

Implications of the orphan C-type lectin receptor in the resolution of inflammation by facilitating neutrophil turnover.
Keywords: Clecsf8, Klebsiella pneumoniae, sepsis, efferocytosis, hyperinflammation
Abstract
Pneumonia is frequently associated with sepsis, characterized by a nonresolving hyperinflammation. However, specific host components of the pulmonary milieu that regulate the perpetuation of inflammation and tissue destruction observed in this immune disorder are not clearly understood. We examined the function of Clec4d, an orphan mammalian CLR, in Gram negative pneumonic sepsis caused by KPn. Whereas the WT mice infected with a sublethal dose of bacteria could resolve the infection, the Clec4d−/− mice were highly susceptible with a progressive increase in bacterial burden, hyperinflammatory response typical of sepsis, and severe lung pathology. This correlated with a massive accumulation of neutrophils in lungs of infected Clec4d−/− mice, which was in contrast with their WT counterparts, where neutrophils transiently infiltrated the lungs. Interestingly, the Clec4d−/− neutrophils did not exhibit any defect in bacterial clearance. These results suggest that Clec4d plays an important role in resolution of inflammation, possibly by facilitating neutrophil turnover in lungs. This is the first report depicting the physiological function of Clec4d in a pathological condition. The results can have implications not only in sepsis but also in other inflammatory diseases, where nonresolving inflammation is the root cause of disease development.
Introduction
Sepsis poses a major health-care burden, with 750,000 cases annually in the United States and a mortality rate of 20–50% [1]. Currently, there are no effective therapies to treat this deadly immune disorder. Pneumonia is the most frequent source of sepsis [2]. In that regard, nosocomial infections caused by the opportunistic pathogen KPn account for 5–20% of Gram-negative sepsis cases. In light of the constant occurrence of antibiotic-resistant strains of this pathogen, an understanding of functioning of host innate immune components might provide targets for modulation of the host immune system in a beneficial manner [3, 4].
Sepsis is now perceived as interplay of pathogen-associated molecular patterns (PAMPs) as well as endogenous host factors (termed alarmins) [5]. CLRs are emerging as pathogen recognition receptors (PRRs) that can shape immune responses by recognizing a variety of PAMPs as well as alarmins [6]. However, their function in development of sepsis is largely unexplored. In that regard, Clec4e (Mincle) of the Dectin-2 subfamily has been shown recently to function as an activating receptor for host endogenous factors released from dead cells and PAMPs of bacterial and fungal origin [7]. Clec4d, another Dectin-2 family member, is localized close to Mincle in the NK gene complex region of chromosome 12p13, and its function is yet to be defined [8, 9]. Based on the close vicinity of this receptor with Mincle, we hypothesized that it likely plays a role in regulating immune responses during pathogenic conditions.
In this study, we tested the role of Clec4d in Gram-negative sepsis induced by pulmonary infection with KPn. Our results show that Clec4d-mediated responses are required for the resolution of pneumonia and to mitigate mortality in pulmonary KPn-induced sepsis. This may have implications for other immune disorders associated with nonresolving, persistent inflammation.
MATERIALS AND METHODS
Infection of mice and survival
Six- to 8-week-old female WT C57BL/6 and Clec4d−/− mice (obtained from the Consortium of Functional Genomics), bred in the animal facility of the University of North Dakota, were used according to institutional and federal guidelines. Mice were infected intranasally with a sublethal dose (5×104 bacteria in 20 μl saline, determined experimentally) of KPn (ATCC strain 43826). The bacteria were grown to log phase in LB medium. Mock-infected mice received saline only. Mice were monitored daily for signs of disease, which typically included piloerection, hunched gait, lethargy, weight loss, and increased respiratory rate. The survival of infected mice was recorded for up to 2 weeks p.i.
Bacterial burden and multianalyte profile analysis
Lungs and livers from infected and mock mice at various times p.i. were removed immediately after perfusion and homogenized aseptically in cold PBS with Complete protease inhibitor cocktail (Roche Diagnostics, Germany), all described by us previously [10, 11]. For the bacterial burden analyses, the organ homogenates and blood were serially diluted in PBS and plated on LB agar. The immune mediators in lung homogenates were determined commercially by Myriad RBM (Austin, TX, USA), using a multiplexed, flow-based system: RodentMAP multianalyte profile analysis technology.
Quantitative RT-PCR
Lungs from infected and mock mice at various times p.i. were removed immediately after perfusion and were used to extract total RNA by Trizol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Real-time PCR analysis of the samples was performed, as described previously [10], using SYBR Green (Applied Biosystems, Foster City, CA, USA) to measure the expression levels of Clec4d-specific mRNAs by using specific primers Clec4d (sense) 5′-GAA CAA ATT CTT GCC GTC CTG ACC-3′ and (antisense) 5′-TCC ATC ACA AGG ACC ACT TTC TGA G-3′ (Integrated DNA Technologies, Coralville, IA, USA). The target gene expression levels were normalized to levels of the housekeeping 18S gene (sense) 5′-CATGTGGTGTTGAGGAAAGCA-3′ and (antisense) 5′-GTCGTGGGTTCTGCATGATG-3′ in the same sample. Expression of Clec4d in infected samples was determined as fold change over that in control samples, as calculated by using the formula 2−(ΔΔCt).
Histological analysis
Lungs from infected and mock mice at various times p.i. were isolated after perfusion and were snap-frozen in OCT resin. For pathological analysis, 10-μm-thick serial horizontal sections of frozen lungs were stained with H&E, as described previously [11].
Flow cytometry
For enumeration of neutrophils by flow cytometry, lungs from mock control and infected animals, isolated 3 dp.i., were processed to get total cellular infiltrates, as described previously by us [11]. Ly6G+CD11b+ neutrophils were quantified using Pacific Blue anti-mouse CD11b and APC anti-mouse Ly6G antibodies (BioLegend, San Diego, CA, USA).
Bacterial phagocytosis and killing activity of neutrophils
For assessing KPn phagocytosis, peritoneal neutrophils were isolated by a previously described method [12] and were incubated with GFP-labeled KPn (kindly provided by Dr. Steven Clegg, University of Iowa, Iowa City, IA, USA) at a multiplicity of infection of 50, with or without opsonization in 10% normal mouse serum. After 1 h, the cells were washed twice with ice-cold PBS, followed by two washes with FACS buffer (PBS+10% FBS). The perecent-positive cells containing fluorescent bacteria were determined by flow cytometry using the BD LSR II (Becton Dickinson Immunocytometry, San Jose, CA, USA). For bacterial killing activity, intracellular CFUs in Clec4d−/− and WT neutrophils were enumerated at 1 h and 3 h p.i. by gentamycin protection assay, as described previously [13].
Statistics
Statistical analysis of survival studies was performed by the Kaplan-Meir log-rank test and bacterial burdens by the nonparametric Mann-Whitney test. All other statistical analyses were performed using the Student's t-test (SigmaPlot 8.0; Systat Software, Chicago, IL, USA).
RESULTS
To investigate the role of Clec4d in KPn pneumonia, we first examined whether the Clec4d−/− and WT mice respond to pulmonary KPn infection differentially. For this, we initially infected the mice intranasally with a lethal dose of 1 × 105 CFUs (determined experimentally). One hundred percent of the WT mice died within 5 dp.i. at this dose, whereas all of the Clec4d−/− mice reproducibly succumbed to infection, 1–2 days earlier than the WT mice (data not shown). To finalize a dose at which the WT mice would display minimal morbidity and mortality, whereas the Clec4d−/− mice would all succumb to infection, mice were infected with a sublethal dose of 5 × 104 CFUs. As shown in Fig. 1A, 70–80% of WT mice infected with 5 × 104 CFUs of KPn survived the infection with transient signs of disease (ruffled fur, lethargy), early during infection, and appeared healthy later. The Clec4d−/− mice, in contrast, were extremely susceptible to this dose. The majority of these mice died within 5 dp.i., and 100% of mice succumbed to infection by Day 7 with progressive development of disease and overt signs of infection (piloerection, hunched gait, lethargy, increased respiratory rate). Importantly whereas the disease signs appeared at similar times (2 dp.i.) in WT and Clec4d−/− mice, they became divergent by 3 dp.i. Whereas the WT mice appeared to enter the recovery phase at that time, the Clec4d−/− mice continued to display disease progression. The recovery phase of WT mice strongly correlated with the expression level of Clec4d in their lungs, where Clec4d mRNA was transcribed maximally by 3 dp.i. (Fig. 1B). This was followed by a down-regulation in the Clec4d transcript that was reduced to only marginal levels by 5 dp.i. These results indicated a protective role of this receptor in the resolution of KPn pneumonia.
Figure 1. Clec4d−/− mice display increased mortality accompanied with overwhelming bacterial burden in systemic organs during Gram-negative pneumonia.

(A) Sixteen WT (C57BL/6) and 17 Clec4d−/− mice were infected intranasally with 5 × 104 CFUs of KPn in 20 μl sterile PBS and were assessed daily for disease severity. The increased susceptibility of Clec4d−/− mice compared with WT mice is statistically significant, as determined by the Kaplan-Meier survival curve statistical analysis (P<0.001). (B) Total RNA was extracted by the Trizol method from lungs harvested at the indicated times after infection of WT mice with 5 × 104 CFUs of KPn. The mRNA levels of Clec4d were analyzed by real-time PCR, as described in Materials and Methods, and are expressed as fold changes over the levels in mock control mice calculated by using the formula 2−(ΔΔCt). Data shown are the averages of six to eight mice at each time point in two independent experiments. (C) WT and Clec4d−/− mice were infected intranasally with 5 × 104 CFUs of KPn. At indicated times p.i., the mice were sacrificed, and systemic organs were isolated, homogenized, and plated, as described in Materials and Methods. Bacterial burden was enumerated after incubating the plates overnight at 37°C. The data shown are from one representative experiment (five animals at each time-point) out of three performed with similar results. Significant differences in bacterial burden (using the nonparametric Mann-Whitney test) in WT and Clec4d−/− mice are denoted by asterisks (**P<0.005; ***P<0.001).
Next, serial dilutions of homogenized lungs, liver, and blood from infected Clec4d−/− and WT mice were plated on LB. For the initial 2 days, the Clec4d−/− and WT animals displayed similar bacterial burdens in their lungs (Fig. 1C). Overwhelming loads were detected in lungs of Clec4d−/− mice at 3 dp.i., which remained high at 5 dp.i., the time when the majority of mice had become moribund. In contrast, the WT mice displayed three to five logs lower bacterial burden at 3 dp.i., and the counts continued to drop through 5 dp.i., indicating a resolution of the infection in these mice. The Clec4d−/− mice also displayed a higher and earlier systemic dissemination of bacteria, as depicted by significantly higher bacterial load in liver, even at 2 dp.i., and a more severe bacteremia (Fig. 1C). In contrast, no viable bacteria were detected in the blood of WT mice by 5 dp.i. These data indicated that Clec4d-mediated responses, directly or indirectly, influenced bacterial clearance in pneumonic infection with KPn.
To examine whether the inability of Clec4d−/− mice to clear bacteria was a result of a defect in a mounting inflammatory response, proinflammatory cytokines in infected mice lungs were analyzed. In both strains, mock-infected mouse lungs displayed similar low basal levels of inflammatory cytokines tested (TNF-α, IFN-γ, IL-6, IL-17; Fig. 2A). Upon KPn infection, WT mice exhibited increased levels of these cytokines at 1 dp.i., which started to drop by 3 dp.i. and were reduced to minimum by 5 dp.i. (Fig. 2A). This was consistent with the reduced bacterial burden in these mice at these times p.i. In contrast, infection of Clec4d−/− mice resulted in a progressive increase in levels of these cytokines through the course of infection, showing a massive up-regulation at 3 days and 5 dp.i. Interestingly, at 1 dp.i., the levels of these cytokines were slightly lower than those in the WT mice at that time p.i.; however, the differences were not statistically significant. Of note, unlike the proinflammatory cytokines tested, the amount of IL-10, a regulatory cytokine, was reduced significantly at 5 dp.i. compared with its level at 3 dp.i. in Clec4d−/− mice. This was in contrast with other cytokines and chemokines in the Clec4d−/− mice whose levels continued to increase or stayed at similar high levels through 5 dp.i. in these mice. In light of a recent study implicating STAT-1-regulated IL-10 production in resolution of KPn pneumonia [14], a reduced level of this cytokine, either resulting from a depletion of IL-10-producing cells in Clec4d−/− mice or because of the role of this receptor in continuous production of this cytokine in a positive-feedback loop manner, possibly plays a role in the lack of resolution in these mice. Notwithstanding the mechanism, our results show that Clec4d−/− mice do not display a defect in their ability to mount an inflammatory response but rather, display a hyperinflammatory phenotype akin to the cytokine storm typically associated with sepsis. Moreover, levels of sepsis marker mediators, such as Factor VII, C-reactive protein, and fibrinogen, were also increased massively in these mice (data not shown). Importantly, in the absence of Clec4d, these mice are unable to resolve the infection and the resulting inflammation. This further indicates a protective role of this receptor in resolution of KPn pneumonia.
Figure 2. Pneumonic Clec4d−/− mice exhibit a hyperinflammatory response and severe lung pathology characterized by massive neutrophil accumulation.
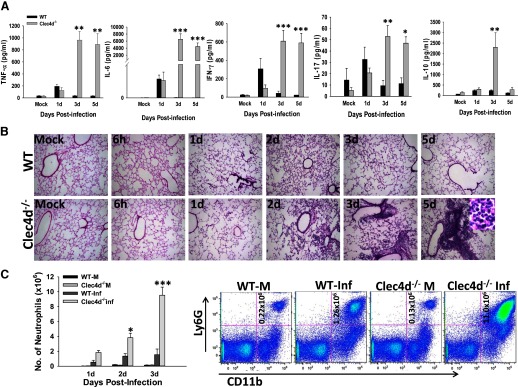
(A) The lungs from mock control and KPn-infected WT and Clec4d−/− mice were harvested at indicated time-points p.i., homogenized in PBS with protease inhibitors, and analyzed for rodent multianalyte profile (Myriad RBM). Results shown are the average of three infected and three mock control mice from three independent experiments. Significant differences are denoted by asterisks (*P<0.05; **P<0.005; ***P<0.001). (B) H&E staining of lung cryosections from mock control and KPn-infected WT and Clec4d−/− mice isolated at indicated times p.i. Original magnification, 100×. The inset in the right panel in the Clec4d−/− row shows a highly magnified area (1000×) of a lesion in infected Clec4d−/−, depicting neutrophils, as indicated by characteristic, multilobed nuclear morphology. (C) Flow cytometry analysis of neutrophils in mock control (WT-M and Clec4d−/− M) and KPn-infected (WT-Inf and Clec4d−/− Inf) mice. Total lung cells were isolated from mice by collagenase treatment at indicated times p.i. The cells were stained with anti-Ly6G-APC and anti-CD11b-Pacific Blue antibodies as markers for neutrophils. Appropriate isotype-matched negative controls were used to set the gates. The bar graph shows the average total number of neutrophils in lungs of three mock control and three KPn-infected WT and Clec4d−/− mice from three independent experiments (total of nine mice/group). Dot plots shown on the right are from one representative mouse in each group. Statistical significance between WT and Clec4d−/− mice is denoted by asterisks (*P<0.05; ***P<0.001).
Next, H&E staining was performed to determine immunopathological changes in WT and Clec4d−/− mice. As shown in Fig. 2B, mock control WT and Clec4d−/− mice display similar, normal lung-tissue morphology. A moderate peribronchial and perivascular infiltration of immune cells was observed in WT mice, which was reduced substantially by 5 dp.i. The overall architecture of the lungs was largely preserved in the WT animals throughout the infection. The Clec4d−/− mice, on the other hand, displayed a moderate immune-cell infiltration for the initial 2 days after infection. However, at later time-points, these mice exhibited much more severe pathological changes in their lungs, with a progressive increase in leukocyte infiltration through 3 dp.i. By day 5 p.i., a time when the mice had become moribund, extensive foci of consolidation were visible, with massive accumulation of neutrophils (as depicted by multilobed nuclear morphology in high magnification in the inset in the right panel in the Clec4d row of Fig. 2B) around alveolar spaces. Flow cytometry analysis of infiltrating cells in lungs showed that the numbers of Ly6G+CD11b+ neutrophils were significantly higher in the Clec4d−/− lungs at 3 dp.i. compared with the WT (Fig. 2C). Interestingly, the Clec4d−/− mice showed a significantly increased number of neutrophils, even at 2 p.i., a time when the bacterial burden was similar in the Clec4d−/− and WT lungs. To further determine whether these neutrophils were defective in bacterial clearance in the absence of Clec4d, phagocytosis of GFP-labeled KPn was compared between WT and Clec4d−/− neutrophils by flow cytometry (as described in Materials and Methods). As shown in Fig. 3A, Clec4d deficiency had no effect on phagocytosis of opsonized or nonopsonized bacteria. Furthermore, these neutrophils showed a similar bacterial killing ability as their WT counterparts (Fig. 3B). This indicated that Clec4d likely does not play a direct role in bacterial clearence, and the increased neutrophil accumulation in pneumonic Clec4d−/− mice is not a secondary effect, resulting from a defective bacterial clearance in the absence of this CLR. On the other hand, neutrophil chemoattractants (CXCL1 and CXCL6), the neutrophil survival mediator (GM-CSF), and neutrophil activation markers (MMP9, MPO) were significantly higher in Clec4d−/− mice compared with their WT counterparts (Fig. 3C). These results show that Clec4d is likely involved in neutrophil turnover, and the absence of Clec4d results in greater and prolonged accumulation of neutrophils in lungs of mice. The persistent activation of these neutrophils is leading to greater lung pathology observed in Clec4d−/− mice compared with their WT counterparts, where the neutrophils infiltrate transiently and are cleared off as the infection resolves. These observations clearly suggest that whereas Clec4d deficiency does not impair infiltration, bacterial phagocytosis, and activation of neutrophils, this CLR is required for resolution of inflammation by way of facilitating neutrophil turnover.
Figure 3. Clec4d deficiency does not impair neutrophil phagocytosis and killing of bacteria but leads to their unbridled accumulation and activation.

(A) Peritoneal neutrophils from WT and Clec4d−/− mice were incubated with GFP-labeled KPn with (opsonized) or without (nonopsonized) 10% normal mouse serum for 1 h, followed by quantitation of phagocytosis by flow cytometry. The results are expressed as percent cells positive for fluorescent bacteria. (B) Bacterial uptake and killing capacity of Clec4d−/− and WT neutrophils were determined at 1 h and 3 h by assessing intracellular CFUs in these cells as described in Materials and Methods. The experiment was repeated three times with similar results. (C) The lungs from mock control and KPn-infected WT and Clec4d−/− mice were harvested at indicated time-points p.i., homogenized in PBS with protease inhibitors, and analyzed commercially for neutrophil-associated immune mediators using the rodent multianalyte profile (Myriad RBM). Results shown are the average of three infected and three mock control mice from three independent experiments. Significant differences are denoted by asterisks (*P<0.05; **P<0.005; ***P<0.001).
DISCUSSION
Resolution of inflammation is an active and tightly controlled process that is necessary for restoration and maintenance of tissue homeostasis. Deregulation of this process is the root cause of many diseases. Our results indicate that Clec4d deficiency, in an otherwise sublethal pulmonary KPn infection, leads to a nonresolving, hyperinflammatory response that culminates in the death of infected KO animals. This report, for the first time, shows that Clec4d likely plays a role in resolution of inflammatory response. Neutrophil-mediated responses are essential for combating pneumonic bacterial infection, and their protective role in sepsis and KPn infection, in particular, has been described elegantly [13, 15]. However, persistent accumulation of neutrophils can lead to bystander tissue destruction, owing to their tissue-destructive cargo. Accumulation of large numbers of neutrophils in the lungs of KO mice and higher amounts of their associated inflammatory mediators suggest that there is a defect in neutrophil turnover in these mice. As Clec4d was found not to be directly involved in bacterial uptake and killing by neutrophils, it seems more likely that as a result of the widespread tissue damage, owing to persistent neutrophil activity in the Clec4d−/− mice, other immune cells important for bacterial clearance are depleted, leading to this increase in local bacterial burden, as well a systemic spread of infection. Thus, an increased bacterial burden (secondary to the defect in neutrophil turnover) is likely a factor contributing to the increased mortality of these mice eventually. As the neutrophil turnover and resolution of neutrophil-associated responses are complex processes involving apoptosis and clearance of their contents (efferocytosis) and class-switching of lipid mediators, we are currently investigating the role of Clec4d in these processes.
This study shows that Clec4d plays an important role in mitigating the inflammation during a pneumonic infection, possibly by facilitating neutrophil turnover. This study opens up new avenues of research on the role of Clec4d in the resolution of inflammatory responses. This can have major implications in the therapeutic measurements of inflammation-associated disorders.
ACKNOWLEDGMENTS
This study was supported by grants 10BGIA4300041 from the American Heart Association and 1R21AI101644-01 from the U.S. National Institutes of Health to J.S.
The authors thank Dr. Steven Clegg (University of Iowa) for providing GFP-labeled KPn and Dr. Umamahesh Gundra (New York University) for technical help with flow cytometry.
SEE CORRESPONDING EDITORIAL ON PAGE 387
- APC
- allophycocyanin
- Clec4d−/−
- Clec4d-deficient
- CLR
- C-type lectin receptor
- Ct
- comparative threshold
- dp.i.
- days postinfection
- KO
- knockout
- KPn
- Klebsiella pneumoniae
- Mincle
- macrophage-inducible C-type lectin
- MMP9
- matrix metalloproteinase 9
- p.i.
- postinfection
DISCLOSURES
The authors declare no financial conflict of interest.
REFERENCES
- 1. Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 2. Hoogerwerf J. J., van der Windt G. J., Blok D. C., Hoogendijk A. J., De Vos A. F., van 't Veer C., Florquin S., Kobayashi K. S., Flavell R. A., van der Poll T. (2012) Interleukin-1 receptor-associated kinase M-deficient mice demonstrate an improved host defense during Gram-negative pneumonia. Mol. Med. 18, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nordmann P., Cuzon G., Naas T. (2009) The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet 9, 228–236 [DOI] [PubMed] [Google Scholar]
- 4. Eddens T., Kolls J. K. (2012) Host defenses against bacterial lower respiratory tract infection. Curr. Opin. Immunol. 24, 424–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Denk S., Perl M., Huber-Lang M. (2012) Damage- and pathogen-associated molecular patterns and alarmins: keys to sepsis? Eur. Surg. Res. 48, 171–179 [DOI] [PubMed] [Google Scholar]
- 6. Rabinovich G. A., van. Kooyk Y., Cobb B. A. (2012) Glycobiology of immune responses. Ann. N. Y. Acad. Sci. 1253, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miyake Y., Ishikawa E., Ishikawa T., Yamasaki S. (2010) Self and nonself recognition through C-type lectin receptor, Mincle. Self Nonself 1, 310–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arce I., Martinez-Munoz L., Roda-Navarro P., Fernandez-Ruiz E. (2004) The human C-type lectin CLECSF8 is a novel monocyte/macrophage endocytic receptor. Eur. J. Immunol. 34, 210–220 [DOI] [PubMed] [Google Scholar]
- 9. Graham L. M., Gupta V., Schafer G., Reid D. M., Kimberg M., Dennehy K. M., Hornsell W. G., Guler R., Campanero-Rhodes M. A., Palma A. S., Feizi T., Kim S. K., Sobieszczuk P., Willment J. A., Brown G. D. (2012) The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J. Biol. Chem. 287, 25964–25974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mishra B. B., Gundra U. M., Teale J. M. (2011) STAT6(−)/(−) mice exhibit decreased cells with alternatively activated macrophage phenotypes and enhanced disease severity in murine neurocysticercosis. J. Immunol. 232, 26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharma J., Li Q., Mishra B. B., Pena C., Teale J. M. (2009) Lethal pulmonary infection with Francisella novicida is associated with severe sepsis. J. Leukoc. Biol. 86, 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Torchinsky M. B., Garaude J., Martin A. P., Blander J. M. (2009) Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature 458, 78–82 [DOI] [PubMed] [Google Scholar]
- 13. Batra S., Cai S., Balamayooran G., Jeyaseelan S. (2012) Intrapulmonary administration of leukotriene B(4) augments neutrophil accumulation and responses in the lung to Klebsiella infection in CXCL1 knockout mice. J. Immunol. 188, 3458–3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Poe S. L., Arora M., Oriss T. B., Yarlagadda M., Isse K., Khare A., Levy D. E., Lee J. S., Mallampalli R. K., Chan Y. R., Ray A., Ray P. (2013) STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 6, 189–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kovach M. A., Standiford T. J. (2012) The function of neutrophils in sepsis. Curr. Opin. Infect. Dis. 25, 321–327 [DOI] [PubMed] [Google Scholar]