

Abstract
Triazoles are known for their non-toxicity, higher stability and therapeutic activity. Few nucleoside (L1, L2 and L3) and non-nucleoside 1,2,3-triazoles (L4–L14) were synthesised using click chemistry and they were screened for tumor cell cytotoxicity and proliferation. Among these triazole ligands studied, nucleoside ligands exhibited higher potential than non-nucleoside ligands. The nucleoside triazole analogues, 3′-Phenyl-1,2,3- triazole-thymidine (L2) and 3′-4-Chlorophenyl-1,2,3-triazole-thymidine (L3), demonstrated higher cytotoxicity in tumor cells than in normal cells. The IC50 value for L3 was lowest (50 µM) among the ligands studied. L3 terminated cell cycle at S, G2/M phases and enhanced sub-G1 populations, manifesting induction of apoptosis in tumor cells. Confocal studies indicated that nucleoside triazole ligands (L2/L3) cause higher DNA fragmentation than other ligands. Preclinical experiments with tumor-induced mice showed greater reduction in tumor size with L3. In vitro DNA synthesis reaction with L3 exhibited higher DNA synthesis inhibition with quadruplex forming DNA (QF DNA) than non quadruplex forming DNA (NQF DNA). Tm of quadruplex DNA increased in the presence of L3, indicating its ability to enhance stability of quadruplex DNA at elevated temperature and the results indicate that it had higher affinity towards quadruplex DNA than the other forms of DNA (like dsDNA and ssDNA). From western blot experiment, it was noticed that telomerase expression levels in the tissues of tumor-induced mice were found to be reduced on L3 treatment. Microcalorimetry results emphasise that two nucleoside triazole ligands (L2/L3) interact with quadruplex DNA with significantly higher affinity (Kd≈10−7 M). Interestingly the addition of an electronegative moiety to the phenyl group of L2 enhanced its anti-proliferative activity. Though IC50 values are not significantly low with L3, the studies on series of synthetic 1,2,3-triazole ligands are useful for improving and building potential pro-apoptotic ligands.
Introduction
Drugs used for the control of cancer are broadly classified into two groups, cytotoxic (cell killing) and cytostatic (cell stabilizing). Several nitrogen-containing ligands have been successfully tested as cytotoxic drugs for cancer treatment. Among them, triazole derivatives were found to be ideal because they are non-toxic, more likely to be water soluble and highly stable [1]. They are not naturally occurring scaffolds but their roles in analgesic [2], anti-inflammatory [3], [4], antiviral [5], antimicrobial [6], [7], antifungal [8]–[10], antibacterial [11], antitubercular [12] and antitumor [13] activities are well documented. Further, several triazole derivatives were tested for their anticancer activity [14]. Considering the broad spectrum of activities that triazoles exhibit, 2 different classes of triazole scaffolds (with and without nucleoside) were synthesized and their physico-chemical, biological characteristics as well as their efficacy in reducing tumor size and control tumor cell proliferation was examined.
During 1960s, Zidovudine (AZT), an azido derivative, was designed to use in chemotherapy for leukaemia [15]. But its use as an antitumor drug was discontinued as it has failed to act specifically on tumor cells. Further, azides are known for their instability, explosive and toxic nature [16]–[18]. Therefore, there is an ongoing search for ideal molecules that reduce tumor cell proliferation and the azido moiety was converted to more stable, safe and non-toxic triazole scaffold. Earlier, similar or same molecules were successfully tested as inhibitors for reverse transcriptase, Mycobacterium tuberculosis thymidine monophosphate kinase (TMPKmt) and human mitochondrial thymidine kinase 2 (TK-2) [19]–[22]. Nitrogen-containing ligands like TMPyP4, BRACO-19, Telomestatin etc., were found to be efficient in stabilizing quadruplex DNA and controlling tumor cell proliferation [23]–[27]. We have earlier reported the interaction and stabilization of quadruplex DNA by several nitrogen-containing ligands [28]–[31].
To the best of our knowledge, we are the first group to demonstrate the potential of Phenyl-1,2,3-triazole nucleosides such as 3′-Phenyl-1,2,3-triazole-thymidine (L2) and 3′-4-Chlorophenyl-1,2,3-triazole-thymidine (L3) in controlling telomerase expression levels in the tissues of tumor-induced mice, specifically stabilize telomere G-quadruplex DNA and reduce tumor cell proliferation. Recently, it was reported that 1,2,3-triazole ligands selectively bind and stabilize quadruplex DNA [32]–[34]. A mixed pyrimidine moiety having phenyl-1,2,3-triazole when included in a synthetic oligonucleotide, enhanced stability of DNA by π-π stacking of phenyl-1,2,3-triazole moiety in the major groove [35]. Recent studies indicate that quadruplex DNA stabilizing ligands have inherent ability to control telomerase and function as anti-proliferative ligands [36].
Quadruplexes are unique, non-canonical DNA structures, found in the human genome, preferably in telomeres and regions slightly upstream of the oncogene promoter. In human somatic cells, telomeres have about 500–3000 [TTAGGG] repeats, which get shortened with age. Recently, it was estimated that the human genome contains about 300000 sequences potentially capable of forming quadruplex structures [37]. G-quadruplex structures play a crucial role in regulating transcription, and therefore, have influence on cellular activities. Stabilization of G-quadruplex structure was reported to control gene expression and subsequently control oncoprotein expression in tumor cells [38].
In an attempt to improve the efficacy of synthetic azido ligand in controlling tumor cell proliferation, under less toxic conditions, an array of 1,2,3-triazole molecules were synthesised and their potentials were studied using various in vivo and in vitro experiments. We have shown that the addition of an electronegative group to the phenyl group enhance the potential of phenyl-1,2,3-triazole-thymidine ligand.
Materials and Methods
Human cell lines
Normal human cell lines CRL-2115 and CRL-2120 were obtained from American Type Culture Collection (Rockville, MD). Human tumor cell lines NCM-460, He La, MCF-7, B16F10 and A549 were obtained from National Centre for Cell Sciences, India and Lonza Cologne GmbH (Germany). The cell lines were grown according to the supplier's recommendation.
DNA and triazole ligand synthesis
Human quadruplex forming DNA [HQF DNA- 5′- TTAGGGTTAGGGTTAGGGTTAGGG-3′ or d(TTAGGG)4] from telomere region, quadru- plex forming DNA (QF DNA) and non-quadruplex forming DNA (NQF DNA) was synthesized by β-cyanoethyl phosphoramidite chemistry and purified by HPLC. Oligonucleotide stock solutions were prepared by dissolving them in BPES buffer solution (30 mM potassium phosphate, pH 7.0, with 100 mM KCl). Oligonucleotide solutions were extensively dialyzed against the BPES buffer using a 1000 Da molecular weight cut off membrane at 4°C. The final concentration was determined by UV absorbance at 254 nm with molar extinction coefficient determined by using the nearest neighbour calculation for single stranded DNA [39] and the absorbance of thermally denatured constructs extrapolated back to 25°C by total phosphate analysis technique [40]. Extinction coefficient of HQF DNA (ε254), QF DNA (ε260) and NQF DNA (ε260) was found to be 254064 M−1 cm−1, 484500 M−1 cm−1 and 219100 M−1 cm−1 respectively. The dsDNA (poly-dA-dT and poly-dT-dA) and ssDNA (poly-dT) were purchased from Sigma Aldrich (USA).
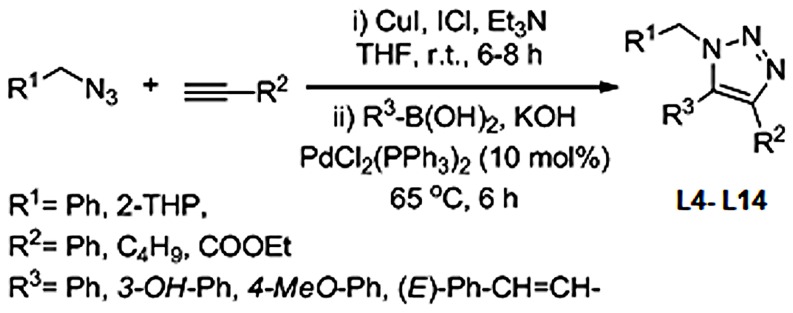
All the ligands except, 1-(4-azido-5-(hydroxylmethyl)tetrahydrofuran-2-yl)-5-methyl- pyrimidine-2,4-(1H,3H)-dione, popularly known as 3′-Azido-3′-deoxythymidine (L1), were synthesized using ‘click chemistry’. L1, a well known, commercially available, telomerase inhibitor, was used as source material for the synthesis of L2 and L3. Among triazole scaffolds, 1-(5-(hydroxyl-methyl)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione, referred in this study as 3′-Phenyl-1,2,3-triazolo-thymidine (L2) and 1-(4-(4-(4-chlorophenly)-1H-1,2,3-triazole-1-yl)-5(hydroxyl-methyl)tetra -hydrofuran-2-yl)-5-methylpyrimidine-2,3(1H,3H)-dione, referred as 3′-4-Chloro-phenyl-1,2, 3-triazolo-thymidine (L3), were synthesized using the method reported by Poecke et al [22]. The stock solutions of all the ligands used in the present study were prepared in 1∶1 water∶methanol. L2 and L3 ligands differ only with respect to the pharmacophores attached to 3′ of thymidine. The remaining non-nucleoside triazole scaffolds differ from each other in the pharmacophores attached to the 1st, 4th, 5th positions (-R1, -R2 and –R3) of 1,2,3-triazole ring. Synthesis details of nucleoside 1,2,3-triazole ligands (L2 and L3) and non-nucleoside 1,2,3-triazole ligands (L4–L14) are documented in the Figure 1 and 2 respectively.
Figure 1. Synthesis of L2 and L3 from L1.

Figure 2. One-pot synthesis of L4–L14.
MTT assay
Normal human cell lines (CRL-2115, CRL-2120) as well as human cancer cell lines (NCM-460, He La, MCF-7, B16F10 and A549) were cultured according to the supplier's recommendations. To study the cytotoxicity of the ligands, after 80% confluence, cells were trypsinized with 0.1% trypsin-EDTA and harvested by centrifugation at 500× g. Serial dilutions of cells were made from 1×106 to 1×103 cells per ml. The cells were seeded in triplicate in a 96 well plate. The suspended cells were treated with 50 µM, 100 µM, 150 µM, 200 µM, 250 µM and 300 µM of each ligand, at two different time points (24 h and 48 h). Normal as well as tumor cells to which no ligand was added served as control. After incubation, MTT (100 µl, 5 mg mL−1) solution was added to each well and the cell viability was determined by measuring the ability of cells to transform MTT to a purple coloured formazan dye. The absorbance of samples at 570 nm was measured using a UV-Visible spectrophotometer. Percentage of viable cells was calculated using the formula given below.
Where the OD570 (sample) corresponds to absorbance obtained from the wells treated with ligands and the OD570 (control) represents the absorbance from the wells in which no ligand was added.
Cell count assay
Normal and tumor cell lines were seeded at 20000–40000 cells/cm2 with 0.2 mL/cm2 media and incubated for 48 h at 37°C, 6% CO2, 95% relative humidity. Cells were treated with 200 µM, 125 µM and 50 µM of L1, L2, L3 respectively (at their respective IC50 value). After 24 h and 48 h of incubation, cells were washed with 300 µl PBS or 0.02% EDTA, trypsinized, followed by harvesting at 37°C. 200 µl cell suspension was transferred into 1.5 ml microcentrifuge tube. To the cell suspension, 300 µl PBS followed by 500 µl of 0.4% trypan blue solution was added. To get the homogenised solution, the microcentrifuge tube was gently tapped for 5 min, 20 µl of cell suspension was loaded in to a chamber of the haemocytometer.
Flow cytometry, Confocal microscopy studies
Since MTT and cell culture assays yielded encouraging results with B16F10 cells they were used for confocal and flow cytometry experiments. After 90% confluence, the cells were treated with 200 µM, 125 µM and 50 µM of L1, L2, L3 respectively, for 3 h. 1×106 cells were washed with 2× binding buffer and resuspended in 100 µl binding buffer and Annexin-V-FITC (1.0 µg). The cells were then incubated at room temperature for 10 min, followed by the addition of 400 µl of binding buffer containing 1 µl of Propidium Iodide (PI). Stained cells were analyzed using FACS Calibur Flow Cytometer from B.D. Biosceinces (USA). Annexin-V-FITC and PI labelled cells were excited using 488 nm solid-state laser and fluorescence emission intensity was captured using 530/30 and 585/42 band pass filters respectively.
For confocal microscopic studies, B16F10 cells were rinsed with 1× PBS and incubated with 2.5% formaldehyde in PBS for 10 min at room temperature, followed by permeabilization with 0.5% Triton X-100 for 5 min. The fixed cells were incubated with 10% FCS in PBS for 1 h at room temperature. The culture plate was then washed with 1× PBS and mounted in mounting medium (Vectashield), containing anti-fade reagent with 300 nM DAPI and incubated for 1–5 min. The cells were rinsed with PBS several times and viewed using a fluorescent microscope.
Stop assay
For this assay, synthetic oligonucleotides rich in guanine that have potential to form G-quadruplex DNA structure (QF-template and QF-primer) as well as oligonucleotides with less guanines, which do not form G-quadruplex DNA structure (NQF-template and NQF-primer) were considered. Sequences of the oligonucleotides used are shown in Table 1. The G-quadruplex structure forming region of QF oligonucleotide is shown in bold letters.
Table 1. Sequences of QF and NQF oligonucleotides used in stop assay.
| S.No | Oligonucleotide name | Oligonucleotide Sequence |
| 1 | QF DNA Primer | 5′-ACGACTCACTATAGCAATTGCG-3′ |
| 2 | QF DNA Template | 5′AGGGTTAGGGTTAGGGTTAGGGGCCACCGCAATTGCTATAGTGAGTCGT-3′ |
| 3 | NQF DNA Primer | 5′-AATGAA-3′ |
| 4 | NQF DNA Template | 5′-ACTTACTTACT TACTTACTTACTTACTTACTTACTT-3′ |
To the PCR reaction mixture (25 µl) containing 10 mM Tris-HCl, pH 8.3, with 50 mM KCl, 1.5 mM Mg(OAc)2, 5 µM of each oligonucleotide, 1 mM dNTPs, 5 units of Taq polymerase, L1, L2, L3 were added at various concentrations (50 µM, 100 µM, 150 µM, 200 µM), and PCR reactions were performed. PCR mixture without any ligand was considered as control. The primers were 5′ end-labelled with [γ-32P] ATP using 1 µl of polynucleotide kinase and 5 µl of 10× kinase buffer and the contents were incubated at 37°C for 5 min. Stop assay was performed using an ABI thermal cycler. Initial denaturation was done at 94°C for 3 min, followed by 20 repeated cycles each having 94°C for 30 sec, 58°C for 30 sec, and 72°C for 30 sec. After PCR, 5 µl of the amplified product was loaded on a 12% non-denaturing polyacrylamide gel with 1×TBE. The in vitro DNA synthetic band intensity was measured using Bio Rad Gel Doc XR+system (USA). The band intensity of the PCR product obtained with no ligand was taken as control for all the quadruplex forming DNA samples (QF DNA). Percentage of DNA synthesis inhibition was calculated using equation 1, whereas, inhibition constant, Ki (using Cheng-Prusoff equation) for each ligand was calculated by using equation 2.
| (1) |
C - Band intensity with control.
L - Band intensity with ligand.
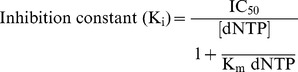 |
(2) |
IC50-Half maximal inhibitory concentration.
[dNTP] – Concentration of dNTPs used.
Km - Michaelis constant.
Preclinical studies
C57BL6/J mice were used from the animal house facility located at CCMB (Hyderabad, India). All animal experimental studies were carried out in strict accordance with the recommendations in the guide for the care and use of laboratory animals of Centre for Cellular and Molecular Biology (CCMB). All the animal experimental protocols were approved by the Institutional Animal Ethical Committee (IAEC) of Centre for Cellular and Molecular Biology (CCMB with Permit number: CPCSEA 20/1999, Ministry of Environment and Forest, Government of India).The protocol was approved by the Institutional Animal Ethical Committee (IAEC) of CCMB (Protocol number: IAEC 16/2012). All the surgery was performed under sodium pentobarbital anaesthesia, and all efforts were made to minimize suffering. C57BL6/J mice, 6–8 weeks old, were subcutaneously injected with 2×105 B16F10 melanoma cells, in the intraperitoneal region. After the tumor attained a size of 500–1000 mm3 (≈9 days following cancer cell implantation), mice were grouped and treated as follows.
Group (i) was treated with vehicle control (4 mice), group (ii) with 200 µM of L1 (4 mice), group (iii) with 125 µM of L2 (4 mice) and group (iv) with 50 µM of L3 (4 mice). These experiments were repeated thrice and mean values were considered. Size of tumor was measured every day at a fixed time. The tumor sizes were calculated using the formula (0.5 ab2), where ‘a’ was the largest dimension and ‘b’ was the smallest dimension of the tumor. The experiment was terminated when the average tumor volume of the control/treated group was 10000–12000 mm3. To validate the effect of ligands on mice survival, in the same manner, three groups of mice were taken and 200 µM, 125 µM and 50 µM of L1, L2 and L3 were injected to each mouse on the alternate days for 2 weeks and their activity was observed, till they die naturally.
Western blotting
Western blot analysis was carried out to study the expression of telomerase in ligand treated as well as untreated tissues of tumor-induced mice. Total proteins were extracted from the tumor tissues into solubilisation buffer (7 M urea, 2 M thiourea, 4% CHAPS, 18 mM Tris–HCl, 14 mM Trizma base, 2 tablets EDTA protease inhibitor, 0.2% Triton-X, 50 mM DTT) following homogenization and sonication. Extracted proteins were quantified against BSA standard using an Amido black assay method [41]. 40 µg of total protein obtained from different time points (48 h, 72 h and 216 h) were electrophoresed on a 10% SDS-PAGE and transferred onto a PVDF membrane using wet-transfer method. Immunoblot analysis of the telomerase reverse transcriptase (TERT) expression was performed using anti-TERT antibody (Pierce, Thermo Scientific) as primary antibody and anti-rabbit HP conjugated secondary antibodies (Pierce, Thermo Scientific). The immuno- blots were scanned using the ECL detection method to estimate the telomerase expression levels in the tissues of tumor-induced mice. The endogenous β-actin expression level was considered as control.
Binding and interaction studies
To further corroborate the efficiency of various synthetic ligands in interacting and stabilizing human telomeric quadruplex DNA, melting studies of DNA with and without ligands was carried out following SYBR Green fluorescence as reported earlier [42], using Eppendorf realplex4 mastercycler (Hamburg, Germany). SYBR Green (SG) was reported to intercalate to quadruplex DNA and enhance fluorescence [43]. When quadruplex DNA melts to form ssDNA structure, SG fluorescence intensity gradually reduces as SG has no binding to ssDNA. Each time, 5 µM of HQF DNA [d(T2AG3)4)], 5 µM of dsDNA (prepared by mixing equal concentration of poly-dA-dT and poly-dT-dA), 5 µM ssDNA ( poly dT alone) and 1 µM of each ligand was incubated at 4°C for 16 h, and used for melting studies. For melting studies, 1 µM of ligands was considered instead of 200 µM, 125 µM, 50 µM of L1, L2 and L3 respectively, because the aim of the experiment is to find the extent of stability provided to quadruplex DNA by each ligand at low concentration. Affinity, stoichiometry and thermodynamic parameters for quadruplex-ligand complex were studied with 60 µM of ligand and 10 µM of HQF DNA using Microcal VP-ITC (Northampton, MA, USA). Ligand addition to G-quadruplex DNA was made every 180 seconds. Titration experiments were performed thrice and blank correction was made each time to minimize error.
Statistical Analysis
The results obtained from different experiments were subjected to statistical analysis. SPSS (Version 18.0, Chicago, IL) was used for all the statistical analysis. The statistical parameters like mean value and standard deviation were calculated using descriptive statistics. Data was considered significant if p value≤0.05. The set of data with p values≤0.001, 0.01 and 0.05 were marked with symbols *, # and **/no symbol, respectively in each graphical representation.
Results and Discussion
MTT assay
This assay indicates the rate of tumor cell proliferation and useful to assess the potential of synthetic ligands in the induction of apoptosis. MTT assay was performed by following the protocol described by Wilson et al [44]. Structure of synthetic ligands used for MTT assay was shown in Figure 3A. From the assay it is clear that among the ligands used in the present study, nucleoside ligands exhibited lower half maximal inhibitory concentration (IC50) than the non-nucleotide ligands. Figure 3B indicates that the half maximal inhibitory concentration (IC50) for nucleoside ligands (L1, L2 and L3) with B16F10 cells were around 200 µM, 125 µM and 50 µM respectively. Whereas cells not treated with any ligand (control cells) continue to proliferate without any inhibition. Though the cytotoxicity of L3 ligand (50 µM), is not very significant, a comparative study of various nucleoside and non-nucleoside triazole molecules will provide insights into the role of pharmacophores attached, effect of size and structure of 1,2,3-triazole ligands on the anti-proliferative activity as well as for building suitable triazole molecules for potential antitumor activity. Triazoles are known for safety and therapeutic applications for a prolonged period. The drugs having triazole scaffold like Vorozole, Letrozole and Anastrozole are under use for cancer treatment for several years [45]. From MTT assay it is evident that among nucleoside ligands, 1,2,3-triazole ligands (L2/L3) are comparatively more efficient than azido thymidine (L1) and non-nucleoside triazole ligands (L4–L14) in bringing down tumor cell proliferation. Hence more emphasis is laid on the relatively new and higher potential, nucleoside 1,2,3-triazole ligands like L2/L3, while a well known azido derivative L1 is considered as control throughout the study.
Figure 3. Structure of ligands used and the MTT assay.

A. Structure of nucleoside 1,2,3-triazole ligands L1, L2, L3 and the basic structure of non-nucleoside 1,2,3-triazole ligands (L4–L14). The position of various pharmacophores on ligands (L4–L14) are labelled with -R1, -R2 and -R3. B. MTT assay with B16F10 cells on treatment with 50 µM, 100 µM, 150 µM, 200 µM, 250 µM and 300 µM concentration of L1, L2 and L3 for 24 h and 48 h. The set of data with reducing order of significance (p value≤0.001, 0.01 and 0.05) were marked with *, # and ** respectively.
Cell count assay
This assay was carried out to verify the efficiency of ligands in imparting cytotoxicity among different cell lines (2 normal and 5 tumor cell lines) in the presence of synthetic 1,2,3-triazole scaffolds at different periods of treatment (24 h and 48 h). Supporting the MTT data, the cell count assay results indicate that among the nucleoside and non-nucleoside triazole derivatives, the number of viable cells was low with nucleoside 1,2,3-triazole ligands indicating that they impart higher cytotoxicity in tumor cells. Nucleoside triazole ligands inhibited tumor cell proliferation more than the normal cell lines probably due to higher uptake of the ligands by rapidly dividing tumor cells, and their structural similarity with the natural nucleosides. Results emphasise that among the triazole ligands studied, L3 exhibited greater inhibition of tumor cell proliferation. This was followed by L2 and L1. The bar graphs shown in Figure S1 indicate that proliferation of tumor cells decreases gradually from L1 to L3 both in the case of 24 h and 48 h of ligand treatment. The extent of decrease of tumor cell proliferation was comparatively more with B16F10 and A549 cell lines. Details of various pharmacophores attached to 1,2,3-triazole moiety at 1st, 4th and 5th positions, are tabulated in Table S1 in Supplementary Data S1. The results obtained in cell count assays with both nucleoside and non-nucleoside synthetic triazole ligands on 7 different cell lines (both normal and tumor) are shown in Table 2.
Table 2. Effect of nucleoside and non-nucleoside ligands on normal and tumor human cell lines.
| S.No | Ligand | Number of viable cells after 24/48 h of ligand treatment | ||||||
| CRL-2115 | CRL-2120 | NCM-460 | He La | MCF-7 | B16F10 | A549 | ||
| 1. | L1 | +/++ | +/++ | +/++ | +/++ | +/++ | ++/+++ | +/++ |
| 2. | L2 | +/++ | +/++ | +/++ | +/++ | +/++ | ++/+++ | ++/++ |
| 3. | L3 | +/++ | +/++ | +/++ | ++/++ | ++/++ | ++/+++ | ++/+++ |
| 4. | L4 | +/++ | +/+ | +/++ | +/++ | ++/++ | ++/++ | +/++ |
| 5. | L5 | +/++ | +/++ | +/++ | ++/++ | ++/++ | ++/++ | +/++ |
| 6. | L6 | +/++ | +/++ | ++/++ | ++/++ | +/++ | +/++ | ++/++ |
| 7. | L7 | +/+ | +/++ | +/++ | +/+ | +/++ | +/++ | +/+ |
| 8. | L8 | +/+ | +/+ | +/++ | +/++ | +/+ | +/++ | +/++ |
| 9. | L9 | +/++ | +/+ | +/+ | +/++ | +/+ | +/++ | +/++ |
| 10. | L10 | +/+ | +/+ | +/++ | +/+ | +/++ | +/++ | +/+ |
| 11. | L11 | +/+ | +/+ | +/+ | +/++ | +/++ | +/++ | +/++ |
| 12. | L12 | +/+ | +/+ | +/++ | +/++ | +/++ | +/++ | +/+ |
| 13. | L13 | +/+ | +/+ | +/+ | +/++ | +/+ | +/++ | +/+ |
| 14. | L14 | +/+ | +/+ | +/++ | +/+ | +/++ | +/++ | +/++ |
+ - Viable cells in the range of 80%–90% ; ++ - Viable cells in the range of 50%–60% ; +++ - Viable cells which are equal or less than 30%.
Flow cytometry studies
Flow cytometry experiments provide insight into the effect of ligands on various phases of cell cycle and their influence on apoptosis. Cell cycle analysis suggests that in control samples (untreated B16F10 cells), ≈60% of all cells accumulated in the G0/G1 phase, ≈24% in the S phase, and ≈8.3% in the G2/M phase. Only 1.5% of the cells are in the subG1 phase. B16F10 tumor cell lines on treatment with 50 µM of L3, resulted in reduction of percentage of cells in G0/G1 (4.46%), S (5.36%) and G2/M (0.91%) phases compared to untreated cells, while the percentage of cells in subG1 phase increased to 89.27%, indicating enhanced DNA fragmentation and induction of apoptosis in tumor cells. On the other hand, cells treated with even 200 µM and 125 µM of L1 and L2 exhibited a moderate decrease in the percentage of cells in G0/G1 (21.24% and 6.43%), S (8.36% and 8.77%) and G2/M (5.58% and 1.93%) phases, compared to untreated cells. The subG1 population in these samples increased to 61.10% and 81.5% respectively. The deconvoluted DNA histograms have been displayed in Figure 4A. The histogram depicts that the subG1 cell population has been enhanced to 10–30% with L3 compared to other nucleoside ligands. Flow cytometry data indicates that cell cycle arrest occurs at S and G2/M phases, accompanied by an increase in the subG1 population with L3, indicating its potential to inhibit DNA synthesis followed by induction of apoptosis in tumor cells.
Figure 4. Cell cycle and dot plots obtained with three nucleoside ligands.

A. Untreated, L1, L2 and L3 treated B16F10 cell lines were stained with Propidium Iodide (PI) and cellular DNA content frequency histograms were shown. B. B16F10 cells and L1,L2 and L3 treated cells were stained with Annexin V and Propidium Iodide and analysed for various types of cells like alive (LL), early apoptotic (LR), late apoptotic (UR) and necrotic cells (UL).
From the dot plot shown in Figure 4B, indicate that the percentage of necrotic and viable cells has reduced and the early or late apoptotic cells have increased on treating the cells with L3. This indicates that L3 is more potent in inducing apoptosis in tumor cells than L1 and L2. Under the same conditions, when B16F10 cells were treated with L2, more necrotic cells were observed compared to apoptotic cells. In L2 treated cells, the subG1 population has increased due to the accumulation of necrotic cells rather than that of apoptotic cells. The dot blot results indicate that treatment of B16F10 cells with L3 resulted in more apoptotic cells (81.25%) than the cells treated with L2 (23.89%) and L1 (14.66%).The percentage of cells accumulated in each phase of cell cycle as well as in different stages of apoptosis were shown in Tables 3 and 4 respectively. Among these phenyl-1,2,3-triazole-thymidine ligands, L3 was found to be more ideal as it has higher capability to induce apoptosis in tumor cells.
Table 3. Distribution of cells in various phase of cell cycle in control and L1, L2 and L3 treated B16F10 cells.
| S.No | Sample Name | Sub G1 | G0/G1 | S | G2/M |
| 1 | Control | 01.52% | 66.41% | 23.81% | 08.30% |
| 2 | L1 | 61.10% | 26.94% | 06.38% | 05.58% |
| 3 | L2 | 81.52% | 07.78% | 08.77% | 01.93% |
| 4 | L3 | 89.27% | 04.46% | 05.36% | 00.91% |
Table 4. Percentage of alive, early apoptotic, late apoptotic and necrotic cells in control and L1, L2 and L3 treated B16F10 cells.
| S.No | Sample Name | Cells Alive | Early apoptotic | Late apoptotic | Necrotic | Total apoptotic cells |
| 1 | Unstained Control | 99.67% | 00.04% | 00.27% | 00.02% | 00.31% |
| 2 | Stained Control | 75.72% | 00.46% | 00.47% | 23.35% | 0.93% |
| 2 | L1 | 68.78% | 02.61% | 12.05% | 16.56% | 14.66% |
| 3 | L2 | 22.88% | 04.82% | 19.07% | 53.23% | 23.89% |
| 4 | L3 | 17.79% | 20.99% | 60.26% | 00.96% | 81.25% |
Confocal microscopy
It is evident from the flow cytometry analysis that L3 has enhanced subG1 population of cells, reduced the percentage of cells in the S phase and effectively inducing apoptosis in B16F10 cells. In order to understand the action of nucleoside 1,2,3-triazole ligands, B16F10 cell lines were treated with L1, L2, L3 at their respective half maximal inhibitory concentration (i.e. 200 µM, 125 µM and 50 µM) for 24 h period. Examination of ligand treated cells under the fluorescence microscope indicated higher nuclear condensation and DNA fragmentation in nucleoside triazole ligand treated tumor cells than in untreated cells. The cell surface was altered or shrunken, DNA fragmented was enhanced and the number of apoptotic cells was increased in cells treated with L3 compared to L1/L2. Figure 5 shows the confocal microscopy results with B16F10 cell lines on treatment with nucleoside triazole ligands. Flow cytometry and confocal microscopy results shows that among the nucleoside ligands tested, induction of apoptosis in tumor cells and fragmentation was highest in the presence of L3 followed by L2 and L1.
Figure 5. DNA fragmentation in B16F10 cells on nucleoside ligands treatment.
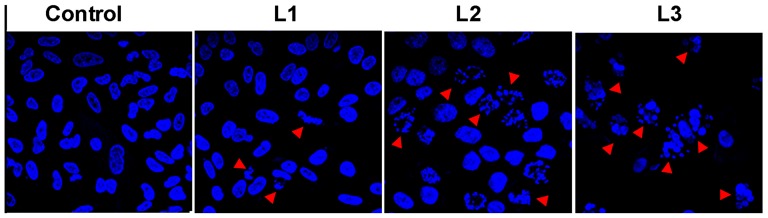
B16F10 cells were treated with 200 µM of L1, 125 µM of L2 and 50 µM of L3 for 24 h. Control cells were treated with equal volume of 1∶1 methanol: water without ligand.
Preclinical studies
Experiments such as flow cytometry, confocal microscopy and MTT assay with normal and tumor cell lines indicate that L3 has the potential to terminate cell cycle at S phase, increase subG1 population, enhance DNA fragmentation and induce apoptosis in tumor cells at comparatively lower concentration (50 µM). However, the effect of synthetic triazole ligands under physiological conditions (under in vivo situation) provides unfeigned data on the actual potential of ligands as anti-proliferative agents. Therefore, it would be interesting to observe variations in tumor sizes among tumor-induced mice after ligand treatment. Figure 6 indicates that in the group of mice treated with L1/L2, the tumor size increased to 6000–8000 mm3, whereas in the group of mice treated with L3, the size of tumor remained at ≈4500 mm3. Thus, in vivo data also support earlier experimental results and indicate that L3 has better tumor controlling capability than L1/L2.
Figure 6. Varation of induced tumor size on treatment with nucleoside ligands.

Tumor was induced in C57BL6/J mice by injecting B16F10 cells subcutaneously and treated with 200 µM of L1, 125 µM of L2 and 50 µM of L3 and an equal volume of 1∶1 methanol: water (without ligand) was treated as vehicle control. Group treated with an equal volume of 1∶1 methanol: water, vehicle control (4 mice) marked •. Group treated with L1, L2 and L3 (4 mice each) were marked ▪, ▴ and ▾ respectively. Tumor volumes were measured at fixed time points and expressed in mm3. Each experiment was repeated thrice and the mean values were plotted. The set of data with p value≤0.001 and 0.01 were marked with * and # respectively. The statistical data with p value≤0.05 was left unmarked.
Further, the survival of mice treated with different ligands was checked and compared with the control group. The details of survival data obtained from 4 different sets of mice are tabulated in Table S2 in Supplementary Data S1. Among the mice treated with nucleoside ligands, the mice treated with L3 survived for longer period (about 6 weeks) indicating that L3 was efficient in controlling tumor cell proliferation without developing harmful effects in tumor-induced mice. Untreated mice, L1 and L2 treated mice died earlier (approximately in 3, 4 and 5 weeks, respectively). Experiments to understand the reason for higher longevity of mice with L3 are in progress.
Stop assay or Inhibition of In vitro DNA synthesis
It is already known that 3′ modified nucleosides inhibit DNA synthesis. Earlier, 3′-Phenyl-1,2,3-triazole-thymidine and similar kind of triazole nucleoside ligands were reported to terminate DNA synthesis in tumor cell lines [46]. In the present study, flow cytometry and confocal studies indicate that the cell cycle was inhibited at S phase and DNA fragmentation occurred on treatment with nucleoside 1,2,3-triazole ligands. Higher the efficiency of inhibition of DNA synthesis, greater will be the effect of the ligand on DNA fragmentation.
The role of nucleoside triazole ligands in inhibiting DNA synthesis by QF DNA and NQF DNA was studied by using the protocol reported earlier [47], [48]. Inhibition of DNA synthesis was studied using 5 µM QF DNA and nucleoside triazole ligands (L1, L2, L3) at different concentrations (50 µM, 100 µM, 150 µM, 200 µM). It was found that the inhibition of DNA synthesis with QF DNA was considerably high (around 80–90%). The in vitro DNA synthesis products obtained with QF DNA and L1, L2, L3 were resolved by loading on 12% nondenaturing polyacrylamide gel as shown in Figure S2. The results reveal that L3 completely inhibited DNA synthesis in the concentration range of 50–100 µM, whereas with L1 and L2, maximum inhibition was observed at higher ligand concentrations (150–200 µM). The results matched with the data provided by MTT assay. However, inhibition of in vitro DNA synthesis with NQF DNA was studied only with L3 ligand, because it was identified as the most potential anti-proliferative ligand among the triazole scaffolds considered in the present study. Competitive inhibition of DNA synthesis by L3 ligand in the presence of natural dNTPs and NQF DNA is shown in Figure S2A. With NQF DNA, inhibition of in vitro DNA synthesis by L3 was moderate (around 33%). From Figure 7A, it is evident that the intensities of synthetic DNA bands were less with L2/L3 than L1, indicating Phenyl triazole ligands were the better DNA synthesis inhibitors than azido scaffolds. The competitive inhibition by various nucleoside triazole ligands and the inhibition constants (Ki) for each of the ligands was calculated using Cheng-Prusoff equation (equation-2, shown under experimental data). They were found to be 2.94 (±0.091) µM, 1.84 (±0.079) µM and 0.735 (±0.087) µM for L1, L2 and L3, respectively. The inhibition constant with L3 was lowest among those tested, indicating its ease to get incorporated into the growing DNA chain.
Figure 7. Effect of ligands on in vitro DNA synthesis inhibition and telomerase expression.

A. Line graph indicating the variation in band intensity with L1, L2 and L3 ligands at various concentrations using QF DNA and L3 alone with NQF DNA. B. Percentage of DNA synthesis inhibition by L1, L2 and L3 with QF DNA and L3 alone with NQF DNA. C. Western blot showing the TERT and β-actin bands. Lane 1,2,3 corresponds to 48 h, 72 h and 216 h of treatment with vehicle control (control samples); lane 4,5,6, lane 7,8,9 and lane 10,11,12 corresponds to 48 h, 72 h and 216 h of L1, L2 and L3 treatment respectively. Top and bottom bands correspond to TERT and β-actin respectively. D. Bar graph showing the comparative intensities of each band in western blot. The numbers marked on the x-axis represents the number of sample in western blot. The set of data with reducing order of significance (p value≤0.001, 0.01 and 0.05) were marked with *, # and ** respectively.
The results obtained from the stop assay indicate that quadruplex complex formation and stabilization of quadruplex complex by suitable ligands would enhance inhibition of DNA synthesis. With NQF DNA, about 33% inhibition was observed, indicating misincorporation of L3 into the growing DNA strand alone was causing inhibition of DNA synthesis. The misincorporation of modified nucleosides probably plays a primary role in terminating the DNA synthesis. However, the inhibition of DNA synthesis was less with NQF DNA compared to QF DNA, emphasising the role of quadruplex formation in DNA synthesis inhibition. Intensity of synthesized DNA bands obtained with different ligands using QF and NQF DNA are shown in Figure S2. The percentage of DNA synthesis inhibition by L1, L2 and L3 with QF DNA at the respective half maximal inhibitory concentration was calculated using equation-1 (shown under experimental section) and was found to be 84%, 91% and 94%, respectively. Percentage of DNA synthesis inhibition with QF and NQF DNA is graphically shown in Figure 7B. Figure S3 illustrates the proposed mechanism by which L3 terminates DNA synthesis in NQF DNA and QF DNA.
Western blot
Telomerase has been reported to play an important role in tumorigenesis [49]. Overproduction of telomerase has been observed in more than 85% of tumor cells than in normal cells [50]. It was reported earlier that an efficient telomerase inhibitor such as 3′-azido-3′-deoxythymidine (L1) [51]–[55] induces apoptosis in tumor cells. To find out the effect of phenyl-triazole ligand treatment on the level of telomerase expression in the tissues of tumor-induced mice, western blot assay was performed. Western blot results show that among the nucleotide triazole ligands, level of telomerase expression was low in tumor tissues treated with L3 than in L1 and L2. Figure 7C shows that telomerase expression levels were higher in control tissue samples (untreated tumor tissue), than in the ligand treated ones. We speculate that the reduction of telomerase expression was due to continuous inhibition of telomerase activity by nucleoside ligands. Levels of telomerase expression reduced gradually in the tumor tissues treated with nucleoside ligands from 48 h to 216 h of treatment (as shown in Figure 7D). Percentages of reduction of telomerase expression on 216 h of treatment with L1, L2 and L3 were calculated using the equation 1, (under materials and methods section) and they were found to be 80%, 88% and 95%, respectively.
Binding and interaction studies
Stop assay results show that stabilization of quadruplex DNA and the inhibition of DNA synthesis were related to each other and it has some role to play in induction of apoptosis. G-quadruplex DNA interaction with 3′ modified nucleoside ligands would throw some light on the nature of DNA-ligand interactions, their potential to stabilize G-quadruplex DNA and role of ligand structure in quadruplex DNA-ligand interaction. ITC and melting studies were carried out to investigate the efficiency of ligands interaction, mode of binding and affinity of ligands towards human telomere DNA (HQF DNA).
Isothermal titration calorimetry is a sensitive microcalorimetric technique for determining binding affinity, stoichiometry and thermodynamic parameters of quadruplex DNA-ligand complex [56]. Interaction between HQF DNA (10 µM) with the nucleoside ligands (60 µM) was performed at 25°C in 100 mM KBPES (pH 7.0) buffer and the data obtained are shown in Table S3 in Supplementary Data S1. On integrating the heats produced per injection, with respect to time and conversion to per mole basis, the binding isotherm (Figure 8A) corresponding to each ligand was obtained. Figure 8A indicates that two ligand molecules would interact with each molecule of quadruplex DNA. After making blank correction, the heat data was fitted with “two-independent sites” model and analyzed using Origin 7.1 to calculate thermodynamic parameters.
Figure 8. Microcalorimetric and DNA melting studies in presence of nucleoside ligands.
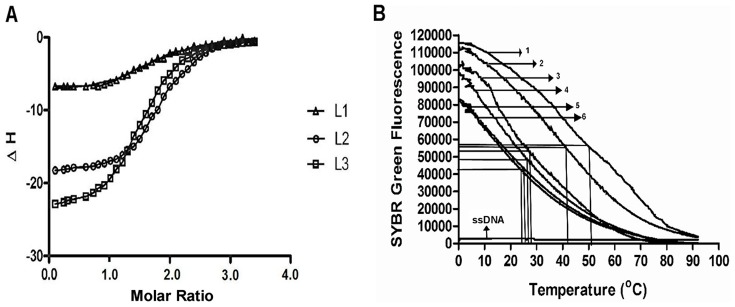
A. ITC binding isotherms obtained when L1, L2 and L3 ligands were titrated with HQF DNA in 100 mM KBPES (pH 7.0) buffer. B. Melting profile of DNA with various ligands. HQF DNA +L3 (marked-1); HQF DNA+L2 (marked 2); HQF DNA+ L1 - (marked 3) ; HQF DNA alone - (marked 4); dsDNA+ L3 ( marked 5); dsDNA alone (marked 6) and ssDNA (poly-dT alone).
The microcalorimetry results indicate that the interaction of ligand takes place in two different modes : one with higher affinity (mode-1, preferred interaction) and the other with lower affinity (mode-2, less preferred interaction). We propose that the higher affinity interaction (mode-1), may be due to the ligand binding externally to quadruplex DNA. The mode-1 interaction occurs with higher binding constant (Kd≈10−7 M) and with more favourable entropy change. The low affinity interaction (mode-2) is due to either π-π stacking interaction of nucleoside triazole ligands with guanines of quadruplex DNA or due to binding of ligands to adenosine bases in the loop (end loop binding with binding constant in the range of Kd≈10−5 M), which occurs with more favourable enthalpy change [57].
Melting studies
In the present study, SYBR Green fluorescence was monitored to find melting of ligand-DNA complexes and to know the efficacy of each triazole ligand in stabilizing the quadruplex complex. Among the different synthetic ligands, the Tm of HQF DNA was 52°C with L3, 42°C with L2 and 28°C with L1. Tm of dsDNA alone, dsDNA with L3, HQF DNA alone and HQF DNA with other non-nucleoside triazole scaffolds (L4–L14) were observed to be 24°C, 25°C, 27°C and 27–33°C, respectively. SYBR Green fluorescence was very low with single stranded DNA, because SYBR Green has less binding to ssDNA. Tm of dsDNA does not alter much in the presence or absence of L3, suggesting that L3 has minimal interaction with dsDNA. These results indicate that L3 specifically interacts with quadruplex DNA and stabilizes the complex. It is interesting to note that attaching an electronegative moiety to the phenyl group of L2 ligand, enhance the stability of quadruplex DNA. From the present study, we clearly demonstrate that quadruplex DNA stabilizers act as efficient inhibitors of DNA synthesis, telomerase expression and act as pro-apoptotic ligands. It was shown earlier that ligands that stabilize quadruplex DNA inhibit telomerase expression [58], [59] and function as antitumor agents.
From the melting assay results, the order of HQF DNA stabilization by triazole and azido ligands is as follows: L3>L2>(L4–L14)>L1. Results indicate that size and shape of the interacting ligand is crucial for efficient stabilization of quadruplex DNA. It was shown earlier that ligands such as TMPyP4 provide higher stability to quadruplex DNA because its molecular shape and size is similar to that of guanine tetrad (diameter of quadruplex DNA complex is ≈13°A) [60], [61]. Previous studies on quadruplex DNA–ligand interactions indicate that molecules having molecular weight in the range of 2000–5000 Da provide better stability to quadruplex DNA [62]. The non-nucleoside ligands L4–L14 fail to stabilize quadruplex DNA probably because of their smaller size or weaker interaction with quadruplex DNA. Though the IC50 values were not significant, studies on nucleoside ligands such as L1, L2 and L3 are useful in building new molecules with improved pro-apoptotic activity, enhance stability of quadruplex DNA, effective inhibition of DNA synthesis. The details of structure of both nucleoside and non-nucleoside ligands, melting temperature of HQF DNA in presence of L1, L2, L3 and L4–L14 and their IUPAC names are provided in Table S4 in Supplementary Data S1 and the melting profiles of HQF DNA with L1, L2, L3, HQF DNA alone, dsDNA, dsDNA with L3, ssDNA are shown in Figure 8B.
By and large, from the results obtained from the present study, the following aspects were deduced about the potential 1,2,3-triazole ligand namely, 3′-Chrolophenyl-1,2,3-triazole-thymidine:
The nucleoside triazole scaffolds like 3′-Phenyl-1,2,3-triazole-thymidine and 3′-4-Chloro-phenyl- 1,2,3-triazole-thymidine ligands (L2 and L3) exhibited higher selectivity and greater stability to quadruplex DNA than the 3′-azidothymidine (L1) and non-nucleoside triazole scaffolds (L4–L14).
Termination of in vitro DNA synthesis by 3′ modified nucleosides is primarily due to its misincorporation into the growing DNA strand. Termination was effective in QF DNA (about 3 times more effective) than in NQF DNA, indicating the role of formation and stabilization of quadruplex DNA complex in the inhibition of DNA synthesis. Higher the quadruplex DNA stabilization, more efficient would be the inhibition of DNA synthesis.
L3 inhibits in vitro DNA synthesis more efficiently than the other two nucleoside triazole ligands. The inhibition constant (Ki) was found to be the lowest (0.735 µM) among the nucleoside triazole ligands used in the present study, indicating higher affinity of L3 towards polymerase enzyme and ease to get incorporated into the growing DNA strand.
L3 reduces telomerase levels at lower concentration (50 µM) than L1/L2. L1 was reported to inhibit telomerase at concentration around 200 µM [63].
DNA fragmentation, inhibition of in vitro DNA synthesis and induction of apoptosis was more with 3′-4-Chlorophenyl-1,2,3-triazole-thymidine (L3) than the 3′-Phenyl-1,2,3-triazole-thymidine (L2). This shows that attaching an electronegative element to the phenyl group, improves the pro-apoptotic activity of the ligand and function as better substrate for enzymatic reactions. Similar observation was made earlier with 3′-Flurophenyl-1,2,3-triazole-thymidine [64].
Conclusion
From the present study, we report that among the library of 1,2,3-triazole ligands, nucleoside ligands are more efficient than non-nucleoside ligands in controlling tumor cell proliferation. Among the nucleoside ligands, 4-Chlorophenyl-1,2,3-trizole-thymidine (L3) exhibits higher potential in bringing down tumor cell proliferation. L3 enhances subG1 population of cells, DNA fragmentation and induces apoptosis in tumor cells. Among the nucleoside 1,2,3-triazole ligands, L3 has profound effect on tumor size reduction, enhancing longevity and lowering telomerase levels in tumor-induced mice. It is interesting to observe that L3 specifically interact and effectively stabilize quadruplex DNA. Stop assay results indicate that quadruplex DNA structure stabilization by synthetic ligands will effectively inhibit in vitro DNA synthesis. L3 binds externally to HQF DNA and stoichiometry of quadruplex DNA: nucleoside ligand is 1∶2. Even though the IC50 values of Phenyl-1,2,3-triazole-thymidine molecules (L2 and L3) are not significantly low to consider them as a candidate molecules for therapeutic purposes, the results from the present study are useful in understanding the role of pharmacorphores on pro-apoptotic activity of ligands, for building suitable molecules to improve quadruplex DNA stability, inhibit in vitro DNA synthesis and reduce expression of telomerase in tumor cells.
Supporting Information
Cell count assay using normal and tumor cell lines. Cell count assay results with normal as well as tumor cell lines after 24 h and 48 h of treatment with nucleoside ligands (L1, L2 and L3). Experiment was repeated thrice and the mean values were plotted. The set of data with reducing order of significance (p value≤0.001, 0.01 and 0.05) were marked with *, # and ** respectively.
(TIF)
Inhibition of in vitro DNA synthesis with NQF and QF DNA in presence of nucleoside ligands. A. Stop assay with L3 and NQF DNA: Lane 1–5 show 5 µM NQF DNA with 0 µM, 50 µM,100 µM,150 µM,200 µM of L3. Figure 7 B,C,D. Stop assay with L1, L2 and L3 and QF DNA: Lane 1–5 show 5 µM QF DNA with 0 µM, 50 µM, 100 µM, 150 µM, 200 µM of L1, L2 and L3 respectively. The control lane for QF DNA is considered common for B,C and D figures.
(TIF)
Possible mechanism of termination of DNA synthesis with NQF and QF DNA in presence of L3. Schematic representation of mechanism of termination of DNA synthesis followed by DNA fragmentation and induction of apoptosis with QF and NQF DNA in presence of L3 and dNTPs.
(TIF)
Contains Tables S1, S2, S3, S4.
(DOCX)
Acknowledgments
We are thankful to the Directors of CSIR-CCMB and CSIR-IICT, for their encouragement and support to carry out these experiments. Special thanks to Dr. T Ramakrishna and Dr. M K Chattopadhyay (Centre for Cellular and Molecular Biology) for critically reading the manuscript.
Funding Statement
Part of the work was partly supported by Indo Swiss Joint Research Programme (ISJRP) with grant number CH: 138844. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kaval N, Drmolat'ev D, Appukkuttan P, Dehaen W, Kappe CO, et al. (2005) The application of “Click Chemistry” for the decoration of 2(1-H)- Pyrazinone scaffold: Generation of templates. J Comb Chem 7: 490–502. [DOI] [PubMed] [Google Scholar]
- 2. Mathew V, Keshavaya J, Vaidya VP, Giles D (2007) Studies on synthesis and pharmacological activities of 3,6-disubstituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and their dihydro analogues. Eur J Med Chem 42: 823–840. [DOI] [PubMed] [Google Scholar]
- 3. Kamotra P, Gupta AK, Gupta R, Somal P, Singh S (2007) Micro-wave assisted synthesis and biological activity of 3-alkyl/aryl-6-(1-chloro-3,4-dihydronaphth-2-yl)-5,6-dihydro-s-triazolo [3,4-b] [1,3,4] thiadiazoles. Indian J Chem 46B: 980–984. [Google Scholar]
- 4. Michael HC, Krishnaswamy S, Hao Q, Duan SX, Patten CJ, et al. (2003) Evaluation of 3′-Azido-3′-deoxythymidine, morphine, and codeine as probe substrates for UDP-Glucurono- syltrasferase 2B7(UGT2B7) in human liver microsomes:specificity and influence of the UGT2B7*2 polymorphism. Drug metabolism and disposition: the biological fate of chemicals 31: 1125–1133. [DOI] [PubMed] [Google Scholar]
- 5. Tomita K, Tsuzuki Y, Shibamori K, Tashima M, Kajikawa F, et al. (2002) Synthesis and structure activity relationships of novel 7-substituted-1,4-dihydro-4-oxo-1-(2thia zolyl)-1,8-aphthyridine-3-carboxylic Acids as antitumor Agents. J Med Chem 45: 5564–5569. [DOI] [PubMed] [Google Scholar]
- 6. Demirbas N, Karaoglu SA, Demirbas A, Sancak K (2004) Synthesis and antimicrobial activities of some new 1-(5-phenylamino-[1,3,4]thiadiazol-2-yl)methyl-5-oxo [1,2,4]triazole and 1-(4-phe-nyl-5 thioxo-[triazol-3yl)methyl-5-oxo [1,2, 4]triazole derivatives. Eur J Med Chem 39: 793–804. [DOI] [PubMed] [Google Scholar]
- 7. Swamy SN, Basappa, Priya BS, Prabhuswamy B, Doreswamy BH, et al. (2006) Synthesis of pharmaceutically important condensed heterocyclic 4,6-disubstituted-1,2,4-triazolo 1,3,4- thiadiazole derivatives as anti- microbials. Eur J Med Chem 41: 531–538. [DOI] [PubMed] [Google Scholar]
- 8. Tsukuda T, Shiratori Y, Watanabe MH, Ontsuka K, Hattori N, et al. (1998) Modeling, synthesis and biological activity of novel antifungal agents. Bioorg Med Chem Lett 8: 1819–1824. [DOI] [PubMed] [Google Scholar]
- 9. Hirpara HM, Sodha VA, Trivedi AM, Khatri BL, Parikh AR (2003) Microwave assisted synthesis and biological activities of some bridge-head nitrogen heterocycles. Indian J Chem 42B: 1756–1759. [Google Scholar]
- 10. Isloor AM, Kalluraya B, Shetty P (2009) Regioselective reaction: synthesis, characterization and pharmacological studies of some new Mannich bases derived from 1,2,4-triazoles. Eur J Med Chem 44: 3784–3787. [DOI] [PubMed] [Google Scholar]
- 11. Wang Z, You T, Xu HY, Haoxin S (1996) Synthesis and biological activities of 2-substituted-5-(b – pyridyl ) -2,3- dihydro- 1,2,4- triazolo [3,4-b] -1,3,4- thiadiazoles. Molecules 1: 68–73. [Google Scholar]
- 12. Udupi RH, Kushnoor A, Bhat AR (1999) Synthesis and biological evaluation of certain pyrazoline derivative of 2-(6-methoxy-naphthyl)-propionic acid. J Indian Chem Soc 76: 461–464. [Google Scholar]
- 13. Shivarama HB, Narayana PK, Sooryanarayana RB, Shivananda MK (2002) New bis-aminomercapto- triazoles and bis-triazolothi-adiazoles as possible anticancer agents. Eur J Med Chem 37: 511–517. [DOI] [PubMed] [Google Scholar]
- 14. Li X, Lin Y, Yuan Y, Liu K, Qian X (2011) Novel efficient anticancer agents and DNA-intercalators of 1,2,3-triazol-1,8-naphthalimides: design, synthesis, and biological activity. Tetrahedron 67: 2299–2304. [Google Scholar]
- 15. Horwitz JP, Chua J, Noel MJ (1964) Monomessylates of 1-(2]-Deoxy-Beta-D-Lyxo- furanosyl) Thymine,. Journal of Organic Chemistry 29 7: 2076–2078 doi: 10.1021/ jo01030a546 [Google Scholar]
- 16. Hassner A, Stern M (1986) Synthesis of Alkyl Azides with a Polymeric Reagent. Angewandte Chemie International Edition in English 25 5: 478–479 doi: 10.1002/anie. 198604781 [Google Scholar]
- 17. Hassner A, Stern M, Gottlieb HE, Frolow F (1990) Synthetic methods. 33. Utility of a polymeric azide reagent in the formation of di- and triazidomethane. Their NMR spectra and the X-ray structure of derived triazoles. J Org Chem 55 8: 2304–2306 doi:10.1021/ jo00295a014 [Google Scholar]
- 18. Marinescu L, Thinggaard J, Thomsen IB, Bols V (2003) Radical Azidonation of Aldehydes. J Org Chem 68 24:: 9453–9455 doi:10.1021/jo035163v [DOI] [PubMed] [Google Scholar]
- 19. Priego E -M, Karlsson A, Gago F, Camarasa M -J, Balzarini J, et al. (2012) Recent advances in Thymidine Kinase 2(TK2) inhibitors and new perspectives for potential applications. Curr Pharml Design 18: 2981–2994. [DOI] [PubMed] [Google Scholar]
- 20. Olomola TO, Klein R, Lobb KA, Sayed Y, Kaye PT (2010) Towards the synthesis of coumarin derivatives as potential dual-action HIV-1 protease and reverse transcriptase inhibitors. Tetrahed- ron Letts 51: 6325–6328. [DOI] [PubMed] [Google Scholar]
- 21. Poecke SV, Lehmann HM, Hel-ynck O, Froeyen M, Calenbergh SV (2011) Synthesis and inhibitory activity of thymidine analogues targeting Mycobacterium tuberculosis thymidine monophos- phate kinase. Bioorg Med Chem 19: 7603–7611. [DOI] [PubMed] [Google Scholar]
- 22. Poecke SV, Negri A, Gago F, Daele IV, Solaroli N, et al. (2010) 3′-[4-Aryl-(1,2,3-triazol-1-yl) 3′deoxy thymidine Analogues as Potent and Selective Inhibitors of Human Mitochondrial Thymidine Kinase. J Med Chem 53: 2902–2912. [DOI] [PubMed] [Google Scholar]
- 23. Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, et al. (2005) The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res 65: 1489–1496. [DOI] [PubMed] [Google Scholar]
- 24. Harrison RJ, Cuesta J, Chessari G, Read MA, Basra SK, et al. (2003) Trisubstituted acridine derivatives as potent and selective telomerase inhibitors. J Med Chem 46: 4463–4476. [DOI] [PubMed] [Google Scholar]
- 25. Caterina ML, Martinez IS, Haider S, Valerie G, Edwin DP, et al. (2010) Structure-based design of selective high-affinity telomeric quadruplex-binding ligands. Chem Commun 46: 9116–9118. [DOI] [PubMed] [Google Scholar]
- 26. Tauchi T, Shin-Ya K, Sashida G, Sumi M, Obake S, et al. (2006) Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: in vitro and in vivo studies in acute leukaemia. Oncogene 25: 5719–5725. [DOI] [PubMed] [Google Scholar]
- 27. Strahl C, Blackburn EH (1996) Effects of reverse transcriptase inhibitors on telomere length and telomerase activity in two immortalized human cell lines. Mol.Cell.Biol 16: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raju G, Srinivas R, Santhosh Reddy V, Idris MM, Kamal A, et al. (2012) Interaction of Pyrrolobenzodiazepine (PBD) ligands with Parallel Intermolecular G-Quadruplex Complex Using Spectroscopy and ESI-MS. PLoS ONE 7 4: e35920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagesh N, Sharma VK, Ganesh Kumar A, Lewis EA (2010) Effect of Ionic Strength on Porphyrin Drugs Interaction with Quadruplex DNA Formed by the Promoter Region of C-myc and BcL3 Oncogenes. J Nucleic Acids 146418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nagesh N, Krishnaiah A (2003) A comparative study on the interaction of acridine and synthetic bis-acridine with G-quadruplex structure. J. Biochem Biophys Methods 57 ((1)): 65–74. [DOI] [PubMed] [Google Scholar]
- 31. Nagesh N, Chatterji D (1995) Ammonium ion at low concentration stabilizes the G-quadruplex formation by telomeric sequence. J Biochem Biophys Methods 30 1: 1–8. [DOI] [PubMed] [Google Scholar]
- 32. Moorhouse AD, Haider S, Gunaratnam M, Munnur D, Neidle S, et al. (2008) Targeting telomerase and telomeres: a click chemistry approach towards highly selective G-quadruplex ligands,. Mol Biosyst 4: 629–642. [DOI] [PubMed] [Google Scholar]
- 33. Dash J, Waller ZAE, Pantos GD, Balasubramanian S (2011) Synthesis and Binding Studies of Novel Diethynyl-pyridine Amides with Genomic Promoter DNA G-Quadruplexes. Chem Eru J 17: 4571–4581. [DOI] [PubMed] [Google Scholar]
- 34. Moses JE, Ritson DJ, Zhang F, Lombardo CM, Haider S, et al. (2010) A click chemistry approach to C 3 symmetric, G-quadruplex stabilising ligands. Org Biomol Chem 8: 2926–2930. [DOI] [PubMed] [Google Scholar]
- 35. Andersen NK, Døssing H, Jensen F, Vester B, Nielsen P (2011) Duplex and Triplex Formation of Mixed Pyrimidine Oligonucleotides with Stacking of Phenyl-triazole Moieties in the Major Groove. J Org.Chem 76: 6177–6187. [DOI] [PubMed] [Google Scholar]
- 36. Paul A, Jain AK, Misra SK, Maji B, Muniyappa K, et al. (2012) Binding of Gemini Bisbenzimidazole Drugs with Human Telomeric G-Quadruplex Dimers: Effect of the Spacer in the Design of Potent Telomerase Inhibitors. PLoS ONE 7 6: e39467 doi: 10.1371/ journal.pone.0039467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huppert JL, Subramanian S (2005) Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33: 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumari S, Bugaut A, Balasubramanian S (2008) Position and Stability Are Determining Factors for Translation Repression by an RNA G-Quadruplex-Forming Sequence within the 5′ UTR of the NRAS Proto-oncogene. Biochemistry 47: 12664–12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plum GE (2000) Determination of Oligonucleotide Molar Extinction Coefficients, Current Protocols in Nucleic Acid Chemistry, Vol. 7.3, John Wiley & Sons, New York, NY, USA. pp.7.3.1–7.3.17.
- 40.Fasman GD (Ed.)(1975) Handbook of Biochemistry and Molecular Biology, Volume 1: Nucleic Acids, 3rd Ed., CRC Press, Cleveland, OH. pp. 589–595.
- 41. Schaffner W, Weissmann C (1973) A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 56: 502–514. [DOI] [PubMed] [Google Scholar]
- 42. Gonzalez JM, Saiz-Jimenez C (2005) A simple fluorimetric method for the estimation of DNA-DNA relatedness between closely related microorganisms by thermal denaturation temperatures. Extremophiles 9: 75–79. [DOI] [PubMed] [Google Scholar]
- 43. Xu H, Gao S, Yang Q, Pan L, Wang C (2010) Fan, Amplified Fluorescent recognition of G-Quadruplex folding with a cationic conjugated polymer and DNA intercalator. Appl Mater Interfaces 2 11: 3211–3216. [DOI] [PubMed] [Google Scholar]
- 44. Wilson JK, Sargent JM, Elgie AW, Hill JG, Taylor CG (1990) A feasibility study of the MTT assay for chemosensitivity testing in ovarian malignancy. Br J Cancer 62: 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clemons M, Coleman RE, Varma S (2004) Aromatase inhibitors in the adjuvant setting: bringing the gold to a standard? Cancer Treat Rev 30 4: 325–332. [DOI] [PubMed] [Google Scholar]
- 46. Agalave SG, Maujan SR, Pore VS (2011) Click Chemistry: 1,2,3 triazoles as pharmacophores. Chem Asian J 6: 2696–2718. [DOI] [PubMed] [Google Scholar]
- 47. Han H, Hurley LH, Salazar M (1999) A DNA polymerase stop assay for G-Quadruplex interactive compounds. Nucleic Acids Res 27: 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sun D, Hurley LH (2010) Biochemical techniques for the Characterization of G-quadruplex structures : EMSA, DMS foot-printing, and DNA polymerase stop assay. Methods Mol Biol 608: 65–79 doi:10.1007/978-1-59745-363-9-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, et al. (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464–468. [DOI] [PubMed] [Google Scholar]
- 50. Raymond E, Sun D, Chen SF, Windle B, Von Hoff DD (1996) Agents that target telomerase and telomeres. Curr Opin Biotechnol 7: 583–591. [DOI] [PubMed] [Google Scholar]
- 51. Kim NW, Piatyszek MA, Powse KR, Harley CB, West MD, et al. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266: 2011–2015. [DOI] [PubMed] [Google Scholar]
- 52. Broccoli D, Young JW, de Lange T (1995) Telomerase activity in normal and Malignant hematopoietic cells. Proc Natl Acad Sci USA 92: 9082–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Counter CM, Gupta J, Harley CB, Leber B, Bacchetti S (1995) Telomerase activity in normal leukocytes and in hematologic malignancies. Blood 85 9: 2315–2320. [PubMed] [Google Scholar]
- 54. Provse KR, Greider CW (1995) Developmental and tissue-specific regulation of mouse telomerease and telomere length. Proc Natl Acad Sci USA 92 11: 4818–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shay JW, Bacchetti S (1997) A survey of telomerase activity in human cancer. Eur J Cancer 33: 787–791 doi:10.1016 /S0959-8049(97)00062-2 [DOI] [PubMed] [Google Scholar]
- 56. Ladbury JE, Chowdhry BZ (1996) Sensing the heat: the application of isothermal titration calorimetry to thermodynamic studies of biomolecular interactions. Chem Biol 3 10: 791–801. [DOI] [PubMed] [Google Scholar]
- 57. Nagesh N, Ruscaglia R, Dettler JM, Lewis EA (2010) Studies on the site and mode of TMPyP4 interactions with Bcl-2 promoter sequence G-Quadruplexes. Biophys J 98 11: 2628–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaiser M, De Cian A, Sainlos M, Renner C, Mergny J-L, et al. (2006) Neomycin capped aromatic platforms: quadruplex DNA recognition and telomerase inhibition. Org Biomol Chem 4: 1049–1057. [DOI] [PubMed] [Google Scholar]
- 59. Cheng M-K, Modi C, Cookson JC, Hutchinson I, Heald RA, et al. (2008) Antitumor Polycyclic Acridines. 20. Search for DNA Quadruplex Binding Selectivity in a series of 8,13-Dimethyl quino [4,3,2-kl]acridiniumSalts:Telomere-Targeted Agents. J Med Chem 51: 963–975. [DOI] [PubMed] [Google Scholar]
- 60. Strahan GD, Keniry MA, Shafer RH (1998) NMR Structure Refinement and Dynamics Of the K+-[d(G3T4G3)]2 Quadruplex via Particle Mesh Ewald Molecular Dynamics Simulations. Biophy J 75: 968–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shi DF, Wheelhouse RT, Sun DY, Hurley LH (2001) Quadruplex-interactive agents as Telomerase inhibitors synthesis of porphyrins and structure-activity relationship for the inhibition of telomerase. J Med Chem 44 26: 4509–4523. [DOI] [PubMed] [Google Scholar]
- 62. Luedtke NW (2009) Targeting G-quadruplex DNA with small molecules. Chimia 63 3: 134–139. [Google Scholar]
- 63. Rha SY, Izbicka E, Lawrence R, Davidson K, Sun D, et al. (2000) Effect of telomere and telomerase interactive agents on human tumor and normal cell lines. Clinical Cancer Res 8: 987–993. [PubMed] [Google Scholar]
- 64. Lin J, Roy V, Wang L, You L, Agrofoglio LA, et al. (2010) 3′-(1,2,3-triazol-1-yl)-3′-deoxy thymidine analogs as substrates for human and Ureaplasma parvum thymidine kinase for structure–activity investigations. Bioorg Med Chem 18 9: 3261–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell count assay using normal and tumor cell lines. Cell count assay results with normal as well as tumor cell lines after 24 h and 48 h of treatment with nucleoside ligands (L1, L2 and L3). Experiment was repeated thrice and the mean values were plotted. The set of data with reducing order of significance (p value≤0.001, 0.01 and 0.05) were marked with *, # and ** respectively.
(TIF)
Inhibition of in vitro DNA synthesis with NQF and QF DNA in presence of nucleoside ligands. A. Stop assay with L3 and NQF DNA: Lane 1–5 show 5 µM NQF DNA with 0 µM, 50 µM,100 µM,150 µM,200 µM of L3. Figure 7 B,C,D. Stop assay with L1, L2 and L3 and QF DNA: Lane 1–5 show 5 µM QF DNA with 0 µM, 50 µM, 100 µM, 150 µM, 200 µM of L1, L2 and L3 respectively. The control lane for QF DNA is considered common for B,C and D figures.
(TIF)
Possible mechanism of termination of DNA synthesis with NQF and QF DNA in presence of L3. Schematic representation of mechanism of termination of DNA synthesis followed by DNA fragmentation and induction of apoptosis with QF and NQF DNA in presence of L3 and dNTPs.
(TIF)
Contains Tables S1, S2, S3, S4.
(DOCX)