

Abstract
Psoriasis is an immune-mediated skin disorder that is inherited as a complex genetic trait. Although genome-wide association scans (GWAS) have identified 36 disease susceptibility regions, more than 50% of the genetic variance can be attributed to a single Major Histocompatibility Complex (MHC) locus, known as PSORS1. Genetic studies indicate that HLA-C is the strongest PSORS1 candidate gene, since markers tagging HLA-Cw*0602 consistently generate the most significant association signals in GWAS. However, it is unclear whether HLA-Cw*0602 is itself the causal PSORS1 allele, especially as the role of SNPs that may affect its expression has not been investigated. Here, we have undertaken an in-depth molecular characterization of the PSORS1 interval, with a view to identifying regulatory variants that may contribute to disease susceptibility. By analysing high-density SNP data, we refined PSORS1 to a 179 kb region encompassing HLA-C and the neighbouring HCG27 pseudogene. We compared multiple MHC sequences spanning this refined locus and identified 144 candidate susceptibility variants, which are unique to chromosomes bearing HLA-Cw*0602. In parallel, we investigated the epigenetic profile of the critical PSORS1 interval and uncovered three enhancer elements likely to be active in T lymphocytes. Finally we showed that nine candidate susceptibility SNPs map within a HLA-C enhancer and that three of these variants co-localise with binding sites for immune-related transcription factors. These data indicate that SNPs affecting HLA-Cw*0602 expression are likely to contribute to psoriasis susceptibility and highlight the importance of integrating multiple experimental approaches in the investigation of complex genomic regions such as the MHC.
Introduction
Psoriasis vulgaris is a chronic inflammatory skin disorder, caused by keratinocyte hyper-proliferation, angiogenesis, and infiltration of immune cells into the dermis and the epidermis [1]. The genetic basis of the disease is well documented by a large body of family- and population-based studies, which have convincingly demonstrated a complex mode of inheritance for this condition [2].
Although genome-wide association scans (GWAS) have identified more than 30 disease susceptibility intervals, up to 50% of the genetic variance is accounted for by a single locus (PSORS1) [3], which consistently generates odds ratios >2.5 [4]–[6]. This region spans approximately 250 kb within the Major Histocompatibility Complex (MHC) and encompasses nine protein-coding genes [7]. Disease associated alleles are specifically found within CDSN, which encodes a keratinocyte structural protein, CCHCR1, which regulates EGFR-STAT3 signalling and HLA-C, which codes for a class I MHC molecule [8]–[10]. Although all three proteins contribute to biological processes that are relevant to disease pathogenesis, HLA-C is widely considered the most likely candidate gene for the PSORS1 locus, as the association with HLA-Cw*0602 is more significant than that observed with any other marker [5], [6]. The functional relevance of this observation, however, remains unclear, as no -Cw6 specific antigen or interacting protein has yet been identified. At the same time, the existence of regulatory polymorphisms co-segregating with HLA-Cw*0602 cannot be excluded. In fact our group has demonstrated that SNPs residing within the HLA-C minimal promoter cause the differential expression of various –Cw alleles, including HLA-Cw*0602 [11].
Here, we have undertaken a systematic analysis of the PSORS1 interval, with the aim of identifying disease associated variants that have the potential to impact on HLA-C expression. By combining the results of high-resolution genetic profiling with an experimental annotation of regulatory sites, we have identified a restricted number of candidate susceptibility alleles mapping to an HLA-C enhancer element.
Results
High-resolution Analysis of Linkage Disequilibrium Conservation Refines the PSORS1 Interval to a 179 kb Region
We sought to refine the boundaries of the PSORS1 interval, by dissecting the pattern of linkage disequilibrium (LD) conservation around the HLA-C locus. We exploited the availability of the high-density SNP data generated by the ImmunoChip Consortium, which genotyped six-hundred and fifteen PSORS1 markers in 4,803 UK controls [3]. By analysing this extended study population, we noted that the PSORS1 locus encompasses several LD bocks (Figure 1). We also observed that HLA-C lies within a 179 kb region of LD conservation, which excludes all other protein coding genes. This refined susceptibility interval became the focus of our subsequent investigations (Figure 1).
Figure 1. High-resolution analysis of the PSORS1 interval reveals multiple regions of LD conservation.

LD decay was measured using the D’ index, which was derived based on the genotypes of 4,803 UK controls. Markers occurring with a minor allele frequency (MAF) <0.1 were excluded from the analysis, as variation at these loci is likely to be the result of recent mutational events. The plot documents the analysis of 147 SNPs, randomly selected from the 373 variants that had a MAF >0.1 (see Methods for details). The top panel shows the position of PSORS1 genes (the HCG27 pseudogene is shaded in grey), with the region of LD conservation around HLA-C highlighted by a double arrow.
Alignment of Multiple MHC Haplotype Sequences Identifies 144 PSORS1 Variants Associated with HLA-Cw*0602
As the latest version of dbSNP (build 137) lists 5,359 common variants (minor allele frequency >1%) mapping to the 179 kb PSORS1 region, we sought to determine the number and distribution of changes unique to haplotypes bearing HLA-Cw*0602. By querying the GenBank database, we identified a number of publicly available MHC sequences which spanned the PSORS1 locus and encompassed a variety of HLA-C alleles (Table 1). Since HLA-Cw*0602 was only found on three sequences, we sought to expand the catalogue of -Cw*0602 associated variants, by characterizing a fourth chromosome. We therefore sequenced the PSORS1 region from the genome of a psoriatic individual who was heterozygous for HLA-Cw*0602. To circumvent the difficulty of mapping short genomic reads to a region as complex as the MHC, we generated a BAC library from the DNA of the patient and sequenced the two clones spanning the critical PSORS1 interval (Figure S1 and Table S1).
Table 1. MHC haplotype sequences analysed in this study.
| HLA-C allele | GenBank Accession n. (reference) |
| HLA-Cw*0602 | KC312698 (this study) |
| HLA-Cw*0602 | DQ249182 [20] |
| HLA-Cw*0602 | DQ249178-DQ249180 [20] |
| HLA-Cw*0602 | GL000252 [37] |
| HLA-Cw*030401 | GL000254 [37] |
| HLA-Cw*030401 | DQ249177 [20] |
| HLA-Cw*0501 | GL000255 [37] |
| HLA-Cw*070101 | GL000251 [37] |
| HLA-Cw*070101 | DQ249172 [20] |
| HLA-Cw*070101 | DQ249174 [20] |
| HLA-Cw*07020103 | reference hg19 genome |
| HLA-Cw*07020103 | DQ249181 [20] |
| HLA-Cw*0802 | DQ249173 [20] |
| HLA-Cw*0802 | DQ249176 [20] |
| HLA-Cw*1601 | GL000253 [37] |
A comparison of the sequences of the four -Cw*0602 bearing haplotypes with those of 11 chromosomes harbouring other HLA-C alleles (Table 1) indicated that only 151/5,359 PSORS1 variants were unique to PSORS1-Cw*0602 haplotypes (i.e. they were present in at least three -Cw*0602 bearing sequences and absent from all other haplotypes). To further develop the panel of PSORS1 chromosomes and increase the power to discriminate risk and non-risk variants, we next imputed HLA-C genotypes for 57 individuals of European descent, previously sequenced by the 1000 Genomes Consortium [12]. To take into account the relative uncertainty of MHC imputation data derived from low-coverage sequence reads, we defined as non-risk only those SNPs that were present in >10 non-Cw*0602 chromosomes. Through this approach we were able to exclude a further seven SNPs from our list of HLA-Cw*0602 associated alleles, bringing the total to count to one hundred and forty-four (Table S2).
The distribution of these candidate susceptibility variants was markedly skewed, with 111/144 markers located within a 72 kb region encompassing HLA-C (Figure 2). Of note, the only two nucleotide substitutions which mapped within the HLA-C coding region (rs1050414 and rs697743) were silent polymorphisms. Thus, neither of these variants is likely to have a pathogenic impact on protein function.
Figure 2. Distribution of PSORS1 candidate variants.
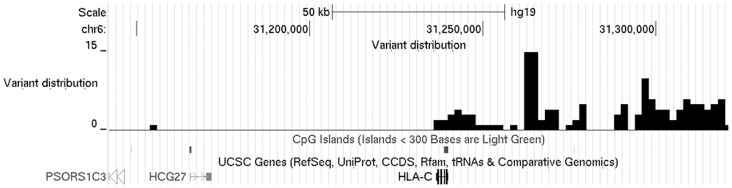
The number of changes that are unique to PSORS1-Cw0602 haplotypes is plotted against GRCh37/hg19 coordinates.
A Molecular Characterization of the Refined PSORS1 Interval Identifies Three Likely Enhancer Elements
We next sought to characterize the regulatory elements that map within the refined PSORS1 locus. We initially queried the University of California Santa Cruz (UCSC) genome browser and assessed the position of open chromatin marks such as unmethylated CpG islands, DNAse hyper-sensitivity sites, histone H3 lysine 4 mono-methylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac). We focused our analysis on primary T lymphocytes, as these cells play a key role in the inflammatory responses underlying the pathogenesis of psoriasis [1]. We found that the refined PSORS1 interval encompassed three DNA segments that presented multiple features associated with open chromatin (Table S3).
As the experimental data annotated in the UCSC genome browser had been generated in cells obtained from healthy individuals, we obtained blood samples from six psoriatic patients, in order to assess the distribution of key epigenetic marks in a context that was more relevant to the pathophysiology of the disease.
We first investigated the methylation status of the four CpG islands (CGIs) that lie within the refined PSORS1 locus (shown in Figure 3A, immediately above the candidate variants track). We analysed the DNAs of four HLA-Cw*0602-positive patients and found that the CGI lying upstream of POU5F1 was partially methylated, whereas the remaining three were completely unmethylated. This matched the modification pattern observed in control individuals (Figure 4).
Figure 3. Epigenetic analysis of the refined PSORS1 locus in patient T-lymphocytes.

A) H3K4me1 and H3K27ac ChiP-Seq peaks are shown for a representative sample. The size of each peak is plotted as a black box immediately below the Chip-Seq track. The figure besides each box indicates the False Discovery Rate (FDR) associated with the identification of the peak (e.g. 0.47 indicates a FDR of 0.47%). B) A detailed view of the HLA-C gene region is shown. The dotted line box highlights the boundaries of the active regulatory region defined by the overlap of an unmethylated CpG island with H3K4me1 and H3K27ac peaks.
Figure 4. Locus specific analysis of whole-blood DNA identified similar methylation patterns in healthy and psoriatic individuals.

(A) The CGI encompassing the minimal promoter and the first two exons of HLA-C was investigated by methyl-specific PCR, following treatment of whole blood DNA with sodium bisulfite. The DNA of a healthy donor was modified with the M.SssI CpG methyl-transferase and used as a positive control (C+). (B) The CGIs lying upstream of HCG27 (top left), HLA-C (top right) and POU5F1 (bottom) were analysed by CoBRA, exploiting the loss of restriction nuclease sites in unmethylated and bisulphite converted samples. A sample that did not undergo bisulfite treatment was used as positive control for the enzymatic digestion (C+). The methylation status of the CGIs was further validated by direct sequencing of cloned PCR products, generated from the DNA of one healthy and one affected individual (data not shown).
We next investigated the distribution of H3K4me1 and H3K27ac binding sites, as these histone modifications are typically found together within active enhancers [13]. We used ChIP-Seq protocols to analyse DNA from the T-lymphocytes of two psoriatic patients who were heterozygous for HLA-Cw*0602 (Table S4). This identified nine H3K4me1 peaks, three of which overlapped with H3K27ac occupancy sites and are therefore likely to represent active enhancers. One of the three elements encompasses HLA-C exons 1–3, as well as the gene upstream region. The other two DNA segments map to a 10 kb region around the HCG27 pseudogene (Figure 3). This pattern matches the distribution of H3K4me1 and H3K7ac sites observed in control T cells (Figure S2).
Three Candidate Susceptibility Alleles Map to Biologically Relevant Transcription Factor Binding Sites within a Likely HLA-C Enhancer
We cross-referenced the position of all 144 SNPs unique to PSORS1-Cw*0602 haplotypes with that of the active regulatory elements identified by epigenetic profiling. While none of the variants map to the HGC27 gene region, nine candidate susceptibility SNPs lie within the putative HLA-C enhancer element (Figure 3). To further explore the functional significance of these variants, we examined the data recently generated by the ENCODE Consortium [14]. We found that SNPs rs1050414 and rs697743 co-localize with experimentally validated binding sites for polymerase II and for POU2F2/Oct2, a well known immune activator [15]. Of note, rs1050414 also maps within a ChIP-Seq peak for NF-κB, a master regulator of inflammatory responses [16] (Figure S3).
To investigate the possibility that any of the nine critical SNPs may lead to the creation of novel transcription factor binding sites, we queried the TRANSFAC database using wild-type and variant sequences. This analysis indicated that SNP rs28993433 may introduce a novel site for SP4 (FDR <5%), a transcription factor involved in the regulation of MHC related genes [17].
Discussion
The evidence supporting PSORS1 as the major genetic determinant for psoriasis is overwhelming [2]. Of note, no secondary association signal has been detected within this susceptibility interval [18], [19], so that the disease risk conferred by PSORS1 is likely due to a single genetic determinant. Although the results of GWAS and preliminary deep-sequencing studies point to HLA-Cw*0602 as a candidate susceptibility allele [6], [20], no aberrant function of the HLA-Cw6 protein has yet been demonstrated. Conversely, a number of studies have reported that HLA-C expression is altered in psoriatic lesions [5], [21] and that regulatory SNPs can affect HLA-C transcript levels [11], [22].
Here, we have undertaken a comprehensive molecular investigation of the PSORS1 locus, with a view to dissecting the pathogenic contribution of SNPs that may affect the expression of HLA-Cw*0602.
We initially sought to refine the localization of the causal susceptibility allele(s), by analysing LD conservation patterns within the susceptibility interval. We exploited the unique dataset generated by the ImmunoChip Consortium, which includes 4,803 individuals, genotyped across the PSORS1 locus at an average density of 1 SNP: 950 bp. The power of this approach is exemplified by the fact that it allowed us to refine the size of the susceptibility interval by nearly a third (from 259 to 179 kb), even though our assessment of LD conservation was rather conservative.
We next compared the sequence of HLA-Cw*0602 bearing chromosomes with that of publicly available non-risk haplotypes. This enabled us to generate a comprehensive catalogue of candidate susceptibility alleles and to exclude a pathogenic role for 97.3% of the common polymorphisms that lie within the refined PSORS1 locus. Importantly, we showed that none of the 144 candidate variants resulted in an amino acid substitution that was unique to risk-bearing chromosomes. This observation argues against the possibility that the association at the PSORS1 locus may be explained by a single amino acid change. Of note, recent studies have demonstrated that single non-synonymous substitutions account for the main MHC association signals underlying rheumatoid arthritis susceptibility and host control of HIV-I infection [23], [24]. Thus, our findings point to a potentially important difference in the make-up of the PSORS1 allele and reinforce the notion that regulatory variants are likely to contribute to its pathogenic effect.
To investigate the biological significance of the 144 critical variants, we sought to identify functional non-coding elements mapping within the refined PSORS1 interval. We initially queried the UCSC genome browser and mined data generated in T lymphocytes, as these cells drive key adaptive immune responses underlying the establishment of chronic inflammation in psoriasis [1]. We retrieved information on a variety of epigenetic marks and identified three likely active enhancers, mapping to the HCG27 and HLA-C gene regions. Importantly, our in-house ChIP-Seq analysis confirmed that this epigenetic profile is also relevant to the transcriptional activity of patient T-lymphocytes. Our experiments were carried out in individuals who were heterozygous for HLA-Cw*0602, as the low frequency of the –Cw*0602/−Cw*0602 genotype hindered the recruitment of homozygous patients. Given the difficulties associated with the isolation of intact chromatin from skin keratinocytes or resident lymphocytes, the ChIP-Seq analysis was undertaken in circulating T-cells. Despite these limitations, the observation of comparable ChIP-Seq profiles in patients and controls indicates that the pathogenic effect of the PSORS1 allele is unlikely to be mediated by the occurrence of major epigenetic alterations.
In the final phase of the study we integrated the results of our genetic and functional investigations and found that 9 candidate SNPs localize to a likely HLA-C enhancer. This element, which encompasses HLA-C exons 1–3 as well as the gene upstream region, spans approximately 5 kb and therefore extends significantly upstream of the canonical gene promoter identified by classic reporter studies [25]. The ChIP-Seq data generated by the ENCODE Consortium [14] indicate that this enhancer harbours binding sites for the NF-κB, STAT2, IRF4 and POU2F2 transcription factors, which all play important roles in inflammatory responses [15], [16], [26], [27]. Two of the PSORS1 critical variants map within ChIP-Seq peaks for NF-κB and POU2F2, with a third predicted to create an SP4 binding site. While further studies will be required to validate the functional impact of individual SNPs, it is noteworthy that our data are entirely consistent with those generated in previous surveys of disease associate regions, which documented a highly significant enrichment of candidate susceptibility alleles within strong enhancer elements [13], [14].
In conclusion, we have carried out an in-depth molecular genetic investigation of the major psoriasis susceptibility locus and identified new regulatory variants that are likely to contribute to disease susceptibility. These findings pave the way for further immunological studies investigating the effects of altered HLA-Cw6 expression on inflammatory responses and psoriasis pathogenesis.
Materials and Methods
Ethics Statement
This study was conducted according to the principles expressed in the Declaration of Helsinki. All participating subjects granted their written informed consent. Ethical approval was obtained from Guy’s and St Thomas Hospital Local Research Ethic Committee.
Subjects
The PSORS1-Cw*0602 sequence was generated from the DNA of an affected patient (JC), selected from an extended pedigree showing co-segregation of psoriasis with chromosome 6p21 markers. The analysis of epigenetic marks was undertaken in ten HLA-Cw*0602 heterozygous individuals (six patients and four controls), selected from a large case-control dataset that has been described elsewhere [6].
High-resolution LD Conservation Analysis
The genotypes of 373 common SNPs (MAF >0.1) mapping to chr6∶31,062,133-31,419,877 were extracted from the data generated by the ImmunoChip consortium for 4,803 unrelated controls, selected from the 1958 British Birth Cohort and National Blood Service donors [3]. The –thin option of PLINK [28] was used to restrict the analysis to a random selection of 147 SNPs and a GOLD heatmap was generated using Haploview 4.2 [29]. The random selection process was repeated three times with comparable results.
Generation of a PSORS1 HLA-Cw*0602 Sequence from Patient DNA
We generated a genomic BAC library using high molecular weight DNA, isolated from the immortalized lymphocytes of patient JC. We screened the library with a set of five probes, which matched unique PSORS1 sequences within the STG, CCHCR1, HCG27 and HLA-C gene regions. After validating the identity of eight positive clones by BAC end sequencing, we genotyped a set of informative SNPs located within the CDSN, CCHCR1, and HLA-C gene regions [30]. This allowed us to distinguish the clones bearing risk alleles from their wild type counterparts and led to the identification of two BACs carrying PSORS1-Cw*0602 sequences. These critical clones were sequenced by Cogenics (Grenoble, France) using a 454 Genome Sequencer FLX analyzer (Roche, Basel, Switzerland). A total of 125,011 reads were generated, 93% of which were mapped to the target sequence and processed with the analysis pipeline described in Figure S4. The resulting sequence was deposited in GenBank (accession n. KC312698).
Identification of Variants Unique to PSORS1-Cw*0602 Haplotypes
Querying the GenBank database identified a total of 16 MHC haplotype sequences spanning the refined PSORS1 locus. Of note, two of these included HLA-Cw*1203, a marker which has previously shown suggestive evidence for association with psoriasis [31], [32]. As the inconclusive nature of these findings prevented a clear definition of risk status for PSORS1-Cw*1203 chromosomes, these samples were excluded from all further analyses. We compared the sequences of the remaining PSORS1 haplotypes using Sequencher v4.9 (Gene Codes Corporation, Ann Arbor, MI) to run the Clustal algorithm [33]. We identified 151 variants (excluding dinucleotide microsatellites and homopolymers exceeding 4 nucleotides in length) that were unique to PSORS1-Cw*0602 haplotypes. We next mined the data generated by the 1000 Genomes Project (available at ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/) and used the genotypes of two critical SNPs (rs887466 and rs4379333 [24]) to predict the HLA-Cw*0602 status of 57 European individuals. We classified these individuals as HLA-Cw*0602 positive (n = 7), HLA-Cw*1203 positive (n = 4; removed from all further analyses) or HLA-Cw*0602/Cw*1203 negative (n = 46). Finally, we interrogated the genotypes of the HLA-Cw*0602/Cw*1203 negative individuals for each of the 151 variants identified in the previous phase. Variants found in >10 HLA-Cw*0602/Cw*1203 negative individuals were excluded from further analyses.
Bioinformatic Analysis of the Refined PSORS1 Interval
We queried the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway) for the annotation relating to the refined PSORS1 locus (GRCh37/hg19 coordinates: 6∶31,142,245-31,321,211). We focused on the experimental data that had been generated in CD4+ T lymphocytes and collated information on open chromatin marks. We looked for DNase I hypersensitive sites (DHS) that were supported by a –log10(pval) >50, a signal value >30, and a score >120. We also annotated less significant peaks, which were found less than 1 kb away from any DHS site that met the above criteria. Finally, we scored the position of unmethylated CGI, H3K4me3 and H3K27ac sites.
We analysed the effect of SNPs that are unique to PSORS1-Cw*0602 and lie within regulatory elements, by querying the TRANSFAC database (BIOBASE Biological Databases, Beverly, MA) with the FIMO motif search tool, which is part of the MEME suite [34].
DNA Methylation Analysis
Genomic DNA was treated with sodium bisulfite and purified using the EZ DNA Methylation-Gold™ Kit (Zymo, Orange, CA). The methylation status of the CGI encompassing the minimal promoter and the first two exons of HLA-C was investigated by methyl-specific PCR. Two independent reactions were carried out using either unmethylated DNA-specific (CpG121.UF1∶5′-GTGTTTTTTGGTTTTAATATTTTGG-3′; CpG121.UR1∶5′-CCACTTCATCTCAATAAACTACATA-3′) or methylated DNA-specific (CpG121.MF1∶5′-TTGTGTTTTTCGGTTTTAATATTTC-3′; CpG121.MR1∶5′-CGCTTCATCTCAATAAACTACGTA-3′) oligonucleotides. The methylation status of the remaining CGIs was investigated by combined bisulphite and restriction analysis (CoBRA). Sodium bisulphite treated DNA samples were amplified for 45 cycles with primers specific for each CpG islands (Table S5). Amplicons were digested using either TaiI or TaqαI (New England Biolabs, Ipswich, MA) and differentially methylated products were resolved on 3% agarose gels.
ChIP-seq Analysis
Peripheral blood mononuclear cells were isolated from whole blood samples by centrifugation on a Ficoll (GE Healthcare, Little Chalfont, UK) layer and T cells were purified by negative selection, using the pan T cell Isolation Kit II Human, (Miltenyi Biotec, Bergisch Gladbach, Germany). Native chromatin was extracted from 25 million cells and fragmented by MNase enzymatic fragmentation. Immuno-precipitation was performed by incubating 3 µg of chromatin with 4 µg of anti H3K4me1 (pAb-037-050 from Diagenode, Liege, Belgium) or anti-H3K27ac (399133 from Active Motif, La Hulpe, Belgium) antibody. Following DNA recovery, ChIP-seq libraries were prepared and amplified (14 cycles) using the NEXTflex ChIP-seq kit from Bio Scientific (Austin, Texas, USA). Each sample was tagged using the NEXTflex ChIP-seq Barcodes. Libraries were normalized to 1 nM, pooled and sequenced on a HiSeq2000 apparatus (Illumina, San Diego, CA).
Pools were de-multiplexed with BCL (Illumina) and reads were mapped to the GRCh37/hg19 reference sequence using NovoAlign (Novocraft Technologies Sdn Bhd, Selangor, Malaysia). PCR duplicates were removed with MarkDuplicates (a utility available at http://picard.sourceforge.net/index.shtml) and poor quality alignments were removed with SAMtools [35]. ChIP-seq peaks were identified based on the depth of uniquely mapped reads, using MACS [36].
Supporting Information
Genomic location of the two BAC clones spanning the PSORS1 locus.
(DOCX)
The PSORS1 epigenetic profile of patient T-lymphocytes is comparable to that observed in controls.
(DOCX)
Candidate susceptibility SNPs co-localize with Pol-II, POU2F2 and NF-κB Chip-Seq peaks.
(DOCX)
Software pipeline used to analyze the sequence of the two BACs spanning the PSORS1 locus.
(DOC)
Sequence coverage statistics for the two BAC clones spanning the refined PSORS1 locus.
(XLSX)
Details of the 144 candidate PSORS1 variants.
(XLSX)
Distribution of PSORS1 active chromatin marks in control T lymphocytes.
(XLSX)
Sequence coverage statistics for the ChIP-Seq analysis of H3K4me1 and H3K27ac marks.
(XLSX)
PCR primers and restriction enzymes used in the CoBRA analysis.
(XLSX)
Acknowledgments
We wish to thank Joseph Costello, Paul Lavender and Jennifer Frost for helpful comments and technical advice.
Funding Statement
The authors acknowledge the use of BRC Core Facilities provided by the financial support from the Department of Health via the National Institute of Health Research (NIHR,www.nihr.ac.uk/) comprehensive Biomedical Research Centre award to Guy’s and St Thomas NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. The British 1958 Birth Cohort DNA collection was funded by Medical Research Council (www.mrc.ac.uk) grant G0000934 and Wellcome Trust (www.wellcome.ac.uk) grant 068545/Z/02, and the UK National Blood Service controls by the Wellcome Trust. This work was supported by the Medical Research Council (grant G0601387 to RCT, JNB, FON and AH), the National Psoriasis Foundation USA (www.psoriasis.org; Discovery grant to FC) and Prime Minister’s Initiative 2 (PMI2)/partnership NPF127, for UK-US partnership between KCL and UCSF. AB’s studentship was funded by The Generation Trust. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nestle FO, Kaplan DH, Barker J (2009) Psoriasis. N Engl J Med 361: 496–509. [DOI] [PubMed] [Google Scholar]
- 2. Capon F, Burden AD, Trembath RC, Barker JN (2012) Psoriasis and Other Complex Trait Dermatoses: From Loci to Functional Pathways. J Invest Dermatol 132: 915–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, et al. (2012) Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 44: 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, et al. (2010) Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet 42: 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, et al. (2009) Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strange A, Capon F, Spencer CC, Knight J, Weale ME, et al. (2010) A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 42: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Capon F, Munro M, Barker J, Trembath R (2002) Searching for the major histocompatibility complex psoriasis susceptibility gene. J Invest Dermatol 118: 745–751. [DOI] [PubMed] [Google Scholar]
- 8.Capon F, Allen MH, Ameen M, Burden AD, Tillman D, et al.. (2004) A synonymous SNP of the corneodesmosin gene leads to increased mRNA stability and demonstrates association with psoriasis across diverse ethnic groups. Hum Mol Genet. [DOI] [PubMed]
- 9. Mallon E, Newson R, Bunker CB (1999) HLA-Cw6 and the genetic predisposition to psoriasis: a meta-analysis of published serologic studies. J Invest Dermatol 113: 693–695. [DOI] [PubMed] [Google Scholar]
- 10. Tervaniemi MH, Siitonen HA, Soderhall C, Minhas G, Vuola J, et al. (2012) Centrosomal localization of the psoriasis candidate gene product, CCHCR1, supports a role in cytoskeletal organization. PLoS One 7: e49920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hundhausen C, Bertoni A, Mak RK, Botti E, Di Meglio P, et al. (2012) Allele-specific cytokine responses at the HLA-C locus: implications for psoriasis. J Invest Dermatol 132: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, et al. (2012) B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J Exp Med 209: 2049–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayden MS, Ghosh S (2008) Shared principles in NF-kappaB signaling. Cell 132: 344–362. [DOI] [PubMed] [Google Scholar]
- 17. Rodriguez-Rodero S, Gonzalez S, Rodrigo L, Fernandez-Morera JL, Martinez-Borra J, et al. (2007) Transcriptional regulation of MICA and MICB: a novel polymorphism in MICB promoter alters transcriptional regulation by Sp1. Eur J Immunol 37: 1938–1953. [DOI] [PubMed] [Google Scholar]
- 18. Feng BJ, Sun LD, Soltani-Arabshahi R, Bowcock AM, Nair RP, et al. (2009) Multiple Loci within the major histocompatibility complex confer risk of psoriasis. PLoS Genet 5: e1000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knight J, Spain SL, Capon F, Hayday A, Nestle FO, et al. (2012) Conditional analysis identifies three novel major histocompatibility complex loci associated with psoriasis. Hum Mol Genet 21: 5185–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NV, et al. (2006) Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet 78: 827–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carlen L, Sakuraba K, Stahle M, Sanchez F (2007) HLA-C expression pattern is spatially different between psoriasis and eczema skin lesions. J Invest Dermatol 127: 342–348. [DOI] [PubMed] [Google Scholar]
- 22. Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, et al. (2011) Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 472: 495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McLaren PJ, Ripke S, Pelak K, Weintrob AC, Patsopoulos NA, et al. (2012) Fine-mapping classical HLA variation associated with durable host control of HIV-1 infection in African Americans. Hum Mol Genet 21: 4334–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, et al. (2012) Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 44: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johnson DR (2003) Locus-specific constitutive and cytokine-induced HLA class I gene expression. J Immunol 170: 1894–1902. [DOI] [PubMed] [Google Scholar]
- 26. Stark GR, Darnell JE Jr (2012) The JAK-STAT pathway at twenty. Immunity 36: 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu WD, Pan HF, Ye DQ, Xu Y (2012) Targeting IRF4 in autoimmune diseases. Autoimmun Rev 11: 918–924. [DOI] [PubMed] [Google Scholar]
- 28. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21: 263–265. [DOI] [PubMed] [Google Scholar]
- 30. Veal CD, Capon F, Allen MH, Heath EK, Evans JC, et al. (2002) Family-based analysis using a dense single-nucleotide polymorphism- based map defines genetic variation at PSORS1, the major psoriasis- susceptibility locus. Am J Hum Genet 71: 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liao HT, Lin KC, Chang YT, Chen CH, Liang TH, et al. (2008) Human leukocyte antigen and clinical and demographic characteristics in psoriatic arthritis and psoriasis in Chinese patients. J Rheumatol 35: 891–895. [PubMed] [Google Scholar]
- 32. Helms C, Saccone NL, Cao L, Daw JA, Cao K, et al. (2005) Localization of PSORS1 to a haplotype block harboring HLA-C and distinct from corneodesmosin and HCR. Hum Genet 118: 466–476. [DOI] [PubMed] [Google Scholar]
- 33.Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics Chapter 2: Unit 2 3. [DOI] [PubMed]
- 34. Bailey TL, Boden M, Buske FA, Frith M, Grant CE, et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Feng J, Liu T, Qin B, Zhang Y, Liu XS (2012) Identifying ChIP-seq enrichment using MACS. Nat Protoc 7: 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Horton R, Gibson R, Coggill P, Miretti M, Allcock RJ, et al. (2008) Variation analysis and gene annotation of eight MHC haplotypes: the MHC Haplotype Project. Immunogenetics 60: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genomic location of the two BAC clones spanning the PSORS1 locus.
(DOCX)
The PSORS1 epigenetic profile of patient T-lymphocytes is comparable to that observed in controls.
(DOCX)
Candidate susceptibility SNPs co-localize with Pol-II, POU2F2 and NF-κB Chip-Seq peaks.
(DOCX)
Software pipeline used to analyze the sequence of the two BACs spanning the PSORS1 locus.
(DOC)
Sequence coverage statistics for the two BAC clones spanning the refined PSORS1 locus.
(XLSX)
Details of the 144 candidate PSORS1 variants.
(XLSX)
Distribution of PSORS1 active chromatin marks in control T lymphocytes.
(XLSX)
Sequence coverage statistics for the ChIP-Seq analysis of H3K4me1 and H3K27ac marks.
(XLSX)
PCR primers and restriction enzymes used in the CoBRA analysis.
(XLSX)