

Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) have long been used to treat pain, fever, and inflammation. However, mounting evidence shows that NSAIDs, such as aspirin, have very promising antineoplastic properties. The chemopreventive, antiproliferative behaviour of NSAIDs has been associated with both their inactivation of cyclooxygenases (COX) and their ability to induce apoptosis via pathways that are largely COX-independent. In this review, the various proapoptotic pathways induced by traditional and novel NSAIDs such as phospho-NSAIDs, hydrogen sulfide-releasing NSAIDs and nitric oxide-releasing NSAIDs in mammalian cell lines are discussed, as well as the proapoptotic effects of NSAIDs on budding yeast which retains the hallmarks of mammalian apoptosis. The significance of these mechanisms in terms of the role of NSAIDs in effective cancer prevention is considered.
1. Introduction
In recent years, there has been a growing interest in aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) because of their promising antineoplastic properties. Epidemiological, clinical, and experimental evidence strongly indicates that NSAIDs exert a significant chemopreventive, antiproliferative effect on several types of cancer cells (see Table 1). Much of the research concerning the antineoplastic effects of NSAIDs has focused on the effect of aspirin in large bowel cancers, with comparatively fewer studies carried out on the chemopreventive effects of other NSAIDs [1]. In fact, aspirin stands as the most widely studied pharmacological agent for the chemoprevention of colorectal malignancies, with numerous clinical trials being carried out to examine its role in the prevention of adenomas, colorectal carcinomas, and inherited colorectal neoplasias such as the Lynch syndrome and familial adenomatous polyposis (FAP) [2]. Such focus on aspirin may be due, in large part, to the increasingly high prevalence and social impact of human colorectal cancer in recent years [2]. It is also due to the simple fact that long-term aspirin use is already widely practised among patients for the prevention of cardiovascular events such as thrombosis and neurovascular events such as stroke, thus providing a convenient opportunity for researchers to study its other long-term chemopreventive effects. In fact, major findings of aspirin's anticancer effects in humans are also derived from clinical trial data originally compiled for the study of its anti-thrombotic effects [3, 4]. Aspirin's affordability and ease of access, together with its relatively reduced side effects with respect to other traditional NSAIDs [5], have also helped to increase its appeal as a potential chemopreventive agent and target in anticancer studies.
Table 1.
Human cancer cell targets of the proapoptotic effects of prominent traditional and modified NSAIDs.
| NSAID type | Target cell type [references] |
|---|---|
| Traditional NSAIDs | |
| Aspirin | Colon cancer cells [34–36], leukaemia cells [37, 38], cervical cancer cells [39, 40], gastric cancer cells [41–44], hepatocellular carcinoma cells [45, 46], endometrial cancer cells [29], neuroblastoma cells [47] |
| Indomethacin | Colon cancer cells [7, 35], Gastric cancer cells [44], Endometrial cancer cells [29] |
| Sulindac | Colon cancer cells [6, 7, 48, 49], prostate cancer cells [48], hepatocellular carcinoma cells [8], lung cancer cells [50] |
| Ibuprofen | Colon cancer cells [51] |
| Diclofenac | Neuroblastoma cells [15], melanoma cells [16] |
| Tolfenamic Acid | Prostate cancer cells [52] |
| Modified NSAIDs | |
| NO-Aspirin | Pancreatic cancer cells [53], colon cancer cells [17, 54–58] |
| NO-sulindac | Colon cancer cells [17], melanoma cells [59] |
| NO-ibuprofen | Colon cancer cells [17] |
| NOSH-aspirin | Colon cancer cells [24] |
| Phosphoaspirin | Colon [19, 21, 56], pancreatic [21], and breast [21] cancer cells |
| Phosphosulindac | Colon, pancreatic, and breast cancer cells [21] |
| HS-aspirin | Colon cancer cells [22], breast cancer cells [60] |
| HS-ibuprofen | Colon cancer cells [22] |
| HS-naproxen | Colon cancer cells [22] |
| HS-sulindac | Colon cancer cells [22] |
Nevertheless, a considerable number of investigations have shown that other NSAIDs including sulindac [6–8], indomethacin [9, 10], ibuprofen [11, 12], naproxen [13], and diclofenac [14–16] also exhibit significant antineoplastic behaviour in mammalian cancer cells. Additionally, recent studies have increasingly focused on the chemopreventive properties of a new NSAID class referred to as modified NSAIDs. These are essentially traditional NSAIDs which can either have nitric oxide- (NO-) donating moieties, hydrogen sulphide- (H2S-) donating moieties, or phosphate moieties covalently attached to the –COOH site via aromatic or aliphatic spacer molecules, as shown in Figure 1. The resulting modified NSAID classes, known as NO-donating NSAIDs (NO-NSAIDs), H2S-donating NSAIDs (HS-NSAIDs), and phospho-NSAIDs, respectively, have all been shown to be far less toxic than their NSAID precursors and several times more potent in terms of antineoplastic efficacy [17–22]. The exceedingly potent anti-neoplastic properties of novel NSAID chimeras, which are characterized by their possession of both NO- and H2S-donating moieties (see Figure 1), have also begun to attract significant attention [23, 24].
Figure 1.
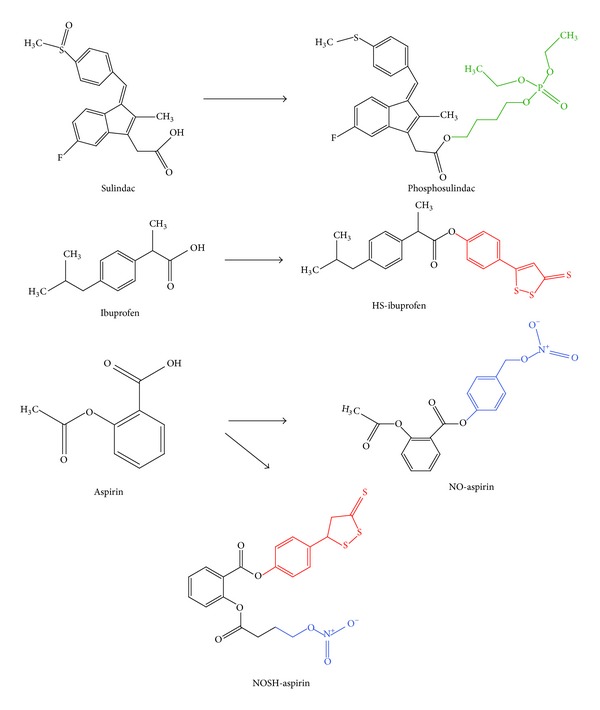
Chemical structures of modified NSAIDs and their traditional NSAID precursors. Phosphosulindac, which exemplifies phospho-NSAIDs, consists of the sulindac molecule linked at the –COOH site to a phosphate group via an aliphatic spacer molecule (both shown in green). In the HS-NSAID known as HS-ibuprofen, an aromatic spacer molecule links an H2S-releasing dithiolethione group (both shown in red) to the ibuprofen structure. Similarly, the NO-NSAID NO-aspirin is composed of an NO-releasing-NO2 group and an aromatic spacer molecule (both shown in blue) which is linked to the –COOH group of aspirin. Finally, the modified NSAID chimera known as NOSH-aspirin is characterized by the aspirin structure linked via two separate spacer molecules to both a H2S-releasing moiety (shown in red) and an NO-releasing moiety (shown in blue).
The mechanistic pathways which mediate the anti-neoplastic effects of traditional and modified NSAIDs are still not fully understood. It has been postulated that the antiproliferative effect of NSAIDs on malignant cells involves the inhibition of proinflammatory COX activity [25] and prostaglandin formation [26]. However, additional evidence shows that NSAIDs can induce apoptotic cell death in tumour cells [27] via pathways that are largely independent of COX [6, 28, 29]. The elucidation of apoptotic mechanisms underlying the chemopreventive effect of NSAIDs has long been the focus of intense research using a broad range of experimental models, including whole mammalian specimens, human cancer cell lines, and Saccharomyces cerevisiae cells.
1.1. Yeast Cells as a Model for the Study of the Proapoptotic Effects of NSAIDs
Yeast cell species, such as Saccharomyces cerevisiae, are among the preferred and extensively used experimental models for the study of programmed cell death associated with ageing, disease, and cancers in living organisms. This is partly because yeast cells retain many core elements of mammalian cell processes such as apoptosis [30]. Additionally, these primitive eukaryotes have a number of important advantages over complex mammalian cell models. Yeast cells are relatively inexpensive and easy to handle, with a relatively brief lifespan that permits rapid generation of experimental results in a shorter span of time. Moreover, the yeast cell genome is very well characterised and relatively easy to manipulate, allowing for the ready availability of yeast mutant strains for experimental studies. Overall, these features collectively account for the successful use of budding yeast as a model organism for the study of molecular pathways underlying mammalian pathologies such as cancer.
These same advantages of the yeast cell experimental model also account for its wide use in the study of proapoptotic pathways underlying the anti-neoplastic behaviour of antitumour drugs such as paclitaxel, bleomycin, valproate, and arsenic [31]. These compounds have been studied extensively in yeast cells in an effort to improve anticancer strategies in human patients. Likewise, S. cerevisiae budding yeast cells have also been used to study the growth inhibitory, proapoptotic effects of NSAIDs such as aspirin [32] and diclofenac [33].
The study of the proapoptotic effects of NSAIDs in yeast models is still a relatively new concept, with far fewer studies having been carried out in yeast with respect to mammalian cells. Regardless, evidence acquired thus far from yeast studies of NSAIDs such as aspirin has already yielded valuable insights into their proapoptotic behaviour, highlighting factors which play key roles in NSAID-induced death (such as reactive oxygen species (ROS) and mitochondrial dysfunction). In fact, compelling evidence has shown that S. cerevisiae cells constitute a powerful model for the screening and development of NSAIDs and other proapoptotic drugs designed for use in human cancer patients, overcoming the problem of cell specificity in the design of antitumour compounds [31], whilst also serving as an inexpensive model to initially test the effect of antitumour drugs before assaying them in more relevant mammalian systems. Therefore, yeast cells clearly serve an important role as a complementary experimental model to mammalian cells in the study and elucidation of NSAID-induced mechanisms of apoptosis.
In this review, important biomolecular pathways triggered by traditional and novel NSAIDs which lead to the induction of apoptosis in mammalian cell lines and in S. cerevisiae yeast cells will be discussed. The significance of these proapoptotic mechanisms, in the context of the role NSAIDs may play in the design of more effective cancer prevention strategies, is also considered.
2. NSAID-Induced COX Inhibition and Apoptotic Cell Death
The cyclooxygenase isoforms COX-1 and COX-2, both of which are key requirements for the synthesis of prostaglandins in mammalian cells, constitute the best defined pharmacological targets of NSAIDs such as aspirin [61, 62]. Thus, it has long been postulated that the chemopreventive effect of NSAIDs is mediated by their ability to inactivate COX-enzymes, causing inhibition of prostaglandin synthesis and consequent suppression of both tissue inflammation and cell proliferation, two conditions heavily associated with tumour formation [25]. In fact, both prostaglandins and COX enzymes, particularly COX-2, are implicated in tumorigenesis [1, 2, 63, 64] and have been observed in high quantities in several types of human tumours, including colorectal carcinomas [65–69]. Moreover, prostaglandins such as PGE2 are known to promote angiogenesis, alter cellular immunity, increase both proliferation and invasiveness of cells, and enhance cellular resistance to apoptosis [2, 68, 70].
It has been shown that deletion of receptors for PGE2 confers resistance to both polyp and cancer formation [71]. Similarly, disruption of COX-2 activity was found to reduce the incidence of polyp [72, 73] and aberrant crypt foci formation [74] in the intestines of rodent models. Inhibition of COX-2 has also been shown to be effective in preventing the formation of human colorectal adenomas [75, 76] and oesophageal squamous cell carcinoma [64]. Thus, inhibition of COX-2-dependent prostaglandin synthesis is thought to mediate, at least in part, the tumour-suppressive, antiproliferative effects of NSAIDs such as the suppression of angiogenesis and the induced lowering of resistance to apoptosis. In fact, inhibition of invasive tumour formation in NSAID-treated mouse models has been associated with decreased cellular levels of PGE2 [26].
The anti-neoplastic behaviour of NSAIDs is also associated with their ability to actively induce apoptosis in malignant cells. However, the means by which COX-2 inhibition could possibly mediate NSAID-induced cancer cell apoptosis has long been the subject of debate [77, 78]. As such, there is no clear evidence implicating direct involvement of prostaglandins in NSAID-induced apoptosis, which suggests that prostaglandins do not directly mediate NSAID-induced death [77–79]. In fact, studies have indicated that aspirin-induced apoptosis in mouse [80] and human [81] cancer cells can occur independently of prostaglandin inhibition. Similarly, Chan and coworkers [7] demonstrated that apoptosis induced by sulindac and indomethacin in mammalian HCT116 and SW480 colon cancer cell lines did not depend on prostaglandin depletion since supplementation of these same cells with prostaglandins failed to protect them from apoptosis. However, this same study did present evidence of COX-2-dependent apoptosis induced by these NSAIDs. The authors suggested that cellular accumulation of the prostaglandin precursor arachidonic acid, brought about by the NSAID-induced inhibition of COX-2, caused cancer cell apoptosis by stimulating sphingomyelinase-mediated synthesis of ceramide [7], which is a proapoptotic molecule [82]. Arachidonic acid accumulation in cancer cells also induces ROS accumulation [83], mitochondrial permeability transition, and cytochrome c release [84], all of which lead to apoptotic cell death.
3. COX-Independent NSAID-Induced Apoptotic Cell Death
The proapoptotic effects underlying the chemopreventive potential of NSAIDs cannot be accounted for by COX inhibition alone. Firstly, NSAIDs have been shown to inhibit proliferation and induce apoptosis in malignant cell lines which do not express either COX-1 or COX-2, as observed in cyclooxygenase-null mouse embryo fibroblasts [85] and human colon cancer cells [28, 81, 86]. Furthermore, NSAID compounds which cannot inactivate COX-2, such as sulindac sulfone, a metabolite of sulindac [87], have been shown to induce apoptosis of gastric tumour cells [88] and inhibit colon tumour formation in rodents [89, 90]. Additionally, NSAID concentrations required to induce apoptosis in cancer cells have often been found to be significantly higher than that required to inhibit COX-2, suggesting the presence of alternative cellular targets of NSAIDs [27, 37, 77, 91]. Indeed, numerous studies have shown that NSAID-induced apoptosis in mammalian tumour cells can be mediated by several largely COX-independent metabolic pathways, the most prominent of which are presented in the following discussion.
3.1. Activation of Caspases and Modulation of Bcl-2 Proteins
NSAIDs can mediate apoptosis by inducing the activation of caspases, a family of proapoptotic cysteine proteinase enzymes which typically exist as latent zymogens in cells. Activation of these proteins by proapoptotic signals initiates a caspase cascade whereby initiator caspases specifically activate other executioner-type caspases. The latter then degrade multiple cellular components so that cells begin to acquire the morphological and biochemical features of apoptosis [92]. Bellosillo and coworkers [37] were among the first to present evidence of NSAID-induced caspase activation. They showed that B-chronic lymphocytic leukemia (B-CLL) cells treated with high doses of aspirin underwent apoptosis characterised by DNA fragmentation and degradation of poly (ADP-ribose) polymerase (PARP), which is a caspase substrate. The apoptotic phenotype was inhibited by application of the pan-caspase inhibitor Z-VAD-FMK, thus confirming the involvement of caspases in aspirin-induced B-cell apoptosis. Similarly, Castaño and coworkers [34] affirmed caspase involvement in aspirin-induced apoptosis of HT-29 human colon adenocarcinoma cells. However, contrary to its effect in B cells, aspirin did not induce PARP degradation in HT-29 cells. This observation is probably one of the earliest to suggest that NSAID-induced activation of caspases can occur via different pathways.
Indeed, it has since been shown that NSAIDs activate caspases both via the mitochondrial-mediated (intrinsic) pathway, which involves mitochondrial cytochrome c release and subsequent activation of caspase 9, and via the death-receptor mediated (extrinsic) pathway, which involves the activation of caspase 8 [41]. Proapoptotic NSAID-induced activation of caspases, mediated by the early release of mitochondrial cytochrome c in mammalian cancer cells, has clearly been demonstrated by studies such as that of Piqué et al. [38]. The authors reported that cytochrome c release preceded both the disruption of the mitochondrial membrane potential (ΔΨm) and the activation of caspases. The latter observation was confirmed by Zimmermann and coworkers [39] who in turn showed that, in aspirin-induced apoptosis of human cancer cells, the release of mitochondrial cytochrome c was itself triggered by translocation of proapoptotic Bax protein to the mitochondria. Conversely, overexpression of antiapoptotic Bcl-2 protein inhibited both cytochrome c release and apoptosis of aspirin-treated cancer cells. Furthermore, deletion of apoptotic protease-activating factor-1 (APAF-1), a cytosolic molecule which mediates caspase 9 activation after binding to mitochondrial cytochrome c, rendered cells resistant to aspirin-induced apoptosis. These observations indicate that cytochrome c release is a critical mediatory mechanism of apoptotic cell death induced by NSAIDs [39]. In fact, it has also been shown that aspirin-induced apoptosis of S. cerevisiae cells deficient in manganese superoxide dismutase (MnSOD) and cultivated in nonfermentable ethanol medium is preceded by the early release of cytochrome c, followed by a drastic fall in ΔΨm [93].
Extrinsic activation of caspase 8 is also an important mediator of NSAID-induced cancer cell apoptosis [42, 48]. Activated caspase 8 can initiate apoptosis by activating downstream caspases or by cleaving the BH3-domain only protein Bid. Truncated Bid (tBid) can translocate to the mitochondria to trigger cytochrome c release [94, 95] and can also activate Bax [96]. Indeed, aspirin-induced apoptosis of AGS gastric cancer cells was marked by activation of caspase 8 and Bid cleavage, along with the mitochondrial translocation of Bax, activation of downstream caspases, and cleavage of PARP. Specific inhibition of caspase 8 abrogated cleavage of both Bid and PARP and prevented aspirin-induced AGS cell apoptosis, thus implicating extrinsic caspase activation in the initiation of aspirin-induced apoptosis [42]. However, in this same study, the release of cytochrome c and activation of caspase 9 were also observed, thus suggesting the involvement even of mitochondrial-mediated caspase activation. In fact several other studies have since shown that the chemopreventive apoptotic effect of aspirin [41, 43, 45, 46] and modified NSAIDs such as NO-aspirin [97] and phosphosulindac [20] on mammalian cancer cells may involve the initiation of both intrinsic and extrinsic caspase activation pathways, which operate in parallel to one another.
The ability of NSAIDs to activate caspases largely explains the profound influence they have on Bcl-2 proteins such as Bax, Bid, and Bcl-2 [42, 43], the expression and cellular distribution of which can be greatly altered by NSAIDs to mediate apoptosis in cancer cells. NSAIDs such as aspirin and indomethacin have long been shown to induce cancer cell apoptosis by upregulating the expression of proapoptotic Bcl-2 proteins, such as Bax and Bak, and by downregulating expression of anti-apoptotic Bcl-2 proteins such as Bcl-2 and Bcl-XL [29, 44]. More recent work has shown that NSAID-induced downregulation of Bcl-XL can be induced in part by proteasome-mediated protein degradation [98].
NSAID-induced intrinsic and extrinsic activation of caspases, along with modulation of Bcl-2 protein expression, all seem to converge at the mitochondria. Here, these pathways induce events such as the release of cytochrome c and other proapoptotic molecules, including SMAC/Diablo, from the mitochondria [43], which irreversibly commit cells to the full apoptotic phenotype. Thus mitochondria clearly constitute a very important target of NSAID-induced apoptosis, as indicated by the many studies associating NSAID-induced apoptosis of mammalian cells with mitochondrial dysfunction, such as uncoupling of oxidative phosphorylation [99], induced mitochondrial permeability transition [100, 101], and inactivation of mitochondrial enzymes such as aconitase and respiratory chain proteins [45].
Likewise, it has been shown that the mitochondria of S. cerevisiae cells constitute a critical target of NSAIDs such as aspirin [32], the proapoptotic effects of which were shown to be associated with inhibition of the electron transport chain [93]. Similarly, Van Leeuwen and coworkers [33] observed that growth inhibition and apoptosis of S. cerevisiae cells caused by treatment with the NSAID diclofenac were due to mitochondrial dysfunctional events involving the inhibition of the electron transport chain. The fact that yeast cells, which are primitive eukaryotes, share a common mitochondrial target of NSAIDs with mammalian cells is highly significant, because it suggests that mitochondria constitute a unifying, dominant target of NSAID-induced apoptosis in all mammalian cancer cell types. This may certainly help inform the design of more effective NSAIDs for chemopreventive purposes and illustrates the important contribution of yeast cells as a complementary experimental model for the study of NSAID-induced apoptotic mechanisms.
3.2. Depletion of Polyamines
Polyamines such as spermine, spermidine, and putrescine are abundant polycations found in all eukaryotic cells and play an essential role in cellular development and proliferation [102]. High polyamine levels are in fact associated with the induction of cell proliferation [103, 104], whilst lowered polyamine levels have been found to promote cell growth inhibition [105] and apoptosis [106]. Hence, it is no surprise that the polyamine content of cancer cells tends to be significantly higher than that of normal cells [102], thus representing a potential target of anti-neoplastic agents such as NSAIDs, a number of which have been shown to mediate their chemopreventive effect by promoting the catabolic degradation of polyamines in cancer cells [35, 49, 107]. This takes place by virtue of the general ability of NSAIDs to modulate cellular polyamine metabolism, which is regulated by the biosynthetic enzyme ornithine decarboxylase (ODC) and the catabolic polyamine-acetylating enzyme spermidine/spermine N 1-acetyltransferase (SSAT) [49, 107]. For instance, indomethacin-induced growth inhibition of human colon cancer cells has been shown to be associated with downregulation of ODC activity and upregulation of SSAT activity, which concurrently impair the synthesis of polyamines and increase the rate at which they are degraded. The consequent depletion of cellular polyamines was accompanied by apoptotic cell death [35]. Other traditional NSAIDs such as aspirin [108], sulindac sulfone [49], and ibuprofen [107] mainly induce the enhanced degradation and export of polyamines by upregulating gene expression of SSAT in cancer cells, resulting in reduced proliferation and increased apoptosis. This is also true of modified NSAIDs such as phosphosulindac [20], the antiproliferative proapoptotic effect of which can, like its NSAID precursor sulindac [109], be enhanced even further by concurrent treatment of cells with ODC inhibitors such as difluoromethylornithine (DFMO) [20, 110, 111]. Both DFMO and phosphosulindac act synergistically to enhance the depletion of polyamines in colon cancer cells, strongly inhibiting their proliferation and greatly enhancing apoptosis [20].
3.3. Modulation of NF-κB Activity
Nuclear factor kappa B (NF-κB) is a ubiquitous cellular transcription factor which regulates the expression of genes associated with inflammation, immune responses, cell growth, differentiation, and apoptosis. Composed of p65 (RelA) and p50 polypeptides, this complex transcription factor is sequestered in an inactive, heterodimeric form within the cell cytoplasm by I kappa B alpha (IκBα) or beta (IκBβ) inhibitor proteins [112, 113]. Stimulation by appropriate signals (such as proinflammatory cytokines including tumour necrosis factor (TNF)) triggers the IKα or IKβ kinase- (IKK-) mediated phosphorylation of IκB proteins, which consequently undergo ubiquitin-dependent proteasomal degradation. This permits translocation of NF-κB molecules to the nucleus, where they then bind to and promote the transcription of numerous target genes bearing a κB-binding motif [79, 113, 114].
The constitutive activation of NF-κB is a hallmark of several types of cancers [115–117] and is heavily associated with cancer cell resistance to cytotoxic agents, due in part to its induced upregulation of anti-apoptotic proteins [118]. Thus, NF-κB constitutes yet another potential target of chemotherapeutic agents such as NSAIDs, which can modulate NF-κB signalling in cancer cells to promote the onset of apoptosis [36, 54, 60, 107, 111, 114, 119].
Traditional NSAIDs such as aspirin have been reported to inhibit NF-κB activation by preventing the degradation of IκB [119]. Aspirin can inhibit TNF-induced IκBα degradation [120] by modulation of p38 mitogen-activated protein (MAP) kinase pathways [121] and by disrupting the ubiquitin-dependent proteasomal pathway, of which IκBα is a substrate [47]. Aspirin can also block IκBβ degradation through competitive inhibition of IKKβ-ATP molecular binding, thus facilitating selective inhibition of IKK kinase (IKKβ) [114]. All these mechanisms prevent NF-κB activation and subsequent transcription of anti-apoptotic proteins. It has also been shown that the NSAID sulindac specifically inhibits IKKβ activity and NF-κB activation in cancer cells, thus promoting apoptosis [122]. Likewise, the growth inhibitory effect of NSAIDs such as ibuprofen [51], indomethacin, and etoricoxib, a recently developed COX-2 inhibitor [117], is associated with their inhibition of NF-κB signalling in cancer cells.
The growth inhibitory effect mediated by modified NSAIDs such as NO-NSAIDs, on various cancer cell lines, also involves the modulation of NF-κB signalling [54, 55]. The growth inhibitory effect of NO-aspirin, associated with its ability to reduce proliferation and enhance apoptosis of cancer cells, was shown to be significantly correlated to its profound inhibition of the NF-κB signalling pathway, the occurrence of which was suggested to be due to inhibition of NF-κB binding to DNA in the nucleus [55]. Sun and Rigas [56] went on to demonstrate that proapoptotic inhibition of NF-κB signalling in human colon cancer cell lines, treated with NO-aspirin, stemmed from the latter's induced generation of reactive oxygen and nitrogen species (RONS), which may have interacted directly or indirectly (via the redox-sensitive thioredoxin (TRX) system) with NF-κB, impairing its ability to bind to recognition DNA sequences in the nucleus. This is highly conceivable given that NF-κB transcriptional activity is sensitive to redox changes [123]. In fact, it has since been shown that structurally diverse NO-NSAIDs such as NO-aspirin and NO-naproxen can suppress NF-κB signalling in cells via S-nitrosylation of the NF-κB transcription factor. This redox-signalling mechanism is mediated by the released NO group which, on binding to the p65 monomer of NF-κB, impairs the transcription factor's ability to bind to DNA [57]. The redox-induced inhibition of NF-κB signalling is thought also to partly mediate the growth inhibitory effect (including apoptosis, cell cycle arrest, and inhibition of proliferation) of phospho-NSAIDs [19, 21, 111] and HS-NSAIDs in cancer cells [60].
Intriguingly, the effect of NSAIDs on NF-κB activity seems to be cell-type specific, since aspirin-induced apoptosis of HCT 116 colon cancer cells was shown to be mediated by the activation of NF-κB signalling, rather than its inhibition [36]. Additionally, Din and coworkers [124] observed that aspirin-induced IκBα degradation, activation of NF-κB signalling, and apoptosis took place in colorectal cancer cells but not in other malignant cell types. Loss of cellular IκBα, which is indicative of NF-κB activation, has also been reported in aspirin-induced apoptosis of both immortalised human endothelial cells [125] and animal models of human colorectal cancer [126]. Likewise, Babbar and coworkers [107] observed that aspirin caused the activation of NF-κB signalling in Caco-2 colon cancer cells, suggesting even that this event was responsible for the upregulation of SSAT expression and polyamine depletion which led to apoptosis. Besides aspirin, other traditional NSAIDs such as diclofenac have also been reported to induce activation of NF-κB signalling as a means to attenuate cancer cell proliferation and promote apoptosis [127]. It has been argued that the varying effects of NF-κB may be due to the specificity by which this transcription factor binds to DNA and activates target genes. Such specificity is in turn dependent on the dimeric composition of the NF-κB complex and on the transcriptional cofactors that it has recruited, both of which can vary depending on the kinetics of induction [128]. Therefore, different stimuli or even the same stimulus exerted under different conditions can induce different NF-κB complexes and different downstream responses [36, 107].
3.4. Modulation of MAP-Kinase Activity
Mitogen activated protein (MAP) kinases are serine/threonine-specific proteins which respond to extracellular stimuli and regulate various cellular pathways including mitosis, cell proliferation, survival and death [129]. The three principal MAP kinase subgroups include the extracellular signal-regulated kinases ERK1/2 (p42/p44), c-Jun N-terminal kinases/stress-activated protein kinases (JNKs/SAPKs), and the p38 MAP kinases [121]. NSAIDs such as aspirin and its metabolite salicylate have long been shown to modulate MAP kinase signalling in mammalian cells [130, 131]. This is exemplified by the salicylate-induced activation of p38 MAP kinase signalling in FS-4 fibroblasts, which induced apoptosis [130] via a pathway involving the NSAID-induced inhibition of IκBα degradation and NF-κB signalling [121]. Based on these observations, the authors concluded that apoptotic cell death induced by p38 MAP kinase activation played an important role in mediating the anti-neoplastic effects of NSAIDs. The important mediatory role of MAP kinase modulation in the context of the anti-neoplastic effects of NSAIDs was further highlighted by Jones and coworkers [132], who demonstrated that NSAID-induced inhibition of angiogenesis involved the disruption of ERK1/2 kinase signalling, a typically prosurvival pathway [133]. Additionally, inhibition of ERK1/2 signalling was shown to be the mechanism by which aspirin sensitises human cervical cancer cells to apoptosis induced by tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) [40]. Modulation of MAP kinases has also been implicated in the suppressive effect of certain NSAIDs such as aspirin upon the factor activator protein (AP-1), a downstream target of MAP kinases which is critical for inducing neoplasia and activation of genes associated with inflammation and infection [134].
The modulation of MAP kinases has been shown to be critical for the growth inhibitory effect of modified NSAIDs such as NO-aspirin [135]. Aside from its propensity to inhibit NF-κB signalling, NO-aspirin was shown to induce the activation (marked by increased phosphorylation) of all three main MAP kinases in a concentration-dependent manner, in colon cancer cells. This was caused by the NO-aspirin-induced generation of RONS and subsequent oxidation of cellular thioredoxin-1 protein (Trx1p), which facilitated the proapoptotic autophosphorylation and activation of ASK1, a protein involved in the MAP kinase cascade and only kept inactive when attached to reduced Trx1p [56]. The same authors implicated ASK1-Trx1p cleavage in the activation of MAP kinase signalling, which in turn partly mediated the growth inhibitory effect of NO-aspirin, an effect marked by increased apoptosis and inhibition of cell proliferation. Likewise, NO-aspirin-induced cell cycle arrest and apoptosis of pancreatic cancer cells has been shown to occur via ROS-mediated modulation of all three MAP kinase signalling pathways and their downstream effector molecules such as p21 and cyclin D1 [53]. The rapid response of MAP kinases to the presence of RONS is not surprising given their well-established redox sensitivity [136]. The modulation of MAP kinase pathways was also implicated in the chemopreventive effect of NO-sulindac on UVB-induced melanoma cells [59] and in phospho-NSAID-mediated, redox-dependent apoptosis of colon cancer cells [19, 111].
3.5. Inhibition of Wnt/β-Catenin Signalling
The Wnt signalling pathway regulates the biosynthesis of β-catenin, a protein which is required for cell-to-cell adhesion and involved in the expression of genes associated with cancer [137]. Constitutive activation of Wnt/β-catenin signalling has been implicated in the development of numerous human malignancies [138–143]. Aberrant Wnt/β-catenin signalling is associated with the nuclear accumulation of β-catenin and the subsequent activation of the transcription factor known as T-cell factor (TCF). The resulting β-catenin/TCF complex promotes the transcriptional activation of target genes associated with proliferation, such as cyclin D1 [144, 145], hence the implicated role of Wnt/β-catenin signalling in human carcinogenesis.
The NSAIDs aspirin and indomethacin have been shown to attenuate Wnt/β-catenin signalling in colorectal cancer cells, in a dose-dependent manner, by inhibiting the transcription of β-catenin/TCF-responsive genes. This NSAID-induced inhibition did not involve cleavage of the β-catenin/TCF complex but rather the hyperphosphorylation and consequent stabilization of β-catenin, presumably caused by the inactivation of a phosphatase enzyme [146, 147]. Further studies have since shown that aspirin-mediated downregulation of Wnt/β-catenin/TCF signalling can indeed be mediated by its induced inactivation of protein phosphatase 2A [148] but also, in part, by the downregulation of upstream specificity protein (Sp) transcription factors [149]. Other NSAIDs, such as sulindac, can also mediate the antiproliferative degradation of β-catenin in cancer cells partly by proteasomal degradation and partly by caspase-mediated cleavage [150], whilst others such as diclofenac have been shown to suppress Wnt/β-catenin/TCF signalling via the activation of NF-κB [127].
The downregulation of Wnt/β-catenin/TCF signalling induced by NSAIDs has been associated with the profound growth inhibition of various cancer cell types, in a manner that seems in large part to be due to inhibition of cell proliferation rather than by direct induction of apoptosis [127, 142, 146, 150, 151]. However, the proapoptotic effect of NSAIDs such as sulindac [152] in colorectal cancer cell lines has been shown to involve downregulation of Wnt/β-catenin/TCF signalling. Furthermore, Wnt/β-catenin signalling was observed to play a key role in directly mediating the proapoptotic effect of aspirin in human mesenchymal stem cells [153]. Regardless, the high prevalence of β-catenin downregulation reported in studies of NSAID-induced growth inhibition of cancer cells underlines the importance of this pathway as a chemopreventive target of NSAIDs.
This is certainly true for modified NSAIDs such as NO-aspirin, the growth inhibitory effect of which is strongly associated with a number of induced cellular events including the profound concentration-dependent inhibition of β-catenin signalling in colon cancer cells [54, 58, 145, 154]. In this regard, NO-aspirin is far more effective than aspirin in that, at concentrations far below those required for the inhibition of cell proliferation, it actually prevented formation of the β-catenin/TCF complex, whereas aspirin did not [145]. Moreover, at higher concentrations, NO-aspirin can induce caspase-3-mediated cleavage of β-catenin itself, leading to a significant decline of cellularβ-catenin levels and loss of cell-to-cell adhesion amongst colon cancer cells [58]. The significant downregulation of Wnt/β-catenin signalling, mediated at least in part by caspase-mediatedβ-catenin degradation, has also been implicated in the growth inhibitory effect of NO-aspirin on leukaemia [97] and breast cancer cell lines [155]. The same applies for phospho-NSAIDs such as phosphosulindac, the growth inhibitory effect of which was shown to involve β-catenin degradation in breast cancer stem cells [156].
3.6. Oxidative Stress and Disruption of Redox Balance
NSAIDs can mediate apoptosis in both malignant cell lines and budding yeast cells by upregulating the generation of ROS and by inducing oxidative stress. Indeed, it has been argued that ROS generation may constitute a central unifying mechanism by which the anti-neoplastic effects of NSAIDs are mediated, given that oxidative stress is coupled with many proapoptotic signals such as NF-κB inhibition and MAP kinase activation [157, 158]. The fact that ROS accumulation also mediates NSAID-induced apoptosis in primitive eukaryotes such as yeast [33] further corroborates this argument. However, the cellular prooxidant behaviour of NSAIDs has often been the subject of controversy due to conflicting reports that NSAIDs such as indomethacin and sulindac [159, 160] scavenge ROS and exert a cytoprotective antioxidant effect in cells. Likewise, there are numerous reports derived from in vivo studies of rats showing that NSAIDs such as aspirin can exert a cytoprotective antioxidant effect associated with the attenuation of ROS [161, 162] and of lipid peroxidation [163, 164], along with the upregulation of antioxidants such as reduced glutathione (GSH) [165] and superoxide dismutases (SODs) [166]. It has also been shown that low doses of aspirin can confer long-term cytoprotective resistance against H2O2-induced oxidative stress in S. cerevisiae cells [167].
Nevertheless, other investigations have clearly shown that NSAIDs can induce the proapoptotic accumulation of ROS in both yeast [33] and mammalian cells [157]. For instance, apoptotic cell death of respiring S. cerevisiae cells cultivated in the presence of diclofenac was clearly linked to a significant increase in cellular ROS, as measured by the ROS-sensitive fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCDHF-DA) [33]. Similarly, indomethacin-induced apoptosis of gastric epithelial cells, which was abrogated after treatment with antioxidants such as N-acetylcysteine (NAC), was shown to require the generation of ROS [168], an event likewise implicated in the proapoptotic depletion of polyamines induced by indomethacin in colorectal cancer cells [35]. Additionally, Chan and coworkers [7] showed that human colorectal cancer cell apoptosis induced by both indomethacin and sulindac was marked by the accumulation of arachidonic acid, an event which is itself heavily associated with the accumulation of cellular ROS [83, 169]. The accumulation of ROS was also shown to be a critical inducer of mitochondrial cytochrome c release, disruption of ΔΨm, caspase activation, and apoptosis in salicylate-treated mammalian tumour cells [170]. Additionally, the NSAID sulindac and its metabolites have been shown to enhance the antitumour effect of the proteasome-inhibitor bortezomib, primarily through the synergistic generation of ROS [171]. It has been suggested that conflicting reports of NSAID redox behaviour in eukaryotic cells might simply be due to differences in the timing of measurements of ROS and antioxidant changes in experimental setups, given that cellular antioxidant levels are naturally expected to increase in subsequent response to elevated ROS [157].
The molecular mechanisms underlying NSAID-induced proapoptotic generation of ROS in mammalian and yeast cells have not yet been fully elucidated. However, it is well known that mitochondria, a major source of cellular ROS [172] and a central component of the apoptotic machinery, are profoundly affected by NSAIDs in both yeast [33, 93] and mammalian cells [99, 101]. For example, the aspirin metabolite salicylate has been shown to inhibit the mitochondrial electron transport chain in mammalian cells by interacting with an Fe-S cluster of Complex I, through its o-hydroxyl group. This was found to induce ROS accumulation and oxidative stress, which in turn caused proapoptotic events such as mitochondrial permeability transition and cytochrome c release [173]. Likewise, aspirin-induced cell cycle arrest and apoptosis of HepG2 hepatoma cells were shown to be induced by ROS accumulation and increased oxidative stress, accompanied by severe mitochondrial dysfunction such as the inactivation of electron transport chain proteins and aconitase [45]. Van Leeuwen et al. [33] observed that reduced cell growth and viability of S. cerevisiae yeast cells treated with diclofenac are due to mitochondrial dysfunction associated with the inhibition of electron transport chain subunit proteins Rip1p (of Complex III) and Cox9p (of Complex IV). This caused inhibition of cell respiration and subsequent ROS production, resulting in cell death. Inhibition of cellular respiration induced by aspirin was also observed in MnSOD-deficient yeast cells cultivated in ethanol medium [93], and recent studies in our laboratory have established the aspirin-induced proapoptotic generation of mitochondrial superoxide radicals in these cells (unpublished work).
It has been shown that the proapoptotic induction of oxidative stress induced by NSAIDs such as aspirin is strongly associated with the modulation of cellular redox homeostasis. This is exemplified by the observed increase of aspirin-induced apoptosis in HepG2 cells with GSH depletion and compromised redox balance [174]. In addition to this, studies in S. cerevisiae yeast cells have shown that aspirin-treated MnSOD-deficient yeast cells grown in ethanol medium experienced a very significant decrease in cellular reducing power with respect to wildtype cells, as measured by the NADPH/NADP+ concentration ratio. This was accompanied by a significant decrease of the GSH/GSSG concentration ratio, owing to a buildup of GSSG, prior to cell death [175].
The induction of oxidative stress mediated by disruption of cellular redox balance is also central to the apoptotic effect of modified NSAIDs in cancer cells [53, 56, 58, 158]. For instance, oxidative stress induced by NO-aspirin in colon cancer cells was shown to be mediated by the depletion of cellular GSH, caused by the latter's association with the spacer component of NO-aspirin and subsequent formation of a GSH conjugate. The resulting redox imbalance then initiated a number of downstream proapoptotic pathways such as β-catenin cleavage, inhibition of Wnt signaling, and mitochondrial-mediated activation of caspases [58]. Sun and Rigas [56] further demonstrated that redox-induced apoptosis of colorectal cancer cells treated with NO-aspirin involved the generation of RONS, the growth inhibitory effect of which was mediated by oxidative alteration and impairment of the thioredoxin redox system. Oxidised thioredoxin-1 induced MAP kinase activation and NF-κB inhibition, both of which are critical mediatory pathways of NO-aspirin-induced apoptosis [55, 135].
Oxidative stress, marked by RONS accumulation and redox imbalance associated with the suppression of GSH and increased oxidation of Trx-1, also plays a central role in apoptosis induced by phospho-NSAIDs [19–21, 56, 110, 111]. Like NO-NSAIDs, the strong prooxidant effect exerted by phospho-NSAIDs is reported to set in motion a pleiotropic cascade of redox-sensitive signalling events including activation of MAP kinases and inhibition of NF-κB signalling [19, 21, 56] along with the depletion of cellular polyamines, at least in the case of phosphosulindac [20, 110, 111]. The collective initiation of all these antiproliferative, proapoptotic pathways accounts for the very potent growth-inhibitory effects of phospho-NSAIDs, with respect to their traditional NSAID precursors [111]. Likewise, recent studies have shown that both apoptosis and cell cycle arrest of cancer cells treated with HS-NSAIDs, such as HS-aspirin, are induced as a result of oxidative stress and redox imbalance [60].
3.7. Other NSAID-Induced Proapoptotic Pathways
Further mechanisms by which NSAIDs can promote apoptosis in malignant cells include the induced depletion of survivin, an inhibitor of apoptosis protein which regulates the cell cycle and apoptosis in eukaryotic cells. Survivin expression in cancers tends to be very high and is in fact associated with tumour cell chemoresistance, making it an attractive target of antineoplastic treatments [176], including NSAIDs. Lu and coworkers [177] showed that aspirin caused significant and targeted depletion of survivin in breast cancer cells by upregulating its proteasomal degradation, consequently sensitizing the tumour cells to TRAIL-induced apoptosis. Moreover, aspirin acted synergistically with TRAIL to promote apoptosis of the breast tumour cells. Similarly it has been shown that cell cycle arrest and apoptotic cell death induced by the NSAIDs ibuprofen [51] and tolfenamic acid [52] in human colon and prostate cancer cells, respectively, are accompanied by significant depletion of survivin levels.
Another COX-independent proapoptotic pathway induced by NSAIDs is the impairment of proteasome function, as demonstrated by Dikshit and co-workers [47], who observed a time- and dose-dependent decline of proteasomal activity in neuroblastoma cells treated with aspirin. Accompanied by the accumulation of ubiquitylated proteins and profound mitochondrial abnormalities, the aspirin-induced impairment of proteasomal function was shown to activate the intrinsic apoptotic pathway, marked by a release of cytochrome c and the activation of caspase 9.
Finally, NSAIDs such as sulindac have been reported to induce sensitization of cancer cells to mda7/IL24-mediated apoptosis. The mda7 gene, also known as IL24, is of the interleukin (IL) 10 family of cytokines [178, 179]. Ectopic expression of mda7 is known to exert a potent tumour-suppressive effect against a variety of human cancer cells, with little to no effect on normal cells [180–182]. Furthermore, intratumoural administration of adenoviral vectors which express mda7 (Ad-mda7) has been shown to exhibit antitumour and antiangiogenic activity in human lung tumour xenografts [183]. Also, Oida and coworkers [50] demonstrated that Ad-mda7-mediated growth inhibition and apoptosis of human lung cancer cells was greatly enhanced by concurrent administration of sulindac, which increased the half-life of MDA7 protein in the cells. This resulted in the increased expression of MDA7 protein and of its proapoptotic downstream effector proteins including p38MAPK, caspase-9, and caspase-3, consequently sensitizing the lung cancer cells to apoptosis. Therefore, sulindac essentially altered MDA7 protein turnover in lung cancer cells in such a way as to promote apoptotic cell death [50].
4. Concluding Remarks
Ongoing investigations of proapoptotic mechanisms underlying the promising anti-neoplastic properties of NSAIDs such as aspirin remain a top research priority, since an improved understanding of such pathways will help to enhance current anticancer drug treatments [2]. What is certain thus far is that NSAIDs generally exert a dose-dependent pleiotropic effect on cancer cells (see Figure 2), initiating a very complex cascade of signalling events which collectively induce apoptosis. Although much more remains to be elucidated, there is also mounting evidence which suggests that NSAID-induced signalling events associated with the induction of oxidative stress, such as mitochondrial dysfunction and altered redox signalling, may be the dominant pathways underlying all other proapoptotic effects induced by NSAIDs in cancer cells [184].
Figure 2.
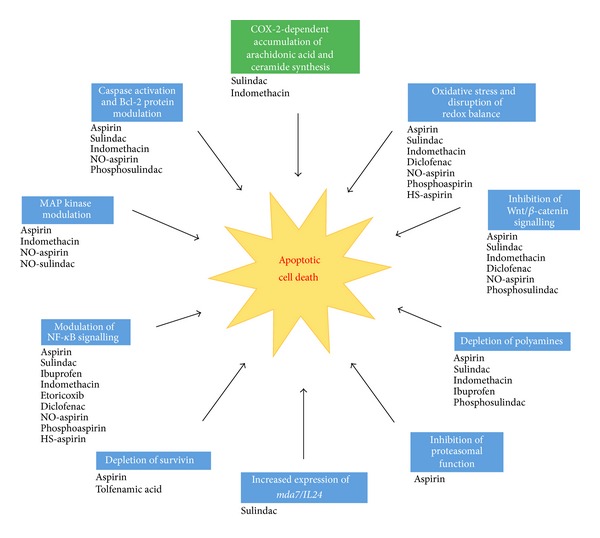
The major proapoptotic pathways induced by NSAIDs. Both traditional and modified NSAIDs have been shown to induce apoptosis in eukaryotic cells by initiating mechanisms which are largely independent of COX inhibition (shown in blue), with the exception being the COX-2-dependent accumulation of arachidonic acid and subsequent synthesis of ceramide induced by sulindac and indomethacin (shown in green). Important COX-independent proapoptotic pathways induced by NSAIDs include caspase activation and modulation of Bcl-2 proteins, depletion of polyamines, modulation of NF-κB signalling and of MAP kinase activity, inhibition of Wnt/β-catenin signalling, inhibition of proteasomal function, depletion of survivin, increased expression of mda7/IL24 and also oxidative stress associated with mitochondrial dysfunction, ROS accumulation and the disruption of cellular redox balance.
Consistent with the growing number of mammalian cell studies implicating oxidative stress as the dominant pathway of NSAID-induced growth inhibition [185], the proapoptotic effects of NSAIDs in yeast cells are likewise associated mainly with mitochondrial dysfunction, ROS generation, and redox imbalance. In particular, yeast cell studies have highlighted the pivotal importance of mitochondrial MnSOD as a cytoprotective defence against NSAIDs such as aspirin [32]. This implies that specific, targeted modulation of mitochondrial MnSOD can be exploited to enhance NSAID-induced oxidative stress and apoptosis in malignant mammalian cells.
It is currently hypothesised that cancer cells constantly experience much higher levels of oxidative stress with respect to normal cells, due to their increased metabolic rate [186]. Because of this, cancer cells are more reliant on antioxidant enzymes such as MnSOD and are thus believed to be far more sensitive to perturbations in redox balance compared to normal cells [16]. In fact, it has been shown that silencing of MnSOD, using anti-sense MnSOD antibodies, amplifies ROS accumulation and apoptosis in squamous cell carcinomas exposed to gamma radiation and anticancer drugs [187]. Moreover, it has recently been shown that silencing of MnSOD messenger RNA, using small interfering RNA (siRNA), amplified apoptosis of melanoma cells induced by the NSAID diclofenac [16]. This same study also demonstrated that diclofenac-induced accumulation of ROS, depletion of MnSOD expression and activity, and apoptosis were specific to melanoma cells.
In light of all these lines of evidence compiled through complementary study of both mammalian and yeast cell models, the induction of oxidative stress and redox imbalance induced by NSAIDs, together with the targeted modulation of mitochondrial MnSOD, merits serious consideration for future investigations.
Acknowledgments
Work in the authors' laboratory is partly funded by the Malta Government Scholarship Scheme (MGSS) Award, fund no. ME 367/07/8, to Gianluca Farrugia and partly by the Research Fund grants to Rena Balzan from the University of Malta.
References
- 1.Baron JA, Sandler RS. Nonsteroidal. anti-inflammatory drugs and cancer prevention. Annual Review of Medicine. 2000;51:511–523. doi: 10.1146/annurev.med.51.1.511. [DOI] [PubMed] [Google Scholar]
- 2.Chan AT, Arber N, Burn J, et al. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prevention Research. 2012;5(2):164–178. doi: 10.1158/1940-6207.CAPR-11-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothwell PM, Wilson M, Elwin C-E, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. The Lancet. 2010;376(9754):1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 4.Rothwell PM, Fowkes FGR, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. The Lancet. 2011;377(9759):31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 5.Trujillo MA, Garewal HS, Sampliner RE. Nonsteroidal antiinflammatory agents in chemoprevention of colorectal cancer: at what cost? Digestive Diseases and Sciences. 1994;39(10):2260–2266. doi: 10.1007/BF02090382. [DOI] [PubMed] [Google Scholar]
- 6.Shiff SJ, Qiao L, Tsai L-L, Rigas B. Sulindac sulfide, an aspirin-like compound, inhibits proliferation, causes cell cycle quiescence, and induces apoptosis in HT-29 colon adenocarcinoma cells. Journal of Clinical Investigation. 1995;96(1):491–503. doi: 10.1172/JCI118060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan TA, Morin PJ, Vogelstein B, Kinzler KW. Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(2):681–686. doi: 10.1073/pnas.95.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahman MA, Dhar DK, Masunaga R, Yamanoi A, Kohno H, Nagasue N. Sulindac and exisulind exhibit a significant antiproliferative effect and induce apoptosis in human hepatocellular carcinoma cell lines. Cancer Research. 2000;60(8):2085–2089. [PubMed] [Google Scholar]
- 9.Pollard M, Luckert PH. Prolonged antitumor effect of indomethacin on autochthonous intestinal tumors in rats. Journal of the National Cancer Institute. 1983;70(6):1103–1105. [PubMed] [Google Scholar]
- 10.Chiu CH, McEntee MF, Whelan J. Discordant effect of aspirin and indomethacin on intestinal tumor burden in Apc(Min/+) mice. Prostaglandins Leukotrienes and Essential Fatty Acids. 2000;62(5):269–275. doi: 10.1054/plef.2000.0154. [DOI] [PubMed] [Google Scholar]
- 11.Andrews J, Djakiew D, Krygier S, Andrews P. Superior effectiveness of ibuprofen compared with other NSAIDs for reducing the survival of human prostate cancer cells. Cancer Chemotherapy and Pharmacology. 2002;50(4):277–284. doi: 10.1007/s00280-002-0485-8. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y-S, Zhu S, Li X-W, et al. Association between NSAIDs use and breast cancer risk: a systematic review and meta-analysis. Breast Cancer Research and Treatment. 2009;117(1):141–150. doi: 10.1007/s10549-008-0228-6. [DOI] [PubMed] [Google Scholar]
- 13.Motawi TM, Bustanji Y, El-Maraghy S, Taha MO, Al-Ghussein MA. Evaluation of naproxen and cromolyn activities against cancer cells viability, proliferation, apoptosis, p53 and gene expression of survivin and caspase-3. Journal of Enzyme Inhibition and Medicinal Chemistry. 2013 doi: 10.3109/14756366.2012.762645. [DOI] [PubMed] [Google Scholar]
- 14.Johnsen JI, Lindskog M, Ponthan F, et al. Cyclooxygenase-2 is expressed in neuroblastoma, and nonsteroidal anti-inflammatory drugs induce apoptosis and inhibit tumor growth in vivo. Cancer Research. 2004;64(20):7210–7215. doi: 10.1158/0008-5472.CAN-04-1795. [DOI] [PubMed] [Google Scholar]
- 15.Cecere F, Iuliano A, Albano F, et al. Diclofenac-induced apoptosis in the neuroblastoma cell line SH-SY5Y: possible involvement of the mitochondrial superoxide dismutase. Journal of Biomedicine and Biotechnology. 2010;2010:11 pages. doi: 10.1155/2010/801726.801726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albano F, Arcucci A, Granato G, et al. Markers of mitochondrial dysfunction during the diclofenac-induced apoptosis in melanoma cell lines. Biochimie. 2013;95:934–945. doi: 10.1016/j.biochi.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Williams JL, Borgo S, Hasan I, Castillo E, Traganos F, Rigas B. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: implications for colon cancer chemoprevention. Cancer Research. 2001;61(8):3285–3289. [PubMed] [Google Scholar]
- 18.Kashfi K, Ryann Y, Qiao LL, et al. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect. Journal of Pharmacology and Experimental Therapeutics. 2002;303(3):1273–1282. doi: 10.1124/jpet.102.042754. [DOI] [PubMed] [Google Scholar]
- 19.Zhao W, Mackenzie GG, Murray OT, Zhang Z, Rigas B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: a redox-dependent effect. Carcinogenesis. 2009;30(3):512–519. doi: 10.1093/carcin/bgp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang L, Zhu C, Sun Y, et al. Phospho-sulindac (OXT-922) inhibits the growth of human colon cancer cell lines: a redox/polyamine-dependent effect. Carcinogenesis. 2010;31(11):1982–1990. doi: 10.1093/carcin/bgq149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang L, Mackenzie GG, Sun Y, et al. Chemotherapeutic properties of phospho-nonsteroidal anti-inflammatory drugs, a new class of anticancer compounds. Cancer Research. 2011;71(24):7617–7627. doi: 10.1158/0008-5472.CAN-11-2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chattopadhyay M, Kodela R, Nath N, et al. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect. Biochemical Pharmacology. 2012;83(6):715–722. doi: 10.1016/j.bcp.2011.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Kodela R, Chattopadhyay M, Kashfi K. NOSH-aspirin: a novel nitric oxide-hydrogen sulfide-releasing hybrid: a new class of anti-inflammatory pharmaceuticals. ACS Medicinal Chemistry Letters. 2012;3(3):257–262. doi: 10.1021/ml300002m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chattopadhyay M, Kodela R, Olson KR, Kashfi K. NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochemical and Biophysical Research Communications. 2012;419(3):523–528. doi: 10.1016/j.bbrc.2012.02.051. [DOI] [PubMed] [Google Scholar]
- 25.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy BS, Rao CV, Rivenson A, Kelloff G. Inhibitory effect of aspirin on azoxymethane-induced colon carcinogenesis in F344 rats. Carcinogenesis. 1993;14(8):1493–1497. doi: 10.1093/carcin/14.8.1493. [DOI] [PubMed] [Google Scholar]
- 27.Qiao L, Hanif R, Sphicas E, Shiff SJ, Rigas B. Effect of aspirin on induction of apoptosis in HT-29 human colon adenocarcinoma cells. Biochemical Pharmacology. 1998;55(1):53–64. doi: 10.1016/s0006-2952(97)00400-0. [DOI] [PubMed] [Google Scholar]
- 28.Yu H-G, Huang J-A, Yang Y-N, et al. The effects of acetylsalicylic acid on proliferation, apoptosis, and invasion of cyclooxygenase-2 negative colon cancer cells. European Journal of Clinical Investigation. 2002;32(11):838–846. doi: 10.1046/j.1365-2362.2002.01080.x. [DOI] [PubMed] [Google Scholar]
- 29.Gao J, Niwa K, Sun W, et al. Non-steroidal anti-inflammatory drugs inhibit cellular proliferation and upregulate cyclooxygenase-2 protein expression in endometrial cancer cells. Cancer Science. 2004;95(11):901–907. doi: 10.1111/j.1349-7006.2004.tb02200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carmona-Gutierrez D, Eisenberg T, Büttner S, Meisinger C, Kroemer G, Madeo F. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death and Differentiation. 2010;17(5):763–773. doi: 10.1038/cdd.2009.219. [DOI] [PubMed] [Google Scholar]
- 31.Almeida B, Silva A, Mesquita A, Sampaio-Marques B, Rodrigues F, Ludovico P. Drug-induced apoptosis in yeast. Biochimica et Biophysica Acta. 2008;1783(7):1436–1448. doi: 10.1016/j.bbamcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Balzan R, Sapienza K, Galea DR, Vassallo N, Frey H, Bannister WH. Aspirin commits yeast cells to apoptosis depending on carbon source. Microbiology. 2004;150(1):109–115. doi: 10.1099/mic.0.26578-0. [DOI] [PubMed] [Google Scholar]
- 33.Van Leeuwen JS, Orij R, Luttik MAH, Smits GJ, Vermeulen NPE, Vos JC. Subunits Rip1p and Cox9p of the respiratory chain contribute to diclofenac-induced mitochondrial dysfunction. Microbiology. 2011;157(3):685–694. doi: 10.1099/mic.0.044578-0. [DOI] [PubMed] [Google Scholar]
- 34.Castaño E, Dalmau M, Barragán M, Pueyo G, Bartrons R, Gil J. Aspirin induces cell death and caspase-dependent phosphatidylserine externalization in HT-29 human colon adenocarcinoma cells. British Journal of Cancer. 1999;81(2):294–299. doi: 10.1038/sj.bjc.6690690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turchanowa L, Dauletbaev N, Milovic V, Stein J. Nonsteroidal anti-inflammatory drugs stimulate spermidine/spermine acetyltransferase and deplete polyamine content in colon cancer cells. European Journal of Clinical Investigation. 2001;31(10):887–893. doi: 10.1046/j.1365-2362.2001.00901.x. [DOI] [PubMed] [Google Scholar]
- 36.Stark LA, Din FV, Zwacka RM, Dunlop MG. Aspirin-induced activation of the NF-kappaB signaling pathway: a novel mechanism for aspirin-mediated apoptosis in colon cancer cells. The FASEB Journal. 2001;15(7):1273–1275. [PubMed] [Google Scholar]
- 37.Bellosillo B, Piqué M, Barragán M, et al. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood. 1998;92(4):1406–1414. [PubMed] [Google Scholar]
- 38.Piqué M, Barragán M, Dalmau M, Bellosillo B, Pons G, Gil J. Aspirin induces apoptosis through mitochondrial cytochrome c release. FEBS Letters. 2000;480(2-3):193–196. doi: 10.1016/s0014-5793(00)01922-0. [DOI] [PubMed] [Google Scholar]
- 39.Zimmermann KC, Waterhouse NJ, Goldstein JC, Schuler M, Green DR. Aspirin induces apoptosis through release of cytochrome c from mitochondria. Neoplasia. 2000;2(6):505–513. doi: 10.1038/sj.neo.7900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Im R, Jang YJ. Aspirin enhances TRAIL-induced apoptosis via regulation of ERK1/2 activation in human cervical cancer cells. Biochemical and Biophysical Research Communications. 2012;424:65–70. doi: 10.1016/j.bbrc.2012.06.067. [DOI] [PubMed] [Google Scholar]
- 41.Power JJ, Dennis MS, Redlak MJ, Miller TA. Aspirin-induced mucosal cell death in human gastric cells: evidence supporting an apoptotic mechanism. Digestive Diseases and Sciences. 2004;49(9):1518–1525. doi: 10.1023/b:ddas.0000042258.41480.30. [DOI] [PubMed] [Google Scholar]
- 42.Gu Q, De Wang J, Xia HHX, et al. Activation of the caspase-8/Bid and Bax pathways in aspirin-induced apoptosis in gastric cancer. Carcinogenesis. 2005;26(3):541–546. doi: 10.1093/carcin/bgh345. [DOI] [PubMed] [Google Scholar]
- 43.Redlak MJ, Power JJ, Miller TA. Role of mitochondria in aspirin-induced apoptosis in human gastric epithelial cells. American Journal of Physiology. 2005;289(4):G731–G738. doi: 10.1152/ajpgi.00150.2005. [DOI] [PubMed] [Google Scholar]
- 44.Zhou XM, Wong BCY, Fan XM, et al. Non-steroidal anti-inflammatory drugs induce apoptosis in gastric cancer cells through up-regulation of bax and bak. Carcinogenesis. 2001;22(9):1393–1397. doi: 10.1093/carcin/22.9.1393. [DOI] [PubMed] [Google Scholar]
- 45.Raza H, John A, Benedict S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. European Journal of Pharmacology. 2011;668(1-2):15–24. doi: 10.1016/j.ejphar.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 46.Hossain MA, Kim DH, Jang JY, et al. Aspirin induces apoptosis in vitro and inhibits tumor growth of human hepatocellular carcinoma cells in a nude mouse xenograft model. International Journal of Oncology. 2012;40(4):1298–1304. doi: 10.3892/ijo.2011.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dikshit P, Chatterjee M, Goswami A, Mishra A, Jana NR. Aspirin induces apoptosis through the inhibition of proteasome function. Journal of Biological Chemistry. 2006;281(39):29228–29235. doi: 10.1074/jbc.M602629200. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y, He Q, Hillman MJ, Rong R, Sheikh MS. Sulindac sulfide-induced apoptosis involves death receptor 5 and the caspase 8-dependent pathway in human colon and prostate cancer cells. Cancer Research. 2001;61(18):6918–6924. [PubMed] [Google Scholar]
- 49.Babbar N, Ignatenko NA, Casero RA, Jr., Gerner EW. Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. Journal of Biological Chemistry. 2003;278(48):47762–47775. doi: 10.1074/jbc.M307265200. [DOI] [PubMed] [Google Scholar]
- 50.Oida Y, Gopalan B, Miyahara R, et al. Sulindac enhances adenoviral vector expressing mda-7/IL-24-mediated apoptosis in human lung cancer. Molecular Cancer Therapeutics. 2005;4(2):291–304. [PubMed] [Google Scholar]
- 51.Greenspan EJ, Madigan JP, Boardman LA, Rosenberg DW. Ibuprofen inhibits activation of nuclear β-catenin in human colon adenomas and induces the phosphorylation of GSK-3β . Cancer Prevention Research. 2011;4(1):161–171. doi: 10.1158/1940-6207.CAPR-10-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sankpal UT, Abdelrahim M, Connelly SF, et al. Small molecule tolfenamic acid inhibits PC-3 cell proliferation and invasion in vitro, and tumor growth in orthotopic mouse model for prostate cancer. Prostate. 2012;72:1648–1658. doi: 10.1002/pros.22518. [DOI] [PubMed] [Google Scholar]
- 53.Zhou H, Huang L, Sun Y, Rigas B. Nitric oxide-donating aspirin inhibits the growth of pancreatic cancer cells through redox-dependent signaling. Cancer Letters. 2009;273(2):292–299. doi: 10.1016/j.canlet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams JL, Nath N, Chen J, et al. Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling, nuclear factor-kappaB, and NO synthase 2 inhibition: Implications for chemoprevention. Cancer Research. 2003;63(22):7613–7618. [PubMed] [Google Scholar]
- 55.Williams JI, Ji P, Ouyang N, Liu X, Rigas B. NO-donating aspirin inhibits the activation of NF-κB in human cancer cell lines and Min mice. Carcinogenesis. 2008;29(2):390–397. doi: 10.1093/carcin/bgm275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Y, Rigas B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: implications for the mechanism of action of anticancer agents. Cancer Research. 2008;68(20):8269–8277. doi: 10.1158/0008-5472.CAN-08-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chattopadhyay M, Goswami S, Rodes DB, et al. NO-releasing NSAIDs suppress NF-κB signaling in vitro and in vivo through S-nitrosylation. Cancer Letters. 2010;298(2):204–211. doi: 10.1016/j.canlet.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 58.Gao J, Liu X, Rigas B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(47):17207–17212. doi: 10.1073/pnas.0506893102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chaudhary C, Singh T, Kapur P, et al. Nitric oxide-releasing sulindac is a novel skin cancer chemopreventive agent for UVB-induced photocarcinogenesis. Toxicology and Applied Pharmacology. 2013;268:249–255. doi: 10.1016/j.taap.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, Kashfi K. Hydrogen sulfide-releasing aspirin suppresses NF-κB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo. Biochemical Pharmacology. 2012;83(6):723–732. doi: 10.1016/j.bcp.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 61.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature: New biology. 1971;231(25):232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 62.Vane JR, Mitchell JA, Appleton I, et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(6):2046–2050. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clinical Cancer Research. 2000;6(1):135–138. [PubMed] [Google Scholar]
- 64.Zhang L, Wu Y-D, Li P, et al. Effects of cyclooxygenase-2 on human esophageal squamous cell carcinoma. World Journal of Gastroenterology. 2011;17(41):4572–4580. doi: 10.3748/wjg.v17.i41.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, Dubois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107(4):1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 66.Sano H, Kawahito Y, Wilder RL, et al. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Research. 1995;55(17):3785–3789. [PubMed] [Google Scholar]
- 67.Kutchera W, Jones DA, Matsunami N, et al. Prostaglandin H synthase 2 is expressed abnormally in human colon cancer: evidence for a transcriptional effect. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(10):4816–4820. doi: 10.1073/pnas.93.10.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lupulescu A. Prostaglandins, their inhibitors and cancer. Prostaglandins Leukotrienes and Essential Fatty Acids. 1996;54(2):83–94. doi: 10.1016/s0952-3278(96)90064-2. [DOI] [PubMed] [Google Scholar]
- 69.Levy GN. Prostaglandin H synthases, nonsteroidal anti-inflammatory drugs, and colon cancer. FASEB Journal. 1997;11(4):234–247. [PubMed] [Google Scholar]
- 70.Shiff SJ, Rigas B. Nonsteroidal anti-inflammatory drugs and colorectal cancer: evolving concepts of their chemopreventive actions. Gastroenterology. 1997;113(6):1992–1998. doi: 10.1016/s0016-5085(97)99999-6. [DOI] [PubMed] [Google Scholar]
- 71.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55(1):115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oshima M, Dinchuk JE, Kargman SL, et al. Suppression of intestinal polyposis in Apc(Δ716) knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87(5):803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 73.Chulada PC, Thompson MB, Mahler JF, et al. Genetic disruption of Ptgs-1, as well as of Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Research. 2000;60(17):4705–4708. [PubMed] [Google Scholar]
- 74.Reddy BS, Rao CV, Seibert K. Evaluation of cyclooxygenase-2 inhibitor for potential chemopreventive properties in colon carcinogenesis. Cancer Research. 1996;56(20):4566–4569. [PubMed] [Google Scholar]
- 75.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. New England Journal of Medicine. 2006;355(9):885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 76.Baron JA, Sandler RS, Bresalier RS, et al. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. Gastroenterology. 2006;131(6):1674–1682. doi: 10.1053/j.gastro.2006.08.079. [DOI] [PubMed] [Google Scholar]
- 77.Dai Y, Wang W-H. Non-steroidal anti-inflammatory drugs in prevention gastric cancer. World Journal of Gastroenterology. 2006;12(19):2884–2889. doi: 10.3748/wjg.v12.i18.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jana NR. NSAIDs and apoptosis. Cellular and Molecular Life Sciences. 2008;65(9):1295–1301. doi: 10.1007/s00018-008-7511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sapienza K, Balzan R. Aspirin and apoptosis. In: Millwood CL, editor. New Research on Aspirin and Health. New York, NY, USA: Nova Science Publishers; 2007. [Google Scholar]
- 80.Chiu C-H, McEntee MF, Whelan J. Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Research. 1997;57(19):4267–4273. [PubMed] [Google Scholar]
- 81.Hanif R, Pittas A, Feng Y, et al. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochemical Pharmacology. 1996;52(2):237–245. doi: 10.1016/0006-2952(96)00181-5. [DOI] [PubMed] [Google Scholar]
- 82.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274(5294):1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 83.Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(21):11280–11285. doi: 10.1073/pnas.200367597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-α apoptotic signaling. Journal of Biological Chemistry. 2001;276(15):12035–12040. doi: 10.1074/jbc.M010603200. [DOI] [PubMed] [Google Scholar]
- 85.Zhang X, Morham SG, Langenbach R, Young DA. Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. Journal of Experimental Medicine. 1999;190(4):451–459. doi: 10.1084/jem.190.4.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Elder DJE, Halton DE, Hague A, Paraskeva C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clinical Cancer Research. 1997;3(10):1679–1683. [PubMed] [Google Scholar]
- 87.Thompson HJ, Briggs S, Paranka NS, et al. Inhibition of mammary carcinogenesis in rats by sulfone metabolite of sulindac. Journal of the National Cancer Institute. 1995;87(16):1259–1260. [PubMed] [Google Scholar]
- 88.Piazza GA, Rahm ALK, Krutzsch M, et al. Antineoplastic drugs sulindac sulfide and sulfone inhibit cell growth by inducing apoptosis. Cancer Research. 1995;55(14):3110–3116. [PubMed] [Google Scholar]
- 89.Charalambous D, O’Brien PE. Inhibition of colon cancer precursors in the rat by sulindac sulphone is not dependent on inhibition of prostaglandin synthesis. Journal of Gastroenterology and Hepatology. 1996;11(4):307–310. doi: 10.1111/j.1440-1746.1996.tb01376.x. [DOI] [PubMed] [Google Scholar]
- 90.Piazza GA, Alberts DS, Hixson LJ, et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Research. 1997;57(14):2909–2915. [PubMed] [Google Scholar]
- 91.Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S. Aspirin and sodium salicylate inhibit proliferation and induce apoptosis in rheumatoid synovial cells. Journal of Pharmacy and Pharmacology. 2002;54(12):1675–1679. doi: 10.1211/002235702261. [DOI] [PubMed] [Google Scholar]
- 92.Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. Journal of Biological Chemistry. 1999;274(29):20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- 93.Sapienza K, Bannister W, Balzan R. Mitochondrial involvement in aspirin-induced apoptosis in yeast. Microbiology. 2008;154(9):2740–2747. doi: 10.1099/mic.0.2008/017228-0. [DOI] [PubMed] [Google Scholar]
- 94.Li H, Zhu H, Xu C-J, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 95.Kruidering M, Evan GI. Caspase-8 in apoptosis: the beginning of “the end”? IUBMB Life. 2000;50(2):85–90. doi: 10.1080/713803693. [DOI] [PubMed] [Google Scholar]
- 96.Roucou X, Montessuit S, Antonsson B, Martinou J-C. Bax oligomerization in mitochondrial membranes requires tBid (caspase-8-cleaved Bid) and a mitochondrial protein. Biochemical Journal. 2002;368(3):915–921. doi: 10.1042/BJ20020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nath N, Labaze G, Rigas B, Kashfi K. NO-donating aspirin inhibits the growth of leukemic Jurkat cells and modulates β-catenin expression. Biochemical and Biophysical Research Communications. 2005;326(1):93–99. doi: 10.1016/j.bbrc.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 98.Bank A, Yu J, Zhang L. NSAIDs downregulate Bcl-XL and dissociate BAX and Bcl-X L to induce apoptosis in colon cancer cells. Nutrition and Cancer. 2008;60(1):98–103. doi: 10.1080/01635580802381261. [DOI] [PubMed] [Google Scholar]
- 99.Petrescu I, Tarba C. Uncoupling effects of diclofenac and aspirin in the perfused liver and isolated hepatic mitochondria of rat. Biochimica et Biophysica Acta-Bioenergetics. 1997;1318(3):385–394. doi: 10.1016/s0005-2728(96)00109-0. [DOI] [PubMed] [Google Scholar]
- 100.Uyemura SA, Santos AC, Mingatto FE, Jordani MC, Curti C. Diclofenac sodium and mefenamic acid: potent inducers of the membrane permeability transition in renal cortex mitochondria. Archives of Biochemistry and Biophysics. 1997;342(2):231–235. doi: 10.1006/abbi.1997.9985. [DOI] [PubMed] [Google Scholar]
- 101.Oh K-W, Qian T, Brenner DA, Lemasters JJ. Salicylate enhances necrosis and apoptosis mediated by the mitochondrial permeability transition. Toxicological Sciences. 2003;73(1):44–52. doi: 10.1093/toxsci/kfg045. [DOI] [PubMed] [Google Scholar]
- 102.Pegg AE. Polyamine metabolism and its importance in neoplastic growth and as a target for chemotherapy. Cancer Research. 1988;48(4):759–774. [PubMed] [Google Scholar]
- 103.Russell D, Snyder SH. Amine synthesis in rapidly growing tissues: ornithine decarboxylase activity in regenerating rat liver, chick embryo, and various tumors. Proceedings of the National Academy of Sciences of the United States of America. 1968;60(4):1420–1427. doi: 10.1073/pnas.60.4.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meyskens FL, Jr., Gerner EW. Development of difluoromethylornithine as a chemoprevention agent for the management of colon cancer. Journal of Cellular Biochemistry. 1995;58(22):126–131. doi: 10.1002/jcb.240590816. [DOI] [PubMed] [Google Scholar]
- 105.Scorcioni F, Corti A, Davalli P, Astancolle S, Bettuzzi S. Manipulation of the expression of regulatory genes of polyamine metabolism results in specific alterations of the cell-cycle progression. Biochemical Journal. 2001;354(1):217–223. doi: 10.1042/0264-6021:3540217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li L, Rao JN, Bass BL, Wang J-Y. NF-κB activation and susceptibility to apoptosis after polyamine depletion in intestinal epithelial cells. American Journal of Physiology. 2001;280(5):G992–G1004. doi: 10.1152/ajpgi.2001.280.5.G992. [DOI] [PubMed] [Google Scholar]
- 107.Babbar N, Gerner EW, Casero RA., Jr. Induction of spermidine/spermine N1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells. Biochemical Journal. 2006;394(1):317–324. doi: 10.1042/BJ20051298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martínez ME, O’Brien TG, Fultz KE, et al. Pronounced reduction in adenoma recurrence associated with aspirin use and a polymorphism in the ornithine decarboxylase gene. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(13):7859–7864. doi: 10.1073/pnas.1332465100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Meyskens FL, Jr., McLaren CE, Pelot D, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prevention Research. 2008;1(1):32–38. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mackenzie GG, Ouyang N, Xie G, et al. Phospho-sulindac (OXT-328) combined with difluoromethylornithine prevents colon cancer in mice. Cancer Prevention Research. 2011;4(7):1052–1060. doi: 10.1158/1940-6207.CAPR-11-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.MacKenzie GG, Sun Y, Huang L, et al. Phospho-sulindac (OXT-328), a novel sulindac derivative, is safe and effective in colon cancer prevention in mice. Gastroenterology. 2010;139(4):1320–1332. doi: 10.1053/j.gastro.2010.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baeuerle PA, Baltimore D. Nf-κB: ten years after. Cell. 1996;87(1):13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 113.Karin M. The beginning of the end: IκB kinase (IKK) and NF-κB activation. Journal of Biological Chemistry. 1999;274(39):27339–27342. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 114.Yin M-J, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IκB kinase-β . Nature. 1998;396(6706):77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 115.Algül H, Adler G, Schmid RM. NF-kappaB/Rel transcriptional pathway: implications in pancreatic cancer. International Journal of Gastrointestinal Cancer. 2002;31:71–78. doi: 10.1385/IJGC:31:1-3:71. [DOI] [PubMed] [Google Scholar]
- 116.Ahmed KM, Cao N, Li JJ. HER-2 and NF-κB as the targets for therapy-resistant breast cancer. Anticancer Research. 2006;26(6 B):4235–4243. [PMC free article] [PubMed] [Google Scholar]
- 117.Setia S, Sanyal SN. Downregulation of NF-kappaB and PCNA in the regulatory pathways of apoptosis by cyclooxygenase-2 inhibitors in experimental lung cancer. Molecular and Cellular Biochemistry. 2012;369:75–86. doi: 10.1007/s11010-012-1370-3. [DOI] [PubMed] [Google Scholar]
- 118.de Groot DJA, de Vries EGE, Groen HJM, de Jong S. Non-steroidal anti-inflammatory drugs to potentiate chemotherapy effects: from lab to clinic. Critical Reviews in Oncology/Hematology. 2007;61(1):52–69. doi: 10.1016/j.critrevonc.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 119.Kopp E, Ghosh S. Inhibition of NF-κB by sodium salicylate and aspirin. Science. 1994;265(5174):956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 120.Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit IκB-α phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. Journal of Immunology. 1996;156(10):3961–3969. [PubMed] [Google Scholar]
- 121.Schwenger P, Alpert D, Skolnik EY, Vilček J. Activation of p38 mitogen-activated protein kinase by sodium salicylate leads to inhibition of tumor necrosis factor-induced IκBα phosphorylation and degradation. Molecular and Cellular Biology. 1998;18(1):78–84. doi: 10.1128/mcb.18.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamamoto Y, Yin M-J, Lin K-M, Gaynor RB. Sulindac inhibits activation of the NF-κB pathway. Journal of Biological Chemistry. 1999;274(38):27307–27314. doi: 10.1074/jbc.274.38.27307. [DOI] [PubMed] [Google Scholar]
- 123.Matthews JR, Wakasugi N, Virelizier J-L, Yodoi J, Hay RT. Thioredoxin regulates the DNA binding activity of N F-κB by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Research. 1992;20(15):3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Din FVN, Dunlop MG, Stark LA. Evidence for colorectal cancer cell specificity of aspirin effects on NFκB signalling and apoptosis. British Journal of Cancer. 2004;91(2):381–388. doi: 10.1038/sj.bjc.6601913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Borthwick GM, Johnson AS, Partington M, Burn J, Wilson R, Arthur HM. Therapeutic levels of aspirin and salicylate directly inhibit a model of angiogenesis through a Cox-independent mechanism. FASEB Journal. 2006;20(12):2009–2016. doi: 10.1096/fj.06-5987com. [DOI] [PubMed] [Google Scholar]
- 126.Stark LA, Reid K, Sansom OJ, et al. Aspirin activates the NF-κB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis. 2007;28(5):968–976. doi: 10.1093/carcin/bgl220. [DOI] [PubMed] [Google Scholar]
- 127.Cho M, Gwak J, Park S, et al. Diclofenac attenuates Wnt/β-catenin signaling in colon cancer cells by activation of NF-κB. FEBS Letters. 2005;579(20):4213–4218. doi: 10.1016/j.febslet.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 128.Sheppard K-A, Rose DW, Haque ZK, et al. Transcriptional activation by NF-κB requires multiple coactivators. Molecular and Cellular Biology. 1999;19(9):6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. Journal of Biological Chemistry. 1995;270(25):14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 130.Schwenger P, Bellosta P, Vietor I, Basilico C, Skolnik EY, Vilček J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):2869–2873. doi: 10.1073/pnas.94.7.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schwenger P, Skolnik EY, Vilček J. Inhibition of tumor necrosis factor-induced p42/p44 mitogen-activated protein kinase activation by sodium salicylate. Journal of Biological Chemistry. 1996;271(14):8089–8094. doi: 10.1074/jbc.271.14.8089. [DOI] [PubMed] [Google Scholar]
- 132.Jones MK, Wang H, Peskar BM, et al. Inhibition of angiogenesis by nonsteroidal anti-inflammatory drugs: insight into mechanisms and implications for cancer growth and ulcer healing. Nature Medicine. 1999;5(12):1418–1423. doi: 10.1038/70995. [DOI] [PubMed] [Google Scholar]
- 133.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 134.Dong Z, Huang C, Brown RE, Ma W-Y. Inhibition of activator protein 1 activity and neoplastic transformation by aspirin. Journal of Biological Chemistry. 1997;272(15):9962–9970. doi: 10.1074/jbc.272.15.9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hundley TR, Rigas B. Nitric oxide-donating aspirin inhibits colon cancer cell growth via mitogen-activated protein kinase activation. Journal of Pharmacology and Experimental Therapeutics. 2006;316(1):25–34. doi: 10.1124/jpet.105.091363. [DOI] [PubMed] [Google Scholar]
- 136.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Reports. 2002;3(5):420–425. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through β-catenin. Science. 2002;296(5573):1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- 138.Hao XP, Pretlow TG, Rao JS, Pretlow TP. β-catenin expression is altered in human colonic aberrant crypt foci. Cancer Research. 2001;61(22):8085–8088. [PubMed] [Google Scholar]
- 139.Chiang JM, Wu Chou YH, Chen TC, Ng KF, Lin JL. Nuclear β-catenin expression is closely related to ulcerative growth of colorectal carcinoma. British Journal of Cancer. 2002;86(7):1124–1129. doi: 10.1038/sj.bjc.6600214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chung EJ, Hwang S-G, Nguyen P, et al. Regulation of leukemic cell adhesion, proliferation, and survival by β-catenin. Blood. 2002;100(3):982–990. doi: 10.1182/blood.v100.3.982. [DOI] [PubMed] [Google Scholar]
- 141.Lu D, Zhao Y, Tawatao R, et al. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):3118–3123. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lu W, Tinsley HN, Keeton A, Qu Z, Piazza GA, Li Y. Suppression of Wnt/β-catenin signaling inhibits prostate cancer cell proliferation. European Journal of Pharmacology. 2009;602(1):8–14. doi: 10.1016/j.ejphar.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Current Opinion in Cell Biology. 2005;17(5):499–508. doi: 10.1016/j.ceb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 144.Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(10):5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits β-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear β-catenin-TCF association. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(22):12584–12589. doi: 10.1073/pnas.2134840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dihlmann S, Siermann A, Von Knebel Doeberitz M. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate β-catenin/TCF-4 signaling. Oncogene. 2001;20(5):645–653. doi: 10.1038/sj.onc.1204123. [DOI] [PubMed] [Google Scholar]
- 147.Dihlmann S, Klein S, Doeberitz Mv MVK. Reduction of beta-catenin/T-cell transcription factor signaling by aspirin and indomethacin is caused by an increased stabilization of phosphorylated beta-catenin. Molecular cancer therapeutics. 2003;2(6):509–516. [PubMed] [Google Scholar]
- 148.Bos CL, Kodach LL, Van Den Brink GR, et al. Effect of aspirin on the Wnt/β-catenin pathway is mediated via protein phosphatase 2A. Oncogene. 2006;25(49):6447–6456. doi: 10.1038/sj.onc.1209658. [DOI] [PubMed] [Google Scholar]
- 149.Pathi S, Jutooru I, Chadalapaka G, Nair V, Lee SO, Safe S. Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (sp) transcription factors. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0048208.e48208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rice PL, Kelloff J, Sullivan H, et al. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Molecular cancer therapeutics. 2003;2(9):885–892. [PubMed] [Google Scholar]
- 151.Wang Y, Chen X, Zhu W, Zhang H, Hu S, Cong X. Growth inhibition of mesenchymal stem cells by aspirin: involvement of the wnt/β-catenin signal pathway. Clinical and Experimental Pharmacology and Physiology. 2006;33(8):696–701. doi: 10.1111/j.1440-1681.2006.04432.x. [DOI] [PubMed] [Google Scholar]
- 152.Li H, Liu L, David ML, et al. Pro-apoptotic actions of exisulind and CP461 in SW480 colon tumor cells involve β-catenin and cyclin D1 down-regulation. Biochemical Pharmacology. 2002;64(9):1325–1336. doi: 10.1016/s0006-2952(02)01345-x. [DOI] [PubMed] [Google Scholar]
- 153.Deng L, Hu S, Baydoun AR, Chen J, Chen X, Cong X. Aspirin induces apoptosis in mesenchymal stem cells requiring Wnt/β-catenin pathway. Cell Proliferation. 2009;42(6):721–730. doi: 10.1111/j.1365-2184.2009.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rao CV, Reddy BS, Steele VE, et al. Nitric oxide-releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Molecular Cancer Therapeutics. 2006;5(6):1530–1538. doi: 10.1158/1535-7163.MCT-06-0061. [DOI] [PubMed] [Google Scholar]
- 155.Nath N, Vassell R, Chattopadhyay M, Kogan M, Kashfi K. Nitro-aspirin inhibits MCF-7 breast cancer cell growth: effects on COX-2 expression and Wnt/β-catenin/TCF-4 signaling. Biochemical Pharmacology. 2009;78(10):1298–1304. doi: 10.1016/j.bcp.2009.06.104. [DOI] [PubMed] [Google Scholar]
- 156.Zhu C, Cheng KW, Ouyang N, et al. Phosphosulindac (OXT-328) selectively targets breast cancer stem cells in vitro and in human breast cancer xenografts. Stem Cells. 2012;30:2065–2075. doi: 10.1002/stem.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Adachi M, Sakamoto H, Kawamura R, Wang W, Imai K, Shinomura Y. Nonsteroidal anti-inflammatory drugs and oxidative stress in cancer cells. Histology and Histopathology. 2007;22(4–6):437–442. doi: 10.14670/HH-22.437. [DOI] [PubMed] [Google Scholar]
- 158.Rigas B. Novel agents for cancer prevention based on nitric oxide. Biochemical Society Transactions. 2007;35(5):1364–1368. doi: 10.1042/BST0351364. [DOI] [PubMed] [Google Scholar]
- 159.Fernandes E, Toste SA, Lima JLFC, Reis S. The metabolism of sulindac enhances its scavenging activity against reactive oxygen and nitrogen species. Free Radical Biology and Medicine. 2003;35(9):1008–1017. doi: 10.1016/s0891-5849(03)00437-4. [DOI] [PubMed] [Google Scholar]
- 160.Costa D, Gomes A, Reis S, Lima JLFC, Fernandes E. Hydrogen peroxide scavenging activity by non-steroidal anti-inflammatory drugs. Life Sciences. 2005;76(24):2841–2848. doi: 10.1016/j.lfs.2004.10.052. [DOI] [PubMed] [Google Scholar]
- 161.El Midaoui A, Wu R, De Champlain J. Prevention of hypertension, hyperglycemia and vascular oxidative stress by aspirin treatment in chronically glucose-fed rats. Journal of Hypertension. 2002;20(7):1407–1412. doi: 10.1097/00004872-200207000-00028. [DOI] [PubMed] [Google Scholar]
- 162.Ishizuka T, Niwa A, Tabuchi M, Ooshima K, Higashino H. Acetylsalicylic acid provides cerebrovascular protection from oxidant damage in salt-loaded stroke-prone rats. Life Sciences. 2008;82(13-14):806–815. doi: 10.1016/j.lfs.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 163.Kumari SS, Varghese A, Muraleedharan D, Menon VP. Protective action of aspirin in experimental myocardial infarction induced by isoproterenol in rats and its effect on lipid peroxidation. Indian Journal of Experimental Biology. 1990;28(5):480–485. [PubMed] [Google Scholar]
- 164.Qiu L-Y, Yu J, Zhou Y, Chen C-H. Protective effects and mechanism of action of aspirin on focal cerebral ischemia-reperfusion in rats. Yaoxue Xuebao. 2003;38(8):561–564. [PubMed] [Google Scholar]
- 165.Tauseef M, Sharma KK, Fahim M. Aspirin restores normal baroreflex function in hypercholesterolemic rats by its antioxidative action. European Journal of Pharmacology. 2007;556(1–3):136–143. doi: 10.1016/j.ejphar.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 166.Kanwar SS, Vaiphei K, Nehru B, Sanyal SN. Antioxidative effects of nonsteroidal anti-inflammatory drugs during the initiation stages of experimental colon carcinogenesis in rats. Journal of Environmental Pathology, Toxicology and Oncology. 2008;27(2):89–100. doi: 10.1615/jenvironpatholtoxicoloncol.v27.i2.20. [DOI] [PubMed] [Google Scholar]
- 167.Yiannakopoulou EC, Tiligada E. Protective effect of salicylates against hydrogen peroxide stress in yeast. Journal of Applied Microbiology. 2009;106(3):903–908. doi: 10.1111/j.1365-2672.2008.04061.x. [DOI] [PubMed] [Google Scholar]
- 168.Kusuhara H, Komatsu H, Sumichika H, Sugahara K. Reactive oxygen species are involved in the apoptosis induced by nonsteroidal anti-inflammatory drugs in cultured gastric cells. European Journal of Pharmacology. 1999;383(3):331–337. doi: 10.1016/s0014-2999(99)00599-3. [DOI] [PubMed] [Google Scholar]
- 169.Giardina C, Inan MS. Nonsteroidal anti-inflammatory drugs, short-chain fatty acids, and reactive oxygen metabolism in human colorectal cancer cells. Biochimica et Biophysica Acta. 1998;1401(3):277–288. doi: 10.1016/s0167-4889(97)00140-7. [DOI] [PubMed] [Google Scholar]
- 170.Chung YM, Bae YS, Lee SY. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radical Biology and Medicine. 2003;34(4):434–442. doi: 10.1016/s0891-5849(02)01301-1. [DOI] [PubMed] [Google Scholar]
- 171.Minami T, Adachi M, Kawamura R, Zhang Y, Shinomura Y, Imai K. Sulindac enhances the proteasome inhibitor bortezomib-mediated oxidative stress and anticancer activity. Clinical Cancer Research. 2005;11(14):5248–5256. doi: 10.1158/1078-0432.CCR-05-0085. [DOI] [PubMed] [Google Scholar]
- 172.Herrero E, Ros J, Bellí G, Cabiscol E. Redox control and oxidative stress in yeast cells. Biochimica et Biophysica Acta. 2008;1780(11):1217–1235. doi: 10.1016/j.bbagen.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 173.Battaglia V, Salvi M, Toninello A. Oxidative stress is responsible for mitochondrial permeability transition induction by salicylate in liver mitochondria. Journal of Biological Chemistry. 2005;280(40):33864–33872. doi: 10.1074/jbc.M502391200. [DOI] [PubMed] [Google Scholar]
- 174.Raza H, John A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0036325.e36325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sapienza K, Balzan R. Metabolic aspects of aspirin-induced apoptosis in yeast. FEMS Yeast Research. 2005;5(12):1207–1213. doi: 10.1016/j.femsyr.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 176.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Molecular Cancer Therapeutics. 2006;5(5):1087–1098. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 177.Lu M, Strohecker A, Chen F, et al. Aspirin sensitizes cancer cells to TRAIL-induced apoptosis by reducing survivin levels. Clinical Cancer Research. 2008;14(10):3168–3176. doi: 10.1158/1078-0432.CCR-07-4362. [DOI] [PubMed] [Google Scholar]
- 178.Wang M, Tan Z, Zhang R, Kotenko SV, Liang P. Interleukin 24 (MDA-7/MOB-5) signals through two heterodimeric receptors, IL-22R1/IL-20R2 and IL-20R1/IL-20R2. Journal of Biological Chemistry. 2002;277(9):7341–7347. doi: 10.1074/jbc.M106043200. [DOI] [PubMed] [Google Scholar]
- 179.Chada S, Sutton RB, Ekmekcioglu S, et al. MDA-7/IL-24 is a unique cytokine-tumor suppressor in the IL-10 Family. International Immunopharmacology. 2004;4(5):649–667. doi: 10.1016/j.intimp.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 180.Su Z-Z, Madireddi MT, Lin JJ, et al. The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(24):14400–14405. doi: 10.1073/pnas.95.24.14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jiang H, Su Z-Z, Jiao Jiao Lin JJL, Goldstein NI, Young CSH, Fisher PB. The melanoma differentiation associated gene mda-7 suppresses cancer cell growth. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(17):9160–9165. doi: 10.1073/pnas.93.17.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Saito Y, Miyahara R, Gopalan B, et al. Selective induction of cell cycle arrest and apoptosis in human prostate cancer cells through adenoviral transfer of the melanoma differentiation- associated-7 (mda-7)/interleukin-24 (IL-24) gene. Cancer Gene Therapy. 2005;12(3):238–247. doi: 10.1038/sj.cgt.7700780. [DOI] [PubMed] [Google Scholar]
- 183.Saeki T, Mhashilkar A, Swanson X, et al. Inhibition of human lung cancer growth following adenovirus-mediated mda-7 gene expression in vivo. Oncogene. 2002;21(29):4558–4566. doi: 10.1038/sj.onc.1205553. [DOI] [PubMed] [Google Scholar]
- 184.Sun Y, Chen J, Rigas B. Chemopreventive agents induce oxidative stress in cancer cells leading to COX-2 overexpression and COX-2-independent cell death. Carcinogenesis. 2009;30(1):93–100. doi: 10.1093/carcin/bgn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rigas B, Sun Y. Induction of oxidative stress as a mechanism of action of chemopreventive agents against cancer. British Journal of Cancer. 2008;98(7):1157–1160. doi: 10.1038/sj.bjc.6604225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Qin Y, Dai W, Wang Y, Gong XG, Lu M. Fe-SOD cooperates with Nutlin3 to selectively inhibit cancer cells in vitro and in vivo. Biochemical and Biophysical Research Communications. 2013;431:169–175. doi: 10.1016/j.bbrc.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 187.Ueta E, Yoneda K, Kimura T, et al. Mn-SOD antisense upregulates in vivo apoptosis of squamous cell carcinoma cells by anticancer drugs and γ-rays regulating expression of the BcL-2 family proteins, COX-2 and p21. International Journal of Cancer. 2001;94(4):545–550. doi: 10.1002/ijc.1513. [DOI] [PubMed] [Google Scholar]