

ABSTRACT
Nosocomial outbreaks of vancomycin-resistant Enterococcus faecium (VREfm) are thought to occur by transmission of VREfm between patients, predicting that infection control interventions will limit cross-transmission. Despite implementation of such strategies, the incidence of VREfm infections continues to rise. We aimed to use genomics to better understand the epidemiology of E. faecium within a large hospital and investigate the reasons for failure of infection control strategies. Whole-genome sequencing was performed on 61 E. faecium (36 VREfm) isolates, predominately from blood cultures collected at a single hospital between 1998 and 2009, and on five vanB-positive anaerobic commensal bacteria isolated from human feces. Phylogenomic analysis and precise mapping of the vanB gene, which contains the Tn1549 transposon, showed that at least 18 of the 36 VREfm isolates had acquired the transposon via independent insertion events, indicating de novo generation of VREfm rather than cross-transmission. Furthermore, Tn1549 sequences found in 15 of the 36 VREfm isolates were the same as the Tn1549 sequence from one of the gut anaerobes. National and international comparator E. faecium isolates were phylogenetically interspersed with isolates from our hospital, suggesting that our findings might be globally representative. These data demonstrate that VREfm generation within a patient is common, presumably occurring in the human bowel during antibiotic therapy, and help explain our inability to reduce VREfm infections. A recommendation from our findings is that infection control practices should include screening patients for specific hospital clones of vancomycin-susceptible E. faecium rather than just VREfm.
IMPORTANCE
Enterococcus faecium is an increasingly important human pathogen causing predominantly antibiotic-resistant infections in hospitalized patients. Large amounts of health care funding are spent trying to control antibiotic-resistant bacteria in hospitals globally, yet in many institutions around the world, vancomycin-resistant E. faecium (VREfm) infections continue to rise. The new findings from this study help explain the failures of our current approaches to controlling vanB VREfm in health care institutions. Given the importance of this bacterium as a cause of hospital-acquired infections and the difficulties faced by infection control units in trying to prevent colonization in their institutions, the novel findings from this study provide evidence that a new approach to controlling VREfm in hospitals is required. In particular, more attention should be given to understanding the epidemiology of hospital-adapted vancomycin-susceptible E. faecium, and patients at higher risk for de novo generation of VREfm need to be identified and optimally managed.
Introduction
Controlling increasing antimicrobial resistance in clinically important bacteria is a key challenge for clinicians and scientists, and there is an urgent need to prevent the emergence and subsequent spread of resistant isolates. Enterococcus faecium is a primary example of one such troublesome pathogen; while innately resistant to many classes of antibiotics, this bacterium has demonstrated a remarkable capacity to evolve new antimicrobial resistances. In fact, E. faecium has been highlighted by the Infectious Diseases Society of America as one of the key problem bacteria, or ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) pathogens, requiring new therapies (1), and there has been a steadily increasing prevalence of E. faecium-related nosocomial infections (2).
Millions of dollars are spent each year by health care systems trying to contain antibiotic-resistant bacteria and prevent cross-transmission. Nosocomial outbreaks of vancomycin-resistant enterococci (VRE) are thought to occur when a patient already carrying VRE in his/her bowel sheds VRE, which are then transmitted by health care workers or via the environment to other patients (3). This model predicts that interventions based on screening and isolation of VRE-colonized patients, improved hand hygiene, and enhanced hospital cleaning will limit cross-transmission (4). Interestingly, interventions at one U.S. hospital based on modified use of antibiotics have also been associated with reducing VRE transmission (5). At the Austin Hospital in Melbourne, Australia (an 800-bed tertiary referral institution), hand hygiene schemes have resulted in a significant reduction in the incidence of methicillin-resistant S. aureus (MRSA); however, the incidence of VRE has continued to rise (6). A similar story has unfolded in other regions, with VRE now being responsible for over 30% of E. faecium bacteremias in some European countries (7) and for outbreaks in other European countries, Asia, and the United States (8–11). In the United Kingdom, concern about increasing VRE incidence led to mandatory reporting of rates of VRE bacteremia, and more than 500 cases per year have been reported since 2004 (http://www.hpa.org.uk/; accessed, 3 December 2012).
Clinically significant VRE strains were first reported from Europe and the United Kingdom in 1986 and in the United States soon after (12–14). Glycopeptide resistance in enterococci is mediated predominantly by mobile gene clusters that confer resistance to vancomycin and teicoplanin (vanA genotype) or vancomycin only (vanB genotype) (15). In the United States, vanA-containing VRE dominate, in Europe, a mix of vanA and vanB-containing VRE is found, while in Australia, vanB-containing VRE dominate (16–18). Nonenterococcal species (predominately anaerobes) may carry the vanB resistance determinant as part of transposon Tn1549 (19) in the human bowel, and in Australia, while carriage of vancomycin-resistant E. faecium (VREfm) in the general community is rare, up to 50% of healthy adults have detectable vanB in their fecal samples as determined by PCR (20, 21). This unexpected finding has also been reported in Canada (22). Transmission of a Tn1549-like element from one human fecal anaerobe, Clostridium symbiosum MLG101, to Enterococcus faecium and Enterococcus faecalis in the digestive tracts of mice has been demonstrated, suggesting that the same process may occur in the human bowel (23).
Recently, the first complete genome sequences of E. faecium have been reported (24, 25), and draft genomes of additional E. faecium isolates have been studied to explore antibiotic resistance mechanisms and phylogenomic relationships (26–29). Hospital-associated ampicillin-resistant E. faecium isolates, which are represented by three dominant sequence types (ST17, ST18, and ST78; ST203 is an ST78 single-locus variant [SLV]), are genetically distinct and replace the normal reservoir of ampicillin-susceptible E. faecium strains in hospitalized patients (24, 25, 30). These three sequence types have been collectively referred to as clonal complex 17 (CC17); however, the growing realization of the significant impact of recombination on the evolution of E. faecium suggests that the CC designation may not be accurate (31, 32) because of the significant genetic variation that has been revealed among these hospital-associated isolates (33). Because of the failure of intense infection control interventions to curtail the rise of VRE, we applied genome sequencing and detailed comparative genomic analysis to a collection of E. faecium isolates to better understand the relationship between vancomycin-susceptible E. faecium (VSEfm) and VREfm isolates and to test the hypothesis that de novo generation of VREfm is a frequent occurrence that is undermining infection control efforts.
RESULTS
In this study, we sequenced and compared the genomes of 61 E. faecium isolates. Fifty-six were bloodstream isolates from unique patients over a 12-year period in a single institute in Melbourne, Australia (32 VREfm and 24 VSEfm isolates), 3 were rectal-screening isolates from Royal Perth Hospital in western Australia (all VREfm), and 2 were clinical isolates from Royal Darwin Hospital, Darwin, Northern Territory, Australia (1 VREfm and 1 VSEfm isolate). Relevant epidemiological information and a summary of sequencing results are presented in Dataset S1 in the supplemental material. The AUS0004 (ST17) (24) and AUS0085 (ST203) (Margaret M. C. Lam, Torsten Seemann, Nicholas J. Tobias, Honglei Chen, Volker Haring, Robert J. Moore, Susan Ballard, M. Lindsay Grayson, Paul D. R. Johnson, Benjamin P. Howden, and Timothy P. Stinear, submitted for publication) genome sequences were used as primary references, and two publicly available international E. faecium blood culture isolates were included for comparison: the ST18 DO (TX16) strain isolated from Texas in 1998 (25) and the ST203 strain 1,231,502 from the Broad Institute collection (26). In addition, five anaerobic bacteria previously isolated from human feces and demonstrated to carry Tn1549 within the vanB operon were also subjected to genome sequencing to allow comparison of the vanB transposon sequences from these anaerobes and the VREfm isolates. The details and sequencing results for these isolates are summarized in Dataset S2 in the supplemental material.
To address the question of whether patients develop VREfm within their bowel or acquire VREfm from an exterior source, we first conducted a detailed analysis of the characteristics of the Tn1549 transposon among the vanB-positive isolates. In our recent comparison of the ST203 VREfm (AUS0085) and ST17 VRE (AUS0004) whole genomes (Margaret M. C. Lam, Torsten Seemann, Nicholas J. Tobias, Honglei Chen, Volker Haring, Robert J. Moore, Susan Ballard, M. Lindsay Grayson, Paul D. R. Johnson, Benjamin P. Howden, and Timothy P. Stinear, submitted for publication), we identified a number of Tn1549 features that would permit identification of unique transposon acquisition events, including transposon sequence type, insertion site, insertion direction, and coupling sequence composition (Fig. 1). Analysis of the 36 VREfm isolates and the anaerobic bacteria in this study found that 15 VREfm isolates harbored a larger 57-kb version of Tn1549 than the other isolates (Fig. 2). The Tn1549 phylogeny, inferred by comparing the 118 variable nucleotides within this element among all isolates (Fig. 3A), revealed two dominant populations of Tn1549, those represented by the sequences from the short, 33-kb element from AUS0069 and those represented by the 57-kb element from AUS0085. Overall, there were five different transposon sequence groups among the 36 VREfm isolates (based on a difference of three or more single nucleotide polymorphisms [SNPs]) (Fig. 3A). Notably, Tn1549 sequences identified in the anaerobic bacteria were similar to those from VREfm isolates. Remarkably, the Tn1549 sequence from Clostridium sp. strain MLG055, isolated from the feces of a dialysis patient in Melbourne, Australia (20), was identical to Tn1549 from a number of VREfm isolates from bloodstream infections, indicating that transfer of this element among the bowel microbiota may occur.
FIG 1 .
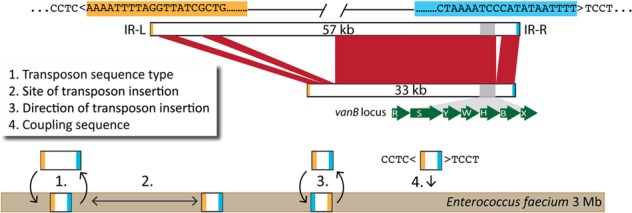
Schematic of the Tn1549 characteristics of E. faecium isolates in this study. Characteristics used to identify unique Tn1549 transmission events in vanB-containing E. faecium isolates, including a unique transposon sequence (1), an insertion site (2), the insertion orientation (3), and the flanking coupling sequence (4). Flanking imperfect left and right inverted repeat (IR-L and IR-R) insertion sites are highlighted in orange and blue, respectively. The vanB locus contains vancomycin resistance genes (green), which are conserved in all vanB sequence types, as well as other genes, which leads to variation in total transposon size, as indicated.
FIG 2 .

Comparison of Tn1549 sequences. DNA-DNA comparison of Tn1549 sequences (33 kb, short version [S]) from AUS0004 and AUS0085 (57 kb, long version [L]), visualized with the Artemis Comparison Tool, showing the location of the vanB locus in both elements (orange shading) and the putative novel conjugation locus (pink shading) in the long version of Tn1549. Dark-blue shading indicates regions of DNA identity. Percentages of GC content (500-bp sliding window) are shown above, followed by heatmap visualizations of results of read mapping normalized to both AUS0004 and AUS0085 for a representative selection of VREfm and vanB-positive bowel anaerobes. Note that changes in read mapping density across Tn1549 are explained by sequencing biases, as these regions correlate with significant changes in GC content.
FIG 3 .

Phylogeny of Tn1549 elements and Enterococcus faecium. (A) SNP-based phylogeny of Tn1549 elements in E. faecium and anaerobes (red dots indicate anaerobes). The color scale represents the Tn1549 sequence type (based on SNP analysis) as well as the transposon insertion site in the E. faecium chromosome, relative to the AUS0004 chromosome sequence. Black bars within the red transposon sequence groups represent single nucleotide differences. The coupling sequence is the 5- to 8-bp sequence introduced from the donor during transposition (“<>” refers to the site of insertion, as shown in Figure 1; * indicates nucleotide deletion). Isolates shaded in pink contain the shorter (33-kb) version of Tn1549, while those in blue contain the longer (57-kb) version of Tn1549. (B) Overview of the seven unique transposon insertion sites for the 36 VREfm clones mapped to the AUS0004 chromosome, as well as the transposon sequence type. The color scheme follows that described in panel A. Outter numbers indicate numbers of isolates with insertions at that position. Inner numbers indicate the insertion position in the AUS0004 chromosome. (C) Maximum-likelihood phylogeny for E. faecium based on SNPs in the core genome, with major multilocus sequence type groups highlighted. Isolates from other institutions are highlighted in yellow and include five isolates from other institutions in Australia (RPH isolates from Perth; isolates 928 and 755 from Darwin) and two isolates from the United States sequenced by others (DO, 1-231-502). Colored bars indicate transposon sequences and insertion sites as defined for panel A. Gray triangles indicate predicted independent VREfm clones based on Tn1549 characteristics and core genome phylogeny. The node represented by the black circle shows the estimated divergence date for these ST17 and ST252 isolates, as inferred by Path-O-Gen.
The chromosomal insertion site of all Tn1549-like elements for the 36 VREfm isolates was investigated, revealing a restricted repertoire of insertions, indicating that Tn1549 insertion is not random (Fig. 3B). Notably, the VREfm isolates contained only a single copy of Tn1549. Seven different chromosomal insertion sites were identified, yet transposons of the same sequence were consistently inserted at the same chromosomal location (Fig. 3A and B). For example, transposons represented by isolate 69 (red in Fig. 3A) all integrated within EFAU004_00592, while transposons represented by isolate 85 (blue in Fig. 3A) all integrated within the signal peptidase I gene (EFAU004_00734). In conjunction with the core genome phylogeny (see below), we then used Tn1549 sequence type, site of insertion, insertion orientation, and coupling sequence to distinguish inherited from independent Tn1549 acquisitions among E. faecium isolates (Fig. 1 and 3).
To understand the population structure of the E. faecium strains included in this study, a core genome E. faecium phylogeny was constructed using 14,988 SNPs identified among the 63 isolates (excluding insertions and deletions). Split decomposition analysis produced a reticulate phylogeny, suggesting that recombination contributes to the phylogenetic signal (data not shown). Because recombination would perturb the phylogeny, we employed an iterative algorithm that searched for a high density of SNPs and excluded chromosome regions likely acquired by recombination (Fig. 4). This analysis predicted 297 recombination events, spanning 1.3 Mb (44% of the chromosome) and accounting for 12,849 SNPs (85.7% of all nucleotide variation). The remaining 2,139 SNPs were aligned among all isolates, and a tree was inferred using maximum likelihood based on these vertically inherited base substitutions alone (Fig. 3C).
FIG 4 .

Recombination in E. faecium. Areas of predicted recombination in the E. faecium chromosome are highlighted, based on SNP density. MLST alleles are indicated by triangles. Only purK and adk are in regions without predicted recombination. Two isolates (69 and 71, circled) are part of the ST252 complex by MLST but cluster with the ST203 isolates based on core genome SNP analysis. Regions in red are recombinant in multiple isolates in the collection, while regions in blue are unique to one strain.
Isolates generally clustered within their previously defined multilocus sequence types (MLSTs); however, there were also some clear exceptions highlighting the impact of recombination events on MLSTs. Five of the seven MLST loci were impacted by recombination (Fig. 4). For example, isolates 69 (SLV of ST252) and 71 (ST252) clustered among the ST203 isolates based on the nonrecombinant core genome analysis (Fig. 4). As expected, the non-CC17 isolates (isolates 10, 92, and 101) were more distantly related to all other isolates. Significantly, CC17 VSEfm isolates were closely related to, and in some cases essentially indistinguishable from, CC17 VREfm isolates at this core genome level, suggesting that the CC17 VSEfm and CC17 VREfm strains were part of the same circulating population rather than that they evolved independently. Thus, the evolution of VREfm strains from colonizing VSEfm strains, and not just the clonal spread of VREfm strains, explains the epidemiology of invasive VREfm disease observed in this study, suggesting that the epidemiology of VSEfm in hospitals is a key factor driving the emergence of clonally related VREfm.
In addition to examining the evolutionary relationship between the isolates based on a core genome, we also assessed an E. faecium pan genome which included plasmids, prophages, and other genomic elements (Dataset S3 in the supplemental material and Fig. 5). The pan genome included more than 5 Mb of sequence bearing >5,000 genes and demonstrated that even among isolates that were found to be very closely related based on their core genomes, variability in gene content was detected. For example, isolates 73 and RPH3 had closely related core genomes; however, isolate 73 harbored a unique region (EFPAN04301 to EFPAN04389) that likely represented a plasmid with a large number of hypothetical proteins and plasmid-associated genes. Similarly, isolates 72 and 78 displayed closely related core genomes, with isolate 72 harboring Tn1549 and isolate 78 lacking this element but possessing a unique prophage (EFPAN02546 to EFPAN 02583).
FIG 5 .

E. faecium pan genome. Blue bar indicates the 5.4-Mb E. faecium pan genome, including 5,000 annotated genes, detected among genomes analyzed in this study. A heat map shows the presence (black) and absence (white) of the 5,000 genes. Dataset S3 in the supplemental material summarizes the variable regions and pan-genome contents for all isolates and includes EFPAN locus tags for all predicted coding sequences.
In some cases, the VREfm phylogeny was suggestive of clonal dissemination of an extant VREfm clone. For example, the ST203 cluster that included isolates 149, 86, 85, 81, 77, 93, and 96 was isolated over a 12-month period from patients in the same hospital (Fig. 3C). Inspection of the pan genomes for these isolates (Fig. 5) suggests that they may have been transmitted directly between patients, except for isolates 86 and 96, where differences in gene content, possibly due to plasmid acquisition, were detected. For example, isolate 86 contained the genes EFPAN04820 to EFPAN04855, encoding a number of hypothetical proteins and a type IV secretion system, while isolate 96 harbored a unique region (EFPAN04913 to EFPAN04927). In contrast, we were able to combine the detailed transposon analysis with the core genome phylogeny to define independent episodes of VREfm evolution (Fig. 3C).
At least 18 distinct episodes of VREfm evolution of the total 36 VREfm isolates were detected, indicating that de novo evolution of VREfm strains is an important source of VREfm infection at our hospital. For example, the ST17 isolates 4 and 21 are closely related at the core genome level, but their transposon sequence and insertion sites are different. Furthermore, the dominant “red-orange” and “blue-aqua” Tn1549 types are dispersed throughout the E. faecium core genome phylogeny and can be explained by instances of independent Tn1549 acquisition (Fig. 3C). In the ST252 cluster, four VREfm bloodstream isolates were sequenced from our hospital, and at least three of these indicate the independent evolution of VREfm strains.
In the 33-month period from March 2007 to November 2009, we had an outbreak of clinical VRE infection at our institution (6). Based on MLST analysis, it appeared that we had a clonal outbreak of VREfm due to the introduction of a new clone of ST203 VREfm. However, analysis of the ST203 VREfm genome data from these isolates demonstrates that at least 9 unique clones of ST203 VREfm have emerged (out of the 22 sequenced). These unique ST203 VREfm isolates highlight the polyclonal nature of VREfm at our hospital, even within a single MLST cluster that was first detected as late as 2007.
In order to determine if these observations were unusual and restricted to our hospital, we included genome sequences from geographically dispersed E. faecium isolates for comparison. The more recent isolates from other Australian cities were closely related to ST203 and ST252 isolates from our institution (highlighted in yellow in Fig. 3C). The older ST18 VREfm isolate from the United States (DO strain, 1998) was more distantly related to the majority of E. faecium isolates in this study. The recent VRE isolate 1-231-502 (2008) was very similar to ST203 isolates from our institution, suggesting global dissemination of this E. faecium clone.
The collection of E. faecium isolates from a single hospital over a 12-year period allowed us to analyze the evolutionary changes and mutation rate in the isolate collection. For this analysis, we used only ST17 and ST252 isolates from our institution, because they were isolated throughout the 12-year study period. This analysis revealed a strong molecular clock signal (Fig. 6) and allowed estimation of a core genome mutation rate of 1.5 × 10−6 substitutions per site per year or ~5 SNPs per genome per year (excluding SNPs introduced via recombination). This analysis suggested that the ST17/ST252 isolates from the collection arose from a common ancestor that existed in the late 1970s.
FIG 6 .
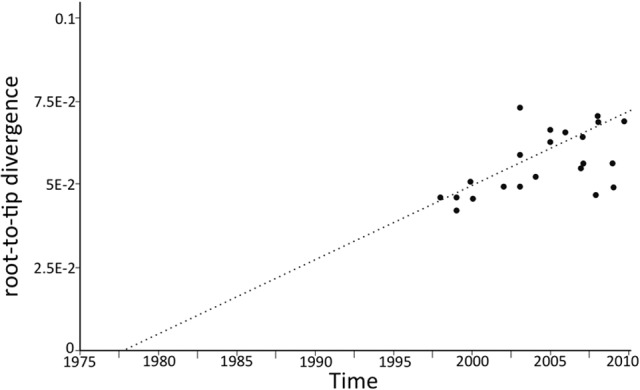
Evolution of E. faecium ST17 and ST252 isolates. Estimate of the divergence date for ST17 and ST252 isolates by branch-to-tip analysis using Path-O-Gen and branch lengths inferred by maximum-likelihood analysis (http://tree.bio.ed.ac.uk/software/pathogen/).
DISCUSSION
Using large-scale comparative genomics, we have demonstrated that hospital vanB VREfm epidemiology is far more complex than previously recognized and is significantly driven by de novo VREfm generation rather than simply nosocomial transmission of extant VREfm strains. We found that many distinct clones of vanB VREfm were responsible for bloodstream infection isolates from patients at our institution. In fact, at least 18 of 36 VREfm isolates sequenced in this study were due to unique VREfm evolution events. This includes a time period where a new sequence type of E. faecium (ST203) emerged in our institution and caused what we thought was a clonal outbreak (6). Additionally, circulating clones of VSEfm and VREfm were phylogenetically intermingled. These findings indicate that unique VREfm evolution events are common, presumably occurring in the human bowel, and taken together suggest that control strategies based on screening for VREfm and isolation of colonized patients might not be completely effective.
We identified a number of Tn1549 features that could be utilized to determine independent VREfm acquisition events (Fig. 1), demonstrating the power of genome sequencing to uncover complex relationships between bacteria. Significantly, the transposon sequences from anaerobic bacteria isolated from human feces were very similar and, in one case, identical to those from VREfm isolates associated with bloodstream infections in humans, suggesting that anaerobic bacteria might form an important component of the Tn1549 reservoir. If the transposon pool is restricted and the VSEfm population is highly clonal, as occurs in hospitalized patients (30), even unique in vivo generation episodes of VREfm may appear like clonal spread of preformed VRE. This issue was highlighted by detailed analysis of three ST203 isolates, 149, 86, and 96. Based on core genome phylogeny and transposon analysis, these isolates appeared clonal; however, analysis of the pan-genome components of these isolates (Fig. 5) demonstrated additional genome diversity, in some cases driven by acquisition of plasmids. While it is possible that individual genome variability evolved in a clonally disseminated preformed VREfm strain, it is also possible that clonally dispersed (but not identical) VSEfm isolates have independently acquired an identical Tn1549 element. If this is the case, then even more than 18 of the 36 VREfm isolates sequenced in this study may have evolved independently.
While we focused our investigation on a single institution with the primary aim of understanding the persistent VREfm outbreak that we had noted, we included isolates from other Australian cities and the United States. Sequences from these isolates were very closely related to VREfm isolates from our institution in Melbourne, Australia, including the United States isolate (1,231,502). Hospital-hospital transfer of colonized patients may explain this observation in Australia; however, such movements are infrequent between Darwin and Melbourne. In our view, the best model which explains our observations is silent circulation internationally of closely related VSEfm isolates, with local Tn1549 transposition events, which then lead to the appearance of new clinical cases of VRE colonization and infection in hospitals.
The E. faecium genome is known to have high rates of recombination, especially compared to other important hospital pathogens, such as Staphylococcus aureus (25, 34, 35). Here we applied whole-genome sequencing and SNP clustering analysis to infer a phylogeny and predict regions of recombination within the 63 E. faecium genomes used in this study (Fig. 3 and 4). A total of 1.3 Mb (44%) of the chromosome was affected by recombination. This includes five of the seven MLST loci (Fig. 4), indicating that MLST analysis of E. faecium based on the currently selected alleles may give misleading results and must be interpreted with care. We also defined an E. faecium pan genome, which includes 1,872 core genes as well as 3,128 accessory genes identified within the 63 E. faecium isolates in this study (Fig. 5). This indicates significant plasticity within the E. faecium genome, with large accessory elements potentially impacting the clinical behavior of these isolates, as previously suggested (25, 33). Clonal complex 17 E. faecium emerged as an important hospital-associated pathogen in the 1980s (36). By analyzing the ST17/ST252 isolates from this study, we found a mutation rate in the core genome of 5 SNPs per genome per year and predicted that the common ancestor strain for E. faecium at our institution existed around 1978 (95% confidence interval [CI], 1973 to 1983), in keeping with the appearance of CC17 E. faecium and VREfm in the 1980s.
Our findings have implications for the control of VREfm in hospitals, where generation of new VREfm clones is a frequent event. Because a high proportion of hospital patients carry nonenterococcal vanB-containing organisms in their bowels (19, 20, 22, 37), we suggest that specific VSEfm clones should also be targets for control in the hospital environment. It remains to be determined if hospital-associated VSEfm is acquired nosocomially by patients, as suggested in European studies, or if in fact these isolates of VSEfm are present in the community in Australia (30, 38). A complete understanding of VSE epidemiology and the biology of the Tn1549 element will be critical to improving strategies to curtail the VRE epidemic. These data dictate that alternative strategies directed at identifying and isolating patients colonized with high-risk VSEfm clones, identifying patients who harbor vanB-containing Tn1549 and who are likely at increased risk of in vivo generation, and identifying and modifying the selective pressures promoting in vivo evolution are needed to prevent the ongoing emergence of VREfm as a public health threat.
MATERIALS AND METHODS
Bacterial isolates.
Isolates used in this study were vancomycin-susceptible and -resistant Enterococcus faecium strains recovered between 1998 and 2009 from blood culture specimens at Austin Health, a tertiary referral hospital in Melbourne, Australia. Additionally, five recent clinical and screening isolates of VSEfm and VREfm from other cities in Australia (Darwin and Perth) were included for comparison, as were five previously identified and characterized human bowel anaerobes containing Tn1549 within their vanB operon that were isolated in Melbourne, Australia (19, 20). All VREfm isolates were found to display the vanB genotype, as determined by PCR (17). Enterococcus faecium isolates were previously analyzed by MLST and were found to belong predominately to clonal complex 17 (CC17) (6). Epidemiological data, including date and location of isolation, were recorded. A summary of isolates and epidemiological data is provided in Dataset S1 (E. faecium) and Dataset S2 (anaerobes) in the supplemental material.
Genome sequencing and analysis.
Genomic DNA was extracted, and all isolates were subjected to whole-genome shotgun sequencing using an Illumina HiSeq-2000 sequencing system and 100-bp-paired-end TruSeq chemistry or Ion Torrent single-end 100-bp or 200-bp sequencing chemistry (39) (Datasets S1 and S2). A read mapping approach was used to align the sequences from these isolates with the recently completed E. faecium ST203 genome, AUS0085 (Margaret M. C. Lam, Torsten Seemann, Nicholas J. Tobias, Honglei Chen, Volker Haring, Robert J. Moore, Susan Ballard, M. Lindsay Grayson, Paul D. R. Johnson, Benjamin P. Howden, and Timothy P. Stinear, submitted for publication), using SHRiMP v2.0 (40). Single nucleotide polymorphisms (SNPs) in the E. faecium core genome were identified using Nesoni v0.93, which aligns the read of each genome to that of the reference to construct a tally of putative differences at each position (http://www.bioinformatics.net.au). In order to exclude regions of suspected recombination from the phylogenomic analysis, regions with high frequencies of polymorphic nucleotides were removed from the alignment, as previously described (41, 42), and a phylogeny was inferred using maximum likelihood with RAxML (43). The temporal signals in the sequence data were analyzed using Path-O-Gen (http://tree.bio.ed.ac.uk/software/pathogen/). SNPs likely introduced via recombination were identified using the method described in Croucher et al. (41).
To investigate the total E. faecium gene content, an E. faecium pan genome was defined after de novo assembly using Velvet v1.2.04 (44) and alignment of contigs to the AUS0085 genome using MUMmer (45). Unaligned contigs were appended to the AUS0085 genome to construct a pan genome, which was annotated using Prokka (http://bioinformatics.net.au/). The proportion of the length of each annotated gene covered by reads was assessed for each isolate and a map summarizing all variable genes and their distribution in each strain produced.
Variable nucleotide positions in Tn1549 for all VREfm isolates and anaerobes were also identified, and a Tn1549 phylogeny was inferred using the neighbor-joining method with uncorrected P distances, as implemented in SplitsTree v4.12.3 (46). In addition, the chromosomal Tn1549 insertion site was determined by aligning the contigs that spanned the left- and right-hand ends of Tn1549 (obtained from de novo assembly of each VREfm clone, as described above) to the AUS0004 reference genome. Other features of the Tn1549 insertion that were determined included the orientation of insertion within the chromosome and identification of the 5- to 8-bp coupling sequence. Separate clones of VREfm (unique VREfm generation events) were defined as taxa with distinct core genome phylogenies, or if they possessed specific Tn1549 characteristics, they were defined by including the variation in their transposon sequence, the insertion site, the insertion orientation, or the coupling sequence. Differences in the pan genomes were also used to help define independent VREfm clones.
Nucleotide sequence accession number.
The sequence reads for all isolates have been submitted to GenBank under BioProject identifier PRJNA205886.
SUPPLEMENTAL MATERIAL
Enterococcus faecium metadata. Enterococcus faecium strain characteristics and sequencing results for isolates included in this study. Download
Anaerobic bacteria metadata. Epidemiological data and sequencing results for anaerobic bacteria included in this study. Download
Enterococcus faecium pan-genome contents of all isolates. Summary of the E. faecium accessory elements used to define the pan genome and presence or absence of these elements in each strain. Download
ACKNOWLEDGMENTS
This research was supported by a grant from the National Health and Medical Research Council (NHMRC) of Australia (number 1027874) and the Austin Hospital Medical Research Foundation. Fellowships from the NHMRC of Australia supported B.P.H. (number 1023526), K.E.H. (number 628930), S.Y.C.T. (number 508829), and T.P.S. (number 1008549).
We thank Matthew Dindinger and Clarence Lee from Life Technologies for sequencing 4 of the 61 E. faecium genomes and Elizabeth Grabsch from Austin Hospital Microbiology for technical assistance.
Footnotes
Citation Howden BP, Holt KE, Lam MMC, Seemann T, Ballard S, Coombs GW, Tong SYC, Grayson ML, Johnson PDR, Stinear TP. 2013. Genomic insights to control the emergence of vancomycin-resistant enterococci. mBio 4(4):e00412-13. doi:10.1128/mBio.00412-13.
REFERENCES
- 1. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update.Clin. Infect. Dis. 48:1–12 [DOI] [PubMed] [Google Scholar]
- 2. Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, National Healthcare Safety Network Team. Participating National Healthcare Safety Network Facilities 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 29:996–1011 [DOI] [PubMed] [Google Scholar]
- 3. Ridwan B, Mascini E, van der Reijden N, Verhoef J, Bonten M. 2002. What action should be taken to prevent spread of vancomycin resistant enterococci in European hospitals? BMJ 324:666–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Austin DJ, Bonten MJ, Weinstein RA, Slaughter S, Anderson RM. 1999. Vancomycin-resistant enterococci in intensive-care hospital settings: transmission dynamics, persistence, and the impact of infection control programs. Proc. Natl. Acad. Sci. U. S. A. 96:6908–6913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quale J, Landman D, Saurina G, Atwood E, DiTore V, Patel K. 1996. Manipulation of a hospital antimicrobial formulary to control an outbreak of vancomycin-resistant enterococci. Clin. Infect. Dis. 23:1020–1025 [DOI] [PubMed] [Google Scholar]
- 6. Johnson PD, Ballard SA, Grabsch EA, Stinear TP, Seemann T, Young HL, Grayson ML, Howden BP. 2010. A sustained hospital outbreak of vancomycin-resistant Enterococcus faecium bacteremia due to emergence of vanB E. faecium sequence type 203. J. Infect. Dis. 202:1278–1286 [DOI] [PubMed] [Google Scholar]
- 7. de Kraker ME, Jarlier V, Monen JC, Heuer OE, van de Sande N, Grundmann H. 3 October 2012. The changing epidemiology of bacteraemias in Europe: trends from the European Antimicrobial Resistance Surveillance System. Clin. Microbiol. Infect. 10.1111/1469-0691.12028 [DOI] [PubMed] [Google Scholar]
- 8. Bourdon N, Fines-Guyon M, Thiolet JM, Maugat S, Coignard B, Leclercq R, Cattoir V. 2011. Changing trends in vancomycin-resistant enterococci in French hospitals, 2001–08. J. Antimicrob. Chemother. 66:713–721 [DOI] [PubMed] [Google Scholar]
- 9. Hsieh YC, Ou TY, Teng SO, Lee WC, Lin YC, Wang JT, Chang SC, Lee WS. 2009. Vancomycin-resistant enterococci in a tertiary teaching hospital in Taiwan. J. Microbiol. Immunol. Infect. 42:63–68 [PubMed] [Google Scholar]
- 10. Zhu X, Zheng B, Wang S, Willems RJ, Xue F, Cao X, Li Y, Bo S, Liu J. 2009. Molecular characterisation of outbreak-related strains of vancomycin-resistant Enterococcus faecium from an intensive care unit in Beijing, China. J. Hosp. Infect. 72:147–154 [DOI] [PubMed] [Google Scholar]
- 11. Martone WJ. 1998. Spread of vancomycin-resistant enterococci: why did it happen in the United States? Infect. Control Hosp. Epidemiol. 19:539–545 [DOI] [PubMed] [Google Scholar]
- 12. Leclercq R, Derlot E, Duval J, Courvalin P. 1988. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med. 319:157–161 [DOI] [PubMed] [Google Scholar]
- 13. Uttley AH, Collins CH, Naidoo J, George RC. 1988. Vancomycin-resistant enterococci. Lancet i:57–58 [DOI] [PubMed] [Google Scholar]
- 14. Frieden TR, Munsiff SS, Low DE, Willey BM, Williams G, Faur Y, Eisner W, Warren S, Kreiswirth B. 1993. Emergence of vancomycin-resistant enterococci in New York City. Lancet 342:76–79 [DOI] [PubMed] [Google Scholar]
- 15. Courvalin P. 2006. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 42(Suppl 1):S25–S34 [DOI] [PubMed] [Google Scholar]
- 16. Werner G, Coque TM, Hammerum AM, Hope R, Hryniewicz W, Johnson A, Klare I, Kristinsson KG, Leclercq R, Lester CH, Lillie M, Novais C, Olsson-Liljequist B, Peixe LV, Sadowy E, Simonsen GS, Top J, Vuopio-Varkila J, Willems RJ, Witte W, Woodford N. 2008. Emergence and spread of vancomycin resistance among enterococci in Europe. Euro Surveill. 13(47):pii=19046 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=19046 [PubMed] [Google Scholar]
- 17. Bell JM, Paton JC, Turnidge J. 1998. Emergence of vancomycin-resistant enterococci in Australia: phenotypic and genotypic characteristics of isolates. J. Clin. Microbiol. 36:2187–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Top J, Willems R, Bonten M. 2008. Emergence of CC17 Enterococcus faecium: from commensal to hospital-adapted pathogen. FEMS Immunol. Med. Microbiol. 52:297–308 [DOI] [PubMed] [Google Scholar]
- 19. Ballard SA, Pertile KK, Lim M, Johnson PD, Grayson ML. 2005. Molecular characterization of vanB elements in naturally occurring gut anaerobes. Antimicrob. Agents Chemother. 49:1688–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stinear TP, Olden DC, Johnson PD, Davies JK, Grayson ML. 2001. Enterococcal vanB resistance locus in anaerobic bacteria in human faeces. Lancet 357:855–856 [DOI] [PubMed] [Google Scholar]
- 21. Graham M, Ballard SA, Grabsch EA, Johnson PD, Grayson ML. 2008. High rates of fecal carriage of nonenterococcal vanB in both children and adults. Antimicrob. Agents Chemother. 52:1195–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mak A, Miller MA, Chong G, Monczak Y. 2009. Comparison of PCR and culture for screening of vancomycin-resistant enterococci: highly disparate results for vanA and vanB. J. Clin. Microbiol. 47:4136–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Launay A, Ballard SA, Johnson PD, Grayson ML, Lambert T. 2006. Transfer of vancomycin resistance transposon Tn 1549 from Clostridium symbiosum to Enterococcus spp. in the gut of gnotobiotic mice. Antimicrob. Agents Chemother. 50:1054–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lam MM, Seemann T, Bulach DM, Gladman SL, Chen H, Haring V, Moore RJ, Ballard S, Grayson ML, Johnson PD, Howden BP, Stinear TP. 2012. Comparative analysis of the first complete Enterococcus faecium genome. J. Bacteriol. 194:2334–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qin X, Galloway-Peña JR, Sillanpaa J, Roh JH, Nallapareddy SR, Chowdhury S, Bourgogne A, Choudhury T, Muzny DM, Buhay CJ, Ding Y, Dugan-Rocha S, Liu W, Kovar C, Sodergren E, Highlander S, Petrosino JF, Worley KC, Gibbs RA, Weinstock GM, Murray BE. 2012. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol. 12:135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Palmer KL, Carniol K, Manson JM, Heiman D, Shea T, Young S, Zeng Q, Gevers D, Feldgarden M, Birren B, Gilmore MS. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J. Bacteriol. 192:2469–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS. 2012. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 3(1):e00318-11. 10.1128/mBio.00318-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-cas. mBio 1(4):e00227-10. 10.1128/mBio.00227-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Paganelli FL, Bierschenk D, Kuipers A, Bonten MJ, Willems RJ, van Schaik W. 2012. Genome-wide identification of ampicillin resistance determinants in Enterococcus faecium. PLoS Genet. 8(6):e1002804. 10.1371/journal.pgen.1002804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weisser M, Oostdijk EA, Willems RJ, Bonten MJ, Frei R, Elzi L, Halter J, Widmer AF, Top J. 2012. Dynamics of ampicillin-resistant Enterococcus faecium clones colonizing hospitalized patients: data from a prospective observational study. BMC Infect. Dis. 12:68. 10.1186/1471-2334-12-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuch A, Willems RJ, Werner G, Coque TM, Hammerum AM, Sundsfjord A, Klare I, Ruiz-Garbajosa P, Simonsen GS, van Luit-Asbroek M, Hryniewicz W, Sadowy E. 2012. Insight into antimicrobial susceptibility and population structure of contemporary human Enterococcus faecalis isolates from Europe. J. Antimicrob. Chemother. 67:551–558 [DOI] [PubMed] [Google Scholar]
- 32. Bonten MJ, Willems RJ. 2012. Vancomycin-resistant Enterococcus—chronicle of a foretold problem. Ned. Tijdschr. Geneeskd. 156(38):A5233. (In Dutch.) [PubMed] [Google Scholar]
- 33. van Schaik W, Top J, Riley DR, Boekhorst J, Vrijenhoek JE, Schapendonk CM, Hendrickx AP, Nijman IJ, Bonten MJ, Tettelin H, Willems RJ. 2010. Pyrosequencing-based comparative genome analysis of the nosocomial pathogen Enterococcus faecium and identification of a large transferable pathogenicity island. BMC Genomics 11:239. 10.1186/1471-2164-11-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Willems RJ, Top J, van Schaik W, Leavis H, Bonten M, Sirén J, Hanage WP, Corander J. 2012. Restricted gene flow among hospital subpopulations of Enterococcus faecium. mBio 3(4):e00151-12. 10.1128/mBio.00151-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leendertse M, Willems RJ, Giebelen IA, Roelofs JJ, Bonten MJ, van der Poll T. 2009. Neutrophils are essential for rapid clearance of Enterococcus faecium in mice. Infect. Immun. 77:485–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Galloway-Peña JR, Nallapareddy SR, Arias CA, Eliopoulos GM, Murray BE. 2009. Analysis of clonality and antibiotic resistance among early clinical isolates of Enterococcus faecium in the United States. J. Infect. Dis. 200:1566–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ballard SA, Grabsch EA, Johnson PD, Grayson ML. 2005. Comparison of three PCR primer sets for identification of vanB gene carriage in feces and correlation with carriage of vancomycin-resistant enterococci: interference by vanB-containing anaerobic bacilli. Antimicrob. Agents Chemother. 49:77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Regt MJ, van Schaik W, van Luit-Asbroek M, Dekker HA, van Duijkeren E, Koning CJ, Bonten MJ, Willems RJ. 2012. Hospital and community ampicillin-resistant Enterococcus faecium are evolutionarily closely linked but have diversified through niche adaptation. PLoS One 7:e30319. 10.1371/journal.pone.0030319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, Hoon J, Simons JF, Marran D, Myers JW, Davidson JF, Branting A, Nobile JR, Puc BP, Light D, Clark TA, Huber M, Branciforte JT, Stoner IB, Cawley SE, Lyons M, Fu Y, Homer N, Sedova M, Miao X, Reed B, Sabina J, Feierstein E, Schorn M, Alanjary M, Dimalanta E, Dressman D, Kasinskas R, Sokolsky T, Fidanza JA, Namsaraev E, McKernan KJ, Williams A, Roth GT, Bustillo J. 2011. An integrated semiconductor device enabling non-optical genome sequencing. Nature 475:348–352 [DOI] [PubMed] [Google Scholar]
- 40. Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. 2009. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 5(5):e1000386. 10.1371/journal.pcbi.1000386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holt KE, Baker S, Weill FX, Holmes EC, Kitchen A, Yu J, Sangal V, Brown DJ, Coia JE, Kim DW, Choi SY, Kim SH, da Silveira WD, Pickard DJ, Farrar JJ, Parkhill J, Dougan G, Thomson NR. 2012. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat. Genet. 44:1056–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690 [DOI] [PubMed] [Google Scholar]
- 44. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12. 10.1186/gb-2004-5-6-p12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23:254–267 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Enterococcus faecium metadata. Enterococcus faecium strain characteristics and sequencing results for isolates included in this study. Download
Anaerobic bacteria metadata. Epidemiological data and sequencing results for anaerobic bacteria included in this study. Download
Enterococcus faecium pan-genome contents of all isolates. Summary of the E. faecium accessory elements used to define the pan genome and presence or absence of these elements in each strain. Download