

Abstract
Diamond-Blackfan anemia (DBA) is a rare inherited bone marrow failure syndrome that is characterized by pure red-cell aplasia and associated physical deformities. It has been proven that defects of ribosomal proteins can lead to this disease and that RPS19 is the most frequently mutated gene in DBA patients. Previous studies suggest that p53-dependent genes and pathways play important roles in RPS19-deficient embryos. However, whether there are other vital factors linked to DBA has not been fully clarified. In this study, we compared the whole genome RNA-Seq data of zebrafish embryos injected with RPS19 morpholino (RPS19 MO), RPS19 and p53 morpholino simultaneously (RPS19+p53 MO) and control morpholino (control). We found that genes enriched in the functions of hematological systems, nervous system development and skeletal and muscular disorders had significant differential expression in RPS19 MO embryos compared with controls. Co-inhibition of p53 partially alleviates the abnormalities for RPS19-deficient embryos. However, the hematopoietic genes, which were down-regulated significantly in RPS19 MO embryos, were not completely recovered by the co-inhibition of p53. Furthermore, we identified the genome-wide p53-dependent and -independent genes and pathways. These results indicate that not only p53 family members but also other factors have important impacts on RPS19-deficient embryos. The detection of potential pathogenic genes and pathways provides us a new paradigm for future research on DBA, which is a systematic and complex hereditary disease.
Introduction
Diamond-Blackfan anemia (DBA) is a congenital anemia and broad developmental disease that develops soon after birth. The anemia results from a failure of erythropoiesis, with normal platelet and myeloid lineages, and it can be managed with steroids, blood transfusions, or stem cell transplantation [1]. DBA is the first known inherited disease that results from a defect in a structural ribosomal protein [2]. In humans, mutations in an increasing number of genes encoding RPs of the small (RPS19, RPS24, RPS17, RPS7, RPS10, RPS26) and large (RPL35A, RPL5, RPL11) ribosomal subunits have been shown to cause DBA. Moreover, RPS19 is the most frequently mutated ribosomal protein gene, which accounts for approximately 25% of DBA patients [3]. However, how the ribosomal protein mutations specifically affect hematopoiesis is still unclear.
The zebrafish (Danio rerio) is an excellent model organism for such studies because of its embryo transparency, high fecundity and fast development of organogenesis. It is notable that many of the key molecular players and events that drive organogenesis in zebrafish are evolutionarily and functionally conserved with other organisms, including mammalians [4]. Several groups have previously used zebrafish for modeling ribosomal protein knockdown in the embryo with morpholino technology or generating ribosomal protein mutants [5]. Recent studies have suggested that p53-mediated cell cycle arrest and/or apoptosis in erythroid cells could be the major factors in DBA development [3], [5], [6], [7]. For example, regarding the RPL11 mutant in zebrafish, Danilova et al propose that the unique phenotype of DBA is the sum of several abnormally regulated molecular pathways mediated by the p53 protein family and p53-independent synergistic impacts on hematological and other cellular pathways affected in DBA [6]. In RPS7-deficient zebrafish embryos, p53 was activated, and its downstream target genes and biological events were induced, including apoptosis and cell cycle arrest. Furthermore, simultaneous knockdown of the p53 protein could partially reverse the abnormal phenotype of the morphants [3]. In the RPS29 mutant, genes that are up-regulated are enriched with genes that are up-regulated by p53 after irradiation and the p53 knockdown almost completely rescues the RPS29 morphological and hematopoietic phenotypes, demonstrating that p53 mediates the effects of rps29 knockdown [5]. Knockdown of two other RP genes, RPS3A and RPL36A, result in severe morphological abnormalities with mild erythroid defects and elicited an activated p53 response. For the RPS19-deficient zebrafish, its phenotype is mediated by dysregulation of deltaNp63 and p53, and suppression of p53 and deltaNp63 alleviates the RPS19-deficient phenotypes [8]. At the same time, co-inhibition of p53 activity rescued the morphological abnormalities but did not alleviate erythroid aplasia in RPS19-deficient zebrafish. Hence, both the p53-independent and p53-dependent pathways could be responsible for the defective erythropoiesis in the zebrafish model of Diamond-Blackfan Anemia because of RPS19 deficiency [7].
In this study, we observed the hemoglobin synthesis defect at 48 hpf in RPS19 knockdown zebrafish embryos, and this defect can only be partially reversed by co-inhibition of p53 activity. To investigate the underlying regulatory mechanisms, we generated three types of zebrafish morphants by MO microinjection, including control morphants (control), RPS19 morphants (RPS19 MO), and RPS19 and p53 morphants (RPS19+p53 MO), and we performed transcriptome analysis of all of the pairs using the RNA-Seq technique. We found significant differentially expressed genes in RPS19 MO and RPS19+p53 MO compared with the controls. These genes are associated with the functions of cell cycle, hematological system and nervous system development and the skeletal and muscular disorders. Additionally, we determined the genome-wide p53-dependent and -independent genes and pathways. Our results demonstrated that members of the p53 network as well as other partners exert important impacts on RPS19-deficient embryos. The detection of potential pathogenic genes and pathways in this study will provide a new research paradigm for the study of DBA.
Results
Zebrafish Phenotypes of RPS19-deficient and Co-inhibition of p53 Activity using MO
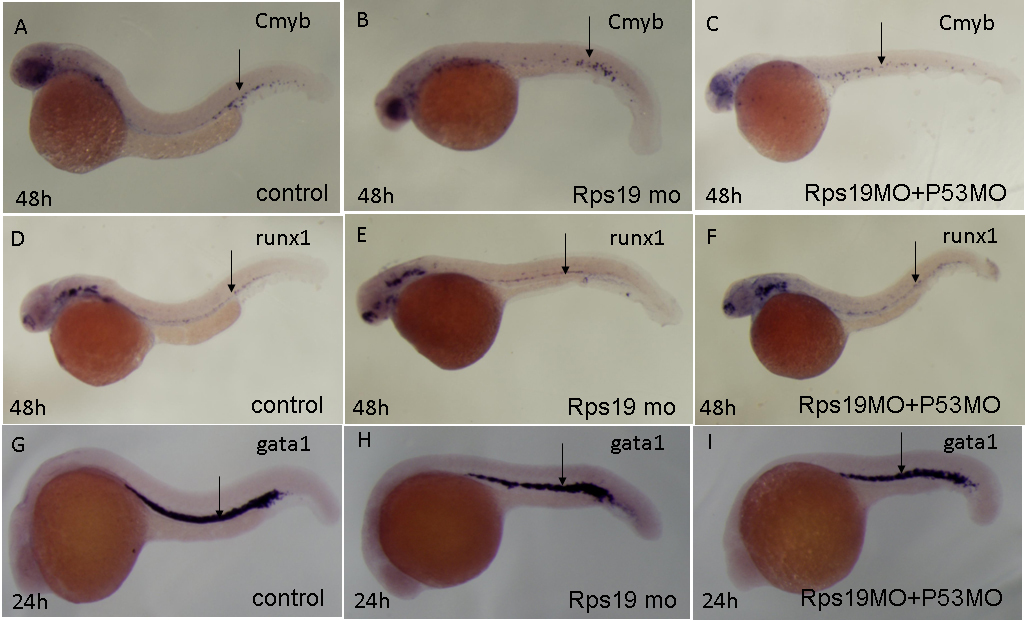
Developmental defects are found in approximately 40% of DBA patients with mutations in RPS19. Zebrafish RPS19 is approximately 87.7% identical to the human homolog. As previously demonstrated, the RPS19 deficiency in zebrafish results in hematopoietic and developmental abnormalities that resemble DBA. The embryos injected with control MO did not display any morphological changes. RPS19 morphants showed an obvious ventrally bent tail and a reduction in the circulating blood cells compared with the control embryos. At 48 hpf, the hemoglobin staining results showed that hemoglobin-stained blood cells in the heart region were markedly decreased in RPS MO, which is partially rescued in RPS19+p53 MO (Fig. 1). Further in situ results showed that the gata1 expression level are comparable in control Mo, RPS MO and RPS19+p53 MO (Fig. S1), which is consistent with previous results [8]. In addition, we used HSCs and definitive hematopoiesis markers cmyb and runx1 to examine the expression after RPS19 morpholino (2 ng) injection or Rps19 (mo) and P53 (mo) co-injection. We did not find significant changes of the expression of cmyb and runx1 (Fig. S1), indicating the divergent functions of RPS19 and RPL22 in hematopoiesis [9]. The effectiveness of translational inhibition by RPS19 MO was confirmed by examining green fluorescent fusion protein under fluorescence microscopy (Fig. 1).
Figure 1. Effectiveness of RPS19 morpholinos and hemoglobin staining of embryos coinjected with Rps19 mRNA and P53 MO using o-dianisidine.

The Rps19:egfp construct was made by inserting a partial sequence of Rps19 cDNA (containing 60 bps from the 5′ UTR ) with the N-terminus of egfp into modified pEGFP-N1 (the ATG codon of EGFP was removed). The sequence of RPS19 MO1 is a compliment of bp 1–24 of Rps19 cDNA. Embryos co-injected with 25 ng Rps19:egfp DNA and 5 ng control MO produced green fluorescent fusion protein (A), and expression of the fusion protein was inhibited by co-injection with 2 ng Rps19 Mo (B). O-staining results show a drastic reduction in the number of hemoglobin-stained blood cells when Rps19 is knockdown (C and D are the control, E and F are Rps19 knockdown) and partially reversed by co-injection of P53 morpholino (G and H). A, B, D, F and H are the lateral view; C, E and G are the ventral view.
Transcriptome Profile Analysis
As previously demonstrated, the RPS19 deficiency in zebrafish results in hematopoietic and developmental abnormalities that resemble DBA. To determine the effect of RPS19 MO on the whole transcriptome and to delineate the function of p53 in RPS19-deficient embryos, we analyzed the transcript profiles using RNA-Seq. Three mRNA-Seq libraries were generated, including zebrafish embryos with control morpholino (control), RPS19 morpholino knockdown (RPS19 MO), and RPS19 and p53 morpholino knockdown simultaneously (RPS19+p53 MO). These libraries were sequenced using the Illumina Hi-seq 2000 Genome Analyzer platform with paired-end 100 base-pair tags to a depth of 35–60 million reads. We mapped the sequencing data to the latest zebrafish genome assembly version 2010 (Zv9). A total of 17–25 million reads could be mapped to the genome, representing 40%–46% of all of the generated reads (Table S1 in Tables S1). FPKM was used to estimate the transcript abundance. Overlapped and distinct gene expression among three samples was shown in Fig. 2. Unsupervised hierarchical clustering of genome-wide expression profiles (FPKM>1) was performed to examine the relationship among these three samples (Fig. 2). We observed that, compared with the control, the pattern of transcriptome profile of RPS19 MO is more similar to that of RPS19+p53 MO. The transcriptome difference between the profiles of RPS19 MO and the control is more significant than that of RPS19+p53 MO and the control. These results implied that simultaneous knockdown of p53 by co-injecting a p53 MO could partially reverse the abnormal phenotype in the RPS19 morphants.
Figure 2. Overview of gene expression profiles of diverse zebrafish embryos.

(A) Venn diagram detailing shared and distinct genes expression among three zebrafish embryo samples: control morpholino (control), RPS19 morpholino knockdown (RPS19 MO), RPS19 and p53 morpholino knockdown simultaneously (RPS19+p53 MO). (B)Unsupervised clustering of genome-wide gene expression (cutoff FPKM<1) for diverse zebrafish embryos (control morpholino (control), RPS19 morpholino knockdown (RPS19 MO), RPS19 and p53 morpholino knockdown simultaneously (RPS19+p53 MO)). Gene expression tracks use red and green to represent over- and under-expression, respectively. The pattern of the transcriptome profile of RPS19 MO is more similar to that of RPS19+p53 MO.
Transcriptome Analysis of RPS19-deficient Embryos
To observe the transcriptome changes of RPS19-deficient embryos, we compared the differences of the whole genome RNA-Seq data between RPS19 MO and the control using the software Cufflinks. There were 47 up-regulated genes and 312 down-regulated genes in RPS19 MO compared with the control sample (fold-change>2.0, p-Value<0.05). These genes were subjected to Ingenuity Pathway Analysis (IPA) to identify the enrichment of genes in specific functional groups and pathways (IPA, http://www.ingenuity.com). The IPA accepts human UniGene IDs as one of the identifiers for data uploading and analysis. Hence, we mapped these differentially expressed genes to their human homologs using the HomoloGene database. Then, human homologs of up- and down-regulated genes of the RPS19-deficient embryos were analyzed by using IPA tools in which the gene sets are enriched for a specific function/pathway and the enrichment is represented as a ratio. The main biological functions that were enriched by differentially expressed genes are associated with genetic disorder, neurological disease, cellular growth and proliferation, cancer and cell death (p-Value<0.05). The number of enriched genes for the above GO terms is more than 50, which accounts for at least 14% of the whole set of differentially expressed genes (Fig. 3). The significantly changed genes are associated with signaling pathways, including the coagulation system, acute phase response signaling, LXR/RXR activation, intrinsic prothrombin activation pathway, and the taurine and hypotaurine metabolism (Table S2 in Tables S1). The networks constructed by differentially expressed genes are associated with cellular development, cellular function and maintenance, development disorder, hematological system development and function, and neurological disease, each of which received an enrichment score of more than 20 (Fig. 4). Since the networks with functions of ‘cellular development’, ‘cellular function and maintenance’, ‘Nervous system development and function’, and ‘cell death and survival’ are inter-connected to form a big and complex regulatory network that is controlled by a few central factors and we observed expression changes in many genes and they could not be attributed to gene mutations (the probability of multi-gene mutations appear at the same time is very low), we assume this change to be a result of abnormal expression of a few central factors that affect the central nodes in the networks. In the network analysis, we found central nodes for which the human homologs are SST, OXA1L, CLDND1, MAL, Hsp70, GCG, PLC, SOX1, VAX1, POMC, and TSH.
Figure 3. The top enriched biological functions of 359 differentially expressed genes in RPS19-deficient embryos (p-value<0.05).

Figure 4. Top networks identified with IPA for the differentially expressed genes in RPS19 MO compared with the control.

The pink or red nodes in the networks indicate a gene that is up-regulated in RPS19 MO, and the green color indicates genes that are down-regulated in RPS19 MO. (A) Network with the functions of embryonic development, organismal development, and cellular development. (B) Network with the functions of digestive system development and function, developmental disorder, and skeletal and muscular disorders. (C) Network with the functions of hematological system development and function, organismal functions, and cellular movement. (D) Network with the functions of nervous system development and function, tissue morphology, and cell-to-cell signaling and interaction.
Transcriptome Analysis of RPS19+p53 MO Embryos
To further study the role of p53 in RPS19-deficient embryos, we analyzed the RNA-Seq data of RPS19+p53 MO. Differential expression analysis is conducted by Cufflinks on the transcriptome data of RPS19+p53 MO and control/RPS19 MO. Compared with the control, RPS19+p53 MO is detected with 34 up-regulated genes and 113 down-regulated genes (fold-change>2.0, p-Value<0.05). These genes were subjected to Ingenuity Pathways Analysis (IPA) to identify the enrichment of genes in specific functional categories and canonical pathways. We found that the main biological functions of these genes are associated with cellular growth and proliferation, hereditary disorder, cellular development, tissue development and small molecule biochemistry (p-Value<0.05) (Fig. 5). The top significant canonical pathways are LXR/RXR activation, acute phase response signaling and coagulation system (Table S3 in Tables S1). The main networks constructed by these differentially expressed genes are involved in small molecule biochemistry, cell cycle, cellular development, and hematological disease. The above networks all obtained enrichment scores of more than 20 (Fig. 6). In addition, the networks with functions of ‘hematological disease’, ‘small molecule biochemistry’, ‘molecular transport’, and ‘cellular development’ are found to be connected. We also found several central nodes in these networks, the human homologs of which are TGM2, A2M, LDL, HDL, Collagen, AGT, SERPINF1, NROB2, CETP, HBZ, hemoglobin, CLDND1, calpain, CD81, and G-protein beta.
Figure 5. The top enriched biological functions of 147 differentially expressed genes in RPS19-deficient embryos with co-inhibition of p53 (p-value<0.05).

Figure 6. Top networks identified with IPA for the differentially expressed genes in RPS19+p53 MO compared with the control.

The pink or red nodes in the networks indicate genes that are up-regulated in RPS19+p53 MO, and the green color indicates genes that are down-regulated in RPS19+p53 MO. (A) Network with the functions of lipid metabolism, molecular transport, and small molecule biochemistry. (B) Network with the functions of cell cycle, cellular development, cellular growth and proliferation. (C) Network with the functions of cancer, hematological disease, and amino acid metabolism. (D) Network with the functions of cellular development, hematological system development and function, and hematopoiesis.
However, by comparing the transcriptome data of RPS19+p53 MO and RPS19 MO, we found only 17 up-regulated genes and 11 down-regulated genes that were significantly expressed in RPS19+p53 MO embryos (fold-change>2.0, p-Value<0.05), which is much less than the number of significant differentially expressed genes of RPS19 versus the control. These results demonstrate that co-inhibition of p53 cannot rescue the abnormalities completely in RPS19-deficient embryos. The main enriched biological functions of these genes, by identified IPA, were associated with cancer, cell death, cellular growth and proliferation, cell signaling and small molecule biochemistry. Moreover, the canonical pathways are enriched on methane metabolism, DNA double-strand break repair by homologous recombination, stilbene, coumarine and lignin biosynthesis (data not shown). These functions might be corrected by p53 MO for RPS19-deficient embryos, which are significantly influenced by the expression of p53.
Changed Hematopoietic Genes in RPS19 MO and RPS19+p53 MO Embryos
To observe the defects of the hematopoietic functions in RPS19-deficiency embryos, we detected the differentially expressed hematopoietic genes by searching the keyword ‘hematopoietic’ while using the software AmiGO in the Gene Ontology database (http://www.geneontology.org/). We observed 12 GO terms that are associated with ‘hematopoietic’ and 177 genes in the latest annotated zebrafish genome. We matched our data to these genes, which have the GO term keyword ‘hematopoietic’. In the RPS19-deficient embryos, we found that no hematopoietic gene was up-regulated significantly, but 5 hematopoietic genes were down-regulated highly and significantly, such as bmper, fzd5, nrp1a, sema3d, and tbx1. The specific bio functions of these 5 genes are ‘hematopoiesis’(bmper), ‘T cell differentiation in thymus’(fzd5), and ‘thymus development’ (nrp1a, sema3d, tbx1). To determine whether co-inhibition of p53 would alleviate hematopoietic defect, we also matched the differential expressed genes in RPS19+p53 MO versus RPS19 MO to the 177 genes associated with hematopoietic in The Gene Ontology database. Interestingly, there was only one hematopoietic gene, fzd5, which was down-regulated in RPS19 MO, but was significantly up-regulated in RPS19+p53 MO (Table S4 in Tables S1). Previous studies showed that in zebrafish, fzd5 was associated with not only eye and retina development, but also canonical Wnt signaling, T cell differentiation in thymus and early liver formation [10], [11] and thus participated in hematopoiesis [12], [13]. As the expression of fzd5 is reversed by co-inhibition of p53, hematopoiesis and other functions of fzd5 are reversed by p53 MO. In combination with our hemoglobin staining results of RPS19+p53 MO, we concluded that the hematopoietic defects could not be remedied completely through co-inhibition of p53 in RPS19-deficient embryos.
p53-dependent and -independent Genes and Pathways
Next, we observed the p53-dependent/independent pathways in RPS19-deficient embryos by comparing the transcriptome profiles of control, RPS19 MO and RPS19+p53 MO, using the software Cufflinks. To determine the p53-dependent genes, we firstly searched the up/down-regulated genes in RPS19 MO (compared with control). We then identified genes that their expression levels were reversed (down/up-regulated) in embryos co-injected with RPS19 MO+p53 MO, and defined them as p53 dependent genes. Here we presume that genes whose expression levels did not return to the normal level (control), but did have partial reverse effect compared with pathological state (RPS19 MO), are p53-dependent genes. To determine the p53-independent genes, we presume genes that are up/down-regulated in RPS19 MO (compared with control), and up/down-regulated in RPS19+p53 MO (compared with control), are p53-independent genes. No matter the degree of changes in these genes in RPS19+p53 MO reach the level of them in RPS19 MO or not, as long as the change trends are the same, we accept them as p53-independent genes. (Fig. 7).
Figure 7. The scheme of detection of p53 dependent and independent genes.

The vertical axis represents the gene expression, which is normalized to FPKM. The red arrows indicate the variation trend of FPKM for abnormal up-regulated genes in RPS19 MO, and the green arrows indicate the down-regulated genes in RPS19 MO. Solid arrows indicate the variation trend of FPKM of genes in RPS19 MO, and dashed arrows indicate the variation trend of FPKM of genes in RPS19+p53 MO. Up−/down-regulated genes are screened with the criterion of a fold-change of >2.0 and a p-value of <0.05. (A) Up−/down-regulated genes in RPS19 MO compared with the control and down−/up-regulated genes in RPS19+p53 MO compared with RPS19 MO; we considered genes that satisfied the above criteria to be genes that are p53 dependent. (B) Up−/down-regulated genes in RPS19 MO compared with the control and up−/down-regulated genes in RPS19+p53 MO compared with the control; we considered genes that satisfied the above criteria to be genes that are independent of p53.
Based on the above strategy, we observed 12 potential p53-dependent genes, in which 1 gene was up-regulated and 11 genes were down-regulated (Table 1). The biological functions enriched by these genes are protein folding, integral to membrane and ATP binding. We also found 110 potential p53-independent genes, of which 21 genes were up-regulated and 89 genes were down-regulated. The up-regulated genes are mainly associated with extracellular regions, structural molecule activity, response to stress and intracellular. The down-regulated genes are mainly associated with negative regulation of endopeptidase activity, negative regulation of peptidase activity, and negative regulation of hydrolase activity (p-Value<0.01, number of gene products>2) (Table 2). There were 20 of the regulated pathways that were determined to be p53-dependent (Table S5 in Tables S1) and 76 that were determined to be p53-independent (Table S6 in Tables S1). Real-time PCR and in situ hybridization results are mostly consistent with the above sequencing results except for mt2. The real-time PCR results of mt2, fzd5, hsp70l, fn1b, optc, klf11a and srf1 are shown in Fig S2. Interestingly, the mt2 expression in sequencing results is different from the real-time PCR results. We postulated that the discrepancy may arise from: a) the design and optimization of gene-specific real-time primer pairs for mt2; b) the PCR amplification steps bear inherent biases; c) the associated tools for computational analysis are in their infancy, thus may contain errors. It is crucial to identify all existing isoforms in the long run for accurate quantification of a transcriptome. Notably, most of the genes we tested are consistent with the initial RNA-seq experiment data and thus we are confident about the quality of our data. The in situ hybridization data of fzd5, alas2, nrp1a and tagln are shown in Fig S3.
Table 1. List of p53-dependent genes.
| Gene ID | Regulation | Annotation |
| mt2 | up | metallothionein 2 |
| dnajb1b | down | DnaJ (Hsp40) homolog, subfamily B, member 1b |
| fzd5 | down | frizzled homolog 5 |
| hsp70 | down | NA |
| hsp70l | down | heat shock cognate 70-kd protein, like |
| hsp90aa1.2 | down | heat shock protein 90, alpha (cytosolic), class A member 1, tandem duplicate 2 |
| LOC562935 | down | NA |
| mal | down | mal, T-cell differentiation protein |
| pdcd4b | down | NA |
| si:dkeyp-35b8.5 | down | NA |
| zgc:136410 | down | zgc:136410 |
| zgc:174006 | down | zgc:174006 |
Table 2. List of p53-independent genes.
| Gene ID | Regulation | Annotation |
| c6 | up | complement component 6 |
| cldn1 | up | claudin 1 |
| cyp24a1 | up | cytochrome P450, family 24, subfamily A, polypeptide 1 |
| cyr61l1 | up | cysteine-rich, angiogenic inducer 61 like 1 |
| fn1b | up | fibronectin 1b |
| hspb11 | up | heat shock protein, alpha-crystallin-related, b11 |
| hspb9 | up | heat shock protein, alpha-crystallin-related, 9 |
| LOC554386 | up | NA |
| mustn1 | up | NA |
| optc | up | opticin |
| pdlim3b | up | PDZ and LIM domain 3b |
| popdc1 | up | popeye domain containing 1 |
| pth1a | up | parathyroid hormone 1a |
| rn7sk | up | NA |
| scpp5 | up | NA |
| srfl | up | serum response factor like |
| tagln | up | transgelin |
| ydjc | up | NA |
| zgc:158463 | up | zgc:158463 |
| zgc:194878 | up | zgc:194878 |
| zgc:92480 | up | zgc:92480 |
| a2ml | down | alpha-2-macroglobulin-like |
| acsl5 | down | acyl-CoA synthetase long-chain family member 5 |
| akr1a1a | down | aldo-keto reductase family 1, member A1a (aldehyde reductase) |
| ambpl | down | alpha-1-microglobulin/bikunin precursor, like |
| ampd1 | down | adenosine monophosphate deaminase 1 (isoform M) |
| apom | down | NA |
| barx1 | down | BarH-like homeobox 1 |
| bhlhe23 | down | basic helix-loop-helix family, member e23 |
| calrl2 | down | calreticulin, like 2 |
| cfh | down | complement factor H |
| col10a1 | down | collagen, type X, alpha 1 |
| crx | down | cone-rod homeobox |
| cyp2aa3v1 | down | NA |
| cyp4v8 | down | cytochrome P450, family 4, subfamily V, polypeptide 8 |
| elavl4 | down | ELAV (embryonic lethal, abnormal vision, Drosophila)-like 4 (Hu antigen D) |
| f7 | down | coagulation factor VII |
| f7i | down | coagulation factor VIIi |
| fabp10a | down | fatty acid binding protein 10a, liver basic |
| gnb3a | down | NA |
| habp2 | down | hyaluronan binding protein 2 |
| hmga1b | down | high mobility group AT-hook 1b |
| hmp19 | down | HMP19 protein |
| hpda | down | 4-hydroxyphenylpyruvate dioxygenase a |
| klf11a | down | Kruppel-like factor 11a |
| krml2 | down | NA |
| lhx9 | down | LIM homeobox 9 |
| lipf | down | lipase, gastric |
| lmo3 | down | LIM domain only 3 |
| LOC561143 | down | NA |
| LOC796447 | down | NA |
| mab21l1 | down | mab-21-like 1 |
| matn1 | down | matrilin 1 |
| msrb2 | down | methionine sulfoxide reductase B2 |
| ndrg1b | down | NA |
| neurod | down | neurogenic differentiation |
| neurod6a | down | neurogenic differentiation 6a |
| neurod6b | down | neurogenic differentiation 6b |
| nfixb | down | nuclear factor I/Xb |
| nr0b2a | down | nuclear receptor subfamily 0, group B, member 2a |
| nr2e3 | down | nuclear receptor subfamily 2, group E, member 3 |
| otx2 | down | orthodenticle homolog 2 |
| otx5 | down | orthodenticle homolog 5 |
| oxct1b | down | 3-oxoacid CoA transferase 1b |
| pbx3b | down | pre-B-cell leukemia transcription factor 3b |
| pcdh1g22 | down | protocadherin 1 gamma 22 |
| pcdh1g26 | down | protocadherin 1 gamma 26 |
| pcdh8 | down | protocadherin 8 |
| pde6g | down | phosphodiesterase 6G, cGMP-specific, rod, gamma |
| pmepa1 | down | prostate transmembrane protein, androgen induced 1 |
| pygl | down | phosphorylase, glycogen; liver (Hers disease, glycogen storage disease type VI) |
| rbp2a | down | retinol binding protein 2a, cellular |
| rgs5a | down | regulator of G-protein signaling 5a |
| samd7 | down | NA |
| sardh | down | sarcosine dehydrogenase |
| sema3d | down | semaphorin 3d |
| sept4a | down | septin 4a |
| serpinc1 | down | serine (or cysteine) proteinase inhibitor, clade C (antithrombin), member 1 |
| serpinf1 | down | serine (or cysteine) peptidase inhibitor, clade F, member 1 |
| sfrp2 | down | secreted frizzled-related protein 2 |
| sh3bgrl2 | down | SH3 domain binding glutamic acid-rich protein like 2 |
| si:ch211-102c2.6 | down | NA |
| si:ch211-284e20.8 | down | si:ch211-284e20.8 |
| si:dkey-22i16.3 | down | NA |
| si:dkey-39n1.2 | down | NA |
| si:dkey-52k20.7 | down | si:dkey-52k20.7 |
| si:dkeyp-86e4.1 | down | NA |
| six7 | down | sine oculis homeobox homolog 7 |
| si:xx-by187g17.1 | down | NA |
| slc13a2 | down | solute carrier family 13 (sodium-dependent dicarboxylate transporter), member 2 |
| slc17a5 | down | solute carrier family 17 (anion/sugar transporter), member 5 |
| slc25a36a | down | solute carrier family 25, member 36a |
| spon2b | down | spondin 2b, extracellular matrix protein |
| tfap2b | down | transcription factor AP-2 beta |
| tuba2 | down | tubulin, alpha 2 |
| uox | down | urate oxidase |
| vasa | down | vasa homolog |
| vgll4 | down | vestigial like 4 (Drosophila) |
| vmhc | down | ventricular myosin heavy chain |
| vsx1 | down | visual system homeobox 1 homolog, chx10-like |
| vtna | down | vitronectin a |
| vtnb | down | vitronectin b |
| zfpm2b | down | zinc finger protein, multitype 2b |
| zgc:103710 | down | zgc:103710 |
| zgc:158288 | down | NA |
| zgc:158291 | down | zgc:158291 |
| zgc:162825 | down | zgc:162825 |
| zgc:194131 | down | zgc:194131 |
| zgc:77439 | down | zgc:77439 |
| zgc:92411 | down | zgc:92411 |
Discussion
An Overview of Our Work
In this study, we provided a transcriptome profile of the zebrafish model of DBA, in which the DBA gene RPS19 is knocked-down by MO, for genome-wide analysis using the RNA-Seq technique. We determined differentially expressed genes with statistical significance and enriched pathways and networks by these genes among RPS19 MO, RPS19+p53 MO and control embryos, and we identified p53-dependent and p53-independent genes and pathways by comparing each pair of the three samples. Our data illustrate that not only p53 is the key factor contributing to the significant abnormalities of RPS19-deficient embryos but also some other important factors and pathways participate in regulating the abnormal phenotypes of RPS19-deficient embryos.
The Reliability of RNA-Seq Data
These samples were sequenced using the Illumina Hi-Seq 2000 Genome Analyzer platform with paired-end 100 base-pair tags to a depth of 35–60 million reads, which is sufficient sequence coverage for transcriptome profiling [14]. We mapped these reads to the zebrafish genome assembly version 2010 (Zv9) using TopHat. Approximately 17–25 million reads could be mapped to the genome, which represents 40%–46% of all of the generated reads. Thus, we believe that the whole genome RNA-Seq data of RPS19-deficient zebrafish transcriptome is of high quality and the genome-wide effects of p53 in RPS19-deficient embryos is reasonable and of scientific significance. It should be noted that the sorted erythrocytes could be a better source for RNA-seq sinc DBA is a red cell disease. However, we could not have enough amount of RNA from blood cells for RNA-seq at the time of experiment. It will be interesting to revisit this when single cell RNA-seq technology is available that are currently being developed.
The Effects of RPS19 MO and RPS19+p53 MO on Zebrafish Embryos
Zebrafish response to RPS19 deficiency has many features resembling DBA, including impaired ribosome biogenesis, increased apoptosis, developmental abnormalities, and defective hematopoiesis [8]. With the results of the mapping, we finished the gene annotation and differential expression analysis using Cufflinks. For a comparison of the transcriptome of RPS19 MO and control embryos, we observed that the number of down-regulated genes is much more than that of the up-regulated genes in RPS19 MO. This result suggests that in RPS19-deficient embryos, more genes have been inhibited and cannot function properly. IPA provides us more information about gene ontology clusters, enriched pathways and networks of these differentially expressed genes. We found that these genes are mainly associated with cell death, developmental disorder, skeletal and muscular disorders, hematological system development and function, nervous system development and function, and tissue morphology. These results are in agreement with previous studies on ribosomal protein defects and the phenotype of RPS19-deficient zebrafish in this study [8], [15]. Overall, our results indicate that, as a ubiquitous protein, ribosomal protein RPS19 deficiency has very strong effects on zebrafish, and many core factors and pathways are involved in the abnormalities.
The p53 pathway is a key pathway in ribosomal protein defect embryos, with additional roles in regulating other signaling molecules. p53 is always transcriptionally up-regulated when ribosomal protein suffers haplo-insufficiency [5], [6], [16], [17]. However, it is still unclear why the mutations in the ubiquitously expressed RPS19 gene specifically affect erythropoiesis, and whether the transcriptome could return to its original state after p53 knockdown in RPS19-deficient embryos. To clarify the influence of the co-inhibition of p53 in RPS19-deficient embryos, we used RPS19 control Mo, Rps19 Mo, and Rps19 Mo+P53 Mo three groups for the comparative studies. We deduced the effects of p53 MO alone by comparing Rps19 Mo+P53 Mo with Rps19 Mo since we used the same dosage of Rps19 Mo. Our O-staining results showed that the phenotype of Rps19 Mo we obtained in the study can be rescued by the P53 and RPS19 double knockdown, confirming the effectiveness of the P53 Mo knockdown. This indicates that our current study and analysis method do not affect the robustness of the conclusions we made. We observed from the heatmap, which is the clustering of the transcriptome of the three samples, that there is a large difference among the profiles of these samples. Compared with RPS19 MO, the distance between RPS19+p53 MO and the control is smaller, suggesting that, after p53 knockdown, the abnormalities of RPS19-deficient embryos have not been retrieved completely but instead have been alleviated partially. Additionally, the number of differentially expressed genes for RPS19+p53 versus the control is much smaller, and the down-regulated genes are still the majority. IPA analysis demonstrated that the differentially expressed genes are mainly associated with functions of hematological system development and function, cell cycle, cellular development, cellular growth and proliferation, lipid metabolism, molecular transport, and small molecule biochemistry. These results suggest that these functions, regulated by some other important factors, could not rescued by the inhibition of p53 in RPS19-deficienct embryos.
Less Alleviation Effect of p53 on Hematological Defects
To further study the alleviation effects of p53 on hematological defects caused by RPS19 deficiency, we screened all the genes that are associated with hematopoiesis in the database (The Gene Ontology), and performed matching analysis. We found that, in the differentially expressed genes between RPS19 MO and RPS19+p53 MO, only one gene associated with hematopoiesis was up-regulated in RPS19+p53 MO (fold-change>2.0, p-value<0.05). We supposed that this gene was alleviated by p53 MO.While in RPS19-deficient embryos there are more abnormally down-regulated genes, that are not rescued by p53 MO (Table S4 in Tables S1).This observation indicates that p53 knockdown could not completely rescue the developmental and functional defects of the hematological system in RPS19-deficiency zebrafish. This is expected since hematopoiesis is governed by multiple genes and some of them are not fully controlled by p53. Both our hemoglobin staining observation and RNA-Seq data analysis support the notion that the hematopoiesis is not fully functional in RPS19+p53 MO.
p53-dependent and -independent Genes and Pathways
In this study we developed a strategy to determine the p53-dependent and -independent genes and pathways from the information on the transcriptome using RNA-Seq data of RPS19 MO, RPS19+p53 MO and the control. Our data shows that the number of p53-independent genes and pathways is significantly more than the number of p53-dependent genes and pathways. This result is in agreement with our hypothesis that many genes and pathways are outside the control of p53. The up-regulated genes link to the functions of structural molecule activity, the extracellular region, response to stress, the intracellular region and more. At the same time, down-regulated genes are associated with the regulation of metabolic processes, the regulation of transcription, sequence-specific DNA binding transcription factor activity and more.
Conclusions
In conclusion, our study provides an outline of the transcript changes in the zebrafish model of DBA. We can conclude from this study that DBA is a systemic and complex disease due to ribosomal protein defects. DBA causes certain downstream phenotypes, including dysfunction of basic biological and physiological process such as transcription, translation, cellular metabolism, and pathways and networks related to many other disorders. The p53 network is one of the primary pathways in DBA disease. Meanwhile, there are many other p53-independent factors and pathways playing important roles in ribosomal protein defect. Our work has laid foundation in zebrafish DBA model and it will be interesting to see the clinical relevance of zebrafish DBA model in comparison with human DBA data.
Materials and Methods
Zebrafish Care
Breeding wild-type zebrafish (Danio rerio) (AB type) were maintained, and embryos were raised under standard library conditions [18]. Zebrafish embryos were kept in a 28.5°C incubator, and the stages (hours post-fertilization) in this study were as described [19]. All of the studies using zebrafish were approved by the Animal Care and Use Committee of Huazhong University of science and technology.
Microinjection of Morpholino RNA
The rps19 Morpholino (5-CACTGTTACACCACCTGGCATCTTG-3), P53 Morpholino (5- GCGCCATTGCTTTGCAAGAATTG-3), and RPS19 control MO (5-CACTcTTAgACgCACCTGcCATgTTG-3) were obtained from Gene-Tools, LLC. Zebrafish embryos at the one-cell stage were injected with the MOs using a Microinjector (WPI SYS-PV830). Based on the literature and our initial injection trials, 2 ng MO and control Mo was chosen as the optimal concentration. Injected embryos were grown at 28.5°C and were observed under a microscope. The effectiveness of translation inhibition by RPS19 MO (2 ng/embryo) was used by examining in vivo the rps19:egfp green fluorescent fusion protein under fluorescence microscopy. We found that at this dosage of 2 ng/embryo, most of the GFP expression was knocked-down.
Hemoglobin Staining
Hemoglobin in zebrafish embryos was analyzed using o-dianisidine (Sigma) as described [20]. Low-power images were collected using an Olympus microscope with a digital camera (OLYMPUS IX71). Images were imported into Adobe Photoshop CS2 9.0.2 for orientation and figure preparation. O-dianisidine staining results confirmed the effectiveness of the morpholino knockdown while the embryo morphology looks normal.
RNA Preparation
Immediately after harvesting, 40∼50 pooled embryos at 48 hpf from different experiment replicates were snap-frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted from the pooled embryos using TRIzol (Invitrogen) according to the manufacturer’s instructions. RNA concentrations were determined using NanoDrop 2000 (Thermo Scientific). The integrity of RNA samples was determined using 1.2% Agarose gel electrophoresis, followed by removal of the residual genomic DNA with RNase-free DNaseI (Ambion).
Library Preparation and Sequencing
mRNA libraries were constructed using the Illumina mRNA-Seq library preparation kit according to the manufacturer’s instructions. The concentration and size distribution of the libraries were determined on an Agilent Bioanalyzer DNA 2000 chip (Agilent Technologies) followed by sequencing on the Illumina Hiseq 2000 Genome Analyser platform in pair-end mode by a 100 bp length. A total of 35–60 million reads were collected for further analysis.
RNA-Seq Data Analysis
Reads were processed and aligned to the UCSC zebrafish reference genome (build Zv9/danRer7, Jul. 2010) using TopHat (version 1.3.3) [21]. TopHat incorporates the Bowtie v0.12.7 algorithm to perform the alignment. Briefly, TopHat initially removes a portion of reads based on quality information accompanying each read, and maps the qualified reads to the reference genome. The reference index was built using Bowtie with a fasta file for the whole genome of zebrafish downloaded from UCSC (http://genome.ucsc.edu/). The parameters were set by default, but the number of threads to align the reads was set to 6. The aligned read files processed by TopHat were used by Cufflinks (version 1.2.1) software for further analysis, including assembling transcripts, estimating their abundances, and testing for differential expression and regulation in RNA-Seq samples [22]. To calculate gene expression intensity, the read counts were normalized to fragments per kilobase of transcript per million mapped reads (FPKM) according to the gene length and the total mapped reads [23]. Confidence intervals for FPKM estimates were calculated using a Bayesian inference method [24]. Once all of the short read sequences were assembled with Cufflinks, the output.GTF files were sent to Cuffcompare along with a reference.GTF annotation file downloaded from UCSC. This process classified each transcript as known or novel. Cuffcompare produces a combined.GTF file that is passed to Cuffdiff along with the original alignment (.BAM) files produced by TopHat. Cuffdiff then re-estimates the abundance of transcripts listed in the.GTF file using alignments from the.BAM file, and concurrently tests for differential expression. The expression testing is performed at the level of transcripts, primary transcripts and genes [25].
Cluster Analysis
Average linkage hierarchical clustering of gene expression intensity was performed in this study. The distance (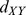 ) between genes and samples was measured by the Pearson correlation coefficient (
) between genes and samples was measured by the Pearson correlation coefficient (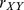 ), which is
), which is 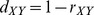 . The Pearson correlation coefficient is calculated through
. The Pearson correlation coefficient is calculated through
where 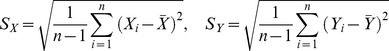 and n is the count of samples, Xi and Yi are observed values of the two variables, and
and n is the count of samples, Xi and Yi are observed values of the two variables, and 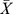 and
and 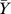 are mean values of the two variables. Computation and visualization were achieved using the heatmap plus package in R. Genes with FPKM greater than 1 in at least one of the 6 samples were chosen to perform cluster analysis.
are mean values of the two variables. Computation and visualization were achieved using the heatmap plus package in R. Genes with FPKM greater than 1 in at least one of the 6 samples were chosen to perform cluster analysis.
IPA Analysis
Gene interaction networks and signal pathways were generated using Ingenuity Pathway Analysis software (http://www.ingenuity.com/). IPA is biological data analysis software from Ingenuity® Systems; it analyzes data from a variety of experimental platforms and provides accurate biological insight into the interactions between genes, proteins, chemicals, pathways, cellular phenotypes, and disease processes.
The differentially expressed genes processed by Cuffdiff and screened with our criteria (fold change>2, p-Value<0.05, we set the p-value to 0.05, which means that the error of discovery of our differential expression genes is 0.05 for statistical significance. It is a meaningful measurement similar to FDR and routinely used in such studies.) Genes identified to be significant were then submitted to IPA for biological function, canonical pathway, and interaction network analysis. Because the current edition of IPA cannot accept and identify the gene ID of zebrafish yet, we must match these differentially expressed genes of zebrafish to their homologs of human genes. The homolog genes database that we used was HomoloGene, which was downloaded from NCBI. After converting to human homolog genes, we submitted these homologs to IPA. IPA identified a set of genes that are enriched for a specific function or pathway, and the enrichment is represented as a ratio. To find the significance of the enrichment in a specific function, IPA calculates the significance value based on the measure of involvement of the gene in the input data set to their respective molecular function/signaling pathways [26]. The significance of the networks is calculated using Fisher’s exact test, and the p-value is the executed negative logarithmic transformation.
Quantitative PCR (qPCR)
2 µg total RNA was used for reverse transcription with TransScript First-strand cDNA Synthesis SuperMix (TransGen). cDNA was then diluted 1∶5 with RNase free H2O and 2 µL of this cDNA was used in the qPCR reaction. Quantitative PCR (qPCR) was performed using GoTaq® qPCR Master Mix (Promega) on an Applied Biosystems 7900 Real-Time PCR System (ABI 7900). The data were normalized against zebrafish β-actin. The sequences of the primers used for qPCR are given in Table S7 in Tables S1.
Whole Mount in situ Hybridization
For in situ hybridization, the following genes were used as probes: nrp1a, alas2, tagln and fzd5. cDNAs were amplified by RT-PCR and the products were cloned into pTA2 vector (TOYOBO). The sequences of the primers used for probe are given in Table S8 in Tables S1. Antisense probes were synthesized using T7 RNA polymerases (Promega) and labeled with DIG RNA labeling Mix (Roche). Embryos at 48 hpf were fixed in 4% PFA/PBS overnight at 4°C, dehydrated in 100% methanol and stored at −20°C before use. Rehydrated embryos were prehybridized for 3 h at 65°C followed by overnight hybridization with DIG-labeled probes. After washing, embryos were incubated with 2% blocking solution, followed by incubation with anti-Digoxlgenin-Ap (Roche), 1∶4000 in blocking solution. After extensive washing, samples are visualized by incubating with NBT/BCIP AP stock solution (Roche) in AP buffer. Photographs were taken by using a stereo fluorescence microscope (OLYMPUS SZX2-ILLB).
Data Access
The raw sequence data can be accessed from the Gene Expression Omnibus, accession number is GSE45699 (available upon request or online when paper get accepted), and all of the genomic resources generated in this study are provided as Supplementary Files.
Supporting Information
RPS19 is not required for HSC formation and RPS19 knockdown embryos have normal number of gata1-positive cells. (A–C) The expression of cmyb (black arrow) was comparable in RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (D–F) The expression of runx1 was comparable in RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (G–I) The expression of gata1 was comparable in RPS19 morphants and RPS19 and P53 double morphants at 24 hpf. A–I is lateral view.
(JPG)
{kind=link}
Real-time PCR results of the mt2, fzd5, hsp70l, klf11a, srf1 and fn1b.
(TIF)
The in situ results of fzd5, alas2, nrp1a and tagln in zebrafish embryos at 48hpf. (A–C) The expression of fzd5 (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (D–F) The expression of alas2 (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (G–I) The expression of nrp1a (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (J–L) The expression of tagln (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. A-L is lateral view.
(TIF)
File includes Tables S1-S8.
(DOC)
Acknowledgments
We would like to thank Drs Qing Wang, Mugen Liu and Ning Wang (Huazhong University of Science and Technology) for useful advices on this study.
Funding Statement
This work was supported by the following funds from the Ministry of Science and Technology (2012CB966600, 2011AA020118, 2011CB964800); NSFC (81090410, 81130074, 31171387, 31000640, 81170465); the National Key Scientific Instrument 23/35 and Equipment Development Projects of China (2011YQ03013404); the “Strategic Priority Research Program” of the Chinese Academy of Sciences, Stem Cell and Regenerative Medicine Research (XDA01040405); Hubei National Natural Science Foundation Grant 2010CDB02402 and State Key Laboratory of Experimental Hematology Pilot Project Grant ZK12-05. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gazda HT, Sieff CA (2006) Recent insights into the pathogenesis of Diamond-Blackfan anaemia. Br J Haematol 135: 149–157. [DOI] [PubMed] [Google Scholar]
- 2. Dianzani I, Loreni F (2008) Diamond-Blackfan anemia: a ribosomal puzzle. Haematologica 93: 1601–1604. [DOI] [PubMed] [Google Scholar]
- 3. Duan J, Ba Q, Wang Z, Hao M, Li X, et al. (2011) Knockdown of ribosomal protein S7 causes developmental abnormalities via p53 dependent and independent pathways in zebrafish. Int J Biochem Cell Biol 43: 1218–1227. [DOI] [PubMed] [Google Scholar]
- 4. Gomez G, Lee JH, Veldman MB, Lu J, Xiao X, et al. (2012) Identification of vascular and hematopoietic genes downstream of etsrp by deep sequencing in zebrafish. PLoS One 7: e31658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor AM, Humphries JM, White RM, Murphey RD, Burns CE, et al.. (2012) Hematopoietic defects in rps29 mutant zebrafish depend upon p53 activation. Exp Hematol 40: 228–237 e225. [DOI] [PMC free article] [PubMed]
- 6. Danilova N, Sakamoto KM, Lin S (2011) Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br J Haematol 152: 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Torihara H, Uechi T, Chakraborty A, Shinya M, Sakai N, et al. (2011) Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br J Haematol 152: 648–654. [DOI] [PubMed] [Google Scholar]
- 8. Danilova N, Sakamoto KM, Lin S (2008) Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 112: 5228–5237. [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y, Duc ACE, Rao SY, Sun XL, Bilbee AN, et al. (2013) Control of Hematopoietic Stem Cell Emergence by Antagonistic Functions of Ribosomal Protein Para logs. Developmental Cell 24: 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poulain M, Ober EA (2011) Interplay between Wnt2 and Wnt2bb controls multiple steps of early foregut-derived organ development. Development 138: 3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burns CJ, Zhang JM, Brown EC, Van Bibber AM, Van Es J, et al. (2008) Investigation of Frizzled-5 during embryonic neural development in mouse. Developmental Dynamics 237: 1614–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mendt M, Cardier JE (2012) Stromal-Derived Factor-1 and Its Receptor, CXCR4, Are Constitutively Expressed by Mouse Liver Sinusoidal Endothelial Cells: Implications for the Regulation of Hematopoietic Cell Migration to the Liver During Extramedullary Hematopoiesis. Stem Cells and Development 21: 2142–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luis TC, Ghazvini M, Naber BAE, de Haas EFE, van Dongen JJM, et al. (2010) Canonical Wnt Signaling Regulates Hematopoiesis in a Dosage-Dependent Fashion. Experimental Hematology 38: S95–S95. [DOI] [PubMed] [Google Scholar]
- 14. Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, et al. (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321: 956–960. [DOI] [PubMed] [Google Scholar]
- 15. Uechi T, Nakajima Y, Chakraborty A, Torihara H, Higa S, et al. (2008) Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum Mol Genet 17: 3204–3211. [DOI] [PubMed] [Google Scholar]
- 16. Chakraborty A, Uechi T, Higa S, Torihara H, Kenmochi N (2009) Loss of ribosomal protein L11 affects zebrafish embryonic development through a p53-dependent apoptotic response. PLoS One 4: e4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, et al. (2011) Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 117: 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westerfield M, Doerry E, Douglas S (1999) Zebrafish in the Net. Trends Genet 15: 248–249. [DOI] [PubMed] [Google Scholar]
- 19. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 20. Detrich HW (1995) Intraembryonic hematopoietic cell migration during vertebrate development. Proc Natl Acad Sci U S A 92: 10713–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L (2011) Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol 12: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology 28: 511–U174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang H, Wong WH (2009) Statistical inferences for isoform expression in RNA-Seq. Bioinformatics 25: 1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Twine NA, Janitz K, Wilkins MR, Janitz M (2011) Whole Transcriptome Sequencing Reveals Gene Expression and Splicing Differences in Brain Regions Affected by Alzheimer’s Disease. PLoS One 6. [DOI] [PMC free article] [PubMed]
- 26.Hegde A, Qiu NC, Qiu XH, Ho SHK, Tay KQY, et al.. (2008) Genomewide Expression Analysis in Zebrafish mind bomb Alleles with Pancreas Defects of Different Severity Identifies Putative Notch Responsive Genes. PLoS One 3. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RPS19 is not required for HSC formation and RPS19 knockdown embryos have normal number of gata1-positive cells. (A–C) The expression of cmyb (black arrow) was comparable in RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (D–F) The expression of runx1 was comparable in RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (G–I) The expression of gata1 was comparable in RPS19 morphants and RPS19 and P53 double morphants at 24 hpf. A–I is lateral view.
(JPG)
Real-time PCR results of the mt2, fzd5, hsp70l, klf11a, srf1 and fn1b.
(TIF)
The in situ results of fzd5, alas2, nrp1a and tagln in zebrafish embryos at 48hpf. (A–C) The expression of fzd5 (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (D–F) The expression of alas2 (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (G–I) The expression of nrp1a (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. (J–L) The expression of tagln (black arrow) in control, RPS19 morphants and RPS19 and P53 double morphants at 48 hpf. A-L is lateral view.
(TIF)
File includes Tables S1-S8.
(DOC)