

Abstract
Duchenne muscular dystrophy (DMD) is an incurable, X-linked progressive muscle degenerative disorder that results from the absence of dystrophin protein and leads to premature death in affected individuals due to respiratory and/or cardiac failure typically by age of 30. Very recently the exciting prospect of an effective oligonucleotide therapy has emerged which restores dystrophin protein expression to affected tissues in DMD patients with highly promising data from a series of clinical trials. This therapeutic approach is highly mutation specific and thus is personalised. Therefore DMD has emerged as a model genetic disorder for understanding and overcoming of the challenges of developing personalised genetic medicines. One of the greatest weaknesses of the current oligonucleotide approach is that it is a mutation-specific therapy. To address this limitation, we have recently demonstrated that exons 45–55 skipping therapy has the potential to treat clusters of mutations that cause DMD, which could significantly reduce the number of compounds that would need to be developed in order to successfully treat all DMD patients. Here we discuss and review the latest preclinical work in this area as well as a variety of accompanying issues, including efficacy and potential toxicity of antisense oligonucleotides, prior to human clinical trials.
1. Introduction
Muscular dystrophy is a group of genetic diseases characterized by generalized progressive muscle atrophy and weakness and histopathology that reveals degeneration and regeneration of muscle fibres. In particular, Duchenne muscular dystrophy (DMD), the most common form of muscular dystrophy, is caused by lack of dystrophin protein [1]. In recent years, a most promising therapeutic approach to restoring dystrophin is antisense-oligonucleotide- (AO-) based exon skipping therapy that targets splicing or other elements in the dystrophin pre-mRNA [2]. This approach has been shown to be practical for up to 90% of DMD patients with deletion mutations, while read-through therapy with PTC124 is applicable for up to 10% of DMD patients with nonsense mutations [3, 4]. The principle underlying exon-skipping therapy for DMD has been demonstrated in cultured mouse, canine or human cells in vitro [5–11]. In addition, in vivo studies in murine or canine animal models have provided preclinical evidence for the therapeutic potential of AO-based exon-skipping strategies for DMD [12–23]. Finally, this therapeutic strategy has demonstrated efficacy in a number of recent phase I/II exon skipping trials which target exon 51 of the DMD gene [24, 25]. Currently phases II b and III exon skipping trials are ongoing by Sarepta Therapeutics and Prosensa/GlaxoSmithKline (GSK) with two different AO chemistries, phosphorodiamidate morpholino oligomers (PMO) and 2′O-methylated phosphorothioates (2′OMePs), respectively (ClinicalTrials.gov identifier: NCT01396239 and NCT01254019). It appears highly likely that AO-based therapeutic agents will receive regulatory approval for use in DMD within the next few years. Although promising, this therapeutic approach is highly mutation specific and thus is personalised. Therefore DMD has emerged as a model genetic disorder for understanding and overcoming the challenges associated with developing personalised genetic medicines [2]. This review highlights novel findings from a DMD mouse model utilizing systemic multiexon skipping targeting exons 45–55, discusses the major hurdles and limitations impeding the translation of this approach into clinical therapies along with the potential solutions, and provides a perspective on the future of clinical exon 45–55 skipping in DMD patients.
2. Duchenne Muscular Dystrophy
DMD, an incurable, X-linked progressive muscle degenerative disorder, presents with walking difficulties around 3–5 years of age [26]. Skeletal muscle involvement is chronically progressive, resulting in wheelchair-bound patients by the age of 13 years and being bed-bound thereafter. DMD patients usually die around the age of 30 years, typically from respiratory or cardiac failure due to dilated cardiomyopathy. DMD is most frequently caused by a frame-shift, nonsense, or duplication mutation in the DMD gene, which encodes dystrophin [27]. Dystrophin protein is localized at the subsarcolemma of muscle fibres and forms a dystrophin glycoprotein complex (DGC) with dystroglycans, sarcoglycans, syntrophins/dystrobrevins, sarcospan, and neuronal nitric oxide synthase (nNOS) complexes [28]. The DGC provides a mechanical and signaling link between the actin cytoskeleton and the extracellular matrix [29]. The absence of dystrophin leads to recurrent muscle fibre damage during contraction, and muscle fibres are eventually replaced by adipose and fibrotic tissues. Interestingly, a related allelic disorder, Becker muscular dystrophy (BMD), can exhibit a much milder phenotype. Many people with BMD live into their 70s and 80s [30]. BMD typically results from shortened, but in-frame, transcripts of the DMD gene that allow for expression of limited amounts of an internally truncated, but partially functional, protein (the reading frame rule) [31]. Interestingly, the severity of BMD can vary considerably from almost asymptomatic to a slightly less severe DMD-like condition [32–34].
3. Antisense Oligonucleotides Frequently Used in Exon Skipping
The development of AOs for manipulating alternative splicing has led to studies that focused on the therapeutic potential of AO-based exon skipping therapy for DMD. AOs can be designed to hybridize to a specific mRNA target and mediate its destruction via Ribonuclease H (RNase H), an enzyme that destroys mRNA in a DNA/RNA complex. However, the requirements for AOs that alter splicing are different from AOs used to achieve gene downregulation [35]. In this context, AOs can be used to modulate the ratio of splicing variants or to correct splicing defects, which has greater implications for treating a variety of diseases. In particular, the AOs should not activate RNase H, which would destroy the pre-mRNA before it can be spliced. In addition, the AOs must access the target pre-mRNAs within the cell nuclei to efficiently compete with splicing factors. Several types of modified synthetic AOs fit these criteria: AOs with backbones based on PMO [24, 36] and with modifications to the 2′ position of RNA, such as 2′OMePs [25, 37], are RNase H inactive and display high nuclease resistance and an affinity for target sequences. Similar characteristics are found in AOs with backbones based on phosphorothioate (PS) [38–40], peptide nucleic acid (PNA) [41, 42], locked nucleic acid (LNA) [43], and tricyclo-DNA [44]. However, a number of limiting factors have slowed the progress of AO drugs in the clinical area, including off target effects and low efficacy, partly due to delivery difficulty. The recent developments of second generation PMO have addressed many of the delivery issues and can, therefore, represent an effective strategy for reducing dose levels and frequencies, as well as delivery to nonleaky fibres [45, 46]. Vivo morpholino (vPMO), a nonpeptide transporter, comprises a dendritic structure assembled around a triazine core which serves to position eight guanidinium head groups in a conformation effective at penetrating cell membranes [47]. More recently, arginine rich, PMO-internalization-peptide- (Pip-) conjugated PMO has been developed [48]. Pips are characterized by a central hydrophobic motif flanked by arginine rich domains. Pip-PMO with improved cardiac exon skipping activity demonstrates highly efficient dystrophin protein expression in various muscles, resulting in up to 50% of wild-type levels of dystrophin protein in the heart in mdx mice [49].
4. Proof of Principle of AO-Based Exon Skipping for DMD Patients
The exciting prospect of an effective AO-based exon skipping to restore dystrophin protein expression to affected tissues in DMD patients has been backed up by highly promising data from a series of clinical trials. Firstly, efficient in-frame dystrophin expression and no clinically adverse events following an exon 51-skipping approach have been successfully demonstrated in human subjects using local intramuscular injection of 2′OMePs or PMO [50, 51]. Recently, systemic trials using both AOs have revealed dose-dependent efficacy without any severe adverse effects [24, 25]. Especially it is very promising that systemically injected 2′OMePs have shown benefits in walking distance [25, 52]. These study results are an important milestone, and Sarepta Therapeutics and Prosensa/GSK are now conducting a double-blind, placebo-controlled, II b and III exon skipping trials with PMO (ClinicalTrials.gov identifier: NCT01396239) and 2′OMePs (ClinicalTrials.gov identifier: NCT01254019).
5. Rationale of Exons 45–55 Skipping in DMD
Although AO-based exon skipping is very promising, exon 51-skipping, which targets a single exon in the DMD gene and whose skipping can restore the open reading frame to the largest proportion of DMD mutations, is still applicable to only some 10% of DMD patients (examples of possible target mutations are as follows: deletions of exons 48–50, 45–50, 49-50, 50–52, 47–50, 43–50, and 52–63) [13, 53]. In addition, a phase I/IIa trial targeting exon 44 is currently underway (ClinicalTrials.gov identifier: NCT01037309) and development of new AO drugs targeting exons 45, 52, 53 and exon 55 is ongoing by Prosensa/GSK and Nippon Shinyaku Co., Ltd (UMIN000010964). By skipping these exons, approximately 28% of DMD patient mutations would be potentially treatable [54]. On the other hand, genotype and phenotype data from clinical databases suggest that simply restoring the reading frame may not necessarily result in restoring dystrophin protein function [55]. Two major hurdles of the current oligonucleotide approach are that (1) they are mutation specific and (2) the function or stability of each resulting in-frame dystrophin is uncertain. The prospect of overcoming both of these hurdles simultaneously has been inspired by the observation that patients with a deletion of exons 45–55 mostly show almost asymptomatic skeletal muscle involvement or exceptionally mild clinical symptoms with high blood creatine kinase concentrations, often associated with a late onset [56, 57]. It has also been noted that exons 45–55 cover the main mutation “hotspot” of the DMD gene so that, theoretically, up to 40–45% of DMD patients could be treated if it was possible to skip the entire exon 45–55 region [58]. Therefore, skipping of exons 45–55, that is, exons 45–55 skipping, at the mutation hotspot of the DMD gene with the second- generation peptide conjugated AOs would address these issues [27, 28]. Although multiexon skipping is technically very challenging, the existence of extremely mild patients with a spontaneous deletion of exons 45–55 implies that exons 45–55 skipping can be effective [12, 14]. Further the feasibility of exons 45–55 skipping can be explained by the order and timing of DMD intron removal. The DMD introns are extremely large compared to the mean size of human introns, 3,300 nucleotides [38, 39]. Figure 1 shows the intron sizes around exons 45–55 of the DMD gene. The intron 44 is 248,400 nucleotides. Subsequent introns are shorter (between 2,300 to 54,200 nucleotides), up until intron 55, which is 120,200 nucleotides. Interestingly, we and another group have detected very minimal spontaneous exons 45–55 skipping at low frequency in mouse and human cells, which may indicate that the acceptor splice sites of introns 44 and 55 can compete [17, 18, 59]. If the smaller introns (45 through 54) are indeed spliced out prior to intron 44 and intron 55, this would result in exons 45–55 skipping.
Figure 1.
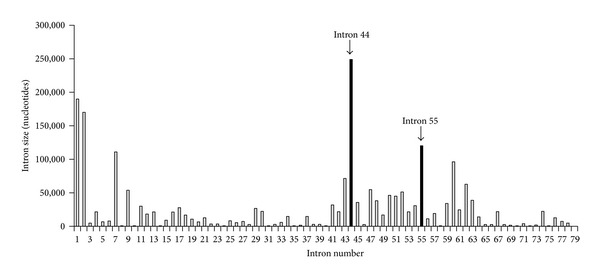
Intron sizes around exons 45–55 of the DMD gene. The intron size of exon 44 (intron 44) is 248,400 nucleotides; subsequent introns are shorter from 2,300 to 54,200 nucleotides, until intron 55, which is 120,200 nucleotides. Interestingly, we and another group have detected very minimal spontaneous exons 45–55 skipping at low frequency in mouse and human cells.
6. Dystrophin Molecular Structure and Function following Exons 45–55 Skipping
The molecular structure of dystrophin is composed of an actin-binding domain 1 at the N-terminus (ABD1), a central rod domain containing 24 spectrin-like repeats (R1–24), four hinge domains, a 20-amino acid insertion between spectrin-like repeats 15 and 16, a cysteine-rich domain, and a C-terminal domain [29]. Hinges are proline-rich, nonrepeat segments that possibly confer flexibility to dystrophin protein [60]. AO-based exon-skipping therapy for DMD is intended to exclude specific exons from out-of-frame dystrophin transcripts, thereby correcting the translational reading frame, resulting in the production of “BMD-like” in frame dystrophin, which would suppress nonsense-mediated mRNA decay and reconstruct the binding domain of dystrophin to DGC [11, 25]. The molecular structure of in-frame dystrophin as a result of exons 45–55 skipping leads to a truncation at the middle of two rod spectrin repeats (rod repeats 17 and 22) [16]. Interestingly, the number of rod repeats in this in-frame dystrophin between the remaining adjacent hinge domains H2 and H4 (16 spectrin repeats) is exactly the same as that between H2 and H3 in full-length dystrophin, which might indicate a requirement for such spacing for protein function or stability [55, 61]. It has been anticipated that the truncated dystrophin induced by exons 45–55 skipping would disturb the site-responsible anchoring of nNOS, which normally binds to spectrin-like repeats 16 and 17 of the dystrophin encoded by exons 42–45 and possibly render its subsarcolemmal localization unstable [62, 63]. It is encouraging to note that mild BMD patients with a deletion of exon 45–55 also lack nNOS at the subsarcolemma but have mild clinical symptoms (Table 1) [56, 58, 62, 64]. For example, it is reported that three patients with exon 45–55 deletions (28, 42, and 69 years old) showed no symptoms except for high blood CK level [56, 64]. That is, the loss of subsarcolemmal nNOS does not lead to a severe phenotype or that lack of nNOS does not appear to be critical. However, animal models should nonetheless be used to investigate the molecular function of in-frame dystrophin following exons 45–55 skipping [62].
Table 1.
Summary of clinical features of patients with an exon 45–55 deletion mutation of the DMD gene.
| Patient ID | Age at onset of symptoms | Symptoms at onset | Clinical status | Dilated cardiomyopathy | Ambulant | Reference |
|---|---|---|---|---|---|---|
| 1 | 2 | Weakness | Mild BMD | not available | + | [58] |
| 2 | 3.5 | Weakness | Exercise intolerance | − | + | [58] |
| 3 | 3.5 | By chance | Asymptomatic | + | + | [58] |
| 4 | 4 | Weakness | Mild BMD | not available | + | [58] |
| 5 | 4 | Weakness | Mild BMD | not available | + | [58] |
| 6 | 4 | Fatigue, cramps after exercise | Mild BMD | − | + | [62] |
| 7 | 4 | Fatigue, mild difficulty in running | Mild BMD | − | + | [62] |
| 8 | 6 | Hyper-Ck-emia | Asymptomatic | − | + | [58] |
| 9 | 8 | Hyper-Ck-emia | Asymptomatic | − | + | [58] |
| 10 | 9 | Myoglobinuria | Mild BMD | − | + | [62] |
| 11 | 12 | Myalgia | Myalgia | not available | + | [58] |
| 12 | 12 | Exercise intolerance | Mild BMD | − | + | [58] |
| 13 | 13 | Muscle pain | Mild BMD | − | + | [58] |
| 14 | 13 | Hyper-Ck-emia | Mild BMD | − | + | [58] |
| 15 | 19 | Weakness | Mild BMD | − | + | [58] |
| 16 | 26 | Exertional dyspnea | No weakness and atrophy | + | + | [64] |
| 17 | 36 | Exertional dyspnea | No weakness and atrophy | + | + | [64] |
| 18 | 40 | Calf hypertrophy | Mild BMD | + | + | [58] |
| 19 | 45 | Weakness | Mild BMD | not available | + | [58] |
| 20 | 49 | Weakness | Mild BMD | not available | + | [58] |
| 21 | 55 | Walking difficulties | Mild BMD | − | + | [62] |
| 22 | 59 | Weakness | Mild BMD | − | + | [64] |
| 23 | 69 | Hyper-Ck-emia | Asymptomatic | − | + | [56] |
7. Preclinical Exons 45–55 Skipping in the Exon 52 Deficient Muscular Dystrophy Mouse
To date, a proof of concept for systemic exons 6 and 8 skipping by targeting two exons has been provided in dystrophic dogs [17, 18, 65]. However, exons 45–55 skipping with a mixture of 2′OMePs in DMD myoblast cells has proved technically challenging, and the levels of exons 45–55 skipping have typically been low and highly variable [59, 66]. Recently Takeda's group tested the feasibility of exons 45–55 skipping using both mouse myoblast cells and systemic injections with a mixture of 10 vPMOs in mdx52 mice, which harbour a deletion mutation in exon 52 [16, 67, 68] (Figure 2). The mixture of vPMOs was newly designed, paying specific attention to the avoidance of formation of self- or heteroduplex of the AOs, which might diminish the efficacy of exons 45–55 skipping. With this aim, we used OligoAnalyzer 3.1 to design the mixture in which most of combinations of ΔG maximum binding forces of two AOs were above −5 kcal/mole [69]. Efficient skipping of all of 10 exons was demonstrated with mixtures of 10 vPMOs in vitro in conditionally immortalized muscle cells called H2K-mdx52 myotubes. Moreover, on systemic injections into mdx52 mice with the mixtures (a total of 12 mg/kg dose), extensive dystrophin-positive fibres and an average of some 8–15% of wild-type levels of exons 45–55 truncated dystrophin protein as determined by Western blotting analysis in a range of skeletal muscles were observed, whereas the dystrophin expression level in the heart was only 2% of wild-type levels. The in-frame dystrophin restored all of the DGC except nNOS at the subsarcolemma. The pathology of skeletal muscles was ameliorated, but the skeletal muscle function was only marginally recovered, probably due to the incomplete restitution of the dystrophin. Although the sequences are specific to the mouse, these data validate the principle that carefully designed AOs may be used to realize exons 45–55 skipping and, by this means, generate effective amounts of in-frame dystrophin of near-optimal structure in 40–45% of DMD patients.
Figure 2.
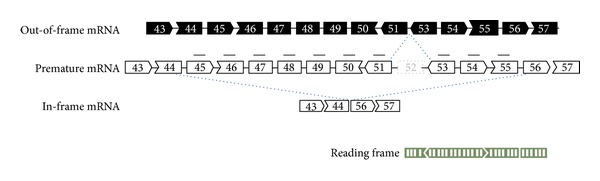
Exons 45–55 skipping with a mixture of 10 vPMOs in mdx52 mouse. Mdx52 mouse lacks exon 52 in the mRNA of the murine DMD gene, leading to out-of-frame products. Exons 45–55 skipping with mixture vPMOs (black line) restores the reading frame of DMD mRNA.
8. Towards Clinical Application: Hurdles and Limitations
Although results from preclinical trials appear encouraging, several issues pose challenges for the use of an AO “mixture approach” as an effective and affordable therapy for DMD. The most significant issue is in developing new approaches to toxicity testing and clinical trial regulatory procedures that are relevant and appropriate for sequence-specific mixture drugs [70]. It is important to note that mixtures of AOs might be considered one drug by drug regulatory agencies including those of the EU, USA, and Japan, even if the mixture contains several different AOs. Otherwise, required pre-clinical toxicology studies to develop this “mixture approach” will be too complicated and the costs for development of each of the 10 AOs and the subsequent clinical trials will be enormous. The second significant issue to translate the “mixture approach” with vPMOs to a therapy for DMD is the lack of adequate delivery to skeletal muscle and especially to the heart muscle. Higher dose administration of vPMOs would likely cause acute toxicity due to their narrow therapeutic range. Transient reduced activity, bradypnea, or seizure was sometimes observed following systemic injection of the mixture with vPMOs at a total of 15–25 mg/kg/dose in mdx52 mice. Importantly, cardiomyopathy is the second leading cause of death in DMD patients, accounting for 10–40% of deaths in DMD populations [71–73]. While some BMD patients with an exon 45–55 deletion show X-linked dilated cardiomyopathy [56, 58, 64, 74], their prognoses are quite favorable and only a few of them show mild heart failure symptom [62]. These imply that cardiac dystrophin correction due to exons 45–55 skipping would effectively rescue cardiac symptoms. The causes of low efficiency of cardiac dystrophin restoration remain unclear but are likely related to the inadequate ability of vPMO to penetrate the heart.To further validate and develop this exons 45–55 skipping approach, there are two critical steps, first to understand the order of DMD intron removal and the minimum combinatorial requirements for multiexon skipping of exons 45–55 (i.e., what is the fewest number of AOs that can be used to generate effective skipping). Secondly, this approach must now be combined with advanced generation peptide conjugated AO chemistry to achieve the maximum therapeutic outcome without toxicity for skeletal and cardiac muscles.
9. Conclusion
Recently, the prospect of successful AO-based therapies has moved a step closer to clinical applicability, in particular for DMD. The first clinical trials in DMD patients demonstrated the proof of principle of exon 51 skipping in humans, resulting in very encouraging results. However, multiple exon skipping, including exons 45–55 skipping using a “mixture approach”, is still in the preclinical stage. A variety of issues accompany this approach, including low efficacy and potential high toxicity of AOs, prior to human clinical trials. Development of an advanced exons 45–55 skipping approach with fewer new generation AOs that have improved cardiac exon skipping activity may reduce the therapeutic dose and interval of administrations, minimising the potential toxicity, off-target effects, and the cost burden for DMD.
Acknowledgments
The authors thank Shin'ichi Takeda, Tetsuya Nagata, Akinori Nakamura, and Andrew Douglas for providing the scientific advice. This work was supported by MRC Confidence in Concept award (grant to Yoshitsugu Aoki and Matthew J. A. Wood), Parent Project Muscular Dystrophy (USA), The Friends of Garrett Cumming Research Fund, Muscular Dystrophy Canada, HM Toupin Neurological Science Research Fund, and Women and Children's Health Research Institute (WCHRI).
References
- 1.Arahata K, Ishiura S, Ishiguro T, et al. Immunostaining of skeletal and cardiac muscle surface membrane with antibody against Duchenne muscular dystrophy peptide. Nature. 1988;333(6176):861–863. doi: 10.1038/333861a0. [DOI] [PubMed] [Google Scholar]
- 2.Wood MJA. Toward an oligonucleotide therapy for duchenne muscular dystrophy: a complex development challenge. Science Translational Medicine. 2010;2(25):25–ps15. doi: 10.1126/scitranslmed.3000512. [DOI] [PubMed] [Google Scholar]
- 3.Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation. 2009;30(3):293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman EP, Bronson A, Levin AA, et al. Restoring dystrophin expression in duchenne muscular dystrophy muscle: progress in exon skipping and stop codon read through. The American Journal of Pathology. 2011;179(1):12–22. doi: 10.1016/j.ajpath.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClorey G, Moulton HM, Iversen PL, Fletcher S, Wilton SD. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Therapy. 2006;13(19):1373–1381. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- 6.Saito T, Nakamura A, Aoki Y, et al. Antisense PMO found in dystrophic dog model was effective in cells from exon 7-deleted DMD patient. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0012239.e12239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunckley MG, Manoharan M, Villiet P, Eperon IC, Dickson G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Human Molecular Genetics. 1998;7(7):1083–1090. doi: 10.1093/hmg/7.7.1083. [DOI] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A, De Winter CL, Janson AAM, et al. Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: indication for steric hindrance of SR protein binding sites. Oligonucleotides. 2005;15(4):284–297. doi: 10.1089/oli.2005.15.284. [DOI] [PubMed] [Google Scholar]
- 9.Arechavala-Gomeza V, Graham IR, Popplewell LJ, et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Human Gene Therapy. 2007;18(9):798–810. doi: 10.1089/hum.2006.061. [DOI] [PubMed] [Google Scholar]
- 10.Wilton SD, Lloyd F, Carville K, et al. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscular Disorders. 1999;9(5):330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 11.van Deutekom JCT, Bremmer-Bout M, Janson AAM, et al. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Human Molecular Genetics. 2001;10(15):1547–1554. doi: 10.1093/hmg/10.15.1547. [DOI] [PubMed] [Google Scholar]
- 12.Matsuo M, Masumura T, Nishio H, et al. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy Kobe. The Journal of Clinical Investigation. 1991;87(6):2127–2131. doi: 10.1172/JCI115244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aoki Y, Nakamura A, Yokota T, et al. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Molecular Therapy. 2010;18(11):1995–2005. doi: 10.1038/mt.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nature Medicine. 2006;12(2):175–177. doi: 10.1038/nm1345. [DOI] [PubMed] [Google Scholar]
- 15.Lu QL, Rabinowitz A, Chen YC, et al. Systemic delivery of antisense oligoribonucleotide restorers dystrophin expression in body-wide skeletal muscles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(1):198–203. doi: 10.1073/pnas.0406700102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aoki Y, Yokota T, Nagata T, et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(34):13763–13768. doi: 10.1073/pnas.1204638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yokota T, Lu Q-L, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in duchenne dystrophy dogs. Annals of Neurology. 2009;65(6):667–676. doi: 10.1002/ana.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokota T, Nakamura A, Nagata T, et al. Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acids Research. 2012;22(5):306–315. doi: 10.1089/nat.2012.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin H, Moulton HM, Betts C, et al. Functional rescue of dystrophin-deficient mdx mice by a chimeric peptide-PMO. Molecular Therapy. 2010;18(10):1822–1829. doi: 10.1038/mt.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu B, Lu P, Benrashid E, et al. Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Therapy. 2010;17(1):132–140. doi: 10.1038/gt.2009.120. [DOI] [PubMed] [Google Scholar]
- 21.Mitrpant C, Fletcher S, Iversen PL, Wilton SD. By-passing the nonsense mutation in the 4CV mouse model of muscular dystrophy by induced exon skipping. Journal of Gene Medicine. 2009;11(1):46–56. doi: 10.1002/jgm.1265. [DOI] [PubMed] [Google Scholar]
- 22.Fletcher S, Honeyman K, Fall AM, Harding PL, Johnsen RD, Wilton SD. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligunucleotide. Journal of Gene Medicine. 2006;8(2):207–216. doi: 10.1002/jgm.838. [DOI] [PubMed] [Google Scholar]
- 23.Goyenvalle A, Babbs A, Powell D, et al. Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Molecular Therapy. 2010;18(1):198–205. doi: 10.1038/mt.2009.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. The Lancet. 2011;378(9791):595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goemans NM, Tulinius M, van Den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. The New England Journal of Medicine. 2011;364(16):1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- 26.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. The Lancet Neurology. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida M, Hama H, Ishikawa-Sakurai M, et al. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Human Molecular Genetics. 2000;9(7):1033–1040. doi: 10.1093/hmg/9.7.1033. [DOI] [PubMed] [Google Scholar]
- 29.Ozawa E. Our trails and trials in the subsarcolemmal cytoskeleton network and muscular dystrophy researches in the dystrophin era. Proceedings of the Japan Academy Series B. 2010;86(8):798–821. doi: 10.2183/pjab.86.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heald A, Anderson LVB, Bushby KMD, Shaw PJ. Becker muscular dystrophy with onset after 60 years. Neurology. 1994;44(12):2388–2390. doi: 10.1212/wnl.44.12.2388. [DOI] [PubMed] [Google Scholar]
- 31.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 32.Baumbach LL, Chamberlain JS, Ward PA, Farwell NJ, Caskey CT. Molecular and clinical correlations of deletions leading to Duchenne and Becker muscular dystrophies. Neurology. 1989;39(4):465–474. doi: 10.1212/wnl.39.4.465. [DOI] [PubMed] [Google Scholar]
- 33.Gillard EF, Chamberlain JS, Murphy EG, et al. Molecular and phenotypic analysis of patients with deletions within the deletion-rich region of the Duchenne muscular dystrophy (DMD) gene. American Journal of Human Genetics. 1989;45(4):507–520. [PMC free article] [PubMed] [Google Scholar]
- 34.Koening M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus becker muscular dystrophy: correlation of severity with type of deletion. American Journal of Human Genetics. 1989;45(4):498–506. [PMC free article] [PubMed] [Google Scholar]
- 35.Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Molecular Cancer Therapeutics. 2002;1(5):347–355. [PubMed] [Google Scholar]
- 36.Iversen PL. Phosphorodiamidate morpholino oligomers: favorable properties for sequence-specific gene inactivation. Current Opinion in Molecular Therapeutics. 2001;3(3):235–238. [PubMed] [Google Scholar]
- 37.Heemskerk H, De Winter C, van Kuik P, et al. Preclinical PK and PD studies on 2′-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Molecular Therapy. 2010;18(6):1210–1217. doi: 10.1038/mt.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pramono ZAD, Takeshima Y, Alimsardjono H, Ishii A, Takeda S-I, Matsuo M. Induction of exon skipping of the dystrophin transcript in lymphoblastoid cells by transfecting an antisense oligodeoxynucleotide complementary to an exon recognition sequence. Biochemical and Biophysical Research Communications. 1996;226(2):445–449. doi: 10.1006/bbrc.1996.1375. [DOI] [PubMed] [Google Scholar]
- 39.Takeshima Y, Wada H, Yagi M, et al. Oligonucleotides against a splicing enhancer sequence led to dystrophin production in muscle cells from a Duchenne muscular dystrophy patient. Brain & Development. 2001;23(8):788–790. doi: 10.1016/s0387-7604(01)00326-6. [DOI] [PubMed] [Google Scholar]
- 40.Takeshima Y, Yagi M, Wada H, Matsuo M. Intraperitoneal administration of phosphorothioate antisense oligodeoxynucleotide against splicing enhancer sequence induced exon skipping in dystrophin mRNA expressed in mdx skeletal muscle. Brain & Development. 2005;27(7):488–493. doi: 10.1016/j.braindev.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Yin H, Lu Q, Wood M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Molecular Therapy. 2008;16(1):38–45. doi: 10.1038/sj.mt.6300329. [DOI] [PubMed] [Google Scholar]
- 42.Yin H, Betts C, Saleh AF, et al. Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse. Molecular Therapy. 2010;18(4):819–827. doi: 10.1038/mt.2009.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aartsma-Rus A, Kaman WE, Bremmer-Bout M, et al. Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cells. Gene Therapy. 2004;11(18):1391–1398. doi: 10.1038/sj.gt.3302313. [DOI] [PubMed] [Google Scholar]
- 44.Renneberg D, Bouliong E, Reber U, Schümperli D, Leumann CJ. Antisense properties of tricyclo-DNA. Nucleic Acids Research. 2002;30(13):2751–2757. doi: 10.1093/nar/gkf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morcos PA, Li Y, Jiang S. Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. BioTechniques. 2008;45(6):613–623. doi: 10.2144/000113005. [DOI] [PubMed] [Google Scholar]
- 46.Wu B, Li Y, Morcos PA, Doran TJ, Lu P, Lu QL. Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Molecular Therapy. 2009;17(5):864–871. doi: 10.1038/mt.2009.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morcos PA, Li Y, Jiang S. Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. BioTechniques. 2008;45(6):613–623. doi: 10.2144/000113005. [DOI] [PubMed] [Google Scholar]
- 48.Ivanova GD, Fabani MM, Arzumanov AA, et al. PNA-peptide conjugates as intracellular gene control agents. Nucleic Acids Symposium Series. 2008;(52):31–32. doi: 10.1093/nass/nrn016. [DOI] [PubMed] [Google Scholar]
- 49.Betts C, Saleh AF, Arzumanov AA, et al. Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Molecular Therapy. Nucleic Acids. 2012;1(8):p. e38. doi: 10.1038/mtna.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Deutekom JC, Janson AA, Ginjaar IB, et al. Local dystrophin restoration with antisense oligonucleotide PRO051. The New England Journal of Medicine. 2007;357(26):2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 51.Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. The Lancet Neurology. 2009;8(10):918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goemans N, Klingels K, van den Hauwe M, et al. Test-retest reliability and developmental evolution of the 6-min walk test in Caucasian boys aged 5-12 years. Neuromuscular Disorders. 2013;23(1):19–24. doi: 10.1016/j.nmd.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 53.Arechavala-Gomeza V, Graham IR, Popplewell LJ, et al. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Human Gene Therapy. 2007;18(9):798–810. doi: 10.1089/hum.2006.061. [DOI] [PubMed] [Google Scholar]
- 54.Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Human Mutation. 2009;30(3):293–299. doi: 10.1002/humu.20918. [DOI] [PubMed] [Google Scholar]
- 55.Yokota T, Takeda S, Lu Q-L, Partridge TA, Nakamura A, Hoffman EP. A renaissance for antisense oligonucleotide drugs in neurology: exon skipping breaks new ground. Archives of Neurology. 2009;66(1):32–38. doi: 10.1001/archneurol.2008.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreiro V, Giliberto F, Noelia Muñiz GM, et al. Asymptomatic Becker muscular dystrophy in a family with a multiexon deletion. Muscle and Nerve. 2009;39(2):239–243. doi: 10.1002/mus.21193. [DOI] [PubMed] [Google Scholar]
- 57.Tselikas L, Rodrigues E, Jammal M, et al. Late onset Becker muscular dystrophy. A case report and literature review. La Revue de Médecine Interne. 2011;32(3):181–186. doi: 10.1016/j.revmed.2010.10.353. [DOI] [PubMed] [Google Scholar]
- 58.Béroud C, Tuffery-Giraud S, Matsuo M, et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Human Mutation. 2007;28(2):196–202. doi: 10.1002/humu.20428. [DOI] [PubMed] [Google Scholar]
- 59.Aoki Y, Yokota T, Nagata T, et al. Bodywide skipping of exons 45–55 in dystrophic mdx52 mice by systemic antisense delivery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(34):13763–13768. doi: 10.1073/pnas.1204638109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. Journal of Biological Chemistry. 1990;265(8):4560–4566. [PubMed] [Google Scholar]
- 61.Yokota T, Duddy W, Partridge T. Optimizing exon skipping therapies for DMD. Acta Myologica. 2007;26(3):179–184. [PMC free article] [PubMed] [Google Scholar]
- 62.Anthony K, Cirak S, Torelli S, et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: implications for clinical trials. Brain. 2011;134(12):3547–3559. doi: 10.1093/brain/awr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gentil C, Leturcq F, Ben Yaou R, et al. Variable phenotype of del45–55 Becker patients correlated with nNOSμ mislocalization and RYR1 hypernitrosylation. Human Molecular Genetics. 2012;21(15):3449–3460. doi: 10.1093/hmg/dds176. [DOI] [PubMed] [Google Scholar]
- 64.Nakamura A, Yoshida K, Fukushima K, et al. Follow-up of three patients with a large in-frame deletion of exons 45–55 in the Duchenne muscular dystrophy (DMD) gene. Journal of Clinical Neuroscience. 2008;15(7):757–763. doi: 10.1016/j.jocn.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 65.Yokota T, Hoffman E, Takeda S. Antisense oligo-mediated multiple exon skipping in a dog model of duchenne muscular dystrophy. Methods in Molecular Biology. 2011;709:299–312. doi: 10.1007/978-1-61737-982-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Vliet L, de Winter CL, van Deutekom JCT, van Ommen G-JB, Aartsma-Rus A. Assessment of the feasibility of exon 45–55 multiexon skipping for duchenne muscular dystrophy. BMC Medical Genetics. 2008;9, article 105 doi: 10.1186/1471-2350-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Araki E, Nakamura K, Nakao K, et al. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in duchenne muscular dystrophy. Biochemical and Biophysical Research Communications. 1997;238(2):492–497. doi: 10.1006/bbrc.1997.7328. [DOI] [PubMed] [Google Scholar]
- 68.Partridge TA. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. The FEBS Journal. 2013 doi: 10.1111/febs.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Owczarzy R, Tataurov AV, Wu Y, et al. IDT SciTools: a suite for analysis and design of nucleic acid oligomers. Nucleic Acids Research. 2008;36:W163–W169. doi: 10.1093/nar/gkn198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koo T, Wood M. Clinical trials using antisense oligonucleotides in duchenne muscular dystrophy. Human Gene Therapy. 2013;24(5):479–488. doi: 10.1089/hum.2012.234. [DOI] [PubMed] [Google Scholar]
- 71.Melacini P, Vianello A, Villanova C, et al. Cardiac and respiratory involvement in advanced stage duchenne muscular dystrophy. Neuromuscular Disorders. 1996;6(5):367–376. doi: 10.1016/0960-8966(96)00357-4. [DOI] [PubMed] [Google Scholar]
- 72.Melacini P, Fanin m. FM, Danieli GA, et al. Cardiac involvement in Becker muscular dystrophy. Journal of the American College of Cardiology. 1993;22(7):1927–1934. doi: 10.1016/0735-1097(93)90781-u. [DOI] [PubMed] [Google Scholar]
- 73.Bushby K, Muntoni F, Bourke JP. 107th ENMC International Workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th-9th June 2002, Naarden, the Netherlands. Neuromuscular Disorders. 2003;13(2):166–172. doi: 10.1016/s0960-8966(02)00213-4. [DOI] [PubMed] [Google Scholar]
- 74.Miyazaki D, Yoshida K, Fukushima K, et al. Characterization of deletion breakpoints in patients with dystrophinopathy carrying a deletion of exons 45–55 of the Duchenne muscular dystrophy (DMD) gene. Journal of Human Genetics. 2009;54(2):127–130. doi: 10.1038/jhg.2008.8. [DOI] [PubMed] [Google Scholar]