

Abstract
In short, manipulation of cytokine pathways shows promise as a mean to tilt the balance of immunity toward tolerance. Effective and regulatory T cells vary in their response to a variety of cytokines. In particular, the ability of certain cytokines, for example, IL-2, to provide vital survival signals to regulatory cells and to trigger death of effector T cells or impede IL-15 driven expansion of memory cells has spurred several trials. The ability of IFNγ, IL-4, TNFα, and lymphotoxin to exert selective effects upon crucial lymphocyte subset populations in vivo may also enable translation into potent therapies.
Introduction
Remission of autoimmune disease activity can be produced when the therapy restrains the capacity of self-reactive tissue destroying T cells to create tissue damage. Many conventional immunosuppressives including calcineurin inhibitors and corticosteroids inhibit the expansion and/or effector function of both tissue destructive effector and tissue protective immunoregulatory T cells. Hence, the remissions are not sustained long following withdrawal of the drug. Immune tolerance is obtained only when the functional supremacy of autoimmune immunoregulatory T cells remains dominant following the cessation of therapy. This review will focus upon the possibility that cytokines or cytokine antagonists can be used to treat patients with autoimmune disorders and obtain remissions of the disease or, even better, to restore self-tolerance. One such strategy is based on the knowledge that cytokine related activation induced (AICD) or passive cell death (PCD) of activated effector T cells can be a requirement for the acquisition of immune tolerance [1,2]. Cytokines including some T cell growth factors can trigger apoptotic AICD cell death of effector T cells and the absence of T cell growth factor renders activated T cell susceptible to PCD. As effector and immunoregulatory T cells differ in their life or death type responses to cytokines, manipulation of cytokine pathways can alter the balance of effector to regulatory cells.
Main text
High affinity interleukin (IL) -2, -15, and -21 receptors are expressed on recently activated T cells but not upon resting T cells. The family of T cell growth factors with pleiotropic and redundant effects includes interleukins-2, -4, -7, -9, -15, and -21, each with a similar bundle helix structure [3•]. Each of these growth factors interacts with a growth factor specific receptor complex that consists of the common gamma chain receptor protein, a crucial element for spawning cell activating intracellular signals and a cytokine specific alpha chain [3•]. In addition the receptor complexes for IL-2 and IL-15 include an identical beta chain. In some settings, however, IL-2 and IL-15, both potent T cell growth factors, exert opposing effects. IL-15, provided in large measure by stromal cells, triggers proliferation of CD8+ T memory cells, a phenomenon that is enhanced by the absence or inhibition of IL-2 while IL-2 indirectly inhibits proliferation of CD8+ memory cells [4]. The ability of IL-2 to inhibit proliferation of memory T cells is though an indirect effect that is mediated partly by regulatory T cells [4,5•]. IL-2 is the only T cell growth factor that following several rounds of T cell proliferation incites massive cell death among activated effector type T cells. By contrast IL-15 acts as an antidote to IL-2 triggered cell death [6•]. In the presence of both IL-2 and IL-15 the apoptotic death of activated effector T cells is avoided and proliferation of activated effector type T cells continues.
In addition IL-15 can replace the requirement for CD4+ T cells in promoting the longevity of antigen activated CD8+ T cells via avoidance of TRAIL mediated apoptosis [7•]. Can IL-2, a death factor for effector T cells, be used as a therapeutic for autoimmune disorders? While IL-2 serves as a death factor for effector T cells, it serves a very different function in respect to effects upon regulatory T cells. Lethal autoimmunity is manifest in IL-2Rβ deficient mice, but transgenic thymic specific expression of IL-2Rβ in the IL-2Rβ deficient mice prevents this syndrome [8••]. Moreover, IL-2 administration in concert with transfer of CD4+ regulatory T cells serves to prevent lethal autoimmunity [9••]. Administration of IL-2 in the absence of infusions of regulatory cells can prevent autoimmunity in several mouse models while neutralization of IL-2 can incite autoimmune gastritis [10]. Why? IL-2, primarily produced by non-regulatory T cells, is essential for the expansion and maintenance of regulatory T cells [10,11]. In reviewing this body of work, several have hypothesized that manipulation of the availability of IL-2 or IL-15 to activated effector type and to regulatory T cells may provide a means to govern the balance of cytopathic to regulatory T cells in vivo. AICD and PCD are routine downstream consequences of T cell activation by antigen [signal 1] plus costimulation [signal 2] [3•,12], and the integrity of these apoptotic pathways is required for the induction of peripheral transplant tolerance across MHC barriers with treatments that do not directly kill T cells [1,2].
Blockade or neutralization of IL-15 is an attractive strategy for treatment of rheumatoid arthritis. Baslund et al. have reported favorable response of patients with rheumatoid arthritis to neutralizing anti IL-15 mAb [13•]. An antagonist form of IL-15 was created through subtle changes in the C terminus of IL-15 that rendered the molecule unable to activate the common gamma chain receptor without impairing the affinity to the trimolecular IL-15R complex [14•].This antagonist type IL-15 protein proved highly effective in blocking DTH responses [14•] and disease progression in the RA-like murine collagen-induced authentic model [15•].
By fusing immunoglobulin heavy chain genes to the cytokine related genes, the fusion proteins possess normal cytokine related bioactivities and extremely long circulating half lives. A strategy aimed at producing massive apoptosis of recently activated pathogenic, but not regulatory, T cells has been developed. The strategy is based on the knowledge that (i) the fate of activated effector and memory T cells, T cells that unlike resting T cells express high affinity receptors for T cell growth factors, is dictated by their response to T cell growth factors and (ii) IL-2 triggers AICD. Whereas IL-15 counteracts this signal and protects activated effector T cells from PCD. Rapamycin, an mTOR inhibitor, blocks the proliferative but not proapoptotic effects of IL-2 and enhances PCD. In short the power mix regimen tilts the expansion of donor reactive T cells from a pattern in which effector T cells are predominant to one in which regulatory T cells predominate in different transplant and immune disease models (Figure 1).
Figure 1.
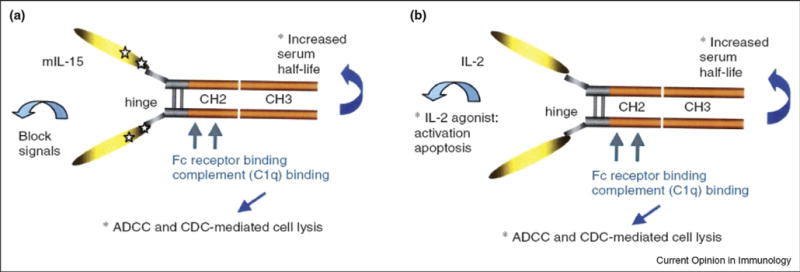
(A) Cytolytic interleukin (IL)-2/Fc Structure. (B) Cytolytic mutant IL-15/Fc structure.
Using this approach, tolerance and/or unprecedented very long-term engraftment of allografts was observed in a variety of daunting allograft models [16•]. While many treatments have succeeded in preventing diabetes in the autoimmune NOD model, this tripartite protocol is the one of very few short-term therapies that permanently restores euglycemia and islet self-tolerance in new onset frankly diabetic NOD mice [17••].
The long loved IL-2.Ig molecule given as a monotherapy prevents type 1 diabetes mellitus (T1DM) in the NOD passive transfer model [18•]. While the best outcomes in the NOD model were obtained using both the IL-2 and IL-15 Ig related fusion proteins plus rapamycin, either fusion protein plus rapamycin produced remissions in many treat hosts [17••]. Indeed, the use of wild-type IL-2 plus rapamycin proved effective in preventing spontaneous and recurrent autoimmune diabetes NOD mice [19•]. The protection is associated with a shift from Th1-to Th2-, and Th3-type cytokine-producing cells, possibly due to deletion of autoreactive Th1 cells [20•]. On the basis of the results of these preclinical studies, The Immune Tolerance Network is conducting a trial of IL-2 and sirolimus in human subjects with new onset type 1 diabetes mellitus. In light of these findings, Tang et al. [20•] have assessed the role of IL-2 in the causation of T1DM in the NOD model. In the NOD model survival of regulatory T cells in pancreatic lymph nodes (PNL) is compromised. As IL-2 is an important TReg survival factor, they administered IL-2 and found an expanded PLN TReg population. Administration of low-dose IL-2, as previously reported, promoted Treg cell survival and protected mice from developing diabetes. Treg cell dysfunction secondary to defective IL-2 production may lead to progressive breakdown of self-tolerance and the development of diabetes in non-obese diabetic mice [20•].
Gamma interferon also promotes T cell AICD. Administration of a fragment of the T1DM related glutamic acid decarboxylase [GAD] autoantigen restored normoglycemia and clearance of islet infiltrating leukocytes. As interferonγ inhibits Th17 cells, it is fascinating that GAD administration to NODs induced expression of IFNγ by the spleen and a marked decrease in the frequency of Th17 cells [21]. A role for defective IL-4 expression in causation of T1DM in the NOD has been supported by the work of Creusot et al. [22] Administration of IL-4 or introduction of an IL-4 transgene in prediabetic mice creates disease resistance to T1DM in NODs, but it does not reverse established diabetes. Hence, the translation of this finding to frankly diabetic humans is uncertain.
Type 1 diabetes mellitus (T1DM) results from T cell dependent destruction of insulin-producing beta cells in the islets of Langerhans [23]. While activated T cells are required for the initiation of the disease in the NOD mouse, macrophages and their ability to release inflammatory cytokines [24] such as TNFγ, IL-1β, and IL-12 [25] have also been implicated. These cytokines have been shown to enhance Th1 mediated inflammatory responses, which seem to be responsible, at least partly, for beta cell destruction [26]. The marked susceptibility of islets to injurious effects of activated macrophages and proinflammatory cytokines, cytokines that are often expressed as the product of activated macrophages, are well known. Proinflammation cytokines clearly exert noxious effects upon beta cells. In addition, lymphotoxin, a protein proinflammatory cytokine, promotes formation of spleen cell lymphoid follicles. Similar follicles are present in the pancreas of diabetic NODs. In keeping with these observations administration of soluble lymphotoxin receptors can prevent diabetes in NOD mice despite the presence of insulitis [27].
Faustman and her colleagues report that treatment of frankly diabetic NOD mice with complete Freund's complete adjuvant (CFA) induced expression of TNFα combined with matched histocompatibility complex class I and self-peptide bearing splenocytes permanently reversed the diabetes in 67% of the treated mice [28••,29••]. Several previous reports demonstrated that CFA administration to young NOD mice prevents development of T1DM through effects directed at autoimmune T cells. The mechanism by which the Faustman regimen provides therapeutic effects was deduced to be twofold. First the adjuvant through induced expression of TNFα modulates the autoimmune state and the transplanted islets maintain normoglycemia long enough for the spleen cell preparation to aid regeneration of the insulin-producing beta cells. Hence, the promise of this therapy is exceptional. Both the underlying autoimmune state is ablated and a fresh source of beta cell precursor cell is introduced.
By contrast, efforts to replicate the experiments reported by Kodama et al. [28••] paper by three independent groups failed in some crucial respects [30••–32••]. The results from all three groups were consistent; they reported that splenocytes did not contribute to islet regeneration. All three studies documented restoration of normoglycemia in a subset of treated subjects; however, recovery of host beta cells rather than spleen cells were responsible for this result. All three groups concluded that the adjuvant-dependent dampening of the autoimmune attack, coupled with the recovery of residual host beta cells were responsible for the reversal of hyperglycemia. The incidence of reversal of diabetes albeit higher than controls did not match that observed by Faustman and her co-workers. Clearly, the potential utility of TNFα as a potential therapy for T1DM is controversial. Faustman and her colleagues have suggested that systemic administration of TNFα may have potential for clinical application. In a more recent work, Faustman's lab has probed for the sensitivity of human autoimmune potentially diabetes causing auto-reactive T lymphocytes to TNFα [33••]. The hypothesis that TNFα or agonists of TNFα selectively kill some, not all, autoreactive T cells, while sparing normal T cells was tested. A subpopulation of CD8, but not CD4, T cells in patients' blood was identified as vulnerable to TNFα or TNFα agonist-induced death [33••]. The subpopulation of T cells susceptible to TNF or TNFR2 agonist-induced death was traced specifically to autoreactive T cells to insulin, a known autoantigen. As other activated and memory T cell populations were resistant to TNF-triggered death. The implications for clinical application are uncertain [33••].
Following interaction of T cell receptors with antigen (signal 1) and co-stimulation (signal 2), naïve CD4+ T cells are activated. Depending upon the fine texture of the inflammatory milieu in which antigen activation takes place, these newly activated T cells commit to one of several CD4+ subset phenotypes. Naïve CD4+ cells T commit to the tissue destructive, gamma interferon expressing Th1 program when signals 1 and 2 are delivered in a milieu rich in IL-12 [34]. By contrast, antigen activation conducted in an IL-4 rich environment leads to commitment to the Th2 phenotype [34]. In keeping with dogma that CD4+ T cells take cues from the cytokine environment, a TGFβ dominant environment leads naïve CD4+ T cells to commit to the regulatory phenotype [35••]. Indeed, this commitment is obtained by the TGFβ triggered expression of the lineage unique Foxp3 lineage specification factor [36••,37••]. Remarkably, TGFβ, in mice the presence of IL-6 [38••] or IL-21 [39••], promotes commitment of naïve murine and human CD4+ T cells to the highly cytopathic Th17 phenotype. In man other proinflammatory cytokines, including TNFα and IL-1β in addition to IL-6 elicit a similar effect [40••]. The presence of these proinflammatory cytokines precludes commitment of naïve CD4+ T cells to the regulatory phenotype [38••–42••]. Th17 cells express variety of potent proinflammatory cytokines including but not limited to IL-17A, IL-17F, and osteopontin. IL-23, while not necessary for the commitment to the Th17 phenotype, is essential to stabilize this commitment [43••]. Th17 cells are potent effector cells, perhaps more potent than Th1 cells, in several, but not all, autoimmune states [41••]. A vicious cycle in respect to Th17 dependent tissue destruction is formed through ability of Th17 cells to stimulate antigen-presenting cells to express IL-6 and by the ability of IL-6 to stimulate commitment of naïve T cells to the Th17 phenotype. Th17 cells participate in extremely inflamed forms of T cell dependent tissue injury. Within these inflamed environments the ability of Foxp3+ T cells to restrain effector T cells from executing tissue injury is severely compromised [39••,41••]. Owing to the violence of Th17 dependent tissue injury, a means to selectively target Th17 for therapy is a potentially important unmet need. Preliminary experiments suggest that targeting IL-17A, one of several proinflammatory cytokines expressed by Th17 cells will not prove effective. As IL-23 is required to maintain and expand Th17 cells, it is notable that IL-23 orchestrates intestinal inflammation [44••–47••]. Moreover, certain IL-23R alleles are linked to disease expression of Crohn's or ulcerative colitis [48]. Clearly the IL-23 axis emerges as an attractive target for immunotherapy.
In contrast to efforts targeting a single cytokine, we tested the hypothesis that inflammatory mechanisms directly trigger the loss of immune tolerance to islets and beta cell destructive insulitis in the NOD mouse model. We demonstrated that treatment with alpha1 antitrypsin (AAT), an agent that dampens expression of proinflammatory, but not ant-inflammatory, cytokines and does not directly inhibit T cell activation, ablates invasive insulitis and restores euglycemia, immune tolerance to beta cells, normal insulin signaling and insulin responsiveness in NOD mice with recent onset T1DM by favorable changes in the inflammatory milieu [49••]. These results support the notion that the inflammatory context in which T cells are activated by antigen dictates the nature, aggressive or benign of the immune response.
Conclusion
In short, manipulation of cytokine pathways shows promise as a mean to tilt the balance of immunity toward tolerance. Effective and regulatory T cells vary in their response to a variety of cytokines. In particular, the ability of certain cytokines, for example, IL-2, to provide vital survival signals to regulatory cells and to trigger death of effector T cells or impede IL-15 driven expansion of memory cells has spurred several trials. The ability of IFNγ, IL-4, TNFα and lymphotoxin to exert selective effects upon crucial lymphocyte population in vivo may also enable translation into the clinic. Fascinating is the observation that IL-23/IL-23R axis is key to intestinal inflammation. The likelihood is that translational investigators can explore these differences to fashion potent therapies.
Acknowledgments
The authors are supported by grants from the National Institutes of Health (NIH) and the Juvenile Diabetes Research Foundation (JDRF). The authors declare no competing financial interests.
Footnotes
Addresses: Harvard Medical School, Beth Israel Deaconess Medical Center, Center for Life Sciences Building, 3 Blackfan Circle, Boston, MA 02115, United States
Current Opinion in Immunology 2008, 20:676–681
This review comes from a themed issue on Autoimmunity
Edited by Ciriaco Piccirillo and Roberta Pelanda
References and recommended reading
Papers of particular interest published within the period of review have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, Nuñez G, Tang A, Sayegh M, Hancock WW, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5:1303–1307. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Li XC, Zheng XX, Wells AD, Turka LA, Strom TB. Blocking both signal 1 and signal 2 of T-cell activation prevents apoptosis of alloreactive T cells and induction of peripheral allograft tolerance. Nat Med. 1999;5:1298–1302. doi: 10.1038/15256. [DOI] [PubMed] [Google Scholar]
- 3•.Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity. 2001;2(14):105–114. A thorough and thoughtful review of the biology of T cell growth factors and their receptors. Both the redundancies and cytokine specific unique effects mediated by this family are outlined. [PubMed] [Google Scholar]
- 4.Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–678. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 5•.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/s1074-7613(00)80564-6. The potent effects of IL-15 in promoting survival of CD8+ memory T cells and of IL-2 in regulating expansion of these memory T cells is revealed, in references [4,5•], that IL-2 and IL-15 exert polar opposing effects is of considerable interest. [DOI] [PubMed] [Google Scholar]
- 6•.Li XC, Demirci G, Ferrari-Lacraz S, Groves C, Coyle A, Malek TR, Strom TB. IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat Med. 71:2001. 114–118. Again, IL-2 promotes AICD of activated effect on T cells while IL-15 protects such cells from IL-2 triggered death. Again, IL-2 and IL15 exert interesting and opposing effects upon T cells. Taken together with the data in references [4,5•], it appears that IL-2 may hinder while IL-15 may promote cytopathic immunity. [Google Scholar]
- 7•.Oh S, Perera LP, Terabe M, Ni L, Waldmann TA, Berzofsky JA. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc Natl Acad Sci U S A. 2008;105(13):5201–5206. doi: 10.1073/pnas.0801003105. The article refines one mechanism by which IL-15 supports cytopathic immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Malek TR, Porter BO, Codias EK, Scibelli P, Yu A. Normal lymphoid homeostasis and lack of lethal autoimmunity in mice containing mature t cells with severely impaired IL-2 receptors. J Immunol. 2000;164:2905–2914. doi: 10.4049/jimmunol.164.6.2905. [DOI] [PubMed] [Google Scholar]
- 9••.Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 2002;17:167–178. doi: 10.1016/s1074-7613(02)00367-9. The data, in references [7•,8••,9••], conclusively demonstrate an important and non-redundant role for IL-2 in supporting immunoregulatory T cells and thereby maintaining self-tolerance. [DOI] [PubMed] [Google Scholar]
- 10.Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3+ CD25+ CD4+ regulatory T cells by interleukin IL-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;2015:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shevach EM. Certified professionals: CD4+ CD25+ suppressor T cells. J Exp Med. 2001;193(11):F41–F45. doi: 10.1084/jem.193.11.f41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marrack P, Bender J, Hildeman D, Jordan M, Mitchell T, Murakami M, Sakamoto A, Schaefer BC, Swanson B, Kappler J. Homeostasis of αβTCR+ T cells. Nat Immunol. 2000;1:107–111. doi: 10.1038/77778. [DOI] [PubMed] [Google Scholar]
- 13•.Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J, Petersen LJ, Beurskens FJM, Schuurman J, van de Winkel JGJ, et al. Targeting Interleukin-15 in patients with rheumatoid arthritis. Arthritis Rheum. 2005;9:2686–2692. doi: 10.1002/art.21249. [DOI] [PubMed] [Google Scholar]
- 14•.Kim YS, Maslinski W, Zheng XX, Stevens AC, Li XC, Tesch GH, Kelley VR, Strom TB. Targeting the IL-15 receptor with an antagonist IL-15 mutant/Fcg2a protein blocks delayed-type hypersensitivity. J Immunol. 1998;160:5742–5748. [PMC free article] [PubMed] [Google Scholar]
- 15•.Ferrari-Lacraz S, Zanelli E, Neuberg M, Donskoy E, Kim YS, Zheng XX, Hancock WW, Maslinski W, Li XC, Strom TB, et al. Targeting IL-15 receptor-bearing cells with an antagonist mutant IL-15/Fc protein prevents disease development and progression in murine collagen-induced arthritis. J Immunol. 2004;173(9):5818–5826. doi: 10.4049/jimmunol.173.9.5818. References [13•–15•]–These papers collectively point to neutralization of IL-15 or blockade of their receptors as a means to treat autoimmune disorders. [DOI] [PubMed] [Google Scholar]
- 16•.Zheng XX, Sánchez-Fueyo A, Sho M, Domenig C, Sayegh MH, Strom TB. Favorably tipping the balance between cytopathic and regulatory T cells to create transplantation tolerance. Immunity. 2003;19(4):503–514. doi: 10.1016/s1074-7613(03)00259-0. [DOI] [PubMed] [Google Scholar]
- 17••.Koulmanda M, Budo E, Bonner-Weir S, Qipo A, Putheti P, Degauque N, Shi H, Fan Z, Flier JS, Auchincloss H, et al. Modification of adverse inflammation is required to cure new-onset type 1 diabetic hosts. Proc Natl Acad Sci U S A. 2007;104(32):13074–13079. doi: 10.1073/pnas.0705863104. The data, in references [15,16], give support to the concept that manipulating the balance of IL-2 and IL-15 triggered immunity can be used to achieve tolerance in very difficult preclinical models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18•.Zheng XX, Steele AW, Hancock WW, Kawamoto K, Li XC, Nickerson PW, Li Y, Tian Y, Strom TB. IL-2 receptor-targeted cytolytic IL-2/Fc fusion protein treatment blocks diabetogenic autoimmunity in nonobese diabetic mice. J Immunol. 1999;163(7):4041–4048. [PubMed] [Google Scholar]
- 19•.Rabinovitch A, Suarez-Pinzon WL, Shapiro AMJ, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes. 2002;51:638. doi: 10.2337/diabetes.51.3.638. [DOI] [PubMed] [Google Scholar]
- 20•.Tang Q, Adams JS, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;285:687–697. doi: 10.1016/j.immuni.2008.03.016. References [18•–20•] collectively point to IL-2 administration as a means to treat patients with diabetogenic autoimmunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jain R, Tartar DM, Gregg RK, Divekar RD, Bell JJ, Lee HH, Yu P, Ellis JS, Hoeman CM, Franklin CL, et al. Innocuous IFNψ induced by adjuvant-free antigen restores normoglycemia in NOD mice through inhibition of IL-17 production. J Exp Med. 2008;2051:207–218. doi: 10.1084/jem.20071878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Creusot RJ, Yaghoubi SS, Kodama K, Dang DN, Dang VH, Breckpot K, Thielemans K, Gambhir SS, Fathman CG. Tissue-targeted therapy of autoimmune diabetes using dendritic cells transduced to express IL-4 in NOD mice. Clin Immunol. 2008;127:176–187. doi: 10.1016/j.clim.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tisch R, McDevitt H. Insulin dependent diabetes mellitus. Cell. 1996;85(3):291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 24.Yoon JW, Jun HS, Santamaria P. Cellular and molecular mechanisms for the initiation and progression of β-cell destruction resulting from the collaboration between macrophages and T cells. Autoimmunity. 1998;127:109–112. doi: 10.3109/08916939809008041. [DOI] [PubMed] [Google Scholar]
- 25.Alleva DG, Pavlovich RP, Grant C, Kaser SB, Beller DI. Aberrant macrophage cytokine production is a conserved feature among autoimmune-prone mouse strains. Diabetes. 2000;49:1106–1115. doi: 10.2337/diabetes.49.7.1106. [DOI] [PubMed] [Google Scholar]
- 26.Haskins K, Wegmann D. Diabetogenic T-cell clones. Diabetes. 1996;45:1299–1305. doi: 10.2337/diab.45.10.1299. [DOI] [PubMed] [Google Scholar]
- 27.Wu Q, Salomon B, Chen M, Wang Y, Hoffman LM, Bluestone JA, Fu YX. Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J Exp Med. 2001;193(11):1327–1332. doi: 10.1084/jem.193.11.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28••.Kodama S, Kuhtreiber W, Fujimura S, Dale EA, Faustman DL. Islet regeneration during the reversal of autoimmune diabetes in NOD mice. Science. 2003;302:1223–1227. doi: 10.1126/science.1088949. [DOI] [PubMed] [Google Scholar]
- 29••.Ryu S, Kodama S, Ryu K, Schoenfeld DA, Faustman DL. Reversal of established autoimmune diabetes by restoration of endogenous cell function. J Clin Invest. 2001;108(1):63–72. doi: 10.1172/JCI12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Chong AS, Shen A, Tao J, Yin D, Kuznetsov A, Hara N, Philipson LH. Reversal of diabetes in non-obese diabetic mice without spleen cell-derived β cell regeneration. Science. 2006;311:1774–1775. doi: 10.1126/science.1123510. [DOI] [PubMed] [Google Scholar]
- 31••.Nishio J, Gaglia JL, Turvey SE, Campbell C, Benoist C, Mathis D. Islet recovery and reversal of murine type 1 diabetes in the absence of any infused spleen cell contribution. Science. 2006;311:1775–1778. doi: 10.1126/science.1124004. [DOI] [PubMed] [Google Scholar]
- 32••.Suri A, Calderon B, Esparza TJ, Frederick K, Bittner P, Unanue ER. Immunological reversal of autoimmune diabetes without hematopoietic replacement of β cells. Science. 2006;311:1778–1780. doi: 10.1126/science.1123500. [DOI] [PubMed] [Google Scholar]
- 33••.Ban L, Zhang J, Wang L, Kuhtreiber W, Burger D, Faustman DL. Selective death of TNF or TNF receptor 2 agonism. Proc Natl Acad Sci USA. 2008;105(36):13644–13649. doi: 10.1073/pnas.0803429105. References [28••–33••]–The notion that spleen cells help to generate new islets in patients with T1DM is not supported in most studies. These articles reveal significant controversy as to whether administration of TNFα or TNF inducers may provide a rational means by which autoimmune disorders may be treated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- 35••.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 36••.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a Jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37••.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. References [35••–37••] are superb reviews of the immunobiology of regulatory T cells by leaders in the field. [DOI] [PubMed] [Google Scholar]
- 38••.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 39••.Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448(7152):484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40••.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 41••.Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 cells: effector T cells with inflammatory properties. Semin Immunol. 2007;19:362–371. doi: 10.1016/j.smim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.Awasthi A, Carrier Y, Peron JPS, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing antiinflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 43••.Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–333. doi: 10.1038/nri1807. References [38••–43••] are superb papers that demonstrate the decisive role played by the various cytokines in dictating and maintaining the commitment of antigen activated CD4+ cells to difference Th functional and molecular phenotypes. [DOI] [PubMed] [Google Scholar]
- 44••.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–1316. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45••.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-HogenovaH, Cua DJ, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 46••.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, Cua DJ, Powrie F, Cheever AW, Maloy KJ, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47••.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. References [44••–47••] point to the IL-23 axis as central to the expression of intestinal autoimmunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGovern D, Powrie F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut J. 2006;56(10):1333–1336. doi: 10.1136/gut.2006.115402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49••.Koulmanda M, Bhasin M, Hoffman L, Fan Z, Qipo A, Degauque N, Shi H, Bonner-Weir S, Putheti P, Auchincloss H, et al. Curative and beta cell regenerative effects of alpha1 antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci. 2008;105(42):16242–16247. doi: 10.1073/pnas.0808031105. The paper demonstrates that self-tolerance and euglycemia can be restored in NOD mice with frank diabetes through a treatment that alters the inflammatory milieu but does not directly influence T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]