacteria, but the effect of this intervention on fungi is not commonly considered [28], [33]–[35].
Here we characterize the bacterial and fungal microbiota of mice during antibiotic treatment and subsequent recovery after cessation, analyzing both the amounts and types of microbes present. We found that fungi indeed grew out upon antibiotic treatment. After cessation of antibiotic treatment, fungal and bacterial communities approached their pre-antibiotic states, but increased abundance of Candida persisted in the gut at the last time point studied eight weeks later. To our surprise, we also found that the fungal communities changed radically over time in both control and treated mice. For each condition, specific fungi colonized multiple mice in the same cage, then gave way to different fungal colonists over time, and different patterns were seen in different cages.
Results
Longitudinal Analysis of the Murine Gut during Antibiotic Treatment
An antibiotic cocktail containing vancomycin, ampicillin, neomycin, and metronidazole was given to twenty C57B6 mice in water. After 2 weeks, antibiotic treatment was stopped for ten of the mice. These mice did not receive any antibiotics during the remaining nine weeks of the study (“AbxShortTerm” mice). The remaining ten mice under antibiotic treatment continued to receive antibiotics for the duration of the study (“AbxContinuous” mice). Ten control mice received no antibiotics over the course of the study (“Control” mice). Fecal samples were collected over one week prior to initiating the study, then at the indicated time points during the study (Figure 1). DNA was purified from stool pellets using a procedure that included bead beating and a high temperature incubation to facilitate lysis of fungal cells [36].
Figure 1. Diagram of the experiment.

The time line for the 76 days of sample collection is shown along the top, and the periods of antibiotic treatment are shown at the bottom. Antibiotic treatment was initiated at time zero.
Analysis of the Numbers of Bacterial 16S and Eukaryotic 18S Gene Copies Present after Antibiotic Treatment
We first investigated the changes in abundance of bacteria and fungi, using stool specimens as a proxy for the lower intestinal microbiome. To assess changes in abundance, we first quantified the relative abundance of bacterial and fungal genomes in the samples per ng of DNA using quantitative PCR. For bacteria, a QPCR assay was used that detected the bacterial 16S rRNA gene, and for fungi, an assay was used detecting the 18S rRNA gene. The primers for the fungal assay were designed to suppress amplification of metazoan DNA originating from the host or food materials [37]. The specificity was confirmed by pyrosequencing products of amplification with these primers (below and Figure S1).
At the start of the study, fecal pellets contained high levels of bacterial 16S rRNA genes per ng DNA (∼106 apparent copies; Figure 2A). After initiation of antibiotic treatment, this fell as low as ∼102 apparent copies per ng DNA. Upon cessation of treatment the community recovered to its former numbers. For fungi, prior to antibiotic treatment, ∼105 apparent 18S rRNA gene copies were detected per ng DNA (Figure 2B). Upon initiation of antibiotic treatment the number climbed to between 3–6×108 apparent copies per ng DNA. Upon cessation of antibiotic treatment, the numbers dropped back to roughly their former levels The abundance of fungi in the Control group showed an unexpected increase at day 22. Further analysis showed that the increase was in only one of the two cages housing the control animals, and correlated with the appearance of a new fungal lineage at high levels in all animals in that cage (described below).
Figure 2. Relative microbial abundance inferred from QPCR.

A) Longitudinal analysis of 16S rRNA gene copies per ng of stool DNA. The groups of mice tested are shown by the color code (key at right). Error bars indicate standard error. B) Longitudinal analysis of 18S rRNA gene copies per ng of stool DNA. The groups of mice tested are shown by the color code (key at right). Error bars indicate standard error. The amplicon used was designed to suppress amplification of DNA from mouse or food materials.
Additional information is required to relate these numbers of rRNA gene copies to the numbers of organisms present. This issue is addressed in the next section.
Assessing the Absolute Abundance of Bacteria and Fungi
Several corrections are required to link the QPCR data to the total number of organisms per stool pellet. One consideration is that bacterial [38], [39] and fungal [40], [41] genomes typically contain multiple rRNA gene copies. From published data on complete genome sequences, we estimated the mean number of 16S rRNA gene copies per bacteria at 5 [38], and 18S copies per fungal genome at 100 [41], though the number for fungi is tentative due to the difficulty of accurately sequencing tandem direct repeats and variability in copy number.
Another concern in assessing possible fungal outgrowth during antibiotic treatment is that the total number of microbes in pellets, and thus total DNA, may go down with treatment, so that fungi could falsely appear to proliferate only because total DNA content went down as bacterial numbers fell. Thus we sought to correct the above assays, which were normalized to weight of DNA, to better reflect the counts of individual organisms by putting the final analysis on a per pellet basis. Average wet weights of pellets were 16.08 mg (SD = 3.329) in the presence of antibiotic (n = 20) and 18.64 (SD = 2.685) in the control mice (n = 19), a slight but significant difference (p = 0.0129, Mann-Whitney U test). A comparison of dry weights showed no significant difference between antibiotic treated (n = 5, mean = 10.615 mg, SD = 1.635791) and control mice (n = 5, mean = 10.875 mg, SD = 0.781025). Thus in what follows we treated the starting weights as equal.
DNA yields per pellet differed substantially (Table 1). Quantification of yields after 15 or 76 days of antibiotic treatment showed drops of 4.7 and 5.7 fold (p = 5.7×10−5, Mann-Whitney U test). After withdrawal of antibiotic treatment (AbxShortTerm, Day 76), the total DNA yield returned to within a factor of two of the starting value. Evidently bacterial DNA is the predominant source of DNA in mouse pellets, and the community mostly returned to its former size after cessation of antibiotic treatment. Thus the analysis of the numbers of microbial genomes needs to take into account the drop in total DNA. In addition, we also corrected for inefficiencies in the Taqman detection of 16S rRNA gene copies, which arise because some 16S rRNA gene sequences contain mismatches within the probe binding sites (described in the Methods).
Table 1. DNA yields and numbers of genomes inferred from data on ribosomal gene copies.
| Sample | DNA yieldper pellet;average (ng) | DNA yieldper pellet(ng); (SD) | Number ofBacterialgenomes/pellet;average | Number ofBacterial genomes/pellet;(SD) | Number of micro-eukaryotegenomes/pellet; average | Number of micro-eukaryotegenomes/pellet; SD | MicroeukaryoteProportion |
| Control (Baseline) | 1.08E+03 | 4.47E+02 | 2.17E+09 | 1.18E+09 | 2.95E+06 | 1.25E+06 | 0.001 |
| Control (Day 15) | 6.27E+02 | 4.02E+02 | 6.03E+08 | 5.48E+08 | 1.98E+06 | 1.30E+06 | 0.003 |
| Control (Day 76) | 4.50E+02 | 2.72E+02 | 6.51E+08 | 4.97E+08 | 1.12E+06 | 9.43E+05 | 0.002 |
| AbxShortTerm (Baseline) | 4.51E+02 | 1.56E+02 | 5.79E+08 | 3.43E+08 | 7.11E+05 | 4.43E+05 | 0.001 |
| AbxShortTerm(Day 15) | 1.13E+02 | 7.42E+01 | 5.43E+04 | 4.92E+04 | 3.56E+07 | 1.38E+07 | 0.998 |
| AbxShortTerm(Day 76) | 3.94E+02 | 1.88E+02 | 3.85E+08 | 3.12E+08 | 5.35E+05 | 4.16E+05 | 0.001 |
| AbxContinuous (Baseline) | 4.39E+02 | 1.79E+02 | 5.55E+08 | 3.68E+08 | 4.62E+05 | 1.96E+05 | 0.001 |
| AbxContinuous(Day 15) | 1.48E+02 | 7.32E+01 | 9.43E+04 | 6.53E+04 | 7.86E+07 | 3.85E+07 | 0.999 |
| AbxContinuous(Day 76) | 1.08E+02 | 1.32E+01 | 2.09E+05 | 1.86E+05 | 2.43E+07 | 7.94E+06 | 0.991 |
Taking these factors into account, we found that in the absence of antibiotic treatment, a typical stool pellet contained 5×108–2×109 bacteria, and this dropped to ∼5×104 bacteria after 15 days of antibiotic treatment (Figure 3A and Table 1). Below we show that 5×104 is in fact an overestimate, because most of the 16S DNA was in fact derived from bacterial DNA in sterile mouse food. Bacteria returned to their former numbers after cessation of antibiotic treatment. Fungal genomes were much less abundant initially, only in the range of 5×105–2×106 per pellet (Figure 3B and Table 1). After 15 days of treatment with antibiotics, the numbers increased to ∼5×107, or an increase of 25–50 fold. Fungal genome numbers remained high for the period of antibiotic treatment. Eight weeks after cessation of antibiotic treatment, counts in the ABXShortTerm groups returned to the pretreatment level. Thus changes in fungal cell abundance were substantial, though less than suggested by the analysis in Figure 2B, which was normalized to the total weight of DNA, because total DNA went down with antibiotic treatment.
Figure 3. Numbers of organisms per stool pellet.

Values from QPCR were corrected to yield an estimate of the true numbers of organisms by accounting for differential DNA yield, numbers of rRNA gene copies per genome, and efficiency of detection. A) Estimated numbers of bacterial genomes per pellet. Note that during antibiotic treatment (Day 15), most of the 16S rRNA gene copies were derived from food and do not correspond to intact organisms. (B) Estimated numbers of fungal genomes per pellet. The x-axis shows the time point studied, and the y-axis shows the inferred numbers of genomes. Each study group is indicated by the color code to the right of the figure panels.
Analysis of Bacterial Lineages using 454/Roche Deep Sequencing
To assess the representation of microbial lineages present and changes with antibiotic therapy, we analyzed the longitudinal DNA samples using 454/Roche pyrosequencing. DNA was purified from stool from 13 time points (Figure 1). Bacterial sequences were amplified using primers matching the 16S rRNA gene V1V2 region [36], [42]. Sequencing yielded 239,867 reads, which were condensed into OTUs at 97% similarity and taxonomy assigned using the RDP classifier [43].
Prior to antibiotic treatment, communities were dominated by the Firmicute lineage Lachnospiraceae and the Bacteriodetes lineage Bacteroidales, along with a substantial number of less abundant lineages (Figure 4; Figure S2 A-I presents time points for each mouse individually). After one day of antibiotic treatment, the previously dominant lineages decreased sharply in abundance, and Lactococcus became the dominant community member. At later times under antibiotic treatment Lactococcus was the predominant or sole lineage detectable.
Figure 4. Longitudinal analysis of bacterial lineages inferred from 16S rRNA gene sequencing.

Bacterial lineages detected are summarized in heat map format. Each column shows the average for the ten mice in each group and at the time point indicated. Sequence samples were rarefied to a standard 200 reads per individual before averaging. The periods of antibiotic treatment are indicated at the top in salmon color, the periods off antibiotic by light blue. The day of treatment is indicated at the bottom. The color code to the right indicates the proportions.
Five aliquots of sterile mouse chow were analyzed by amplification with the V1V2 primers and 454/Roche pyrosequencing, revealing that a single Lactococcus OTU was the predominant phylotype in both stool from antibiotic treated animals and in chow (Figure S3). We thus conclude that Lactococcus DNA is present in sterile mouse food, and that the antibiotic treatment eliminated the great majority of live bacteria, i. e. the 5×104 bacteria detected per pellet in Table 1 represents mostly bacterial DNA in food.
After antibiotic treatment was stopped for the ABXShortTerm group, major groups that were predominant before antibiotic treatment returned to their former levels, but at different rates. An OTU classified as Lachnospiraceae and several OTUs classified as Clostridium returned within one week. Several other clades, including Ruminococcaceae and other Firmicutes increased in proportion by two weeks after cessation of treatment. Bacteroidales did not fully return until the end of the experiment at eight weeks. Enteroccocus, Escherichia, and Paenibacillus, which were not dominant members of the communities in the Control or antibiotic treated groups, had elevated proportions over the recovery period but decreased in relative abundance after eight weeks off antibiotics. Table S1 presents a statistical analysis of the bacterial lineages detected and their behavior over the time course studied.
Changes in the types of bacterial lineages were paralleled by changes in the species richness (Figure 5). Prior to antibiotic treatment, 54.6 (SD = 6.9) phylotypes were detected after data from each mouse was normalized to 200 reads. After 2 days of antibiotic treatment, this fell to 7 (SD = 2.0) and persisted for the remainder of the antibiotic treatment (p<2.2×10−16 for comparison to the pretreatment group, Friedman test). Upon cessation of antibiotic treatment, the community slowly returned to its former richness reaching 49.4 (SD = 5.6) lineages over 61 days, still less than the corresponding Control group which averaged 57.2 (SD = 17) lineages on the same day (p = 0.02 Mann-Whitney U test).
Figure 5. Abundance analysis of observed bacterial lineages.
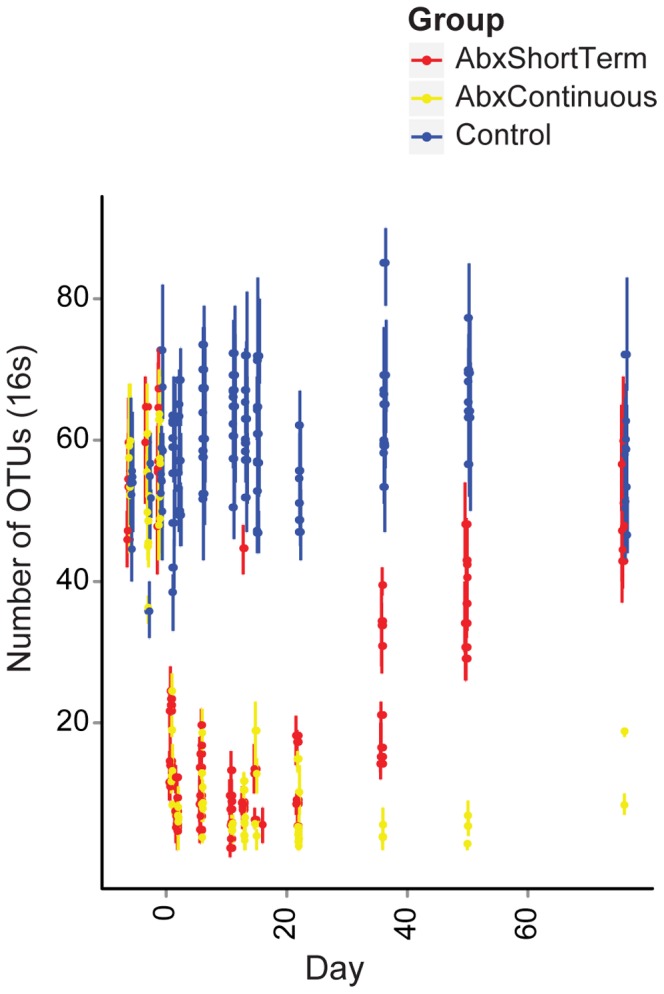
Each sequence set for each animal was rarefied to 200 sequences per sample 10 times, and the number of different OTUs assessed. Means are indicated by points, error bars indicate the range observed. The groups studied are indicated by the key at the right.
Analysis of Microeukaryotes using 454/Roche Deep Sequencing
To characterize microeukaryotes, we sequenced selected samples using 18S and ITS amplicons. To compare samples from the different treatment groups, 134,677 ITS sequences and 26,355 18S sequences were generated, OTUs were formed, and taxonomic attribution was preformed with BROCC [44]. The 18S amplicon is more universal than the ITS amplicon [44], while the ITS amplicon provides greater resolution for some fungal lineages [45], so both were used [37]. To check that the two amplicons were yielding consistent information, we compared sequence samples from 15 mice amplified using both amplicons. Sequence samples were characterized by generating pairwise UniFrac distances, then the distance matrices for each were compared using Procrustes analysis. This showed high correlation between the two (p<0.0001, no better fits after 104 permutations) and compositional comparison also showed similar profiles (Figure S1).
The longitudinal behavior of fungal communities was explored in detail using the ITS amplicon, which revealed strong effects of both antibiotic treatment and caging history of the animals (Figure 6; the full set of time points, with each animal shown individually, is in Figure S4A-I). In the control animals (five mice in each of two cages), although the composition of the bacterial community remained relatively stable, the fungal community changed dramatically. For four samples taken over the first eight days, most of the ten mice in the two cages showed colonization by diverse fungal lineages, and no lineage predominated. By day 15, however, the situation had changed radically, with both cages dominated by a phylotype annotated as Wickerhamomyces. This changed by day 22, with cage 1 dominated by Debaryomyces, and the second cage showing more diverse colonization, where Debaryomyces was present but not predominant. The outgrowth of Debaryomyces in cage 1 correlated with the increase in abundance of total DNA in the 18S QPCR analysis in the control animals at day 22 (Figure 3), which also occurred only in cage 1. By day 76 the controls had changed again. At this time both cages were dominated by Eurotiales, though the abundance was greater in cage 1 than cage 2. These findings document radical changes in murine gut fungi 1) in a single mouse facility 2) for mice on a homogeneous diet, 3) in the absence of any intervention, and 4) differing between cages.
Figure 6. Longitudinal analysis of fungal lineages inferred from ITS rRNA gene sequencing.

Fungal lineages detected are summarized in heat map format. Data for four days are shown (days 1, 15, 22, and 76). Each column indicates a single mouse. The groups tested are indicated at the top of the columns. The ten mice in each of the three treatment groups were each housed in two cages of five each. The distribution of mice in cages is indicated at the bottom of the columns. The day of treatment is indicated at the top. The color codes at the top right indicate the proportions and Phyla of origin.
For the antibiotic treated animals, the communities were diverse prior to initiation of treatment, then under treatment showed cage-specific patterns of divergence. By day 2, Clavispora and Cyberlindnera were predominant in both cages in the ABXContinuous group and cage 1 in the ABXShortTerm group. In cage 2 in the short term group the mice were more heterogenous, with Candida, Cyberlindnera, and Pichia dominating in different mice. By day 6 this had changed, with Cyberlinderna dominating in three cages (both ABXContinuous and ABXShortTerm cage 1), and Candida dominating in all mice in ABXShortTerm cage 2. This pattern persisted at day 15, but by day 22 one ABXContinuous cage was dominated by Candida, and by day 76 Candida was the only fungus detectable in both ABXContinuous cages. For the ABXShortTerm mice, Sporopachydermis dominated in cage 1 and Candida in cage 2 on day 22, then the fungal populations returned to a more diverse mixture by day 76, but Candida was relatively abundant in most of the short term treated mice at the last time point studied. Thus these data emphasize the heterogeneity of the community responses in individual cages, but also the robust persistence of the Candida community. Table S2 presents a statistical analysis of the fungal lineages detected and their behavior over the time course studied. A few samples were also analyzed with the 18S rRNA gene amplicon and generally yielded similar results (Figure S1). An analysis of mouse chow DNA using the ITS amplicon showed no obvious relationship to the major lineages detected in pellets (Figure S3).
Initially an average of 30.7 (SD = 9.9) phylotypes were detected in the ITS data per animal (Figure 7), but these numbers fell to 5.2 (SD = 2.9) per animal during antibiotic treatment as the Candida overgrew the community (p = 8.3×10−12, Friedman test). Upon cessation of antibiotic treatment the number of phylotypes returned to their former levels (31.1, SD = 8.0, p = 0.673, Mann-Whintey U). Thus the fungal community returned to its original richness, but despite this, the contribution of Candida was higher than before treatment.
Figure 7. Abundance analysis of observed fungal lineages.
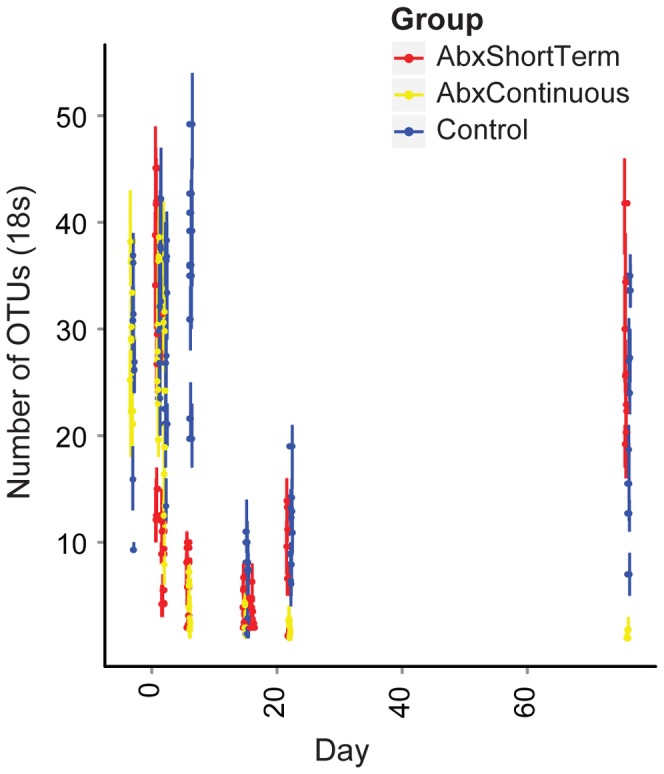
Each sequence set was rarefied to 200 sequences per sample, and the number of different OTUs assessed. Means are indicated by points, error bars indicate the range observed. The groups studied are indicated by the key at the right.
Community Comparisons using Unifrac
To compare community structures over the course of antibiotic treatment, the 454 data for the 16S and ITS tags were analyzed using weighted [46] and unweighted [47] Unifrac (Figure S5). We tested for differenced in community composition between the control group and the treatment groups at each time point by comparing distances within the control group to distances between control and treatment groups. Significance was determined using the Mann-Whitney test.
The ABXShortTerm and ABXContinuous groups were not significantly different from the Control group before antibiotic treatment for either the bacterial or fungal communities (Figure S5). After one day of treatment, both groups were significantly different from the Control in both weighted and unweighted Unifrac and remained distinct throughout the antibiotic treatment (Figure S5). One week after the ShortTermABX group stopped receiving antibiotics, both the bacterial and fungal communities remained significantly different from the other two (Figure S5). On day 76, 61 days after antibiotic cessation, differences between the bacterial communities were slight but still achieved significance in the weighted and unweighted analysis, and some but not all comparisons were significant for the ITS analysis. Thus by the last time point the communities had approached but not completely returned to their pre-treatment states (p<0.005).
Discussion
We report a longitudinal study of the bacterial and fungal communities in the mouse gut after treatment with a cocktail of four antibiotics. Our main observations were that 1) bacterial communities dropped sharply in abundance, then recovered to near to their starting state after cessation of antibiotic treatment, 2) fungal communities increased in abundance with the fall in bacterial abundance, and 3) waves of fungal colonization swept through all of the cages in our study, including the untreated controls (Figure 8). We discuss each of these findings in turn below.
Figure 8. Longitudinal variation in fungal abundance in the six cages studied.

Each cage is shown as a row labeled at left, the types of fungi detected are shown at the right. The background shading indicates the presence or absence of antibiotics (key at bottom).
Multiple studies have examined the effects of antibiotic treatment on the gut microbiome in vertebrates. Dethlefsen et al. studied the gut microbiome of three humans treated with ciprofloxacin [48], and found that while diversity was reduced upon treatment, the response of bacterial lineages differed among the individuals studied. Communities mostly returned to their initial states with cessation of treatment, though detectable differences remained. Hill et al. studied mice subjected to an extreme antibiotic treatment (ampicillin, gentamicin, metronidazole, neomycin, and vancomycin) designed to eradicate gut bacteria [26], and found that gut could be cleared out to such an extent that only food sequences were detectable, paralleling findings here. A profound and reproducible change was observed in the composition of the microbiota at both luminal and tissue associated sites. Antonopoulos et al. studied mice exposed to a cefoperazone regimen followed by a 6 week recovery period [49]. They observed bacterial counts drop by 3 orders of magnitude during antibiotic treatment and saw significant compositional changes and decreased diversity in bacterial communities after cessation of antibiotics. Peterfreund et al. studied bacteria and fungi of the hamster gut following treatment with clindamycin and C. difficile infection [29], and found that after treating C. difficile with an antitoxin antibody the community reached an altogether new stable state.
The bacterial communities studied here were initially dominated by Bacteriodetes and Lachnospiraceae,but changed quickly upon initiation of antibiotic treatment, reaching a stable state by day two. At this time, the number of inferred organisms had dropped >3 orders of magnitude, to the point that the 16S rRNA gene sequences present in stool were mostly transients from food, documenting near complete clearance of bacteria from the gut. Upon cessation of antibiotic treatment, the bacteria returned to nearly their original state, though perturbations remained. Taken together with previous studies, these findings emphasize that of the response of gut bacteria to antibiotics vary quite substantially with the choices of host organism, antibiotic regimen, and other experimental features.
The bacterial populations in the control animals remained relatively stable over the time course, dominated by Bacteriodetes and Lachnospiraceae throughout. A study by Schloss et al. showed that the bacteria of the gut of new borne pups varied over time initially, but stabilizes later in life [50], paralleling studies of human infants [51]. Our data showed that the animals studied here had indeed achieved a stable adult bacterial profile, allowing us to attribute changes to antibiotic treatment. However, the same was not true for fungi.
Fungal communities changed radically over the time course studied in association with the caging history of the mice. The amounts of fungi detected here prior to Abx treatment were comparable to previous studies. Qin et al. [52] suggested that the proportion of eukaryotic DNA in human stool was <0.1%, while FISH analysis of murine gut biofilms [53] suggested 0–10% fungi, with a median of 2%. Our data in the absence of Abx suggested fungal proportions in the range of 0.1–0.3%.
Unexpectedly, even the Control group showed waves of succession that differed in each cage (Figure 8). Cage 1 was first heterogenous, then dominated by Wickerhamomyces, then Debaryomyces, then Eurotiales. Cage 2 was heterogenous through the first six days, then dominated by Wickerhamomyces, then again heterogeneous. These data document a quite surprising degree of fungal variation in healthy laboratory mice. Previous studies have shown that colonization by specific bacteria in mice can strongly influence the outcome of immunological assays [54]. Given the recently reported importance of fungi in mouse models of IBD [31], our data suggest that researchers will need to take care to monitor and control fungal populations to obtain meaningful data. The mice in this study were housed on a conventional SPF environment. It would be useful to compare housing in a barrier facility where food, water, and bedding have all been sterilized. Colitis phenotypes in genetically-determined mouse models are known to vary in different facilities, suggesting that it will be useful to assess the role of variability in fungal colonization.
In the presence of antibiotics, the fungal community showed several waves of succession, which again differed between cages. During Abx treatment, the fungal proportions exceeded 99% of all microbes. Depending on the cage and time point, the communities could be heterogeneous, or dominated by Clavispora, Cyberlindnera, Sporopachydermia or Candida. In cases where communities in a cage were dominated by a single fungal lineage, this was true of all mice in the cage at that time point. One likely explanation is that coprophagia resulted in all mice in a single cage acquiring the same fungal colonists. Some of these fungal lineages were seen sporadically in contamination controls (Figure S6), suggesting that gut fungi may have been acquired episodically from the environment.
Candida was a particularly robust colonist in the presence of antibiotics. By day 76, all the ABXContinuous mice were colonized at a high level exclusively by Candida. Further supporting the robustness of Candida, results from cage two in the ABXShortTerm group by chance provides a competition experiment. Of the five mice analyzed on day two, two were colonized with Candida, one with Cyberlindnera, one with Pichia, and one with both Candida and Cyberlindnera (Figure S4, part C). Coprophagia would presumably allow the three species to compete for colonization opportunities. By day six, all mice were colonized with Candida, and this persisted through the cessation of antibiotic treatment by day 22. At the end of the experiment on day 76, Candida was still more abundant in the ABXShortTerm group than prior to treatment, all emphasizing that Candida was favored by the antibiotic treatment and persisted subsequently. These data motivate more careful studies of fungal blooms, and particularly Candida, in human subjects undergoing antibiotic treatment.
Methods
Mouse Husbandry
Thirty C57B6 eight week old female mice were purchased from Jackson Laboratory and placed on a standardized diet for two weeks prior to the study to stabilize their flora. Mice were housed by treatment group with five mice per cage. Mice treated with antibiotics received ampicillin, neomycin, vancomycin, and metronidazole in water. Water was spiked with aspartame in both the treated groups and controls. Mice were fed AIN-76A Rodent Diet from Research Diet (D10001) for the course of the study, which includes 15% casein lactic. The Institutional Review Board of the University of Pennsylvania approved all IACUC protocols (protocol #803408). The animal care facility is operated by the University Laboratory Animal Resources, which is fully accredited by the American Association for Accreditation of Laboratory Animal Care. Laboratory animals are maintained in accordance with the applicable portions of the Animal Welfare Act and their guidelines prescribed in the DHHS publication, “Guide for the Care and Use of Laboratory Animals”. Mice are euthanized using C02 inhalation. Methods for euthanasia are consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
DNA Extraction
DNA was extracted from 1–2 mouse pellets per mouse per time point. Samples were homogenized for 80 seconds on a Mini-BeadBeater-16 (BioSpec) in Lysing Matrix E tubes (MP Biomedical). Samples were then incubated at 95°C for 15 minutes and then cooled on ice for 60 seconds. Then samples were extracted with the PSP DNA extraction kit and using a protocol described previously [44]. Sequences of oligonucleotides used in this study are presented in Table S3.
DNA Sequencing and QPCR
Extracted DNA was quantified with the Picogreen system. Typical volumes produced, used in the calculations in Table 1, were 250 microliters. DNA was amplified using primers annealing to the V1V2 region of the 16S bacterial gene or the ITS1 fungal rRNA gene spacer, and amplified with AccuPrime taq with Buffer 2 (Invitrogen). Thermocycler protocols for 16S [36] and ITS and 18S [44] amplicons were described previously. PCR amplicons were purified with Agencourt AMPure XP beads. Sequencing was performed on a 454 Junior using Titanium chemistry. For both 16S and ITS amplicons, DNA free water was subjected to the same purification procedure and analyzed by 454/Roche pyrosequencing (Figure S6). A subset of samples showed recoverable sequences, but stool samples showed distinct community composition, so we conclude that environmental contamination made a minimal contribution to the samples analyzed.
16S qPCR was performed using the Taqman method as described previously [26]. The Taqman method was chosen because it provides good specificity for bacterial sequences in complex mixtures, but reconstruction experiments show that the Taqman values were on average 11-fold lower than for parallel Syber Green assays, and follow up studies suggested the Syber Green values were reliable. Thus a correction was added for the quantitative comparison in Figure 3A and Table 1. 18S qPCR was performed with the Applied Biosystems SYBR Green Fast chemistry. Ten µL SYBRGreen FAST 2×master mix, 1 µL of each primer diluted to 20 pM, and 8 µL DNA were added to each reaction. Thermocycling was performed as follows: samples were initially denatured once for 2 minutes at 50°C then 10 minutes at 95°C. Then samples were cycled 40 times with a dissociation step at 95°C for 15 seconds and an annealing and extension step at 60°C for 1 minute. Primer sequences for these assays can be found in Table S3. All DNA sequences generated in this study have been submitted to the Sequence Read Archive under accession number SRP020503.
Bioinformatic Analysis
Bioinformatic analysis was performed with the QIIME software package [55] using default parameters except where indicated, and using R. Fungal reads were queried against the nt database using NCBI’s blastn tool and then classified with the BROCC classifier [44].
Eukaryotic PCoA analysis was performed using taxonomic relationship corresponding to the NCBI Taxonomy [56]. Because de novo tree construction using the ITS1 region is not feasible due to length variation inherent in the ITS gene [44], [57]–[59], we chose to asses Unifrac distances between eukaryotic communities using the NCBI Taxonomy to generate taxonomic trees. To transform the taxonomy into a phylogenetic tree, all edges between taxa were assigned equal weight. Classifications were curated manually for parsimony. While the fungal taxonomy is imperfect and in a state flux [45], [60]–[62], we note that in practice the Unifrac metric is relatively robust to the method used in creating phylogenetic trees [63]. Comprehensive data on OTU mean proportions and counts are in Tables S4–S7. Statistical significance for treatment groups was determined using the nonparametric Mann-Whitney test in R. Permanova tests and Procrustes analysis were performed in QIIME.
Supporting Information
Comparison of microeukaryote lineages specified by the ITS and 18S amplicons. A) Heat maps comparing selected samples analyzed using both the 18S and ITS amplicons. Each column shows the average for mice in the group and at the time point indicated rarefied to 200 reads per individual. The color code to the right indicates the scale. B) Procrustes analysis comparing results for the 18S and ITS analysis. Data from the 18S and ITS amplicons for each mouse are shown by balls connected by a line.
(TIF)
Heat maps showing the composition of bacterial communities inferred from 16S sequence data for each time point, with each mouse shown individually. The scale of relative proportions is shown on the far right.
(TIF)
16S and ITS sequences recovered from five samples of mouse chow. A) Sequences from the 16S analysis. B) Sequences from the ITS analysis. The scale of relative proportions is shown on the far right.
(TIF)
Heat maps showing the composition of fungal communities inferred from ITS sequence data for each time point, with each mouse shown individually. The scale of relative proportions is shown on the far right.
(TIF)
PCoA analysis distances measures for bacterial (16S sequence data) and fungal (ITS sequence data) communities. Distances matrices were calculated using weighted or unweighted UniFrac, then the pairwise distances between a treatment group and the control group compared to the distances within the control group on that day. Asterisks above each box and whisker plot indicate whether the comparison was significantly different.
(TIF)
Comparison of contamination controls to experimental samples for the 16S A) and ITS B) amplicons. “Extraction control” indicates sequences derived from blank purifications using DNA-free water. Each column showing mouse data is an average over all reads at that time point.
(TIF)
Bacterial lineages detected in this study and statistical analysis of longitudinal changes. Each time course was separated into intervals, and species abundance was compared between the time intervals indicated in the column headings. Mean values were compared for each lineage using the Friedman test (non-parametric with repeated measures). P-values that survived Bonferroni correction are highlighted and bolded. Intervals were compared as follows: -Baseline: Comparison between groups among baseline time points. -Early ABX vs Control: The first two abx timepoints samples are compared to Control samples from the same time point. -Late ABX vs Control: All time points after the first two abx days when ABXShortTerm are still receiving abx. Control and abx treated samples are compared. -Recovery vs Control: All time points after abx were stopped for the ABXShortTerm group. Control and ABXShortTerm are compared. -Last Day Recovery vs Control: The last day of the study only. Control and ABXShortTerm are compared. -Early ABX vs Late ABX: The first two abx time points are compared to later (days 6–15) abx time points. ABXShorterm and ABXContinuous are lumped in together.
(XLSX)
Fungal lineages detected in this study and statistical analysis of longitudinal changes. Each time course was separated into intervals, and species abundance was compared between the time intervals indicated in the column headings. Mean values were compared for each lineage using the Friedman test (non-parametric with repeated measures). p-values that survived Bonferroni correction are highlighted and bolded. Comparisons where a species did not appear in both time periods or groups being compared are marked n/a. Intervals were compared as follows: -Baseline: Comparison between groups among baseline time points. -Early ABX vs Control: The first two abx timepoints samples are compared to Control samples from the same time point. -Late ABX vs Control: All time points after the first two abx days when ABXShortTerm are still receiving abx. Control and abx treated samples are compared. -Recovery vs Control: All time points after abx were stopped for the ABXShortTerm group. Control and ABXShortTerm are compared. -Last Day Recovery vs Control: The last day of the study only. Control and ABXShortTerm are compared. -Early ABX vs Late ABX: The first two abx time points are compared to later (days 6–15) abx time points. ABXShorterm and ABXContinuous are lumped in together.
(XLSX)
Oligonucleotides used in this study.
(XLSX)
Means, variances, and standard deviations for bacterial OTUs within treatment groups and time points rarefied to 200 reads per sample.
(XLSX)
Means, variances, and standard deviations for fungal OTUs within treatment groups and time points rarefied to 200 reads per sample.
(XLSX)
OTU table of bactieral 16S sequences rarefied to 200 reads per sample.
(XLSX)
OTU table of fungal ITS sequences rarefied to 200 reads per sample.
(XLSX)
Acknowledgments
We are grateful to members of the Bushman laboratory for help and suggestions. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, National Institutes of Health, or Pennsylvania Department of Health.
Funding Statement
This work was supported by Project UH2DK083981, the Penn Genome Frontiers Institute; National Institutes of Health (NIH) AI39368 (GDW); Penn Digestive Disease Center (P30 DK050306); The Joint Penn-CHOP Center for Digestive, Liver, and Pancreatic Medicine; S10RR024525; UL1RR024134, and K24-DK078228; and the University of Pennsylvania Center for AIDS Research (CFAR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chen KY, Ko SC, Hsueh PR, Luh KT, Yang PC (2001) Pulmonary fungal infection: emphasis on microbiological spectra, patient outcome, and prognostic factors. Chest 120: 177–184. [DOI] [PubMed] [Google Scholar]
- 2. Samonis G, Gikas A, Anaissie EJ, Vrenzos G, Maraki S, et al. (1993) Prospective evaluation of effects of broad-spectrum antibiotics on gastrointestinal yeast colonization of humans. Antimicrobial agents and chemotherapy 37: 51–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mulligan ME, Citron DM, McNamara BT, Finegold SM (1982) Impact of cefoperazone therapy on fecal flora. Antimicrobial agents and chemotherapy 22: 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abbott J (1995) Clinical and microscopic diagnosis of vaginal yeast infection: a prospective analysis. Annals of emergency medicine 25: 587–591. [DOI] [PubMed] [Google Scholar]
- 5. Oksala E (1990) Factors predisposing to oral yeast infections. Acta odontologica Scandinavica 48: 71–74. [DOI] [PubMed] [Google Scholar]
- 6. Leyden JJ, Marples RR (1973) Ecologic principles and antibiotic therapy in chronic dermatoses. Archives of dermatology 107: 208–211. [PubMed] [Google Scholar]
- 7. Ben-Ami R, Olshtain-Pops K, Krieger M, Oren I, Bishara J, et al. (2012) Antibiotic exposure as a risk factor for fluconazole-resistant Candida bloodstream infection. Antimicrobial agents and chemotherapy 56: 2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bow EJ, Louie TJ (1987) Changes in endogenous microflora among febrile granulocytopenic patients receiving empiric antibiotic therapy: implications for fungal superinfection. CMAJ : Canadian Medical Association journal = journal de l’Association medicale canadienne 137: 397–403. [PMC free article] [PubMed] [Google Scholar]
- 9.Cheong JW, McCormack J (2012) Fluconazole resistance in cryptococcal disease: emerging or intrinsic? Medical mycology : official publication of the International Society for Human and Animal Mycology. [DOI] [PubMed]
- 10. Moris DV, Melhem MS, Martins MA, Souza LR, Kacew S, et al. (2012) Prevalence and antifungal susceptibility of Candida parapsilosis complex isolates collected from oral cavities of HIV-infected individuals. Journal of medical microbiology 61: 1758–1765. [DOI] [PubMed] [Google Scholar]
- 11. Ampel NM (1996) Emerging disease issues and fungal pathogens associated with HIV infection. Emerging infectious diseases 2: 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chamilos G, Luna M, Lewis RE, Bodey GP, Chemaly R, et al. (2006) Invasive fungal infections in patients with hematologic malignancies in a tertiary care cancer center: an autopsy study over a 15-year period (1989–2003). Haematologica 91: 986–989. [PubMed] [Google Scholar]
- 13. Viscoli C, Girmenia C, Marinus A, Collette L, Martino P, et al. (1999) Candidemia in cancer patients: a prospective, multicenter surveillance study by the Invasive Fungal Infection Group (IFIG) of the European Organization for Research and Treatment of Cancer (EORTC). Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 28: 1071–1079. [DOI] [PubMed] [Google Scholar]
- 14. Gallis HA, Berman RA, Cate TR, Hamilton JD, Gunnells JC, et al. (1975) Fungal infection following renal transplantation. Archives of internal medicine 135: 1163–1172. [PubMed] [Google Scholar]
- 15. Trenschel R, Peceny R, Runde V, Elmaagacli A, Dermoumi H, et al. (2000) Fungal colonization and invasive fungal infections following allogeneic BMT using metronidazole, ciprofloxacin and fluconazole or ciprofloxacin and fluconazole as intestinal decontamination. Bone marrow transplantation 26: 993–997. [DOI] [PubMed] [Google Scholar]
- 16. Koo S, Kubiak DW, Issa NC, Dietzek A, Boukedes S, et al. (2012) A targeted peritransplant antifungal strategy for the prevention of invasive fungal disease after lung transplantation: a sequential cohort analysis. Transplantation 94: 281–286. [DOI] [PubMed] [Google Scholar]
- 17.Karchmer AW, Samore MH, Hadley S, Collins LA, Jenkins RL, et al.. (1995) Fungal infections complicating orthotopic liver transplantation. Transactions of the American Clinical and Climatological Association 106: 38–47; discussion 47–38. [PMC free article] [PubMed]
- 18. Charlson ES, Diamond JM, Bittinger K, Fitzgerald AS, Yadav A, et al. (2012) Lung-enriched Organisms and Aberrant Bacterial and Fungal Respiratory Microbiota after Lung Transplant. Am J Respir Crit Care Med 186: 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lionakis MS, Kontoyiannis DP (2003) Glucocorticoids and invasive fungal infections. Lancet 362: 1828–1838. [DOI] [PubMed] [Google Scholar]
- 20. Sasse C, Dunkel N, Schafer T, Schneider S, Dierolf F, et al. (2012) The stepwise acquisition of fluconazole resistance mutations causes a gradual loss of fitness in Candida albicans. Molecular microbiology 86: 539–556. [DOI] [PubMed] [Google Scholar]
- 21. Burgel PR, Baixench MT, Amsellem M, Audureau E, Chapron J, et al. (2012) High prevalence of azole-resistant Aspergillus fumigatus in adults with cystic fibrosis exposed to itraconazole. Antimicrobial agents and chemotherapy 56: 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arikan-Akdagli S (2012) Azole resistance in Aspergillus: global status in Europe and Asia. Annals of the New York Academy of Sciences 1272: 9–14. [DOI] [PubMed] [Google Scholar]
- 23. Perfect JR, Cox GM (1999) Drug resistance in Cryptococcus neoformans. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy 2: 259–269. [DOI] [PubMed] [Google Scholar]
- 24. Samonis G, Anastassiadou H, Dassiou M, Tselentis Y, Bodey GP (1994) Effects of broad-spectrum antibiotics on colonization of gastrointestinal tracts of mice by Candida albicans. Antimicrobial agents and chemotherapy 38: 602–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mason KL, Erb DownwardJR, Mason KD, Falkowski NR, Eaton KA, et al. (2012) Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infection and immunity 80: 3371–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, et al. (2010) Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol 3: 148–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noverr MC, Falkowski NR, McDonald RA, McKenzie AN, Huffnagle GB (2005) Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infection and immunity 73: 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, et al. (2012) Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336: 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peterfreund GL, Vandivier LE, Sinha R, Marozsan AJ, Olson WC, et al. (2012) Succession in the Gut Microbiome following Antibiotic and Antibody Therapies for Clostridium difficile. PLoS One 7: e46966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mason KL, Erb DownwardJR, Falkowski NR, Young VB, Kao JY, et al. (2012) Interplay between the gastric bacterial microbiota and Candida albicans during postantibiotic recolonization and gastritis. Infection and immunity 80: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, et al. (2012) Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336: 1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, et al. (2012) Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488: 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, et al. (2007) Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proceedings of the National Academy of Sciences of the United States of America 104: 10643–10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, et al. (2009) The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 1: 6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, et al. (2012) Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149: 1578–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, et al. (2010) Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol 10: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dollive S, Peterfreund GL, Sherrill-Mix S, Bittinger K, Sinha R, et al. (2012) A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol 13: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. Journal of bacteriology 186: 2629–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee CM, Sieo CC, Abdullah N, Ho YW (2008) Estimation of 16S rRNA gene copy number in several probiotic Lactobacillus strains isolated from the gastrointestinal tract of chicken. FEMS microbiology letters 287: 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Herrera ML, Vallor AC, Gelfond JA, Patterson TF, Wickes BL (2009) Strain-dependent variation in 18S ribosomal DNA Copy numbers in Aspergillus fumigatus. Journal of clinical microbiology 47: 1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Metwally L, Fairley DJ, Coyle PV, Hay RJ, Hedderwick S, et al. (2008) Improving molecular detection of Candida DNA in whole blood: comparison of seven fungal DNA extraction protocols using real-time PCR. Journal of medical microbiology 57: 296–303. [DOI] [PubMed] [Google Scholar]
- 42. Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dollive S, Peterfreund GL, Sherrill-Mix S, Bittinger K, Sinha R, et al. (2012) A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome biology 13: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, et al.. (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed]
- 46. Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and Qualitative {beta} Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Applied and Environmental Microbiology 73: 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71: 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS biology 6: e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, et al. (2009) Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun 77: 2367–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schloss PD, Schubert AM, Zackular JP, Iverson KD, Young VB, et al. (2012) Stabilization of the murine gut microbiome following weaning. Gut Microbes 3: 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, et al. (2011) Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 108 Suppl 14578–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scupham AJ, Presley LL, Wei B, Bent E, Griffith N, et al. (2006) Abundant and diverse fungal microbiota in the murine intestine. Appl Environ Microbiol 72: 793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ivanov, II, Atarashi K, Manel N, Brodie EL, Shima T, et al. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, et al. (2009) Database resources of the National Center for Biotechnology Information. Nucleic acids research 37: D5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ko KS, Jung HS (2002) Three nonorthologous ITS1 types are present in a polypore fungus Trichaptum abietinum. Molecular phylogenetics and evolution 23: 112–122. [DOI] [PubMed] [Google Scholar]
- 58. Rohel E, Couteaudier Y, Papierok B, Cavelier N, Dedryver CA (1997) Ribosomal internal transcribed spacer size variation correlated with RAPD-PCR pattern polymorphisms in the entomopathogenic fungus Erynia neoaphidis and some closely related species. Mycological Research 101: 573–579. [Google Scholar]
- 59. Valente P, Gouveia FC, Lemos GA, Pimentel D, Mendonca-Hagler LC, et al. (1997) PCR-amplified ITS length variation within the yeast genus Metschnikowia. The Journal of general and applied microbiology 43: 179–181. [DOI] [PubMed] [Google Scholar]
- 60. Chang CF, Liu YR, Chen SF, Naumov GI, Naumova ES, et al. (2012) Five novel species of the anamorphic genus Candida in the Cyberlindnera clade isolated from natural substrates in Taiwan. Antonie van Leeuwenhoek 102: 9–21. [DOI] [PubMed] [Google Scholar]
- 61. Kurtzman CP, Robnett CJ, Basehoar-Powers E (2008) Phylogenetic relationships among species of Pichia, Issatchenkia and Williopsis determined from multigene sequence analysis, and the proposal of Barnettozyma gen. nov., Lindnera gen. nov. and Wickerhamomyces gen. nov. FEMS yeast research 8: 939–954. [DOI] [PubMed] [Google Scholar]
- 62. Kurtzman CP (2011) Phylogeny of the ascomycetous yeasts and the renaming of Pichia anomala to Wickerhamomyces anomalus. Antonie van Leeuwenhoek 99: 13–23. [DOI] [PubMed] [Google Scholar]
- 63. Lozupone CA, Knight R (2008) Species divergence and the measurement of microbial diversity. FEMS microbiology reviews 32: 557–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of microeukaryote lineages specified by the ITS and 18S amplicons. A) Heat maps comparing selected samples analyzed using both the 18S and ITS amplicons. Each column shows the average for mice in the group and at the time point indicated rarefied to 200 reads per individual. The color code to the right indicates the scale. B) Procrustes analysis comparing results for the 18S and ITS analysis. Data from the 18S and ITS amplicons for each mouse are shown by balls connected by a line.
(TIF)
Heat maps showing the composition of bacterial communities inferred from 16S sequence data for each time point, with each mouse shown individually. The scale of relative proportions is shown on the far right.
(TIF)
16S and ITS sequences recovered from five samples of mouse chow. A) Sequences from the 16S analysis. B) Sequences from the ITS analysis. The scale of relative proportions is shown on the far right.
(TIF)
Heat maps showing the composition of fungal communities inferred from ITS sequence data for each time point, with each mouse shown individually. The scale of relative proportions is shown on the far right.
(TIF)
PCoA analysis distances measures for bacterial (16S sequence data) and fungal (ITS sequence data) communities. Distances matrices were calculated using weighted or unweighted UniFrac, then the pairwise distances between a treatment group and the control group compared to the distances within the control group on that day. Asterisks above each box and whisker plot indicate whether the comparison was significantly different.
(TIF)
Comparison of contamination controls to experimental samples for the 16S A) and ITS B) amplicons. “Extraction control” indicates sequences derived from blank purifications using DNA-free water. Each column showing mouse data is an average over all reads at that time point.
(TIF)
Bacterial lineages detected in this study and statistical analysis of longitudinal changes. Each time course was separated into intervals, and species abundance was compared between the time intervals indicated in the column headings. Mean values were compared for each lineage using the Friedman test (non-parametric with repeated measures). P-values that survived Bonferroni correction are highlighted and bolded. Intervals were compared as follows: -Baseline: Comparison between groups among baseline time points. -Early ABX vs Control: The first two abx timepoints samples are compared to Control samples from the same time point. -Late ABX vs Control: All time points after the first two abx days when ABXShortTerm are still receiving abx. Control and abx treated samples are compared. -Recovery vs Control: All time points after abx were stopped for the ABXShortTerm group. Control and ABXShortTerm are compared. -Last Day Recovery vs Control: The last day of the study only. Control and ABXShortTerm are compared. -Early ABX vs Late ABX: The first two abx time points are compared to later (days 6–15) abx time points. ABXShorterm and ABXContinuous are lumped in together.
(XLSX)
Fungal lineages detected in this study and statistical analysis of longitudinal changes. Each time course was separated into intervals, and species abundance was compared between the time intervals indicated in the column headings. Mean values were compared for each lineage using the Friedman test (non-parametric with repeated measures). p-values that survived Bonferroni correction are highlighted and bolded. Comparisons where a species did not appear in both time periods or groups being compared are marked n/a. Intervals were compared as follows: -Baseline: Comparison between groups among baseline time points. -Early ABX vs Control: The first two abx timepoints samples are compared to Control samples from the same time point. -Late ABX vs Control: All time points after the first two abx days when ABXShortTerm are still receiving abx. Control and abx treated samples are compared. -Recovery vs Control: All time points after abx were stopped for the ABXShortTerm group. Control and ABXShortTerm are compared. -Last Day Recovery vs Control: The last day of the study only. Control and ABXShortTerm are compared. -Early ABX vs Late ABX: The first two abx time points are compared to later (days 6–15) abx time points. ABXShorterm and ABXContinuous are lumped in together.
(XLSX)
Oligonucleotides used in this study.
(XLSX)
Means, variances, and standard deviations for bacterial OTUs within treatment groups and time points rarefied to 200 reads per sample.
(XLSX)
Means, variances, and standard deviations for fungal OTUs within treatment groups and time points rarefied to 200 reads per sample.
(XLSX)
OTU table of bactieral 16S sequences rarefied to 200 reads per sample.
(XLSX)
OTU table of fungal ITS sequences rarefied to 200 reads per sample.
(XLSX)