

hGAAP promotes cell adhesion and migration by increasing localized Ca2+-dependent activation of calpain, leading to increased focal adhesion dynamics.
Abstract
Golgi antiapoptotic proteins (GAAPs) are highly conserved Golgi membrane proteins that inhibit apoptosis and promote Ca2+ release from intracellular stores. Given the role of Ca2+ in controlling cell adhesion and motility, we hypothesized that human GAAP (hGAAP) might influence these events. In this paper, we present evidence that hGAAP increased cell adhesion, spreading, and migration in a manner that depended on the C-terminal domain of hGAAP. We show that hGAAP increased store-operated Ca2+ entry and thereby the activity of calpain at newly forming protrusions. These hGAAP-dependent effects regulated focal adhesion dynamics and cell migration. Indeed, inhibition or knockdown of calpain 2 abrogated the effects of hGAAP on cell spreading and migration. Our data reveal that hGAAP is a novel regulator of focal adhesion dynamics, cell adhesion, and migration by controlling localized Ca2+-dependent activation of calpain.
Introduction
The first Golgi antiapoptotic protein (GAAP), also known as TMBIM4 (transmembrane Bax [Bcl-2–associated X protein] inhibitor–containing motif protein 4), was found in camelpox virus. Closely related proteins were subsequently found in a few strains of vaccinia virus (VACV) and throughout eukaryotes (Gubser et al., 2007). The related human GAAP (hGAAP), which shares 73% amino acid identity with viral GAAP (vGAAP), is expressed ubiquitously, and it is essential for cell survival (Gubser et al., 2007). All GAAPs, from evolutionary diverse sources, have similar lengths and hydrophobicity profiles, suggesting important and evolutionarily conserved functions. Phylogenetic analysis suggests that GAAPs have ancient origins within eukaryotes, supporting the expansion of some members of the transmembrane BI-1 (Bax inhibitor-1)–containing motif (TMBIM) family from a GAAP-like ancestor ∼2,000 million years ago (Hu et al., 2009). hGAAP is proposed to be a housekeeping gene based on its widespread expression, its requirement for cell viability (Gubser et al., 2007), and from statistical analysis of microarrays (Lee et al., 2007). Furthermore, hGAAP mRNA levels are dysregulated in some human breast tumors, making it a putative oncogene and a possible target for anticancer therapy (van ’t Veer et al., 2002; Gubser et al., 2007).
hGAAP, vGAAP, and BI-1, another widely expressed and conserved antiapoptotic TMBIM protein, have similar secondary structures. Each has six transmembrane domains with short interconnecting loops, a putative reentrant loop toward the charged C terminus (Carrara et al., 2012), and a conserved UPF0005 motif (Reimers et al., 2008; Hu et al., 2009). These features are conserved within the TMBIM family. hGAAP localizes predominantly to Golgi membranes and provides protection from a broad range of apoptotic stimuli (Gubser et al., 2007). Overexpression of hGAAP reduces both the Ca2+ content of the Golgi and ER, and the amplitude of the Ca2+ signals evoked by either staurosporine to trigger apoptosis or histamine to stimulate formation of inositol 1,4,5-trisphosphate (de Mattia et al., 2009). Reducing the expression of endogenous hGAAP has the opposite effects (de Mattia et al., 2009). Overexpression of BI-1 also reduces the Ca2+ content of the ER (Xu et al., 2008), and it increases both polymerization of actin and cell adhesion (Lee et al., 2010a). These observations and the contributions of Ca2+ signals to the control of migration and adhesion (Giannone et al., 2002; Clark et al., 2006; Ying et al., 2009) suggest that GAAPs might also affect these processes via their effects on Ca2+ signaling.
During cell migration, protrusion of the cell membrane is followed by formation of new adhesions at the front of the cell. These establish connections between the substratum and the actin cytoskeleton, generating traction forces that ultimately make the cell move forward as adhesions at the rear disassemble (Petrie et al., 2009). This coordinated assembly and disassembly of cell adhesions is essential for cell migration, and it is associated with spatially organized Ca2+ signals. In many migrating cells, there is a gradient of cytosolic free Ca2+ concentration ([Ca2+]i) from front to rear. The highest [Ca2+]i is at the rear of the cell (Marks and Maxfield, 1990; Brundage et al., 1991), where Ca2+ influx through stretch-activated channels in the plasma membrane is essential for detachment and retraction (Lee et al., 1999). Ca2+ influx also controls migration at the leading edge. Here, Ca2+ influx via stretch-activated TrpM7 (transient receptor potential M7) channels can be amplified by Ca2+ release from intracellular stores mediated by inositol 1,4,5-trisphosphate receptors (Clark et al., 2006; Wei et al., 2009). The resulting polarized local increases in [Ca2+]i, “Ca2+ flickers,” control the direction of migration (Wei et al., 2009). In most cells, depletion of intracellular Ca2+ stores stimulates Ca2+ influx across the plasma membrane via store-operated Ca2+ entry (SOCE; Putney, 2009). SOCE is activated when the ER Ca2+-sensor, STIM1 (stromal interaction molecule 1), detects a decrease in the luminal Ca2+ concentration of the ER and activates Ca2+-permeable Orai channels in the plasma membrane (Lewis, 2011). SOCE also contributes to cell adhesion and migration (Yang et al., 2009) and to tumor metastasis (Yang et al., 2009; Feng et al., 2010; Chen et al., 2011; Prevarskaya et al., 2011).
Disassembly of adhesions, which is required for their turnover, is partially dependent on the activity of intracellular calpain 2 (Franco and Huttenlocher, 2005). This Ca2+-activated cysteine protease cleaves several components of the focal adhesion complex, such as FAK (Chan et al., 2010) and talin (Franco et al., 2004), triggering disassembly of the complex at the rear of the cell. Calpain inhibition impairs cell spreading in several cell types (Rock et al., 2000; Parnaud et al., 2005). However, the mechanisms that control the local Ca2+ signals during cell migration, and their links with dynamic cell movements are poorly understood.
Here, we demonstrate that hGAAP promotes cell adhesion and migration by increasing the turnover of focal adhesions. These effects require activation of calpain 2. hGAAP causes activation of SOCE and thereby Ca2+-mediated stimulation of calpain activity near the plasma membrane. Thus, we report a novel function for a highly conserved Golgi membrane protein in coordinating cell migration via localized activation of calpain 2.
Results
hGAAP increases cell adhesion and the rate of cell spreading
Given the important roles of Ca2+ in cell adhesion and migration, and the ability of hGAAP to modulate Ca2+ fluxes, we explored a possible role for hGAAP in regulating cell adhesion. Two human cancer cell lines, U2-OS (osteosarcoma) and HeLa (cervical cancer), were engineered to overexpress either hGAAP or a mutant in which C-terminal charged amino acids were mutated to alanines (hGAAP Ctmut) to ablate its antiapoptotic function (Carrara et al., 2012). This short C-terminal sequence is also essential for BI-1 both to modulate intracellular Ca2+ and to protect from apoptosis (Kim et al., 2008). Expression of hGAAP protein was confirmed by immunoblotting (Fig. 1, A and B). Levels of overexpressed hGAAP in U2-OS cell lines were comparable to those of vGAAP during VACV infection (Fig. S1). In addition, siRNA was used to achieve knockdown of endogenous hGAAP (Gubser et al., 2007). The effectiveness of the hGAAP-specific siRNAs (siRNA 1 and 2) was verified by RT-PCR (Fig. 1 C) because there is currently no available antibody (Ab) that detects endogenous hGAAP. Concomitant reduction of protein expression by siRNA was confirmed in cells overexpressing HA-tagged hGAAP (Fig. 1, D and E). No difference in the basal level of apoptosis, as measured by caspase 3 and caspase 7 activities, was detected after knockdown of hGAAP within the time frame of these experiments (Fig. S2).
Figure 1.

Overexpression or knockdown of hGAAP affects cell adhesion, cell detachment, and cell spreading. (A and B) Immunoblot (IB) of U2-OS cells with anti-HA Ab (A) or HeLa cells with anti-V5 Ab (B) shows expression of tagged hGAAP and hGAAP Ctmut in stable cell lines but not in cells expressing control plasmids (neo and puro). (C) Levels of endogenous hGAAP mRNA were determined by RT-PCR in U2-OS cells transfected with siRNAs for hGAAP (siRNA 1 and 2) or control siRNA. An analysis without reverse transcription (−RT) was included to control for DNA contamination. (D) U2-OS cells overexpressing hGAAP-HA were transfected with the indicated siRNAs, and hGAAP-HA expression was measured using IB with an anti-HA Ab. (E) Summary results (means ± SEM, from three independent experiments) show relative expression of hGAAP protein quantified from IB as in D. (F and G) Adherence of U2-OS (F) or HeLa cells (G) expressing the indicated hGAAPs was determined after washing cells with PBS at intervals after seeding. Results are means ± SEM, from three independent experiments. *, P < 0.05; **, P < 0.01 (Student’s t test, relative to cells overexpressing hGAAP). (H–J) Adherent cells were quantified 20 min after addition of the indicated concentrations of EDTA to U2-OS (H), HeLa cells (I) overexpressing hGAAPs, or U2-OS cells treated with siRNAs (J). Results (H–J) are representative of three experiments and show means ± SEM. (K and L) U2-OS cells overexpressing hGAAPs (K) or after treatment with siRNAs (L) were seeded onto fibronectin-coated slides. Cell areas were measured after fixing cells at intervals after seeding. Results show means ± SEM for ≥35 cells and are typical of three experiments. (H–L) *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t test, compared with control). (M) Traces show [Ca2+]i after addition of the Ca2+ ionophore, ionomycin (1 µM), in Ca2+-free HBS to populations of fura-2–loaded U2-OS cells expressing the indicated hGAAPs. (N) Summary results (means ± SEM from three independent experiments) show peak increases in [Ca2+]i expressed as fractions of those in matched neo cells. **, P < 0.01 by one-sample Student’s t test.
Several observations suggested that hGAAP might play a role in cell migration or adhesion. Both HeLa and U2-OS cells overexpressing hGAAP were more resistant to trypsin-induced detachment during culture. Knockdown of endogenous hGAAP caused cell elongation (Fig. S3 A), and actin stress fibers became concentrated at the cell periphery, whereas in control cells, they were organized in a typical meshwork that spanned the center of the cell (Fig. 2 A). These observations prompted an analysis of the effects of hGAAP on cell adhesion. Overexpression of hGAAP increased the rates of cell adhesion in both U2-OS and HeLa cells (Fig. 1, F and G). In addition, overexpression of hGAAP rendered cells more resistant to EDTA-induced detachment (Fig. 1, H and I). Knockdown of endogenous hGAAP had the opposite effect (Fig. 1 J). Similar effects were observed with EGTA-induced detachment (unpublished data). In both assays, overexpression of hGAAP Ctmut had no effect (Fig. 1, F and H). To control for possible differences in cell size that could account for differences in the crystal violet staining used to measure cell attachment, cell areas of U2-OS cells seeded for 24 h were measured. No significant differences were found between the areas of control cells and those overexpressing hGAAP or hGAAP Ctmut (Fig. S3 B). To define further the hGAAP-dependent control of adhesion kinetics, cell areas were measured in cells undergoing spreading on fibronectin. Overexpression of hGAAP, but not of hGAAP Ctmut, increased the rate of cell spreading (Fig. 1 K), whereas knockdown of endogenous hGAAP slowed it (Fig. 1 L).
Figure 2.

hGAAP expression alters the number and size of focal adhesions. U2-OS cells transfected with siRNA (24 h) or plasmids encoding hGAAP were seeded onto fibronectin-coated slides. (A) Confocal images show cells transfected with vinculin-GFP (14 h), fixed, and stained with phalloidin–Alexa Fluor 568. (B) Summary results (means ± SEM from ≥25 cells for each condition) show numbers of focal adhesions per cell, determined by counting vinculin-GFP spots. (C) IRM images of U2-OS cells overexpressing hGAAP or hGAAP Ctmut, or transfected with the indicated siRNAs. Images are typical of three independent experiments. (D) Example of the image analysis used to determine focal adhesion number and size. Individual cells were imaged for IRM (a) and phalloidin staining (b). IRM images were then band pass filtered (c), and a threshold was imposed (d) to obtain the final images used to determine the number and size of the adhesions. (E and F) Summary results (means ± SEM from ≥20 cells for each condition) show numbers of adhesions per cell (E) and their areas (F). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t test). Bars, 10 µm.
Given that the C-terminal charged region of hGAAP regulates its function (Carrara et al., 2012), we assessed whether hGAAP Ctmut could deplete Ca2+ from intracellular stores. In U2-OS cells, overexpression of hGAAP reduced the Ca2+ content of the intracellular stores, and this effect was abolished by mutation of the C-terminal of hGAAP (Fig. 1, M and N). The charged C-terminal residues of hGAAP are therefore essential for its effects on intracellular Ca2+ stores, cell adhesion, and spreading. Rates of cell spreading are known to depend on focal adhesion dynamics (Vicente-Manzanares and Horwitz, 2011), suggesting a role for hGAAP-dependent Ca2+ signaling in this process. Subsequent experiments explore this possibility further.
hGAAP regulates focal adhesion dynamics and cell migration
Integrins are the major adhesion receptors that control cell attachment and spreading (Hynes, 2002), and Ca2+ release from intracellular stores is implicated in their activation (McHugh et al., 2010). We therefore assessed whether expression of hGAAP affected integrin activation and cell spreading. A general effect of hGAAP on protein trafficking to the plasma membrane is unlikely because cell surface expression of GFP-tagged VSV-G (vesicular stomatitis virus glycoprotein G; Katz et al., 1977) was unaffected by overexpression or knockdown of hGAAP (Fig. S4, A and B). FACS using antibodies specific for total or active human β1 integrin was used to measure cell surface expression of β1 integrins. Overexpression of hGAAP or knockdown of endogenous hGAAP had no effect on steady-state expression of either active or total populations of β1 integrins at the cell surface (Fig. S5). Mechanisms other than control of β1 integrin activation must, therefore, link hGAAP to regulation of adhesion. We investigated whether hGAAP may instead control focal adhesion assembly and dynamics. We chose vinculin as a general marker of focal adhesions, as this protein is known to associate with all types of cell matrix adhesions in a wide range of cell types. Confocal analysis revealed that the number of vinculin-positive focal adhesions was significantly increased in cells overexpressing hGAAP but not in cells expressing hGAAP Ctmut (Fig. 2 B). Knockdown of endogenous hGAAP had the opposite effect; an almost threefold reduction in the number of focal adhesions (Fig. 2, A and B). Interference reflection microscopy (IRM) allows for analysis of sites of contact between the cell and the substratum to be quantified without the requirement for expression or staining of a specific reporter protein (Holt et al., 2008). We therefore used IRM as an additional, complementary, and “nonbiased” method to analyze the adhesion footprint of cells overexpressing hGAAP (Fig. 2 C). Quantification of IRM images demonstrated that overexpression of hGAAP increased the number of adhesions (Fig. 2 E), consistent with the results obtained using vinculin-GFP. The size of focal adhesions was significantly smaller in cells overexpressing hGAAP compared with control cells or those overexpressing hGAAP Ctmut (Fig. 2 F). Conversely, knockdown of endogenous hGAAP decreased significantly the number of adhesions and increased their size (Fig. 2 , C, E, and F).
To determine whether the effects of hGAAP on the size and number of focal adhesions were caused by changes in adhesion dynamics, the turnover of focal adhesions was analyzed by time-lapse microscopy in live cells plated on fibronectin, using vinculin-GFP as a marker (Fig. 3). Focal adhesions in cells overexpressing hGAAP had shorter lifetimes, and they assembled and disassembled more rapidly than in control cells. Overexpression of hGAAP Ctmut had no significant effect (Fig. 3, A–D; and Videos 1, 2, and 3). Conversely, knockdown of endogenous hGAAP resulted in more stable focal adhesions (Fig. 3, E–H; and Videos 4, 5, and 6). Thus, hGAAP controls the Ca2+ content of intracellular stores, cell adhesion and spreading, and the dynamics of individual focal adhesions. Importantly, all of these effects were abolished by mutation of critical charged residues within the C-terminal region of hGAAP.
Figure 3.

hGAAP expression alters focal adhesion dynamics. (A and E) U2-OS cells overexpressing hGAAP (A) or transfected with siRNAs (E) were transfected with vinculin-GFP and seeded onto fibronectin-coated dishes. After 30 min, individual cells were imaged at 2-min intervals for 2 h. Representative frames are shown, with arrows highlighting a single typical adhesion for each cell line. Bars, 10 µm. (B–D and F–H) Summary results (means ± SEM, n = 6 cells, with 4–10 focal adhesions analyzed in each) show mean lifetimes of focal adhesions (B and F) and rate constants for their disassembly (C and G) and assembly (D and H). **, P < 0.01; ***, P < 0.001 (Student’s t test). FA, focal adhesion.
Given the changes in adhesion dynamics, we sought to determine whether hGAAP also played a role in random cell migration. We therefore performed time-lapse microscopy and cell tracking to determine the migration phenotypes in each cell line. Overexpression of hGAAP in U2-OS cells increased migration speed and decreased directional persistence, whereas overexpressed hGAAP Ctmut had no significant effect (Fig. 4, A–C). Overexpression of hGAAP also increased migration speed significantly in HeLa cells (Fig. S4), suggesting that the hGAAP-dependent phenotypes are not restricted to U2-OS cells. Conversely, knockdown of endogenous hGAAP in U2-OS cells using two independent siRNAs resulted in significantly slower migration speed (Fig. 4, D–F). Together, these results demonstrate that hGAAP promotes focal adhesion turnover (Fig. 3) and increases the speed of random migration (Fig. 4).
Figure 4.

Overexpression of hGAAP increases the speed of cell migration. (A–F) U2-OS cells overexpressing hGAAP, hGAAP Ctmut, or control neo (A–C) or cells transfected with siRNA (D–F) were seeded at low density on fibronectin-coated dishes. Individual cells were imaged at 5-min intervals for 18 h. Tracks of individual cells (n = 30) are shown in A and D. Migration rates (B and E) and persistence (where 1 represents a straight line of migration from start to finish; C and F) are shown as means ± SEM from 30 cells. **, P < 0.01; ***, P < 0.001 (Student’s t test, relative to neo or control siRNA cells). AU, arbitrary unit.
hGAAP enhances speed of migration via increased calpain activation
Focal adhesion dynamics are known to depend on the proteolytic activity of calpain 2, which is in turn regulated by local increases in [Ca2+]i (Franco et al., 2004; Chan et al., 2010). To determine whether enhanced turnover of focal adhesions in hGAAP-overexpressing cells was caused by an increase in the proteolytic activity of calpain, we used a Förster resonance energy transfer (FRET)–based biosensor (Stockholm et al., 2005) to report the spatial distribution of calpain protease activity near the plasma membrane in live cells. The sensor comprises CFP separated from YFP by a peptide sequence that is proteolytically cleaved by calpain, thereby abolishing FRET between the fluorescent proteins. IRM was used in parallel to define the adhesion areas where cells contacted the substrate. Live-imaging analysis revealed a significant increase in calpain activity near the plasma membrane in U2-OS cells overexpressing hGAAP compared with control cells or those expressing hGAAP Ctmut (Fig. 5, A and B). IRM data from the same cells also suggested a temporal and spatial overlap of the increase in calpain activity with the disassembly of focal adhesions (Fig. 5 A, right). This is consistent with previous results showing the importance of calpain 2 activity in the disassembly of focal adhesions (Franco and Huttenlocher, 2005).
Figure 5.

hGAAP stimulates calpain activity. (A) U2-OS cells were transfected with the CFP/YFP calpain FRET biosensor, plated on fibronectin, and imaged live over 20 min in both FRET and IRM channels. Typical examples show stills from videos. Right images show enlargements of boxed areas in left images. Bars, 5 µm. (B) Summary results from the images collected at t = 0 (means ± SEM, for >10 cells) show the percentages of low-FRET pixels (ratio < 1.5, i.e., high calpain activity) within 5 µm of the plasma membrane. (C) Typical IB showing total FAK, calpain 2–dependent cleaved FAK (asterisk, FAK(clv)), and the loading control (α-GAPDH) for U2-OS cells transfected as shown, reseeded on fibronectin for 30 min, and assayed with or without treatment with ALLM. (D) Summary results (means ± SEM, n = 3). (E) IB, typical of three independent experiments, showing total endogenous calpain 2. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student’s t test, relative to neo or hGAAP cells).
Talin is a substrate for calpain 2 at focal adhesions, and cleavage of talin plays a key role in controlling activation of integrins and their linkage to the actin cytoskeleton (Franco et al., 2004). We therefore measured amounts of talin cleavage products in control cells and cells overexpressing hGAAP, but we found no significant differences between these cells (unpublished data). This is consistent with our finding that levels of active integrins are unchanged in cells overexpressing hGAAP (Fig. S5) and further suggests that hGAAP acts downstream of active integrins to regulate adhesion dynamics and migration. FAK was recently shown to be a substrate for calpain 2, and cleavage of FAK can control assembly and disassembly of focal adhesions (Chan et al., 2010). To determine whether hGAAP plays a role in adhesion dynamics through FAK, we measured levels of cleaved FAK in cells seeded on fibronectin for 30 min. To confirm that cleavage was mediated by calpain, we treated cells with the calpain inhibitor ALLM (acetyl-Leu-Leu-Met). Using this approach, we detected increased levels of calpain-cleaved FAK in cells overexpressing hGAAP but not in those expressing hGAAP Ctmut (Fig. 5, C and D). There were no significant differences in the levels of endogenous calpain 2 between the cell lines (Fig. 5 E). This suggests that the increased cleavage of both the FRET-based biosensor and FAK is caused by enhanced activation of calpain by hGAAP.
To determine whether the effects of hGAAP on cell adhesion and migration were associated with hGAAP-dependent control of calpain 1/2 activity, spreading and random migration assays were performed in the presence of the calpain 1/2 inhibitors (ALLM or PD150606) or after calpain 2 knockdown using siRNA. The hGAAP-dependent increases in migration and cell spreading were reduced significantly by the calpain 1/2 inhibitors (Fig. 6, A–F). Similarly, knockdown of calpain 2 (Fig. 6 G) in cells expressing hGAAP restored cell spreading (Fig. 6 H) and migration (Fig. 6, I and J) to the levels observed in control cells or those expressing hGAAP Ctmut. These results demonstrate that activation of calpain 2 plays an important role in mediating hGAAP-dependent effects on cell spreading and migration.
Figure 6.

Inhibition of calpain reduces the effects of hGAAP on cell migration and spreading. (A and B) Cells treated with PD150606 (A) or ALLM (B) were seeded on fibronectin-coated slides and left to adhere and spread for different times. Cells were fixed, and cell size was measured for >50 cells per condition. (C–F) Cells treated with PD150606 (C and D) or ALLM (E and F) were seeded at low density in fibronectin-coated dishes, and cells were imaged every 5 min for 18 h. Individual cell tracks are shown in C and E (n = 25 cells), and cumulative migration speeds from multiple migration tracks are shown in D and F. (G–J) Cells expressing hGAAP, hGAAP Ctmut, or control neo were treated with calpain 2–specific siRNAs. (G) IB showing effectiveness of calpain 2 knockdown in cells expressing hGAAP, hGAAP Ctmut, or control neo, and treated with calpain 2–specific siRNAs. (H) Areas of cells (measured 60 h after calpain 2 (Capn2) siRNA transfection, >50 cells per condition) seeded on fibronectin-coated slides. (I and J) Cells treated with calpain 2 siRNA were seeded at low density in fibronectin-coated dishes, and individual cells were imaged every 5 min for 18 h. Individual cell tracks are shown (n = 25 cells; I), and cumulative migration speeds are shown (J). Data are representative of three experiments and are shown as mean ± SEM. *, P < 0.5; **, P < 0.01; ***, P < 0.001 (Student’s t test, in comparison to neo cells).
hGAAP enhances SOCE
Our evidence that hGAAP both stimulates the turnover of cell adhesions (Fig. 3) and reduces the Ca2+ content of the intracellular stores (Fig. 1 M; de Mattia et al., 2009) suggested that an increase in [Ca2+]i might mediate the effects of hGAAP on cell adhesion and migration. We hypothesized that hGAAP, by reducing the Ca2+ content of the intracellular stores, might activate SOCE and thereby evoke a local increase in [Ca2+]i near focal adhesions, causing stimulation of calpain 2. This would be consistent with evidence that SOCE has been shown to regulate cell migration and the turnover of adhesions via activation of calpain 2 (Chen et al., 2011). Restoration of extracellular Ca2+ to U2-OS cells bathed in nominally Ca2+-free medium evoked a transient increase in [Ca2+]i. This increase was unaffected by expression of hGAAP Ctmut but enhanced in cells overexpressing hGAAP (Fig. 7, A and B). Confirmation that hGAAP stimulated entry of divalent cations across the plasma membrane was provided by adding Mn2+ to the extracellular medium and measuring its rate of entry. Quenching of intracellular fura-2 fluorescence by Mn2+ is used routinely to measure the activity of store-operated Ca2+ channels (Bird et al., 2008). The rate of Mn2+ entry was increased by overexpression of hGAAP, whereas hGAAP Ctmut had no significant effect (Fig. 7, C and D). These results show that overexpression of hGAAP stimulates Ca2+ influx across the plasma membrane.
Figure 7.

hGAAP increases calpain activity by enhancing SOCE. Changes in [Ca2+]i or rates of Mn2+ entry were measured in fura-2–loaded U2-OS cells expressing the indicated hGAAPs or neo control or treated with siRNA. (A) Typical traces show changes in [Ca2+]i after restoration of extracellular Ca2+ (2 mM) to populations of cells bathed in nominally Ca2+-free HBS. (B) Summary results (means ± SEM, n = 3) show the peak increases in [Ca2+]i. (C) Addition of 1 mM Mn2+ to populations of cells in normal HBS quenches cytosolic fura-2 fluorescence as Mn2+ enters cells via channels in the plasma membrane. Typical traces show fluorescence normalized to the initial fluorescence intensity. (D) Summary results (means ± SEM, n = 3) show rates of fluorescence quenching, in arbitrary fluorescence units per second (AFU/s), determined from the gradients of lines fitted by linear regression to intervals of 100–200 s after Mn2+ addition. (E) Effects of 50 µM 2-APB or 1 µM Gd3+ on [Ca2+]i in cells overexpressing hGAAP after restoration of extracellular Ca2+ (2 mM) to populations of cells bathed in nominally Ca2+-free HBS. (F) Summary results (means ± SEM, n = 3) show the normalized peak Ca2+ signals. (G) Effects of 50 µM 2-APB or 1 µM Gd3+ on the quenching of fura-2 fluorescence after addition of 1 mM Mn2+ to populations of cells overexpressing hGAAP. (H) Summary results (means ± SEM, n = 3) show normalized rates of Mn2+-evoked fluorescence quenching. (I) Typical traces show changes in [Ca2+]i after addition of 1 µM thapsigargin (Tg) in nominally Ca2+-free HBS to populations of cells overexpressing hGAAP followed by restoration of extracellular Ca2+ (2 mM). The initial thapsigargin-evoked increases in [Ca2+]i were smaller in cells overexpressing hGAAP (139 ± 4 nM, n = 5) than in neo cells (154 ± 4 nM, n = 5; P < 0.05) or cells expressing hGAAP Ctmut (176 ± 8 nM, n = 5). (J) Summary results (means ± SEM, n = 3–4) show normalized peak changes in [Ca2+]i after restoration of extracellular Ca2+ (2 mM) to thapsigargin-treated cells. Effects of 50 µM 2-APB or 1 µM Gd3+ are indicated. Responses are normalized to parallel measurements from cells overexpressing hGAAP. (K) Single-cell analysis shows changes in [Ca2+]i after treatment with 1 µM thapsigargin and restoration of extracellular Ca2+ (2 mM) to U2-OS cells overexpressing hGAAP and transfected with either DN-Orai1 or control pcDNA3.1 plasmid. Transfected cells were identified using cotransfected pmCherry-C1. Typical traces show average responses from 11 transfected cells in each condition. (L) Summary results show inhibition of thapsigargin-evoked SOCE by DN-Orai1 in hGAAP and neo cells, calculated from the differences between parallel measurements of SOCE in cells expressing DN-Orai1 or pcDNA3.1 (means ± SEM, from three independent transfections, with responses from 6–18 transfected cells measured in each). (M) Effect of hGAAP siRNA on rates of Mn2+-evoked quenching of fura-2 fluorescence (means ± SEM, n = 3). (N) U2-OS cells transfected with the calpain FRET biosensor were plated on fibronectin, treated with 2 µM Gd3+ or 50 µM 2-APB, and imaged for 20 min. Typical images, taken from videos, show the IRM and FRET signals recorded 5 min after addition of the inhibitors. Bar, 5 µm. (O) Summary results (means ± SEM, from ≥10 cells) show the percentage of low FRET pixels (<1.5 ratio, i.e., high calpain activity) within 5 µm of the plasma membrane in the image collected at t = 0. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (unpaired Student’s t test).
SOCE is usually mediated by Orai channels (Várnai et al., 2009), and it can be inhibited by Gd3+ and 2-aminoethoxydiphenyl borate (2-APB; Bird et al., 2008). 1 µM Gd3+ or 50 µM 2-APB attenuated the Ca2+ signals evoked by restoration of extracellular Ca2+ (Fig. 7, E and F) and the Mn2+ entry (Fig. 7, G and H) in cells overexpressing hGAAP. SOCE can also be activated by thapsigargin, which irreversibly inhibits the sarcoendoplasmic reticulum/ER Ca2+-ATPase and thereby depletes intracellular Ca2+ stores (Bird et al., 2008). As expected, thapsigargin evoked transient increases in [Ca2+]i in cells bathed in nominally Ca2+-free medium (Fig. 7 I), reflecting the release of Ca2+ from intracellular stores. Overexpression of hGAAP, but not of hGAAP Ctmut, decreased this initial thapsigargin-evoked increase in [Ca2+]i (Fig. 7 I), consistent with hGAAP causing loss of Ca2+ from intracellular stores (de Mattia et al., 2009). Restoration of extracellular Ca2+ to cells in which SOCE had been activated by thapsigargin evoked an increase in [Ca2+]i that was larger in cells overexpressing hGAAP than in either control cells or cells expressing hGAAP Ctmut (Fig. 7, I and J). These Ca2+ signals were inhibited by Gd3+ and 2-APB (Fig. 7 J) and by expression of a dominant-negative (DN) mutant of Orai1 (Orai1-R91W; DN-Orai1; Fig. 7, K and L; Li et al., 2011). The fractional inhibition of SOCE by DN-Orai1 was similar in control cells and those overexpressing hGAAP (Fig. 7 L). This suggests that SOCE via Orai channels mediates both the native Ca2+ entry and the additional Ca2+ entry evoked by overexpression of hGAAP. Knockdown of endogenous hGAAP reduced the rate of Mn2+ entry (Fig. 7 M), suggesting that endogenous hGAAP stimulates a basal Ca2+ influx pathway. These results establish that hGAAP enhances SOCE mediated by Orai channels in the plasma membrane via two mechanisms. First, hGAAP partially depletes intracellular stores of Ca2+ and thereby stimulates SOCE (Fig. 1 M and Fig. 7, A–H), and second, even when the ER is depleted of Ca2+ by treatment with thapsigargin, SOCE is greater in cells in which hGAAP is overexpressed (Fig. 7, I–L).
Finally, to determine whether the effect of hGAAP on calpain activity was a consequence of enhanced SOCE, we tested the effects of inhibitors of SOCE on calpain 2 activity. Analysis of local calpain activity using FRET in live cells demonstrated that inhibition of SOCE by Gd3+ or 2-APB in U2-OS cells overexpressing hGAAP significantly reduced calpain activity at the plasma membrane (Fig. 7, N and O). These results demonstrate that hGAAP enhances SOCE mediated by Orai channels, and the resulting local increases in [Ca2+]i then control the dynamics of focal adhesions and cell migration by stimulating the activity of calpain 2.
Discussion
hGAAP is a highly conserved Golgi protein that regulates apoptosis and Ca2+ release from intracellular stores. Here, we demonstrate a novel role for hGAAP in controlling cell adhesion, spreading, and migration (Fig. 1, Fig. 2, and Fig. 4) by modulating focal adhesion dynamics (Fig. 3). These effects of hGAAP are abolished by mutation of critical residues near the cytosolic C terminus, which prevents hGAAP from depleting intracellular Ca2+ stores (Carrara et al., 2012). We conclude that hGAAP enhances SOCE, which then stimulates localized activation of calpain near the plasma membrane (Fig. 5 and Fig. 7, N and O) and thereby increased cleavage of focal adhesion proteins such as FAK (Fig. 5, C and D). Results with both small molecule inhibitors and siRNA confirm that stimulation of calpain 2 activity is required for hGAAP to promote cleavage of FAK (Fig. 5), cell spreading, and enhanced migration (Fig. 6).
We propose that calpain near focal adhesions at the plasma membrane is activated by local increases in [Ca2+]i resulting from SOCE (Fig. 7, N and O). The SOCE is enhanced by hGAAP via its ability to both deplete intracellular Ca2+ stores and promote more effective activation of SOCE by empty stores (Fig. 7, A–M). The ER and Golgi usually contain relatively high concentrations of Ca2+ (Dolman and Tepikin, 2006; Tang et al., 2011). Targeted aequorins have shown previously that hGAAP reduces luminal Ca2+ concentrations in both the ER and Golgi, in the same U2-OS cells used in our current study (de Mattia et al., 2009). This was shown for overexpression of hGAAP and knockdown of endogenous hGAAP (de Mattia et al., 2009). Constitutive and bidirectional cycling of membranous compartments occurs between the ER and Golgi (English and Voeltz, 2013). Depletion of luminal Ca2+ in one organelle may therefore affect the luminal Ca2+ concentration in the other. It is presently unclear whether the more effective activation of SOCE by empty stores results from more effective coupling of empty stores to SOCE in the presence of hGAAP or changes in the expression of Orai and/or stromal interaction molecule proteins. Nor can we exclude the possibility that hGAAP within the Golgi contributes directly to activation of SOCE. The localized activation of calpain 2 by hGAAP-evoked SOCE is probably facilitated by the association of calpain 2 with Golgi and ER, and its requirement for substantial increases in [Ca2+]i for activity (Hood et al., 2003, 2004). Such interactions may also contribute to the established effects of the orientation and polarity of the Golgi in controlling cell migration (Orlando and Guo, 2009). Calpain cleaves and releases focal adhesion adaptors such as FAK from focal adhesion complexes, leading to increased turnover of focal contacts (Fig. 3; Chan et al., 2010). The hGAAP-evoked increase in calpain activity causes increased cleavage of FAK (Fig. 5 C) but not of another calpain substrate, talin. This suggests that the localized signaling between Golgi/ER, SOCE, and calpain initiated by hGAAP may serve both to expose calpain 2 to the high local [Ca2+]i required for its activity and to direct active calpain to specific substrates.
BI-1 and GAAPs are related members of the TMBIM family of antiapoptotic proteins, and both cause loss of Ca2+ from intracellular stores (Bultynck et al., 2012). The latter has been proposed to be mediated by a channel-like (Bultynck et al., 2012) or Ca2+/H+ exchange activity (Ahn et al., 2009) of BI-1, and it is likely to be similar for hGAAP. BI-1 is up-regulated in prostate cancers (Grzmil et al., 2006) and, like hGAAP (see Introduction), in some human breast cancers (Igney and Krammer, 2002). BI-1 was shown to increase metastasis by increasing cell motility and invasiveness and by altering glucose metabolism (Lee et al., 2010b), and it has been identified as a promising target for anticancer therapy (Yun et al., 2012). The influence of hGAAP via regulation of SOCE on adhesion and migration (Fig. 8), together with the up-regulation of hGAAP mRNA in some human cancers (van ’t Veer et al., 2002), suggests that hGAAP might also contribute to cancer progression and metastasis. This would be consistent with enhanced activation of SOCE, calpain activity, migration, and metastases in cancer models (Yang et al., 2009; Feng et al., 2010; Chen et al., 2011).
Figure 8.
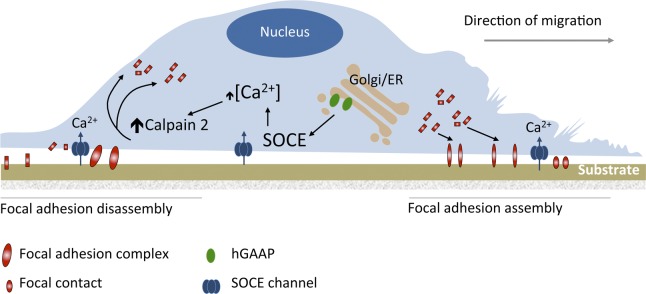
Model of hGAAP-dependent control of cell adhesion and migration. hGAAP both stimulates loss of Ca2+ from the Golgi/ER, thereby activating SOCE, and it promotes activation of SOCE by empty stores. The resulting localized increase in [Ca2+]i near the plasma membrane stimulates calpain 2 activity, leading to increased turnover of focal adhesions. This in turn leads to increased speed of cell spreading and more rapid migration. See text in Results and Discussion for further details.
Apoptosis and survival signals are intimately linked to the ability of cells to adhere either to other cells or to the extracellular matrix (Frisch and Francis, 1994; Gilmore et al., 2009). In many cases, this is orchestrated by integrin-dependent survival signals that signal via FAK (Mitra and Schlaepfer, 2006). The hGAAP-dependent inhibition of apoptosis may, therefore, be directly linked with the new roles for GAAP in cell adhesion that we describe here.
Materials and methods
Cell culture and transfection
U2-OS and HeLa cells were grown in DMEM and MEM (Gibco; Invitrogen), respectively. Media were supplemented with 10% heat-treated (56°C for 1 h) FBS, 50 U/ml penicillin, 50 µg/ml streptomycin, and 2 mM l-glutamine. Plasmid transfections used FuGENE6 (Roche) according to the manufacturer’s instructions. Cells were seeded to achieve 20–30% confluence in 24 h. The siRNA oligonucleotide duplexes (Invitrogen) were transfected using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. Sequences of the siRNAs were reported previously (Gubser et al., 2007): siRNA1 and 2 are specific for hGAAP, whereas an siRNA for GFP (Invitrogen) was used as a control. Sequences of the siRNA1 and 2 for hGAAP were as follows: siRNA1, 5′-CGAUCGAGGAACGACUUCAACU-3′; and siRNA2, 5′-CUGUACGGACAUUUGUACAUG-3′. Cells were used 36–40 h after transfection with these siRNAs. Cells transfected with siRNAs specific for human calpain 2 (Ambion; siRNA ID: s320 and s321 are denoted capn2 siRNA1 and capn2 siRNA2, respectively) were used 60 h after transfection.
Plasmids and stable cell lines
Polyclonal HeLa cell lines stably expressing the control plasmid (puromycin [puro]) or hGAAP with an N-terminal V5 tag were generated using the HIV-1–based lentiviral vector pdlNot′MCS′R′PK derived from pHR-SIN-CSGW (Demaison et al., 2002). Polyclonal U2-OS cells stably expressing the pcDNA3.1 control plasmid (neomycin [neo]), hGAAP, or hGAAP Ctmut with C-terminal mutations (E233AVNKK238 to A233AVAAA238), with a C-terminal HA tag in Invitrogen pcDNA3.1 backbone, were described previously (Gubser et al., 2007; Carrara et al., 2012). The plasmids encoding vinculin-GFP (in Takara Bio Inc. EGFP-C3 vector backbone; Cohen et al., 2005) and the calpain FRET sensor (based around Takara Bio Inc. peCFP-N1 vector containing pECFP/YFP and pTOM; Stockholm et al., 2005) were gifts from S. Craig (Johns Hopkins School of Medicine, Baltimore, MA) and I. Richard (Genethon, Centre National de la Recherche Scientifique UMR8587, Evry, France), respectively. The EGFP–VSV-G plasmid was obtained from Addgene (Presley et al., 1997). The DN-Orai1 plasmid in pcDNA6 backbone vector (Li et al., 2011) was a gift from D. Beech (University of Leeds, Leeds, England, UK).
Measurements of [Ca2+]i and SOCE
For measurements of [Ca2+]i in cell populations, U2-OS cells were seeded in 96-well plates coated with 10 µg/ml fibronectin (Invitrogen) at densities that achieved near confluence after 24 h. Cells were transfected with appropriate siRNAs 12 h before seeding. After 24 h, cells were incubated with 6 µM fura-2/AM (Invitrogen) for 40 min at 37°C in cell culture medium. The cells were then washed with Hepes-buffered saline (HBS; 132 mM NaCl, 5 mM KCl, 2 mM, MgCl2, 10 mM d-glucose, 10 mM Hepes, and 2 mM CaCl2, pH 7.2) and incubated at 20°C for 15 min to allow de-esterification of fura-2/AM. [Ca2+]i was measured at 20°C by measuring fluorescence (excitation at 340 and 380 nm and emission at 510 nm) using a fluorescence plate reader (FlexStation 3; Molecular Devices). Background fluorescence was determined by addition of 1 µM ionomycin with 10 mM MnCl2. Background-corrected fluorescence ratios (F340/F380) were calibrated to [Ca2+]i using Ca2+ standard solutions (Invitrogen) and a Kd for Ca2+ of 236 nM (Groden et al., 1991). In nominally Ca2+-free HBS, CaCl2 was omitted, and in Ca2+-free HBS, it was replaced by 1 mM EGTA. For single-cell imaging, transfected U2-OS cells were seeded onto glass-bottomed culture dishes coated with 10 µg/ml fibronectin. After 24 h, when cells were ∼60% confluent, they were transfected with DN-Orai1 (Orai1-R91W) in pcDNA 6 (Li et al., 2011) or 2 µg control pcDNA 3.1 plasmid (Invitrogen), together with 0.5 µg pmCherry-C1 (Takara Bio Inc.) as a transfection marker, using FuGENE 6 (Promega). After a further 24 h, cells were washed with HBS, loaded with 2 µM fura-2/AM (1 h at 20°C), washed, and incubated for a further 15 min in nominally Ca2+-free HBS. Fluorescence after alternating excitation at 340 and 380 nm was detected using an inverted fluorescence microscope (IX81; Olympus) and 40×, 1.35 NA objective. Images were acquired with a camera (iXon 897; Andor Technology) and processed using Cell^R software (Olympus). Correction for background fluorescence and calibration used the methods described for cell populations.
Cell adhesion and detachment assays
For attachment assays, subconfluent cells were detached with trypsin, washed twice with DMEM containing 1% FBS, resuspended in the same medium, and counted. Cells were seeded in 12-well plates (37°C at 5% CO2), and at specified intervals, they were gently washed twice with warm (37°C) PBS and fixed with 4% PFA. For detachment assays, cells were seeded in 12-well plates, and after 24 h, when cells were ∼60% confluent, they were washed twice with warm PBS and incubated with warm 1 ml PBS for 10 min. 0.2–1 mM EDTA was then added, and after a further 20–30 min at 37°C, cells were washed gently with PBS to remove nonadherent cells. The remaining adherent cells were fixed using 4% PFA and stained with 0.1% crystal violet in 200 mM Hepes for 1 h. The wells were washed three times with water, and the dye was eluted using 10% acetic acid. Samples were transferred to a 96-well plate, and absorbance was measured at 560 nm. To calculate the rate of cell adhesion, absorbance was plotted against time, and the slope of the graph (where the increase of adhesion was linear) was calculated.
Cell spreading assay
U2-OS cells were detached, rinsed twice in DMEM containing 1% FBS, and seeded on 10 µg/ml fibronectin-coated (Invitrogen) slides. After intervals at 37°C, cells were fixed with 4% PFA. Image acquisition was performed using a microscope (LSM 510 META; Carl Zeiss) with a 63×, 1.4 NA oil objective. Cell areas were determined using ImageJ (National Institutes of Health). To inhibit calpains 1 and 2, 50 µM N-ALLM-CHO (EMD Millipore) or 75–100 µg/ml PD150606 (Sigma-Aldrich) was used in DMEM containing 1% FBS for 3.5 h before and then during cell spreading.
Measurements of focal adhesion turnover
These measurements were based on published work (Webb et al., 2004). U2-OS cells in 6-well plates were transfected with a plasmid encoding 1 µg vinculin-GFP. After 24 h, cells were reseeded in glass-bottomed culture chambers (MatTek Corporation) coated with 10 µg/ml fibronectin (Invitrogen) in conditioned DMEM containing 1% FBS. After 1 h, GFP fluorescence was monitored at the edges of cells for ∼2 h at 2-min intervals using a microscope (LSM 510 META) with a 63×, 1.4 NA oil objective contained within an environmental chamber at 37°C. Image acquisition used LSM image browser software (Carl Zeiss). Images were analyzed using ImageJ. The lifespan of each vinculin-GFP–labeled focal adhesion was calculated by determining the interval between the first and last frames in which an individual adhesion was observed. Rates of assembly and disassembly of adhesions were calculated as reported previously, by measuring the incorporation or loss of fluorescent signal of vinculin-GFP over time (Worth et al., 2010). These intensity values were plotted over time on semilogarithmic graphs to provide a profile of intensity ratios over time. These ratios were calculated using the formula In(I/I0) for assembly and In(I0/I) for disassembly (in which I0 is the initial fluorescence intensity value, and I is the intensity value for the relevant time point). Rates were then calculated from the gradient of the line of best fit.
IRM
U2-OS cells on fibronectin-coated glass-bottomed dishes were fixed with 4% PFA and then stained with phalloidin conjugated to Alexa Fluor 488 or Alexa Fluor 546 (1:400; Invitrogen) to identify F-actin. Dishes were then filled with PBS, and IRM images were collected using a confocal microscope (LSM 510 META) and LSM software with a 63×, 1.4 oil objective (Carl Zeiss). Image analysis was performed using ImageJ. With phalloidin defining the cell boundaries, the number and size of adhesions were calculated using the Analyze Particles function (threshold for particles with size between 0.15 and 4 µm), after setting the image threshold so that only cell substrate contact areas were black (Fig. 2 D; Holt et al., 2008).
Immunoblotting
Cells were lysed on ice in lysis buffer (when detection of hGAAP was required) or radioimmunoprecipitation assay buffer (all other analyses). Lysis buffer comprised of 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM EDTA, 1% CHAPS, and protease and phosphatase inhibitor cocktails (Roche). Radioimmunoprecipitation assay buffer comprised of 50 mM Tris, pH 7.5, 500 mM NaCl, 0.5 mM MgCl2, 0.1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM EDTA, and protease and phosphatase inhibitor cocktails (Roche). The lysates were cleared by centrifugation (15,000 g for 15 min), resolved using NuPAGE Novex 4–12% Bis-Tris gels (Invitrogen), and transferred onto a nitrocellulose membrane. Antigen–Ab complexes were detected using HRP-conjugated secondary antibodies (Sigma-Aldrich). The primary antibodies used were: anti-HA (1:10,000; Sigma-Aldrich), anti-V5 (1:5,000; AbD Serotec), antitubulin (1:10,000; EMD Millipore), anti-GAPDH (1:5,000; Cell Signaling Technology), mouse anti-D8 monoclonal AB1.1 (1:1,000; Parkinson and Smith, 1994), anti–calpain 2 (1:5,000; Cell Signaling Technology), and anti-FAK (1:2,500; EMD Millipore).
Random migration assay
U2-OS cells were seeded at low density (30%) on 10 µg/ml fibronectin-coated (Invitrogen) 12-well plates. Individual cells were imaged at 5-min intervals for 8 or 18 h with a wide-field microscope (Observer.Z1; Carl Zeiss) contained within an environmental chamber at 37°C using a 10× objective and a camera (AxioCam HRm; Carl Zeiss). To inhibit calpains 1 and 2, 50 µM ALLM (EMD Millipore) or 75 or 100 µg/ml PD150606 (Sigma-Aldrich) was used in DMEM containing 1% FBS for 1 h before and then during image acquisition. Migration tracks were generated using the ImageJ Manual Tracking plugin, and tracks were analyzed using an in house–written Mathematica 7 notebook (provided by G. Dunn, King’s College London, London, England, UK) to calculate migration rates and persistence.
Measurements of calpain activity with a FRET biosensor
Cells were transfected with a CFP/YFP-based calpain FRET biosensor (Stockholm et al., 2005) and plated onto glass-bottomed imaging chambers (ibidi). Cells were imaged using an inverted confocal microscope (A1R Ti-E; Nikon) contained within an environmental chamber at 37°C (OKOlab). CFP/YFP channels were excited using a 440-nm diode laser and a 514-nm Argon laser, respectively. Images of CFP and FRET channels were acquired every 20 s using a CFI Plan Apochromat VC 60× oil N2, NA 1.4 objective. Videos were exported into ImageJ for analysis using the riFRET plugin (Roszik et al., 2009). Ratio images were generated after correction of FRET channel images for donor (CFP) intensity (NIS Elements software; Nikon). For quantitative analysis, FRET ratio images were masked in ImageJ, and a threshold was applied to permit measurement of the percentage of pixels with more than a 1.5-fold ratio increase within a 5-µm region of the plasma membrane. Data from >10 cells per condition were pooled and presented as percentages of active calpain at the plasma membrane.
Immunofluorescence
Cells were fixed with either methanol (−20°C) on ice or 4% PFA and then stained with phalloidin conjugated to Alexa Fluor 488 or Alexa Fluor 546 to stain F-actin (1:400; Invitrogen) or anti–VSV-G (a gift from D. Lyles, Wake Forest University School of Medicine, Winston Salem, NC) and with secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 (Invitrogen). Coverslips were mounted in Mowiol 4–88 (EMD Millipore) containing DAPI. Cells were imaged by confocal microscopy using a 63× oil objective and a microscope (LSM 5 PASCAL; Carl Zeiss). Images were acquired and exported using LSM image browser software (Carl Zeiss).
FACS analysis of cell surface integrins
Cell surface expression of total and active β1 integrin in U2-OS cells seeded on 10 µg/ml fibronectin-coated dishes (Invitrogen) was determined by FACS (CyAn ADP multiple laser excitation; Dako). Fixed nonpermeabilized cells were stained using antibodies specific for active human β1 integrin (MAB 2247 clone 12G10; EMD Millipore) or total human β1 integrin (MAB2250; EMD Millipore) and detected with Alexa Fluor 488–labeled secondary antibodies.
Assay of caspase activity
U2-OS neo cells were seeded in 96-well plates (103 cells/well). After 24 h, cells were transfected with appropriate siRNAs, and after a further 36 h, caspase activity was determined using Caspase-Glo 3/7 substrate according to the manufacturer’s instructions (Promega).
Infection with VACV
U2-OS cells were infected at 90% density with an MOI of 10 with wild-type, vGAAP deletion (Δ vGAAP), or revertant VACV in which vGAAP was reinserted in the viral genome with a C-terminal HA tag (vGAAP-HA; Gubser et al., 2007). After 6 h, cells were harvested in lysis buffer for immunoblotting.
Online supplemental material
Fig. S1 shows that amounts of hGAAP protein in U2-OS cells overexpressing hGAAP are comparable to amounts of vGAAP expressed in cells infected with VACV. Fig. S2 shows that knockdown of hGAAP does not increase caspase 3 and caspase 7 activities measured after 36 h in U2-OS cells. Fig. S3 shows that loss of hGAAP causes cells to elongate, whereas overexpression of hGAAP does not affect cell length or area. Fig. S4 shows that protein trafficking to the plasma membrane is unaffected by knockdown of hGAAP. Fig. S5 shows that cell surface expression of integrins is unaffected by changes in expression of hGAAP. Videos 1–6 are representative time-lapse videos of U2-OS cells transfected with vinculin-GFP to determine focal adhesion turnover, assembly, and disassembly in control cells (Video 1), cells overexpressing hGAAP (Video 2), cells expressing hGAAP Ctmut (Video 3), cells transfected with control siRNA (Video 4), cells transfected with siRNA1 against hGAAP (Video 5), and cells transfected with siRNA2 against hGAAP (Video 6). Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201301016/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Susan Craig (Johns Hopkins School of Medicine, Baltimore, MD) for the vinculin-GFP plasmid, Professor Graham Dunn (King’s College, London, England, UK) for use of Mathematica 6 notebook, Dr. Isabelle Richard (Genethon, Centre National de la Recherche Scientifique UMR8587, Evry, France) for the calpain FRET biosensor, Prof. Rick Randall (University of St. Andrews, Fife, Scotland, UK) for the HIV-1–based lentivirus system used to generate the HeLa cell lines expressing GAAP, and Prof. David Beech (University of Leeds, Leeds, England, UK) for the DN-Orai1 plasmid. Thanks also to Michael Hollinshead (Imperial College London, London, England, UK) for help with microscopy.
This work was supported by the Medical Research Council and the Wellcome Trust (085295). N. Saraiva was supported by the Portuguese Foundation for Science and Technology (Fundação para a Ciência e a Tecnologia). G.L. Smith is a Wellcome Trust Principal Research Fellow. D.L. Prole was supported in part by a Meres research associateship from St. John’s College, Cambridge. M. Parsons is funded by a Royal Society University Research Fellowship.
Footnotes
Abbreviations used in this paper:
- Ab
- antibody
- 2-APB
- 2-aminoethoxydiphenyl borate
- BI-1
- Bax inhibitor-1
- DN
- dominant negative
- FRET
- Förster resonance energy transfer
- GAAP
- Golgi antiapoptotic protein
- HBS
- Hepes-buffered saline
- hGAAP
- human GAAP
- IB
- immunoblot
- IRM
- interference reflection microscopy
- SOCE
- store-operated Ca2+ entry
- TMBIM
- transmembrane BI-1–containing motif
- VACV
- vaccinia virus
- vGAAP
- viral GAAP
References
- Ahn T., Yun C.H., Chae H.Z., Kim H.R., Chae H.J. 2009. Ca2+/H+ antiporter-like activity of human recombinant Bax inhibitor-1 reconstituted into liposomes. FEBS J. 276:2285–2291 10.1111/j.1742-4658.2009.06956.x [DOI] [PubMed] [Google Scholar]
- Bird G.S., DeHaven W.I., Smyth J.T., Putney J.W., Jr 2008. Methods for studying store-operated calcium entry. Methods. 46:204–212 10.1016/j.ymeth.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage R.A., Fogarty K.E., Tuft R.A., Fay F.S. 1991. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science. 254:703–706 10.1126/science.1948048 [DOI] [PubMed] [Google Scholar]
- Bultynck G., Kiviluoto S., Henke N., Ivanova H., Schneider L., Rybalchenko V., Luyten T., Nuyts K., De Borggraeve W., Bezprozvanny I., et al. 2012. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. J. Biol. Chem. 287:2544–2557 10.1074/jbc.M111.275354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrara G., Saraiva N., Gubser C., Johnson B.F., Smith G.L. 2012. Six-transmembrane topology for Golgi anti-apoptotic protein (GAAP) and Bax inhibitor 1 (BI-1) provides model for the transmembrane Bax inhibitor-containing motif (TMBIM) family. J. Biol. Chem. 287:15896–15905 10.1074/jbc.M111.336149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.T., Bennin D.A., Huttenlocher A. 2010. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 285:11418–11426 10.1074/jbc.M109.090746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.F., Chiu W.T., Chen Y.T., Lin P.Y., Huang H.J., Chou C.Y., Chang H.C., Tang M.J., Shen M.R. 2011. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA. 108:15225–15230 10.1073/pnas.1103315108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Langeslag M., van Leeuwen B., Ran L., Ryazanov A.G., Figdor C.G., Moolenaar W.H., Jalink K., van Leeuwen F.N. 2006. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 25:290–301 10.1038/sj.emboj.7600931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D.M., Chen H., Johnson R.P., Choudhury B., Craig S.W. 2005. Two distinct head-tail interfaces cooperate to suppress activation of vinculin by talin. J. Biol. Chem. 280:17109–17117 10.1074/jbc.M414704200 [DOI] [PubMed] [Google Scholar]
- Demaison C., Parsley K., Brouns G., Scherr M., Battmer K., Kinnon C., Grez M., Thrasher A.J. 2002. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of immunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum. Gene Ther. 13:803–813 10.1089/10430340252898984 [DOI] [PubMed] [Google Scholar]
- de Mattia F., Gubser C., van Dommelen M.M., Visch H.J., Distelmaier F., Postigo A., Luyten T., Parys J.B., de Smedt H., Smith G.L., et al. 2009. Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Mol. Biol. Cell. 20:3638–3645 10.1091/mbc.E09-05-0385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolman N.J., Tepikin A.V. 2006. Calcium gradients and the Golgi. Cell Calcium. 40:505–512 10.1016/j.ceca.2006.08.012 [DOI] [PubMed] [Google Scholar]
- English A.R., Voeltz G.K. 2013. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 5:a013227 10.1101/cshperspect.a013227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M., Grice D.M., Faddy H.M., Nguyen N., Leitch S., Wang Y., Muend S., Kenny P.A., Sukumar S., Roberts-Thomson S.J., et al. 2010. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell. 143:84–98 10.1016/j.cell.2010.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco S.J., Huttenlocher A. 2005. Regulating cell migration: calpains make the cut. J. Cell Sci. 118:3829–3838 10.1242/jcs.02562 [DOI] [PubMed] [Google Scholar]
- Franco S.J., Rodgers M.A., Perrin B.J., Han J., Bennin D.A., Critchley D.R., Huttenlocher A. 2004. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6:977–983 10.1038/ncb1175 [DOI] [PubMed] [Google Scholar]
- Frisch S.M., Francis H. 1994. Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell Biol. 124:619–626 10.1083/jcb.124.4.619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G., Rondé P., Gaire M., Haiech J., Takeda K. 2002. Calcium oscillations trigger focal adhesion disassembly in human U87 astrocytoma cells. J. Biol. Chem. 277:26364–26371 10.1074/jbc.M203952200 [DOI] [PubMed] [Google Scholar]
- Gilmore A.P., Owens T.W., Foster F.M., Lindsay J. 2009. How adhesion signals reach a mitochondrial conclusion—ECM regulation of apoptosis. Curr. Opin. Cell Biol. 21:654–661 10.1016/j.ceb.2009.05.009 [DOI] [PubMed] [Google Scholar]
- Groden D.L., Guan Z., Stokes B.T. 1991. Determination of Fura-2 dissociation constants following adjustment of the apparent Ca-EGTA association constant for temperature and ionic strength. Cell Calcium. 12:279–287 10.1016/0143-4160(91)90002-V [DOI] [PubMed] [Google Scholar]
- Grzmil M., Kaulfuss S., Thelen P., Hemmerlein B., Schweyer S., Obenauer S., Kang T.W., Burfeind P. 2006. Expression and functional analysis of Bax inhibitor-1 in human breast cancer cells. J. Pathol. 208:340–349 10.1002/path.1902 [DOI] [PubMed] [Google Scholar]
- Gubser C., Bergamaschi D., Hollinshead M., Lu X., van Kuppeveld F.J., Smith G.L. 2007. A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog. 3:e17 10.1371/journal.ppat.0030017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M.R., Calle Y., Sutton D.H., Critchley D.R., Jones G.E., Dunn G.A. 2008. Quantifying cell-matrix adhesion dynamics in living cells using interference reflection microscopy. J. Microsc. 232:73–81 10.1111/j.1365-2818.2008.02069.x [DOI] [PubMed] [Google Scholar]
- Hood J.L., Logan B.B., Sinai A.P., Brooks W.H., Roszman T.L. 2003. Association of the calpain/calpastatin network with subcellular organelles. Biochem. Biophys. Res. Commun. 310:1200–1212 10.1016/j.bbrc.2003.09.142 [DOI] [PubMed] [Google Scholar]
- Hood J.L., Brooks W.H., Roszman T.L. 2004. Differential compartmentalization of the calpain/calpastatin network with the endoplasmic reticulum and Golgi apparatus. J. Biol. Chem. 279:43126–43135 10.1074/jbc.M408100200 [DOI] [PubMed] [Google Scholar]
- Hu L., Smith T.F., Goldberger G. 2009. LFG: a candidate apoptosis regulatory gene family. Apoptosis. 14:1255–1265 10.1007/s10495-009-0402-2 [DOI] [PubMed] [Google Scholar]
- Hynes R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- Igney F.H., Krammer P.H. 2002. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer. 2:277–288 10.1038/nrc776 [DOI] [PubMed] [Google Scholar]
- Katz F.N., Rothman J.E., Knipe D.M., Lodish H.F. 1977. Membrane assembly: synthesis and intracellular processing of the vesicular stomatitis viral glycoprotein. J. Supramol. Struct. 7:353–370 10.1002/jss.400070308 [DOI] [PubMed] [Google Scholar]
- Kim H.R., Lee G.H., Ha K.C., Ahn T., Moon J.Y., Lee B.J., Cho S.G., Kim S., Seo Y.R., Shin Y.J., et al. 2008. Bax Inhibitor-1 Is a pH-dependent regulator of Ca2+ channel activity in the endoplasmic reticulum. J. Biol. Chem. 283:15946–15955 10.1074/jbc.M800075200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.H., Ahn T., Kim D.S., Park S.J., Lee Y.C., Yoo W.H., Jung S.J., Yang J.S., Kim S., Muhlrad A., et al. 2010a. Bax inhibitor 1 increases cell adhesion through actin polymerization: involvement of calcium and actin binding. Mol. Cell. Biol. 30:1800–1813 10.1128/MCB.01357-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.H., Yan C., Shin S.J., Hong S.C., Ahn T., Moon A., Park S.J., Lee Y.C., Yoo W.H., Kim H.T., et al. 2010b. BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene. 29:2130–2141 10.1038/onc.2009.491 [DOI] [PubMed] [Google Scholar]
- Lee J., Ishihara A., Oxford G., Johnson B., Jacobson K. 1999. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature. 400:382–386 10.1038/22578 [DOI] [PubMed] [Google Scholar]
- Lee S., Jo M., Lee J., Koh S.S., Kim S. 2007. Identification of novel universal housekeeping genes by statistical analysis of microarray data. J. Biochem. Mol. Biol. 40:226–231 10.5483/BMBRep.2007.40.2.226 [DOI] [PubMed] [Google Scholar]
- Lewis R.S. 2011. Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb. Perspect. Biol. 3:a003970 10.1101/cshperspect.a003970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Cubbon R.M., Wilson L.A., Amer M.S., McKeown L., Hou B., Majeed Y., Tumova S., Seymour V.A., Taylor H., et al. 2011. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 108:1190–1198 10.1161/CIRCRESAHA.111.243352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P.W., Maxfield F.R. 1990. Transient increases in cytosolic free calcium appear to be required for the migration of adherent human neutrophils. J. Cell Biol. 110:43–52 10.1083/jcb.110.1.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh B.J., Buttery R., Lad Y., Banks S., Haslett C., Sethi T. 2010. Integrin activation by Fam38A uses a novel mechanism of R-Ras targeting to the endoplasmic reticulum. J. Cell Sci. 123:51–61 10.1242/jcs.056424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S.K., Schlaepfer D.D. 2006. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18:516–523 10.1016/j.ceb.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Orlando K., Guo W. 2009. Membrane organization and dynamics in cell polarity. Cold Spring Harb. Perspect. Biol. 1:a001321 10.1101/cshperspect.a001321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J.E., Smith G.L. 1994. Vaccinia virus gene A36R encodes a M(r) 43-50 K protein on the surface of extracellular enveloped virus. Virology. 204:376–390 10.1006/viro.1994.1542 [DOI] [PubMed] [Google Scholar]
- Parnaud G., Hammar E., Rouiller D.G., Bosco D. 2005. Inhibition of calpain blocks pancreatic beta-cell spreading and insulin secretion. Am. J. Physiol. Endocrinol. Metab. 289:E313–E321 10.1152/ajpendo.00006.2005 [DOI] [PubMed] [Google Scholar]
- Petrie R.J., Doyle A.D., Yamada K.M. 2009. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10:538–549 10.1038/nrm2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley J.F., Cole N.B., Schroer T.A., Hirschberg K., Zaal K.J., Lippincott-Schwartz J. 1997. ER-to-Golgi transport visualized in living cells. Nature. 389:81–85 10.1038/38001 [DOI] [PubMed] [Google Scholar]
- Prevarskaya N., Skryma R., Shuba Y. 2011. Calcium in tumour metastasis: new roles for known actors. Nat. Rev. Cancer. 11:609–618 10.1038/nrc3105 [DOI] [PubMed] [Google Scholar]
- Putney J.W. 2009. Capacitative calcium entry: from concept to molecules. Immunol. Rev. 231:10–22 10.1111/j.1600-065X.2009.00810.x [DOI] [PubMed] [Google Scholar]
- Reimers K., Choi C.Y., Bucan V., Vogt P.M. 2008. The Bax Inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr. Mol. Med. 8:148–156 10.2174/156652408783769562 [DOI] [PubMed] [Google Scholar]
- Rock M.T., Dix A.R., Brooks W.H., Roszman T.L. 2000. Beta1 integrin-mediated T cell adhesion and cell spreading are regulated by calpain. Exp. Cell Res. 261:260–270 10.1006/excr.2000.5048 [DOI] [PubMed] [Google Scholar]
- Roszik J., Lisboa D., Szöllosi J., Vereb G. 2009. Evaluation of intensity-based ratiometric FRET in image cytometry—approaches and a software solution. Cytometry A. 75:761–767 [DOI] [PubMed] [Google Scholar]
- Stockholm D., Bartoli M., Sillon G., Bourg N., Davoust J., Richard I. 2005. Imaging calpain protease activity by multiphoton FRET in living mice. J. Mol. Biol. 346:215–222 10.1016/j.jmb.2004.11.039 [DOI] [PubMed] [Google Scholar]
- Tang S., Wong H.C., Wang Z.M., Huang Y., Zou J., Zhuo Y., Pennati A., Gadda G., Delbono O., Yang J.J. 2011. Design and application of a class of sensors to monitor Ca2+ dynamics in high Ca2+ concentration cellular compartments. Proc. Natl. Acad. Sci. USA. 108:16265–16270 10.1073/pnas.1103015108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van ’t Veer L.J., Dai H., van de Vijver M.J., He Y.D., Hart A.A., Mao M., Peterse H.L., van der Kooy K., Marton M.J., Witteveen A.T., et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415:530–536 10.1038/415530a [DOI] [PubMed] [Google Scholar]
- Várnai P., Hunyady L., Balla T. 2009. STIM and Orai: the long-awaited constituents of store-operated calcium entry. Trends Pharmacol. Sci. 30:118–128 10.1016/j.tips.2008.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M., Horwitz A.R. 2011. Adhesion dynamics at a glance. J. Cell Sci. 124:3923–3927 10.1242/jcs.095653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb D.J., Donais K., Whitmore L.A., Thomas S.M., Turner C.E., Parsons J.T., Horwitz A.F. 2004. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 6:154–161 10.1038/ncb1094 [DOI] [PubMed] [Google Scholar]
- Wei C., Wang X., Chen M., Ouyang K., Song L.S., Cheng H. 2009. Calcium flickers steer cell migration. Nature. 457:901–905 10.1038/nature07577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth D.C., Hodivala-Dilke K., Robinson S.D., King S.J., Morton P.E., Gertler F.B., Humphries M.J., Parsons M. 2010. αvβ3 integrin spatially regulates VASP and RIAM to control adhesion dynamics and migration. J. Cell Biol. 189:369–383 10.1083/jcb.200912014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Xu W., Palmer A.E., Reed J.C. 2008. BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins. J. Biol. Chem. 283:11477–11484 10.1074/jbc.M708385200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Zhang J.J., Huang X.Y. 2009. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 15:124–134 10.1016/j.ccr.2008.12.019 [DOI] [PubMed] [Google Scholar]
- Ying Z., Giachini F.R., Tostes R.C., Webb R.C. 2009. PYK2/PDZ-RhoGEF links Ca2+ signaling to RhoA. Arterioscler. Thromb. Vasc. Biol. 29:1657–1663 10.1161/ATVBAHA.109.190892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C.H., Chae H.J., Kim H.R., Ahn T. 2012. Doxorubicin- and daunorubicin-induced regulation of Ca2+ and H+ fluxes through human bax inhibitor-1 reconstituted into membranes. J. Pharm. Sci. 101:1314–1326 10.1002/jps.23007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.