

ABSTRACT
In order to survive and persist in an immunocompetent human host, Borrelia burgdorferi controls the human immune attack and blocks the damaging effects of the activated complement system. These Gram-negative spirochetes use CspA (CRASP-1) and four additional immune evasion proteins to bind combinations of human plasma regulators, including factor H, factor H-like protein 1 (FHL-1), complement factor H-related protein 1 (CFHR1), CFHR2, CFHR5, and plasminogen. As many microbial immune evasion proteins have multiple functions, we hypothesized that CspA has additional roles in complement or immune control. Here, we identify CspA as a terminal complement inhibitor. Borrelial CspA binds the human terminal complement components C7 and C9 and blocks assembly and membrane insertion of the terminal complement complex (TCC). CspA inhibits TCC assembly at the level of C7, as revealed by hemolytic assays, and inhibits polymerization of C9. CspA, when ectopically expressed on the surface of serum-sensitive Borrelia garinii, blocks TCC assembly on the level of C7 and induces serum resistance in the transformed bacteria. This CspA-mediated serum resistance and terminal complement pathway inhibition allow B. burgdorferi to survive in the hostile environment of human plasma.
IMPORTANCE
The present study defines a new mechanism by which the pathogenic bacterium Borrelia burgdorferi controls the terminal complement pathway of the human host to survive in human serum. The borrelial CspA binds to terminal pathway proteins C7 and C9 and inhibits the terminal complement pathway at the step of C7 and thereby inhibits terminal complement complex (TCC) assembly and membrane insertion. CspA blocks TCC assembly and insertion when expressed at the bacterial surface. CspA is the first TCC inhibitor cloned and functionally characterized from a Gram-negative bacterium. This identification of a bacterial TCC inhibitor of pathogen origin expands our knowledge of complement evasion of pathogenic bacteria and shows that pathogenic bacteria target the terminal pathway of complement. Thus, CspA as a central microbial virulence factor can represent an interesting biomarker and a target to develop new therapeutics and vaccines against borreliae.
Introduction
Any infectious microbe that enters the human host faces the adverse effects and toxic reactions of the activated complement system, which is a central element of the host innate immune response. Upon infection, invading microbes activate complement, and the activated host defense system initiates a cascade of tightly regulated events that result in C3b surface deposition (opsonization), generation of the inflammatory anaphylatoxins C3a and C5a, and formation of the cytolytic terminal complement complex (TCC), also termed the membrane attack complex (MAC) (1). Infectious microbes activate all three complement pathways, i.e., the alternative, classical, and lectin pathways (1, 2), and each pathway forms a C3 convertase. In the absence of regulators, activation progresses, C5 convertases are formed, and the terminal pathway is initiated. C6 and C7 bind to the newly formed C5b and form the C5b-7 complex, which attaches to and inserts into a target membrane. Further binding of C8 and C9 results in assembly and polymerization of C9 and in formation of the C5b-9 complex (TCC) (3). C5b-9, when inserted into the target membrane, changes the osmotic pressure and causes cell lysis.
Inappropriate control of the complement network results in cascade amplification, generation of toxic effector molecules, and formation of inflammatory or antimicrobial mediators. Host cells and tissues express surface-exposed complement regulators to efficiently control complement action and functions (1). Infectious microbes are normally recognized and eliminated by complement and the host immune system. However, pathogenic microbes survive in an immune competent host, as they express immune evasion proteins that acquire and exploit host complement regulators and consequently block complement (4).
Borrelia burgdorferi is a Gram-negative bacterium that causes Lyme disease in Eurasia and North America (5). These spirochetes are transmitted by ticks of the genus Ixodes (6). If the infection progresses and if left untreated, severe and chronic symptoms develop, including neuroborreliosis, carditis, and arthritis (7–9). In order to persist in an immunocompetent host and to multiply, B. burgdorferi has developed sophisticated strategies to control the human immune response. Borreliae acquire host regulators to modulate complement attack and to disseminate into deeper tissue layers (10). B. burgdorferi expresses CspA and the four additional immune evasion proteins, CspZ, ErpP, ErpC, and ErpA, which represent a group of functionally related but structurally heterologous proteins (10, 11).
These five borrelial proteins bind the human plasma regulators factor H and plasminogen, but the borrelial proteins differ in binding of factor H-like protein 1 (FHL-1) and complement factor H-related proteins (CFHRs) (12). CspA and CspZ bind factor H and FHL-1 but not CFHR1 (13–15). In contrast, ErpP, ErpC, and ErpA bind factor H and CFHR1 but not FHL-1 (13, 15). ErpP and ErpA also bind the human complement proteins CFHR2 and CFHR5 (16). CspA is highly conserved among borrelial species, and this 25.9-kDa protein also binds collagen I, collagen III, collagen IV, fibronectin, laminin, and bone morphogenic protein 2 (17, 18). CspA expression is induced during tick feeding and made during transmission and early mammalian infection and mediates serum resistance of B. burgdorferi (18–20).
Acquisition of soluble complement regulators by pathogenic microbes and inhibition of C3 and C5 convertases is a common evasion strategy and is relatively well characterized. In addition, pathogens express and secrete proteins that bind C3 and C3 cleavage products and thereby inhibit C3 activation or C3 effector functions. At present, microbial evasion of the terminal complement pathway is less understood. So far, five pathogen-encoded TCC inhibitors are, to our knowledge, identified: (i) a CD59-like protein from B. burgdorferi, (ii) streptococcal inhibitor of complement (SIC) from Streptococcus pyogenes, (iii) paramyosin from Schistosoma mansoni and Trichinella spiralis, (iv) Schistosoma C inhibitory protein type 1 (SCIP-1) from Schistosoma mansoni, and (v) galactose-specific adhesin from Entamoeba histolytica (21–26).
Each of the five borrelial complement evasion proteins binds at least two human complement regulators, but the borrelial proteins differ in serum resistance (15, 16, 27). Therefore, we hypothesized that these borrelial proteins have additional roles in immune evasion and we searched for additional complement inhibitory activities. Here, we identify CspA as a terminal complement inhibitor. CspA blocks the terminal complement pathway by inhibiting C5b-9 assembly at the level of C7 and also by blocking C9 polymerization. The TCC inhibitory domain of CspA was located in a stretch of 107 residues ranging from amino acids 109 to 215. Inhibition of the terminal pathway contributes to serum resistance of B. burgdorferi. To our knowledge, CspA is the first terminal pathway inhibitor of B. burgdorferi identified on the gene level.
RESULTS
CspA inhibits the alternative complement pathway independent of factor H acquisition.
The Gram-negative bacterium B. burgdorferi expresses immune evasion proteins that bind human complement regulators (13, 15, 16, 27). In order to define additional roles in immune evasion, we analyzed whether CspA and the other borrelial proteins inhibit hemolytic activity of normal human serum (NHS) using rabbit erythrocytes. CspA and ErpP, but neither CspZ, ErpC, nor ErpA, inhibited alternative pathway-mediated hemolysis (Fig. 1A). CspA was the most potent inhibitor and at 10 µg/ml blocked erythrocyte lysis by 80% (Fig. 1A). CspA displayed this inhibitory activity for serum concentrations ranging from 2.5 to 10% (Fig. 1B). In order to prove that this inhibitory activity is independent of factor H binding, CspA-mediated inhibition was assayed in factor H-depleted, complement-active human serum (HSΔFactor H). Also in this depleted serum, CspA inhibited alternative pathway-mediated hemolysis (Fig. 1C). This inhibitory effect of CspA was specific for human serum and was not detected when mouse, sheep, goat, guinea pig, or dog serum were used as the complement source (Fig. 1D).
FIG 1 .
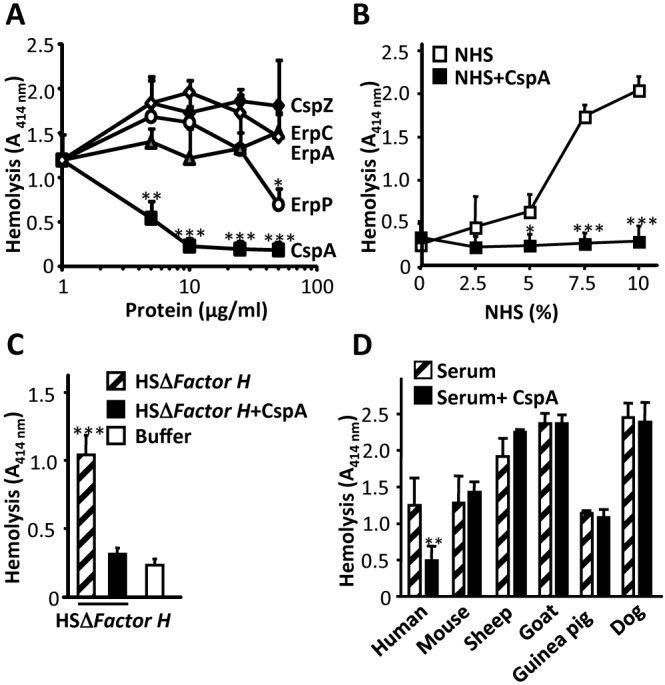
CspA inhibits the alternative pathway of complement independent of factor H. (A) CspA and ErpP, but not CspZ, ErpC, or ErpA, inhibit alternative pathway-mediated hemolysis. The five recombinant borrelial proteins (5 to 50 µg/ml) were added to NHS, and then the mixture was applied to rabbit erythrocytes. Following incubation, erythrocyte lysis was recorded. (B) CspA inhibited alternative pathway-mediated hemolysis in 2.5 to 10% serum. The CspA (10 µg/ml) inhibitory effect on alternative pathway-mediated hemolysis of rabbit erythrocytes using NHS at concentrations ranging from 2.5 to 10% was analyzed. (C) CspA inhibited alternative pathway-mediated hemolysis in complement-active human serum depleted for factor H (HSΔFactor H). CspA (50 µg/ml) was added to HSΔFactor H (15%), and then the mixture was applied to sheep erythrocytes. (D) Lysis of rabbit erythrocytes in serum derived from the indicated species, i.e., mouse, sheep, goat, guinea pig, or dog. Each panel represents the mean values from three separate experiments, and error bars show standard deviations (SD). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
CspA inhibits all three complement pathways.
CspA inhibits the alternative pathway of complement in the absence of factor H. We next asked whether CspA also inhibits the lectin and the classical pathways of complement. To this end, pathway-specific activators and buffers were used to activate each complement pathway using NHS, and C5b-9 deposition was followed after incubation (28). CspA blocked complement activated by each pathway (Fig. 2). CspA used at a concentration of 100 µg/ml blocked the alternative pathway and the lectin pathway by 84% and 77%, respectively (Fig. 2A and B). In addition, CspA blocked the classical pathway, and at 100 µg/ml, C5b-9 deposition was reduced by 34% (Fig. 2C). To confirm this inhibitory effect, CspA was also tested in a hemolytic assay with amboceptor-coated sheep erythrocytes. Again, the effect of CspA was dose dependent, and at 67 µg/ml, CspA inhibited hemolysis by 70% (Fig. 2D). Similarly, CspA inhibited all three complement pathways in the Wielisa complement activation assay (see Fig. S1 in the supplemental material). Thus, CspA is a borrelial complement inhibitor that blocks all three complement pathways.
FIG 2 .
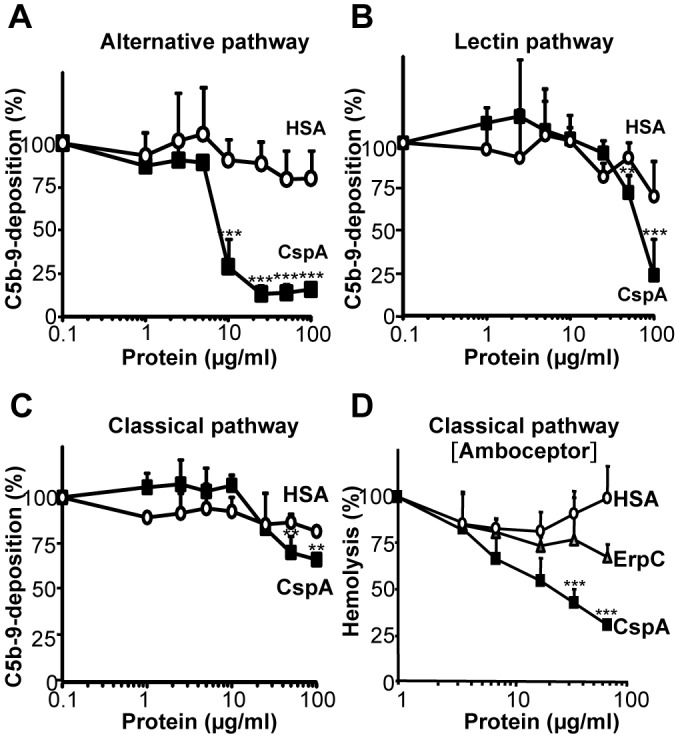
CspA inhibits all three complement pathways. CspA (■) or HSA (○) (1 to 100 µg/ml) was added together with NHS (20%) diluted in MgEGTA buffer (alternative pathway) (A) or NHS (1%) diluted in GVB++ buffer (lectin and classical pathways) (B and C) to pathway-specific activators. Following incubation, C5b-9 deposition was recorded using an anti-C5b-9 MAb and HRP-conjugated rabbit anti-mouse pAb. C5b-9 deposition from NHS was set to 100%. (D) CspA inhibits classical pathway-mediated hemolysis of amboceptor-coated sheep erythrocytes. CspA (■), ErpC (△), or HSA (○) (used at 2 to 67 µg/ml) was added to NHS (1%), and then this mixture was combined with amboceptor-coated sheep erythrocytes. The mean values out of three separate experiments are shown, and error bars show SD. **, P ≤ 0.01; ***, P ≤ 0.001.
CspA inhibits complement downstream of the C3 and the C5 convertases.
In order to define at which step CspA blocks the complement cascade, zymosan-activated NHS was used. First, the effect of CspA on the C3 convertase of the alternative and classical/lectin pathways was analyzed. CspA affected neither C3a (see Fig. S2A and B in the supplemental material), Ba (see Fig. S2C and D), Bb (see Fig. S2D), nor C2b (see Fig. S2E) generation. Thus, CspA did not influence C3 convertase activity of the alternative, classical, or lectin pathway. Furthermore, CspA lacked decay-accelerating activity for the alternative pathway C3 convertase (see Fig. S2F). In addition, CspA did not affect C5 convertase activity and did not influence C5a generation (see Fig. S3).
CspA is a terminal pathway inhibitor.
CspA blocked complement activation in factor H-depleted human serum but did not influence the C3 or C5 convertase. These scenarios suggested an effect distal of the two central enzymatic convertases on the terminal pathway. To analyze a role of CspA on the terminal pathway, first binding of terminal complement components to CspA was assayed. C7 and C9 bound to immobilized CspA, and C5b-6, C8, and C5b-9 bound with lower intensity (Fig. 3A). C7 bound to immobilized CspA with an intensity similar to that of the terminal pathway regulator vitronectin. The analyzed terminal complement proteins bound with rather low intensity to ErpC (Fig. 3A). To demonstrate a functional activity of CspA on the terminal pathway, next the effect of CspA on TCC-mediated lysis of sheep erythrocytes was tested using purified terminal pathway components. CspA combined with C7, C8, and C9 was added to C5b-6-coated sheep erythrocytes, and erythrocyte lysis was recorded after incubation. CspA inhibited erythrocyte lysis, and the effect was dose dependent and at 100 µg/ml reduced lysis by 71% (Fig. 3B). This effect was specific for CspA, as neither ErpC nor factor H, the human C3 convertase regulator, blocked erythrocyte lysis (Fig. 3B).
FIG 3 .

CspA is a terminal complement pathway inhibitor. (A) Terminal complement components C7 and C9 bind to immobilized CspA. C5b-6, C7, C8, C9, and C5b-9 (each at 5 µg/ml) were added, and binding to immobilized CspA (5 µg/ml) was analyzed. Bound components were identified with specific antisera followed by HRP-conjugated anti-goat antibody. (B) CspA blocks terminal pathway-mediated hemolysis of sheep erythrocytes using purified TCC components. CspA (■) (2 to 200 µg/ml) was combined with C7, C8, and C9, and thereafter the mixture was added to C5b-6-coated sheep erythrocytes. Following incubation, hemolysis was recorded. Lysis of sheep erythrocytes in the presence of C5b-9 was set to 100%. Similarly, ErpC (△), factor H (○), and the terminal pathway inhibitor vitronectin (◊) were analyzed for their effect on terminal pathway-mediated hemolysis. (C) CspA inhibits the terminal complement pathway at the level of C7. CspA (used at 50 and 100 µg/ml) was added together with C7, C8, and C9, and this mixture was added to C5b-6-coated sheep erythrocytes. Following incubation, hemolysis was recorded (first set of columns, C7-C9). ErpC (dark gray columns), vitronectin (gray columns), or factor H (white columns) was used at the indicated concentrations. Similarly, CspA was added to C8 and C9, and then the mixture was combined with C5b-7-coated erythrocytes (second set of columns, C8-C9), and CspA added to C9 was then applied to C5b-8-coated erythrocytes (third set of columns, C9). The mean values from three separate experiments are shown, and error bars correspond to SD. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
In order to assay at which level CspA interferes with C5b-9 formation, CspA was added sequentially to terminal complement components. First, CspA was preincubated with C7, C8, and C9 and then added to C5b-6-treated erythrocytes. In this setup, CspA (at 100 µg/ml) reduced erythrocyte lysis by 71% (Fig. 3C). When CspA was combined with C8 and C9 and then added to C5b-7-coated sheep erythrocytes, the inhibitory effect was lower and lysis was reduced by 38% (Fig. 3C). In addition, CspA when combined with C9 and then added to C5b-8-coated erythrocytes had a much lower effect on lysis (19%) (Fig. 3C). Thus, CspA inhibits C5b-9 formation predominantly at the level of C7.
In order to define this inhibitory effect on TCC in more detail, we analyzed whether CspA inhibits binding of C7 to the C5b-6 complex. CspA was preincubated with C7, and thereafter the mixture was added to immobilized C5b-6. C7 bound to C5b-6, and C7 complexed with CspA bound with the same intensity (see Fig. S4A in the supplemental material). Vitronectin slightly reduced binding of C7, and the C7-specific monoclonal antibody (MAb) blocked C7 binding to immobilized C5b-6. Thus, CspA binds to C7 but does not block attachment of C7 to the C5b-6 complex.
The CspA-C7 interaction was further characterized by analyzing binding of C7 to immobilized CspA. C7 bound dose dependently to immobilized CspA (see Fig. S4B). At 20 µg/ml, C7 bound both the borrelial CspA protein and human vitronectin with similar intensity. C7 is a heparin binding protein, and therefore the effect of heparin on the C7-CspA interaction was followed (29). Heparin modulated binding of C7 dose dependently and at 500 µg/ml reduced binding by 42% (see Fig. S4C). In addition, NaCl inhibited binding of C7 to immobilized CspA and at 0.15 M reduced binding by 60% (see Fig. S4D).
CspA binds to C9 and inhibits polymerization.
CspA binds to C7 and also to C9. C9 bound to immobilized CspA, and binding was dose dependent (Fig. 4A). C9 is a heparin binding protein (29), and heparin inhibited binding of C9 to CspA dose dependently (Fig. 4B). Used at 500 µg/ml, heparin inhibited C9 binding to CspA by 46%. In addition, NaCl at 0.15 M inhibited C9 binding to CspA by 35% (Fig. 4C). C5b-9 formation results in C9 polymerization. To determine whether CspA inhibits C9 polymerization, CspA was mixed with C9, and C9 polymerization was induced by ZnCl2. C9 monomers (mC9) and C9 polymers (pC9), when separated by SDS-PAGE, were directly identified by silver staining based on their distinct mobilities. C9 polymerization was induced by ZnCl2 (Fig. 4D, lane 1). In the presence of CspA and ZnCl2, no C9 polymers were identified (Fig. 4D, lanes 2 and 3). ErpC did not influence C9 polymerization, and both C9 monomers and C9 polymers were detected (Fig. 4D, lane 4). Thus, CspA blocks polymerization of C9.
FIG 4 .
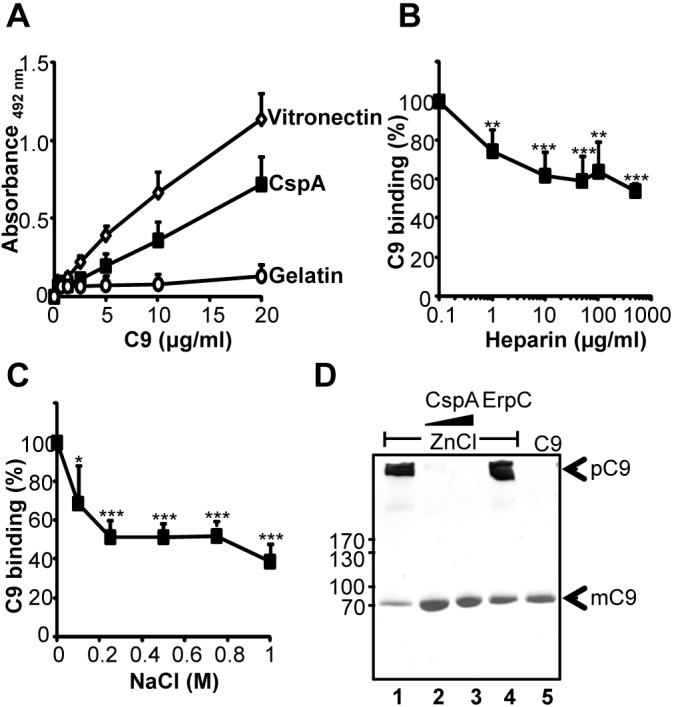
CspA binds to C9 and inhibits C9 polymerization. (A) C9 binds to immobilized CspA, and the effect is dose dependent. Binding of C9 (0.01 to 20 µg/ml) to immobilized CspA (5 µg/ml) was assayed by ELISA, and bound C9 was detected with polyclonal C9 antiserum followed by HRP-conjugated anti-goat antibody. (B) Heparin affects the CspA-C9 interaction. The effect of heparin (1 to 500 µg/ml) on binding of C9 (5 µg/ml) to immobilized CspA (5 µg/ml) was assayed. (C) NaCl inhibits the CspA-C9 interaction. The effect of NaCl (0.1 to 1 M) on binding of C9 (5 µg/ml) to immobilized CspA (5 µg/ml) was assayed. The mean values from three separate experiments are shown, and error bars show SD. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. (D) CspA inhibits polymerization of C9. ZnCl2 induced C9 polymerization, and after incubation the samples were separated by SDS-PAGE. Following silver staining, C9 polymers and C9 monomers were identified by their mobility. C9 polymerizes in the presence of ZnCl2 (lane 1). CspA (2.5 and 5 µg) blocks polymer formation (lanes 2 and 3). The borrelial immune evasion protein ErpC (5 µg) (lane 4) does not influence C9 polymerization. In the absence of ZnCl2, C9 does not form polymers (lane 5). The data shown are representative of three independent experiments.
Localization of the CspA region that mediates TCC inhibition.
In order to localize the region of the CspA protein that mediates TCC inhibition, CspA and CspA deletion mutants were used for binding and functional assays (Fig. 5A, left). First, the binding region of CspA for C7 or C9 was localized. Both C7 and C9 bound to immobilized full-length CspA26–251 and to the CspA26–240, CspA26–215, and CspA61–250 deletion mutants (Fig. 5A). In contrast, neither C7 nor C9 bound to the CspA26–108 deletion mutant. Thus, the major C7 and C9 binding region of CspA was localized within amino acid residues 109 to 215. Binding of CspA to C7 or C9 was confirmed by microscale thermophoresis. CspA bound to labeled C7 with an affinity of 5.1 ± 0.2 µM (Fig. 5B) and to C9 with an affinity of 3.4 ± 0.1 µM (Fig. 5C). The binding of C7 and C9 to the CspA26–108 deletion mutant and to bovine serum albumin (BSA) was significantly reduced compared to binding of full-length CspA.
FIG 5 .
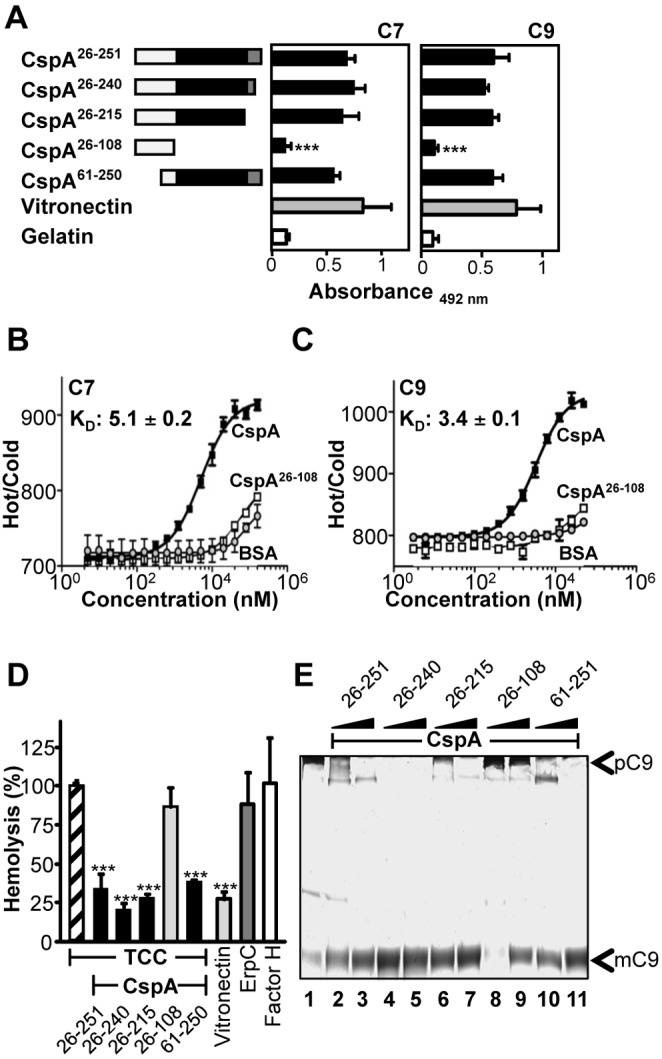
Localization of the TCC inhibitory region in CspA. (A) Full-length CspA26–251 and four deletion mutants were expressed in Escherichia coli and purified. The numbers refer to amino acid residues that are included in each construct (left). Black indicates the C7/C9 binding region, and gray indicates the factor H binding region of CspA (left). Binding of C7 (5 µg/ml) (middle) or C9 (5 µg/ml) (right) to immobilized CspA deletion mutants was assayed by ELISA. Bound C7 was detected with polyclonal C7 antiserum, and bound C9 was detected with polyclonal C9 antiserum followed by HRP-conjugated anti-goat antibody. (B and C) C7 binds to CspA with an affinity of 5.1 ± 0.2 µM and C9 with an affinity of 3.4 ± 0.1 µM. Binding of CspA, CspA26–108, or BSA (0.005 to 160 µM) to NT-647-labeled C7 (12.5 nM) or C9 (12.5 nM) was evaluated in fluid phase by microscale thermophoresis. Thermophoresis was recorded at 80% LED power and 80% MST power for 30 s in a Monilith NT.115 instrument. The relative fluorescence in the thermophoresis phase of the experiment has been plotted against the concentration of CspA. (D) Localization of the region within CspA that mediates TCC inhibition. Full-length CspA26–251 and the four deletion mutants were combined with C7, C8, and C9, and thereafter the mixture was added to C5b-6-coated sheep erythrocytes. Lysis of sheep erythrocytes in the presence of C5b-9 was set to 100%. The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. (E) The amino acid residues 109 to 251 of CspA are relevant for inhibition of C9 polymerization. ZnCl2 induced C9 polymerization, and after incubation the samples were separated by SDS-PAGE; following silver staining, C9 polymers (pC9) and C9 monomers (mC9) were identified by their mobility. C9 polymerizes in the presence of ZnCl2 (lane 1). CspA26–251, CspA26–240, CspA26–215 (lanes 2 to 7), and CspA61–251 (lanes 10 and 11) (2.5 and 5 µg) block polymer formation. CspA26–108 (lanes 8 and 9) does not influence C9 polymerization. The data shown are representative of three independent experiments.
The various CspA deletion mutants were further analyzed for their effect on TCC-mediated hemolysis. Full-length CspA26–251 and the CspA26–240, CspA26–215, and CspA61–250 deletion mutants, but not the CspA26–108 deletion mutant, blocked terminal pathway-mediated hemolysis of sheep erythrocytes, induced by purified terminal pathway components (Fig. 5D). Thus, the TCC inhibitory region of CspA was localized to the 107-amino-acid-long region, i.e., 109 to 215, which also includes the major C7 and C9 binding regions. The CspA mutants were further tested for inhibition of C9 polymerization. Similar to full-length CspA (Fig. 5E, lanes 2 and 3), the CspA26–240 (Fig. 5E, lanes 4 and 5), CspA26–215 (Fig. 5E, lanes 6 and 7), and CspA61–250 (Fig. 5E, lanes 10 and 11) deletion mutants, but not the CspA26–108 deletion mutant (Fig. 5E, lanes 8 and 9), inhibited C9 polymerization.
C7 and C9 bind simultaneously to CspA.
C7 and C9 bind within the same 107-amino-acid-long region of CspA. Therefore, we asked if the two human terminal pathway components bind to the same overlapping region and compete for binding or if they bind to separate sites and therefore bind simultaneously to CspA. C7 in the presence of constant amounts of C9 (10 µg/ml) bound dose dependently to CspA and did not influence C9 binding (see Fig. S5A in the supplemental material). Similarly, C9 bound dose dependently to CspA and did not significantly influence C7 binding (see Fig. S5B). This simultaneous binding suggests that C7 and C9 have distinct binding sites in the 107-amino-acid-long region of CspA.
C7/C9 and factor H attach to different regions in CspA.
As C7, C9, and factor H bind to CspA, we asked if these proteins bind simultaneously to CspA or compete for binding. C7 bound dose dependently to immobilized CspA and did not affect factor H binding (see Fig. S5C). Factor H, when used at increasing concentrations, slightly reduced C7 binding to CspA, and when used at 5 µg/ml, factor H reduced C7 binding only by 17% (see Fig. S5D). As C9 binds to CspA, simultaneous binding of C9 and factor H was analyzed next. C9, when used at increasing concentrations, slightly reduced factor H binding (11%) to CspA (see Fig. S5E). Similarly, in a reverse setting, factor H slightly reduced C9 binding to CspA (see Fig. S5F). Factor H used at 5 µg/ml reduced C9 binding by 23%. Thus, both C7 and C9 bind independently and do not compete with factor H and also bind independently and most likely to different regions of the CspA protein.
CspA inhibits C5b-9/TCC in fluid phase and contributes to borrelial survival.
CspA inhibits TCC-mediated lysis of erythrocytes. Next, we asked whether CspA added in fluid phase contributes to borrelial survival by inhibiting the terminal complement pathway. To this end, CspA was first added to NHS. Following incubation of serum-sensitive Borrelia garinii G1 in this CspA-supplemented serum, surface deposition of C6 and C5b-9 was assayed by confocal microscopy. In the presence of CspA, C6 and low levels of C5b-9 were detected on the bacterial surface (Fig. 6A, top panels). When bacteria that do not express CspA were challenged with NHS, C6 and C5b-9 were deposited on the bacterial surface (Fig. 6B, top panels). In addition, the effect of CspA for survival of serum-sensitive B. garinii G1 was followed using a standard growth inhibition assay. When serum-sensitive B. garinii G1 was cultivated in CspA-supplemented NHS, bacteria remained viable over the whole time period of 7 days (Fig. 6C). In contrast, spirochetes challenged with complement-active NHS did not survive (Fig. 6C). Those bacteria lacked metabolic activity, as evidenced by constant absorbance over the whole experimental period of seven days. Furthermore, CspA, when added to NHS, also blocked complement-mediated killing of the serum-sensitive, Gram-negative bacterium Pseudomonas aeruginosa strain SG137 (Fig. 6D to F). This inhibitory effect was specific for CspA, as the borrelial protein ErpC and also human serum albumin (HSA) did not affect the serum-damaging activity of complement. CspA inhibited complement in NHS when the alternative pathway was activated (Fig. 6D) or when all three complement pathways were active (Fig. 6E). Sbi, a Staphylococcus aureus complement inhibitor, also blocked the complement-mediated damaging effects, resulting in survival of P. aeruginosa (Fig. 6D and E) (30). Similarly, heat-inactivated NHS (hiNHS) lacked such bactericidal activity. To provide additional proof that the TCC inhibitory effect of CspA is independent of the protective effect of factor H, CspA-mediated inhibition was assayed in factor H-depleted, complement-active human serum (HSΔFactor H), and survival of P. aeruginosa was evaluated. CspA inhibited alternative pathway-mediated killing of P. aeruginosa in the absence of factor H, and the effect was dose dependent (Fig. 6F). Thus, CspA inhibits TCC action of complement independent of factor H acquisition and in consequence protects B. garinii and also P. aeruginosa from complement-mediated killing.
FIG 6 .
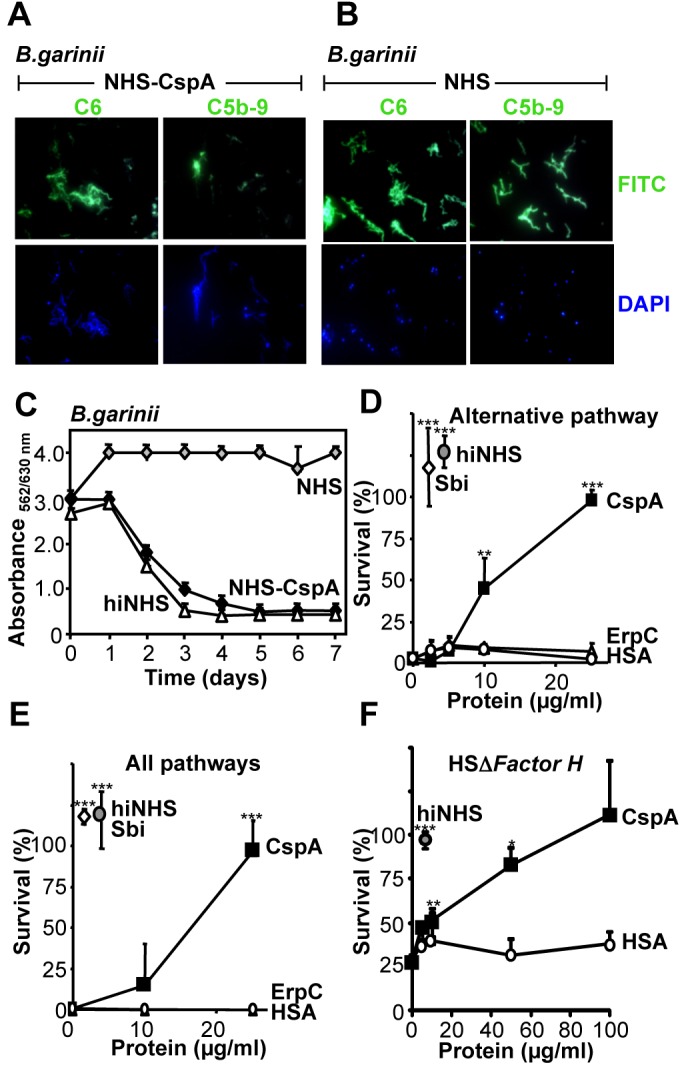
CspA inhibits complement in fluid phase independent of factor H. (A) CspA (200 µg/ml) when added to NHS (25%) (CspA-NHS) inhibited C5b-9 deposition on the surface of the serum-sensitive B. garinii strain G1. C6 or C5b-9 surface deposition was evaluated by confocal microscopy using specific antibodies followed by Alexa 488-conjugated antibodies. DAPI was used to visualize intact spirochetes. (B) B. garinii was challenged with NHS, and C6 and C5b-9 deposition on the surface was evaluated. (C) CspA-NHS blocked complement (■) and resulted in survival of the serum-sensitive B. garinii. Bacterial growth was determined by measuring the ratio of absorbance at 562 versus 630 nm. B. garinii was cultivated in NHS (50%) (◊) or hiNHS (50%) (△). (D and E) CspA, when added to NHS, blocked the alternative pathway or all complement pathways. CspA (■) (2.5 to 25 µg/ml), ErpC (△) (2.5 to 25 µg/ml), HSA (○) (2.5 to 25 µg/ml), and Sbi (◊) (10 µg/ml) were added to NHS (20%) diluted in MgEGTA (alternative pathway) or GVB++ (all pathways) buffer, and this mixture was used to challenge P. aeruginosa strain SG137. (F) CspA, when added to factor H-depleted, complement-active human serum (HSΔFactor H), blocked alternative pathway-mediated killing of P. aeruginosa. CspA (■) (5 to 100 µg/ml) or HSA (○) (5 to 100 µg/ml) was added to HSΔFactor H (20%) diluted in MgEGTA buffer, and this mixture was used to challenge P. aeruginosa strain SG137. After incubation, bacteria were plated on NB agar plates, and the number (CFU) of surviving bacteria was determined. CFU at time point 0 was set to 100%. The mean values from three separate experiments are shown, and error bars show SD. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
CspA ectopically expressed on the surface of serum-sensitive B. garinii contributes to serum resistance and blocks C5b-9 deposition at the level of C7.
CspA is a borrelial surface protein (15, 18). To demonstrate a protective role of CspA directly on the bacterial surface, CspA was ectopically expressed on the surface of the serum-sensitive B. garinii strain G1. B. garinii G1 was transformed with either the plasmid pCspA harboring the cspA gene or with the shuttle plasmid pKFSS1 that lacks the cspA insert. Transformation of the plasmid was confirmed by PCR amplification of the cspA gene and the streptomycin resistance aadA gene. Both the cspA and the aaaA genes were detected in the transformed B. garinii-CspA spirochetes (see Fig. S6A, lane 4, in the supplemental material) but not in the B. garinii G1 strain transformed with a vector without the cspA insert (see Fig. S6A, lane 3). The vector-specific aadA gene was detected in the B. garinii G1 transformants containing the vector with or without cspA (see Fig. S6A, lanes 3 to 5). To confirm surface expression of CspA in the transformed B. garinii (B. garinii-CspA) strain, intact spirochetes were treated with the protease proteinase K or trypsin. Surface-exposed proteins, but neither periplasmic nor intracellular proteins, are susceptible to protease treatment. CspA was susceptible to proteinase K digestion (see Fig. S6B, top, lane 2). The outer membrane of the spirochetes remained intact, and FlaB, a component of the periplasmic flagella (31), was not degraded (see Fig. S6B, bottom). Thus, CspA is expressed at the surface of transformed B. garinii-CspA spirochetes.
Following NHS challenge of transformed B. garinii-CspA spirochetes and also mock-transformed B. garinii, surface deposition of C3 and individual terminal pathway components was analyzed. The CspA-expressing bacteria had C3 and C6 deposited onto their surface, but neither deposition of C7, C8, C9, nor C5b-9 was detected (Fig. 7A). In contrast, B. garinii had C3 and all analyzed TCC components deposited on its surface (Fig. 7B). To confirm that CspA by inhibiting C5b-9 contributes to bacterial survival, B. garinii-CspA spirochetes were challenged with NHS. Bacterial survival was assessed by monitoring the metabolic activity. CspA-expressing spirochetes survived in complement-active NHS, remained viable, and retained their metabolic activity over the whole time period (Fig. 7C). The growth rate of the CspA-expressing B. garinii was comparable to that of wild-type B. burgdorferi B31, which expresses endogenous CspA on the surface (see Fig. S7 in the supplemental material). As expected, B. garinii transformed with the empty shuttle vector (B. garinii) and thus lacking CspA on the surface did not survive this serum challenge, as evidenced by constant absorbance (Fig. 7D). Thus, CspA expressed on the surface of borreliae confers serum resistance and blocks C5b-9/TCC deposition at the level of C7.
FIG 7 .
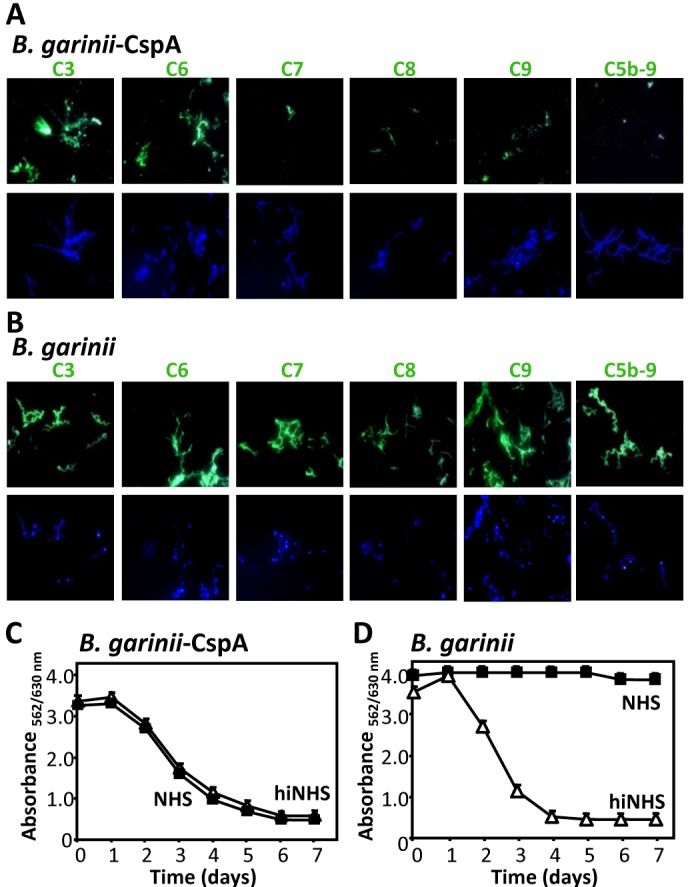
CspA expressed on the surface of B. garinii inhibits deposition of terminal pathway components and confers serum resistance. (A) CspA ectopically expressed on the surface of serum-sensitive B. garinii inhibited deposition of the terminal complement components. B. garinii transfected with a CspA-expressing vector (B. garinii-CspA) was challenged with NHS (25%), and following incubation, deposition of the indicated complement component was followed by immunofluorescence microscopy. DAPI (blue) was used for visualization of intact spirochetes. (B) B. garinii G1 transformed with the shuttle vector (B. garinii) lacking the CspA coding sequence (B. garinii) was treated as described above. (C and D) CspA ectopically expressed on the surface of B. garinii neutralized the toxic effector functions of the activated complement. The B. garinii-CspA strain or B. garinii was challenged with NHS (50%) (■) or hiNHS (50%) (△). Following incubation over 7 days at 33°, bacterial growth was monitored by measuring the ratio of culture medium absorbance at 562 versus 630 nm daily. The mean values from three separate experiments performed in triplicate are shown, and the error bars show SD.
DISCUSSION
Here, we identify CspA of B. burgdorferi as an inhibitor of the terminal complement pathway. CspA is a multifunctional borrelial immune evasion protein that binds several human plasma proteins, including the complement regulators factor H, FHL-1, and plasminogen (17, 18, 32, 33). CspA binds to human TCC proteins, in particular to C7 and C9, and thereby blocks human C5b-9 complex assembly and formation. This inhibitory effect on the TCC is independent of factor H, as CspA blocks C5b-9 complex formation in factor H-depleted human serum, and the inhibitory effect was also detected in hemolytic assays with purified terminal pathway components. Thus, this blockade is independent of enzymatic complement action. CspA inhibits all three complement pathways, i.e., the alternative, the lectin, and the classical pathway. CspA expressed at the borrelial surface blocked C5b-9 complex assembly and formation at the level of C7.
CspA blocks C5b-9 assembly on the borrelial surface and also in solution. Thereby, CspA enhances survival of serum-sensitive B. garinii and, due to the blocking action in fluid phase, also increases survival of the Gram-negative bacterium P. aeruginosa. CspA blocks the terminal pathway of complement. The effect appears to be a general feature for Gram-negative bacteria, as CspA protected B. burgdorferi, B. garinii, and also P. aeruginosa from TCC-mediated lysis. This inhibitory effect is independent of factor H acquisition, and CspA mediates this antibactericidal activity in factor H-depleted, complement-active serum. CspA, when ectopically expressed on the surface of the serum-sensitive B. garinii strain G1, inhibited C5b-9 complex formation at the level of C7. In the presence of CspA, terminal complex assembly proceeds to the level of C6, as C6 is deposited at the surface when bacteria were challenged with NHS supplemented with CspA and when CspA is expressed at the surface of borreliae. In the presence of CspA, neither C7, C8, nor C9 was deposited on the bacterial surface. In consequence, CspA blocked C5b-9 complex formation and in addition polymerization of C9. Thus, CspA protects the bacterium from the damaging effects of the terminal pathway.
CspA binds to terminal pathway components, in particular to C7 and C9. In the C7 protein, the CspA binding site and the attachment site to C5b-6 are distinct, as CspA attached to C7 did not block binding of C7 to C5b-6. CspA binds C7 and C9 with comparable affinity, i.e., 5.1 ± 0.2 µM for C7 and 3.4 ± 0.1 µM for C9. The binding site for both C7 and C9 was localized within the same region of CspA, i.e., amino acid residues 109 to 215. However, the two human plasma proteins bind simultaneously to CspA and do not compete for binding. The TCC inhibitory region of CspA, which spans from amino acids 109 to 215, includes the attachment sites for C7 and C9, inhibits hemolysis of sheep erythrocytes upon challenge with purified terminal pathway components, and also inhibits C9 polymerization. This TCC inhibitory region of CspA is distinct from the factor H binding region, which is located within the C-terminal amino acid residues 241 to 251 (18). Thus, borrelial CspA inhibits complement action in two separate ways and influences two major steps in the complement cascade. As reported here, CspA blocks TCC action by binding C7 and C9 and as described earlier by binding the inhibitors factor H and FHL-1, which block the C3 convertase (15, 34).
For soluble C5b-9, additional functions are proposed, including promotion and modulation of inflammation, release of chemokines, and expression of adhesion molecules (35, 36). Whether the borrelial CspA also influences these additional effector functions of soluble C5b-9 needs to be determined in future assays.
CspA, the new 25.9-kDa borrelial terminal pathway inhibitor, and the previously described borrelial CD59-like protein may represent different proteins. The CD59-like protein is contained in membrane extracts of the serum-resistant B. burgdorferi strains Alcaide, Tir, and B31 and inhibited TCC-mediated lysis of guinea pig erythrocytes (25). CspA has an apparent molecular mass of 25.9 kDa, compared to 80 kDa for the CD59-like protein (25). In addition, CspA lacks reactivity with a MAb specific for CD59 (see Fig. S8 in the supplemental material). In contrast, the CD59-like protein was identified by a CD59-specific antiserum but was never recombinantly expressed. Based on the size and reactivity with the antibodies, CspA and CD59-like protein are most likely different proteins.
At present, a limited number of pathogen-encoded terminal pathway inhibitors are identified. In addition to the two borrelial TCC inhibitors CspA (this study) and CD59-like protein, four other TCC inhibitors are known, i.e., streptococcal inhibitor of complement (SIC) from S. pyogenes, paramyosin from S. mansoni and T. spiralis, schistosome C inhibitory protein type 1 (SCIP-1) of S. mansoni, and the galactose-specific adhesin from E. histolytica (21–24, 26). Paramyosin and galactose-specific adhesin and the borrelial CD59-like protein, but not CspA, react with CD59-specific antibodies (21, 22, 25).
Taken together, CspA is a surface-exposed immune evasion protein of B. burgdorferi that blocks the action of the terminal complement pathway. CspA blocks C5b-9 assembly and TCC effector functions and thus protects borrelia from the effects of the human terminal complement attack. CspA is a multifunctional immune evasion protein that controls human complement at multiple levels. The present study defines how the pathogenic bacterium B. burgdorferi controls the terminal complement pathway of the human host and adds a new level of complement control in microbial complement and immune evasion. The identification of this complement evasion strategy of B. burgdorferi, the characterization of the CspA as a borrelial TCC inhibitor, and the elucidation of the mechanism of how the bacterial pathogen interferes with the human complement add a new aspect to the complex immune interplay of the bacterial pathogen and the human host. CspA is the first TCC inhibitor cloned and functionally characterized from a Gram-negative bacterium. This identification of this borrelial TCC inhibitor expands our knowledge of complement evasion of pathogenic bacteria and shows that B. burgdorferi targets also the terminal pathway of complement. Thus, CspA as a central microbial virulence factor can represent an interesting biomarker and a target to develop new therapeutics and vaccines against borreliae.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
B. burgdorferi B31, B. burgdorferi LW2, and B. garinii transformants G1/pKFSS1 (B. garinii) and B. garinii-CspA were grown at 33°C for 2 to 4 days to mid-exponential phase (1 × 107 to 5 × 107 spirochetes/ml) in modified Barbour-Stoenner-Kelly (BSK) medium or BSK medium supplemented with 20 µg/ml streptomycin (15). P. aeruginosa strain SG137 was routinely cultured in enriched nutrient broth (Serva, Amstetten, Austria) at 37°C to an optical density at 600 nm (OD600) of ≈1.0.
Generation and purification of recombinant proteins and construction of shuttle vectors.
Construction and expression of plasmids harboring the cspA, cspZ, erpP, erpC, and erpA genes encoding CspA, CspZ, ErpP, ErpC, and ErpA, respectively, and purification of the proteins were described (13, 18, 37, 38). Cloning and expression of the CspA deletion mutants were performed as previously described (18, 39). For generation of the CspA deletion mutants, specific primers (Table 1) were used, and the inserts were recloned into the pQE-30 Xa vector (Qiagen). CspA was ectopically expressed on the surface of the serum-sensitive B. garinii strain G1 (40). Briefly, the cspA gene with its flanking regulatory regions was amplified from B. burgdorferi LW2 by PCR using primers with restriction sites BamHI and HindIII (Table 1). The sequence of the cspA gene of B. burgdorferi LW2 is identical to the cspA homolog of B. burgdorferi B31. B. garinii G1 was transformed with either the plasmid pCspA harboring the cspA gene or with the shuttle plasmid pKFSS1 that lacks the cspA insert, and transformants were selected as previously described (40). Cloning and protein expression of Staphylococcus aureus binder of IgG (Sbi) of S. aureus was performed as previously described (30, 41).
TABLE 1 .
Oligonucleotides used in this study
| Primer | Sequence (5′–3′)a | Used in this work |
|---|---|---|
| CspA BamHI | GCTAAAACTTCTCTTTTTTTTAGGATCCCAACCCAAATCC | Cloning in pKFSS1 |
| CspA HindIII | GAAAGAAAAAAAATAAGCTTTTGCACTTGATATTTTTAAAAAG | Cloning in pKFSS1 |
| pGEX-CspA75 | CGGATCTGGTTCCGCGTGGATCCGCACCTTTTAGC | Recloning in pQE-30 Xa |
| BbCspA 175(+) | ACCCAAAATTTTGAAGATAAATCTGGATCCCTTAGCACTTCTGATGAA | Recloning in pQE-30 Xa |
| pGEX(−) | CCGGGAGCTGCATGTGTCAGAGG | Recloning in pQE-30 Xa |
Nucleotides underlined indicate the appropriate restriction sites for cloning of the PCR-amplified cspA gene of B. burgdorferi LW2 into the shuttle vector pKFSS1 or recloning of truncated CspA genes encoding diverse CspA deletion mutants into the pQE-30 Xa expression vector (see Materials and Methods).
Hemolytic assays.
Normal human serum (NHS) was preincubated with recombinant proteins for 15 min at 37°C. Hemolytic assays were used to analyze the alternative pathway (rabbit erythrocytes), the classical pathway (amboceptor-coated sheep erythrocytes), alternative pathway in factor H-depleted serum (HSΔFactor H) (sheep erythrocytes), or the terminal pathway (sheep erythrocytes). Erythrocytes (2 × 108 cells/ml) were incubated with 2.5 to 10% NHS (alternative pathway), 15% HSΔFactor H (alternative pathway), or 1% NHS (classical pathway) with or without (5 to 50 µg/ml) CspA, CspZ, ErpP, ErpC and ErpA, or the CspA deletion mutants (10 µg/ml) in MgEGTA buffer (20 mM HEPES, 144 mM NaCl, 7 mM MgCl2, and 10 mM EGTA, pH 7.4) (alternative pathway) or CspA (2 to 67 µg/ml), ErpC (2 to 67 µg/ml), or HSA (2 to 67 µg/ml) in gelatin Veronal buffer (GVB++) (Complement Technology) (classical pathway) for 30 min at 37°C. The terminal pathway was analyzed by using sheep erythrocytes (5 × 108 cells/ml) preincubated with C5b-6 (1.5 µg/ml) for 10 min at room temperature (RT) in GVB++ buffer. C7 (1 µg/ml), C8 (0.4 µg/ml), and C9 (1 µg/ml) were preincubated with CspA (used at concentrations of 2.0 to 200 µg/ml), CspA deletion mutants (25 µg/ml), ErpC, vitronectin, or factor H for 5 min at RT. Alternatively, C8 and C9 or C9 alone was preincubated for 5 min at RT with 50 to 100 µg/ml CspA, ErpC, vitronectin, and factor H before incubation with the C5b-7- or C5b-8-coated erythrocytes, respectively. After 30 min at 37°C, cells were centrifuged, and lysed erythrocytes were determined by measuring the release of hemoglobin at 414 nm. The effect of CspA on different animal sera (Innovative Research) was assayed using 30% animal serum, i.e., mouse, sheep, goat, guinea pig, and dog serum, and 10 µg/ml CspA.
Complement activation assays.
The activity of CspA on each complement pathway was analyzed by enzyme-linked immunosorbent assay (ELISA). The alternative, lectin, or classical pathway was activated separately by lipopolysaccharide (LPS) (10 µg/ml) (alternative pathway), mannan (100 µg/ml) (lectin pathway), or IgM (2 µg/ml) (classical pathway), and C5b-9 deposition was assayed (28). The positive control (NHS) was set to 100%. For inhibition, CspA (1 to 100 µg/ml) and HSA (1 to 100 µg/ml) were preincubated with 20% NHS (alternative pathway) or 1% NHS (classical/lectin pathways) for 15 min at 37°C and thereafter added to the wells.
ELISA.
Microtiter plates (F96 Maxisorb, Nunc-Immuno module) were coated with CspA (5 µg/ml), CspA deletion mutants (5 µg/ml), ErpC (5 µg/ml), vitronectin (5 µg/ml), or gelatin (5 µg/ml) overnight at 4°C. The plates were washed four times with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBS-Tw) and blocked for 1 h with blocking buffer I (Applichem). After being washed, the plates were incubated for 1 h at RT with C5b-6 (5 µg/ml), C7 (0.01 to 40 µg/ml), C8 (5 µg/ml), C9 (0.01 to 40 µg/ml), or C5b-9 (5 µg/ml). Thereafter, the wells were washed, and bound proteins were detected with anti-C5, anti-C7, anti-C8, or anti-C9 antibody (all goat anti-human polyclonal antibodies [pAbs] from Complement Technology) and horseradish peroxidase (HRP)-conjugated donkey anti-goat pAbs (DakoCytomation). The reaction was developed with 1,2-phenylenediamine dihydrochloride (OPD; DakoCytomation), and the absorbance was measured at 492 nm. In the competition assays, the effect of heparin (1 to 500 µg/ml) or NaCl (0.1 to 1 M) on C7 (10 µg/ml) and C9 (10 µg/ml) binding to immobilized CspA was assayed.
Microscale thermophoresis.
The CspA interaction with C7 and C9 was analyzed by microscale thermophoresis (MST) (42, 43). CspA, CspA26–108, or BSA used at the indicated concentrations was combined with human C7 (10.8 µM) or C9 (15.15 µM), which were both labeled with NT-647 using the protein labeling kit RED-NHS (NanoTemper Technologies). CspA, CspA26–108, or BSA used at 0.005 to 160 µM was added to NT-647-labeled C7 (12.5 nM), and CspA, CspA26–108, or BSA used at 0.006 to 50 µM was added to C9 (12.5 nM). Samples were diluted in MST buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 10 mM MgCl2, and 0.05% Tween 20). Thermophoresis was measured at 80% LED power and 80% MST power for 30 s in a Monilith NT.115 instrument (NanoTemper Technologies GmbH). All measurements were performed at RT using hydrophilic capillaries.
C9 polymerization assay.
The effect of CspA on Zn2+-induced C9 polymerization was assayed (26). CspA (2.5 to 5 µg), CspA deletion mutants (2.5 to 5 µg), or ErpC (5 µg) was preincubated with 3 µg C9 at 37°C in 20 mM Tris-HCl (pH 7.2). After 40 min of incubation, 50 µM ZnCl2 in 20 mM Tris-HCl (pH 7.2) was added for 2 h at 37°C. The samples were subjected to an 8 to 16% SDS-PAGE gradient gel (Bio-Rad, Hercules, CA), and C9 polymerization was determined by silver staining.
Deposition of terminal complement proteins.
Deposition of activated complement components on the surface of the borreliae was analyzed by immunofluorescence microscopy (16). Deposited C3, C6, C7, C8, C9, or C5b-9 were detected with anti-C3 pAb (Calbiochem), anti-human C6 pAb, anti-human C7 pAb, anti-human C8 pAb, anti-human C9 pAb, or mouse anti-human C5b-9 MAb (Quidel, San Diego, CA) followed by Alexa 488-conjugated anti-goat antibody or Alexa 488-conjugated anti-mouse antibody (Molecular Probes). 4′,6-Diamidino-2-phenylindole (DAPI) was used to visualize intact spirochetes. In the inhibition experiments, CspA (200 µg/ml) was preincubated with 25% NHS for 15 min at 37°C, followed by incubation with B. garinii strain G1.
Serum resistance.
Survival of the CspA-expressing B. garinii (B. garinii-CspA) strain and B. garinii transfected with an empty plasmid (B. garinii) in NHS was assessed by a growth inhibition assay as previously described (40). Briefly, highly motile spirochetes (1.25 × 107 cells) diluted in BSK medium containing 240 µg/ml phenol red were incubated with 50% NHS or 50% hiNHS in microtiter plates for 7 days at 33°C. During bacterial growth, the medium acidifies and the pH indicator dye turns from red to yellow. Bacterial growth was monitored by measuring the ratio of culture medium absorbance at 562 versus 630 nm daily. In the inhibition experiments, CspA (200 µg/ml) was preincubated with 50% NHS for 15 min at 37°C, followed by incubation with B. garinii strain G1. In addition, the complement inhibitory effect of CspA was analyzed in a serum resistance assay using a serum-sensitive P. aeruginosa strain (SG137). NHS (20%) was preincubated with CspA (2.5 to 25 µg/ml), ErpC (2.5 to 25 µg/ml), HSA (2.5 to 25 µg/ml), or Sbi (10 µg/ml) for 15 min at 37°C, followed by incubation with the P. aeruginosa strain SG137 (104 CFU) diluted in Mg-EGTA buffer (alternative pathway) or GVB++ buffer (all pathways) at 37°C. CspA or HSA used at different concentrations (0 to 100 µg/ml) was added to factor H-depleted, complement-active human serum (HSΔFactor H) (20%), and the mixture was then added to P. aeruginosa strain SG137 (104 CFU) resuspended in MgEGTA buffer and incubated for 20 min at 37°C. After 0, 20, or 60 min, 10-µl aliquots were removed and spread onto NB plates, and after 18 h of incubation at 37°C, CFU were determined.
Statistical analysis.
Statistics were analyzed by Student’s t test for paired data; P values of ≤0.05 were considered to be statistically significant.
SUPPLEMENTAL MATERIAL
CspA inhibits all complement pathways. The activity of CspA on each complement pathway was analyzed using a Wielisa total complement screen (Wieslab, Malmö, Sweden) described by Seelen et al. (J. Immunol. Methods 296:187–198, 2005). The mean values from three separate experiments are shown, and error bars show SD. **, P ≤ 0.01; ***, P ≤ 0.001. Download
CspA does not inhibit the C3 convertase of the alternative pathway. (A to B) CspA does not affect C3a generation. (A) C3a generation was determined by ELISA. Detection of C3a in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), or HSA (25 µg/ml). (B) Cleavage products generated when the C3 convertase is formed were analyzed by Western blotting using C3a-specific antisera. (C and D) CspA does not affect Ba and Bb generation. (C) Ba generation was determined by ELISA. Detection of Ba in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), or HSA (25 µg/ml). (D) Cleavage products generated when the C3 convertase is formed were analyzed by Western blotting using factor B-specific antisera. (E) CspA does not inhibit C2b generation. CspA does not affect C2b generation in zymosan-activated serum. Detection of C2b in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), HSA (25 µg/ml), or anti-human C4d MAb (12 µg/ml). Cleavage products generated when the C3 convertases are formed were analyzed by Western blotting using a C2-specific antiserum. The immunoblot is a typical experiment of three. (F) CspA lacks C3 convertase decay-accelerating activity. The alternative pathway C3 convertase was assembled on the surface of microtiter plates, and then CspA (black columns) or factor H (white columns) was added. After incubation, C3 convertase levels were analyzed by ELISA using factor B-specific antiserum. The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. Download
CspA does not inhibit the C5 convertase activity. CspA does not affect C5a generation in zymosan-activated serum as determined by ELISA. Detection of C5a in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), or HSA (25 µg/ml). The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. Download
CspA binds C7 and does not inhibit binding of C7 to the C5b-6 complex. (A) Binding of C7 to immobilized C5b-6 was assayed in the presence of CspA, vitronectin, anti-C7 MAb, or ErpC by ELISA. CspA, vitronectin, ErpC (each at 50 µg/ml), or anti-C7 MAb (1:100) was added to C7 (10 µg/ml) and incubated for 15 min at RT, and thereafter the mixture was added to immobilized C5b-6 (5 µg/ml). After being washed, bound C7 was detected with C7-specific antiserum followed by HRP-conjugated anti-goat antibody. (B) C7 binds to immobilized CspA, and binding is dose dependent. Binding of C7 (0.01 to 20 µg/ml) to immobilized CspA was assayed by ELISA. (C) Heparin inhibits the CspA-C7 interaction. The effect of heparin (1 to 500 µg/ml) on binding of C7 (10 µg/ml) to immobilized CspA (5 µg/ml) was assayed. (D) NaCl inhibits the CspA-C7 interaction. The effect of NaCl (0.1 to 1 M) on binding of C7 (5 µg/ml) to immobilized CspA (5 µg/ml) was assayed. The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. Download
C7, C9, and factor H bind simultaneously to CspA. (A) Effect of increasing C7 levels in the presence of a constant concentration of C9. Binding of C7 (used at the indicated concentrations) and C9 (10 µg/ml) to immobilized CspA was analyzed by ELISA. Bound C7 was detected with C7-specific antiserum (black columns), and bound C9 was detected with C9-specific antiserum (white columns). (B) In a reverse setting, the C7 concentration was kept constant (10 µg/ml), and binding of C9 (used at the indicated concentrations) was evaluated. (C) Effect of increasing C7 levels in the presence of a constant concentration of factor H. Binding of C7 (used at the indicated concentrations) and factor H (2.5 µg/ml) to immobilized CspA was analyzed by ELISA. Bound C7 was detected with C7-specific antiserum (black columns), and bound factor H was detected with factor H-specific antiserum (white columns). (D) In a reverse setting, the C7 (10 µg/ml) concentration was kept constant, and binding of factor H (used at the indicated concentrations) was evaluated. (E and F) Similarly, binding of C9 and factor H to immobilized CspA was analyzed by ELISA. Bound C9 was detected with C9-specific antiserum (black columns), and bound factor H was detected with factor H-specific antiserum (white columns). The mean values from three independent experiments and SD are presented. Statistical significance of differences was estimated using Student’s t test. *, P ≤ 0.05. Download
Characterization of B. garinii G1 producing CspA from B. burgdorferi LW2. (A) The B. garinii-CspA strain harbors both the cspA and the plasmid-specific gene aaaA. B. garinii G1, the G1/pKFSS1 and B. garinii-CspA transformed strains, and shuttle vector pCspA were characterized by PCR amplification using cspA-, aadA-, and flaB-specific primers. (B) The B. garinii-CspA strain expresses CspA on the surface of intact spirochetes. Proteinase K degrades surface-exposed CspA but not the periplasmic FlaB. Spirochetes (4 × 108) were incubated with or without proteinase K (100 µg/ml) or trypsin (100 µg/ml) for 2 h at RT. After incubation, phenylmethylsulfonyl fluoride (50 mg/ml) was added to inactivate proteinase K and trypsin, and thereafter spirochetes were lysed by sonication and total proteins were separated by SDS-PAGE and analyzed by Western blotting using a CspA-specific antiserum or an antiflagellin (FlaB) MAb (L41 1C11). A typical experiment of three is demonstrated. Download
CspA-expressing B. burgdorferi strain B31 is serum resistant. A growth inhibition assay was used to investigate susceptibility to human serum of B. burgdorferi strain B31. Spirochetes were challenged with NHS (50%) (■) or heat-inactivated NHS (hiNHS) (50%) (△). Following incubation over 7 days at 33°C, bacterial growth was monitored by measuring the ratio of culture medium absorbance at 562 versus 630 nm daily. The mean values from three separate experiments are shown, and error bars show SD. Download
CD59-specific MAbs do not bind CspA. Binding of anti-human CD59 MAb to immobilized human erythrocytes, CspA (5 µg/ml), CspZ (5 µg/ml), ErpP (5 µg/ml), ErpC (5 µg/ml), and ErpA (5 µg/ml) was assayed by ELISA. Bound anti-CD59 antibody was detected with HRP-conjugated anti-mouse antibody. Anti-CD59 bound to human erythrocytes but to none of the borrelial proteins. The mean values from three separate experiments are shown, and error bars show SD. Download
ACKNOWLEDGMENTS
This work was supported by the Leibniz Institute for Natural Product Research and Infection Biology.
We thank Michael Reuter and Sascha Böhm from the Department of Infection Biology of the Leibniz Institute for Natural Product Research and Infection Biology, Hans Knöll Institute, for providing the recombinant Sbi and SCIN from S. aureus. We also thank Jessica Günnewig for skillful and expert technical assistance and Scott Samuels for providing the plasmid pKFSS1.
Footnotes
Citation Hallström T, Siegel C, Mörgelin M, Kraiczy P, Skerka C, Zipfel PF. 2013. CspA from Borrelia burgdorferi inhibits the terminal complement pathway. mBio 4(4):e00481-13. doi:10.1128/mBio.00481-13.
REFERENCES
- 1. Zipfel PF, Skerka C. 2009. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 9:729–740 [DOI] [PubMed] [Google Scholar]
- 2. Fujita T. 2002. Evolution of the lectin-complement pathway and its role in innate immunity. Nat. Rev. Immunol. 2:346–353 [DOI] [PubMed] [Google Scholar]
- 3. Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, Pangburn MK, Llorca O, Lea SM, Gros P. 2012. Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep. 1:200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blom AM, Hallström T, Riesbeck K. 2009. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol. Immunol. 46:2808–2817 [DOI] [PubMed] [Google Scholar]
- 5. Stanek G, Reiter M. 2011. The expanding Lyme Borrelia complex---clinical significance of genomic species? Clin. Microbiol. Infect. 17:487–493 [DOI] [PubMed] [Google Scholar]
- 6. Steere AC. 2001. Lyme disease. N. Engl. J. Med. 345:115–125 [DOI] [PubMed] [Google Scholar]
- 7. Hildenbrand P, Craven DE, Jones R, Nemeskal P. 2009. Lyme neuroborreliosis: manifestations of a rapidly emerging zoonosis. AJNR Am. J. Neuroradiol. 30:1079–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lelovas P, Dontas I, Bassiakou E, Xanthos T. 2008. Cardiac implications of Lyme disease, diagnosis and therapeutic approach. Int. J. Cardiol. 129:15–21 [DOI] [PubMed] [Google Scholar]
- 9. Puius YA, Kalish RA. 2008. Lyme arthritis: pathogenesis, clinical presentation, and management. Infect. Dis. Clin. North Am. 22:289–300, vi-vii [DOI] [PubMed] [Google Scholar]
- 10. Kraiczy P, Stevenson B. 2013. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: structure, function and regulation of gene expression. Ticks Tick Borne Dis. 4:26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, Kraiczy P, Stevenson B. 2007. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete’s mammal-tick infection cycle. Infect. Immun. 75:4227–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seling A, Siegel C, Fingerle V, Jutras BL, Brissette CA, Skerka C, Wallich R, Zipfel PF, Stevenson B, Kraiczy P. 2010. Functional characterization of Borrelia spielmanii outer surface proteins that interact with distinct members of the human factor H protein family and with plasminogen. Infect. Immun. 78:39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF. 2007. Binding of human factor H-related protein 1 to serum-resistant Borrelia burgdorferi is mediated by borrelial complement regulator-acquiring surface proteins. J. Infect. Dis. 196:124–133 [DOI] [PubMed] [Google Scholar]
- 14. Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF. 2008. FHR-1, an additional human plasma protein, binds to complement regulator-acquiring surface proteins of Borrelia burgdorferi. Int. J. Med. Microbiol. 298:287–291 [Google Scholar]
- 15. Kraiczy P, Skerka C, Brade V, Zipfel PF. 2001. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect. Immun. 69:7800–7809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siegel C, Hallström T, Skerka C, Eberhardt H, Uzonyi B, Beckhaus T, Karas M, Wallich R, Stevenson B, Zipfel PF, Kraiczy P. 2010. Complement factor H-related proteins CFHR2 and CFHR5 represent novel ligands for the infection-associated CRASP proteins of Borrelia burgdorferi. PLoS One 5:e13519. 10.1371/journal.pone.0013519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hallström T, Haupt K, Kraiczy P, Hortschansky P, Wallich R, Skerka C, Zipfel PF. 2010. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. J. Infect. Dis. 202:490–498 [DOI] [PubMed] [Google Scholar]
- 18. Kraiczy P, Hellwage J, Skerka C, Becker H, Kirschfink M, Simon MM, Brade V, Zipfel PF, Wallich R. 2004. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J. Biol. Chem. 279:2421–2429 [DOI] [PubMed] [Google Scholar]
- 19. Brooks CS, Vuppala SR, Jett AM, Alitalo A, Meri S, Akins DR. 2005. Complement regulator-acquiring surface protein 1 imparts resistance to human serum in Borrelia burgdorferi. J. Immunol. 175:3299–3308 [DOI] [PubMed] [Google Scholar]
- 20. von Lackum K, Miller JC, Bykowski T, Riley SP, Woodman ME, Brade V, Kraiczy P, Stevenson B, Wallich R. 2005. Borrelia burgdorferi regulates expression of complement regulator-acquiring surface protein 1 during the mammal-tick infection cycle. Infect. Immun. 73:7398–7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braga LL, Ninomiya H, McCoy JJ, Eacker S, Wiedmer T, Pham C, Wood S, Sims PJ, Petri WA., Jr. 1992. Inhibition of the complement membrane attack complex by the galactose-specific adhesin of Entamoeba histolytica. J. Clin. Invest. 90:1131–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deng J, Gold D, LoVerde PT, Fishelson Z. 2003. Inhibition of the complement membrane attack complex by Schistosoma mansoni paramyosin. Infect. Immun. 71:6402–6410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fernie-King BA, Seilly DJ, Willers C, Würzner R, Davies A, Lachmann PJ. 2001. Streptococcal inhibitor of complement (SIC) inhibits the membrane attack complex by preventing uptake of C567 onto cell membranes. Immunology 103:390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Parizade M, Arnon R, Lachmann PJ, Fishelson Z. 1994. Functional and antigenic similarities between a 94-kD protein of Schistosoma mansoni (SCIP-1) and human CD59. J. Exp. Med. 179:1625–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pausa M, Pellis V, Cinco M, Giulianini PG, Presani G, Perticarari S, Murgia R, Tedesco F. 2003. Serum-resistant strains of Borrelia burgdorferi evade complement-mediated killing by expressing a CD59-like complement inhibitory molecule. J. Immunol. 170:3214–3222 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Z, Yang J, Wei J, Yang Y, Chen X, Zhao X, Gu Y, Cui S, Zhu X. 2011. Trichinella spiralis paramyosin binds to C8 and C9 and protects the tissue-dwelling nematode from being attacked by host complement. PLoS Negl. Trop. Dis. 5:e1225. 10.1371/journal.pntd.0001225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coleman AS, Yang X, Kumar M, Zhang X, Promnares K, Shroder D, Kenedy MR, Anderson JF, Akins DR, Pal U. 2008. Borrelia burgdorferi complement regulator-acquiring surface protein 2 does not contribute to complement resistance or host infectivity. PLoS One 3:3010e. 10.1371/journal.pone.0003010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roos A, Bouwman LH, Munoz J, Zuiverloon T, Faber-Krol MC, Fallaux-van den Houten FC, Klar-Mohamad N, Hack CE, Tilanus MG, Daha MR. 2003. Functional characterization of the lectin pathway of complement in human serum. Mol. Immunol. 39:655–668 [DOI] [PubMed] [Google Scholar]
- 29. Yu H, Muñoz EM, Edens RE, Linhardt RJ. 2005. Kinetic studies on the interactions of heparin and complement proteins using surface plasmon resonance. Biochim. Biophys. Acta 1726:168–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burman JD, Leung E, Atkins KL, O’Seaghdha MN, Lango L, Bernadó P, Bagby S, Svergun DI, Foster TJ, Isenman DE, van den Elsen JM. 2008. Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: indications of a novel mechanism of complement evasion by Staphylococcus aureus. J. Biol. Chem. 283:17579–17593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holt SC. 1978. Anatomy and chemistry of spirochetes. Microbiol. Rev. 42:114–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kenedy MR, Vuppala SR, Siegel C, Kraiczy P, Akins DR. 2009. CspA-mediated binding of human factor H inhibits complement deposition and confers serum resistance in Borrelia burgdorferi. Infect. Immun. 77:2773–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDowell JV, Harlin ME, Rogers EA, Marconi RT. 2005. Putative coiled-coil structural elements of the BBA68 protein of Lyme disease spirochetes are required for formation of its factor H binding site. J. Bacteriol. 187:1317–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. 2001. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconnection and factor H. Eur. J. Immunol. 31:1674–1684 [DOI] [PubMed] [Google Scholar]
- 35. Kilgore KS, Flory CM, Miller BF, Evans VM, Warren JS. 1996. The membrane attack complex of complement induces interleukin-8 and monocyte chemoattractant protein-1 secretion from human umbilical vein endothelial cells. Am. J. Pathol. 149:953–961 [PMC free article] [PubMed] [Google Scholar]
- 36. Woodruff TM, Nandakumar KS, Tedesco F. 2011. Inhibiting the C5-C5a receptor axis. Mol. Immunol. 48:1631–1642 [DOI] [PubMed] [Google Scholar]
- 37. Kraiczy P, Hellwage J, Skerka C, Kirschfink M, Brade V, Zipfel PF, Wallich R. 2003. Immune evasion of Borrelia burgdorferi: mapping of a complement-inhibitor factor H-binding site of BbCRASP-3, a novel member of the Erp protein family. Eur. J. Immunol. 33:697–707 [DOI] [PubMed] [Google Scholar]
- 38. Hartmann K, Corvey C, Skerka C, Kirschfink M, Karas M, Brade V, Miller JC, Stevenson B, Wallich R, Zipfel PF, Kraiczy P. 2006. Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol. Microbiol. 61:1220–1236 [DOI] [PubMed] [Google Scholar]
- 39. Kraiczy P, Hanssen-Hübner C, Kitiratschky V, Brenner C, Besier S, Brade V, Simon MM, Skerka C, Roversi P, Lea SM, Stevenson B, Wallich R, Zipfel PF. 2009. Mutational analyses of the BbCRASP-1 protein of Borrelia burgdorferi identify residues relevant for the architecture and binding of host complement regulators FHL-1 and factor H. Int. J. Med. Microbiol. 299:255–268 [DOI] [PubMed] [Google Scholar]
- 40. Hammerschmidt C, Hallström T, Skerka C, Wallich R, Stevenson B, Zipfel PF, Kraiczy P. 2012. Contribution of the infection-associated complement regulator-acquiring surface protein 4 (ErpC) to complement resistance of Borrelia burgdorferi. Clin. Dev. Immunol. 2012:349657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, Skerka C, Zipfel PF. 2008. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog. 4:e1000250. 10.1371/journal.ppat.1000250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. 2011. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 9:342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seidel SA, Dijkman PM, Lea WA, van den Bogaart G, Jerabek-Willemsen M, Lazic A, Joseph JS, Srinivasan P, Baaske P, Simeonov A, Katritch I, Melo FA, Ladbury JE, Schreiber G, Watts A, Braun D, Duhr S. 2013. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59:301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CspA inhibits all complement pathways. The activity of CspA on each complement pathway was analyzed using a Wielisa total complement screen (Wieslab, Malmö, Sweden) described by Seelen et al. (J. Immunol. Methods 296:187–198, 2005). The mean values from three separate experiments are shown, and error bars show SD. **, P ≤ 0.01; ***, P ≤ 0.001. Download
CspA does not inhibit the C3 convertase of the alternative pathway. (A to B) CspA does not affect C3a generation. (A) C3a generation was determined by ELISA. Detection of C3a in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), or HSA (25 µg/ml). (B) Cleavage products generated when the C3 convertase is formed were analyzed by Western blotting using C3a-specific antisera. (C and D) CspA does not affect Ba and Bb generation. (C) Ba generation was determined by ELISA. Detection of Ba in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), or HSA (25 µg/ml). (D) Cleavage products generated when the C3 convertase is formed were analyzed by Western blotting using factor B-specific antisera. (E) CspA does not inhibit C2b generation. CspA does not affect C2b generation in zymosan-activated serum. Detection of C2b in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), HSA (25 µg/ml), or anti-human C4d MAb (12 µg/ml). Cleavage products generated when the C3 convertases are formed were analyzed by Western blotting using a C2-specific antiserum. The immunoblot is a typical experiment of three. (F) CspA lacks C3 convertase decay-accelerating activity. The alternative pathway C3 convertase was assembled on the surface of microtiter plates, and then CspA (black columns) or factor H (white columns) was added. After incubation, C3 convertase levels were analyzed by ELISA using factor B-specific antiserum. The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. Download
CspA does not inhibit the C5 convertase activity. CspA does not affect C5a generation in zymosan-activated serum as determined by ELISA. Detection of C5a in zymosan-activated NHS with CspA (5 to 25 µg/ml), ErpC (25 µg/ml), SCIN (25 µg/ml), or HSA (25 µg/ml). The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. Download
CspA binds C7 and does not inhibit binding of C7 to the C5b-6 complex. (A) Binding of C7 to immobilized C5b-6 was assayed in the presence of CspA, vitronectin, anti-C7 MAb, or ErpC by ELISA. CspA, vitronectin, ErpC (each at 50 µg/ml), or anti-C7 MAb (1:100) was added to C7 (10 µg/ml) and incubated for 15 min at RT, and thereafter the mixture was added to immobilized C5b-6 (5 µg/ml). After being washed, bound C7 was detected with C7-specific antiserum followed by HRP-conjugated anti-goat antibody. (B) C7 binds to immobilized CspA, and binding is dose dependent. Binding of C7 (0.01 to 20 µg/ml) to immobilized CspA was assayed by ELISA. (C) Heparin inhibits the CspA-C7 interaction. The effect of heparin (1 to 500 µg/ml) on binding of C7 (10 µg/ml) to immobilized CspA (5 µg/ml) was assayed. (D) NaCl inhibits the CspA-C7 interaction. The effect of NaCl (0.1 to 1 M) on binding of C7 (5 µg/ml) to immobilized CspA (5 µg/ml) was assayed. The mean values from three separate experiments are shown, and error bars show SD. ***, P ≤ 0.001. Download
C7, C9, and factor H bind simultaneously to CspA. (A) Effect of increasing C7 levels in the presence of a constant concentration of C9. Binding of C7 (used at the indicated concentrations) and C9 (10 µg/ml) to immobilized CspA was analyzed by ELISA. Bound C7 was detected with C7-specific antiserum (black columns), and bound C9 was detected with C9-specific antiserum (white columns). (B) In a reverse setting, the C7 concentration was kept constant (10 µg/ml), and binding of C9 (used at the indicated concentrations) was evaluated. (C) Effect of increasing C7 levels in the presence of a constant concentration of factor H. Binding of C7 (used at the indicated concentrations) and factor H (2.5 µg/ml) to immobilized CspA was analyzed by ELISA. Bound C7 was detected with C7-specific antiserum (black columns), and bound factor H was detected with factor H-specific antiserum (white columns). (D) In a reverse setting, the C7 (10 µg/ml) concentration was kept constant, and binding of factor H (used at the indicated concentrations) was evaluated. (E and F) Similarly, binding of C9 and factor H to immobilized CspA was analyzed by ELISA. Bound C9 was detected with C9-specific antiserum (black columns), and bound factor H was detected with factor H-specific antiserum (white columns). The mean values from three independent experiments and SD are presented. Statistical significance of differences was estimated using Student’s t test. *, P ≤ 0.05. Download
Characterization of B. garinii G1 producing CspA from B. burgdorferi LW2. (A) The B. garinii-CspA strain harbors both the cspA and the plasmid-specific gene aaaA. B. garinii G1, the G1/pKFSS1 and B. garinii-CspA transformed strains, and shuttle vector pCspA were characterized by PCR amplification using cspA-, aadA-, and flaB-specific primers. (B) The B. garinii-CspA strain expresses CspA on the surface of intact spirochetes. Proteinase K degrades surface-exposed CspA but not the periplasmic FlaB. Spirochetes (4 × 108) were incubated with or without proteinase K (100 µg/ml) or trypsin (100 µg/ml) for 2 h at RT. After incubation, phenylmethylsulfonyl fluoride (50 mg/ml) was added to inactivate proteinase K and trypsin, and thereafter spirochetes were lysed by sonication and total proteins were separated by SDS-PAGE and analyzed by Western blotting using a CspA-specific antiserum or an antiflagellin (FlaB) MAb (L41 1C11). A typical experiment of three is demonstrated. Download
CspA-expressing B. burgdorferi strain B31 is serum resistant. A growth inhibition assay was used to investigate susceptibility to human serum of B. burgdorferi strain B31. Spirochetes were challenged with NHS (50%) (■) or heat-inactivated NHS (hiNHS) (50%) (△). Following incubation over 7 days at 33°C, bacterial growth was monitored by measuring the ratio of culture medium absorbance at 562 versus 630 nm daily. The mean values from three separate experiments are shown, and error bars show SD. Download
CD59-specific MAbs do not bind CspA. Binding of anti-human CD59 MAb to immobilized human erythrocytes, CspA (5 µg/ml), CspZ (5 µg/ml), ErpP (5 µg/ml), ErpC (5 µg/ml), and ErpA (5 µg/ml) was assayed by ELISA. Bound anti-CD59 antibody was detected with HRP-conjugated anti-mouse antibody. Anti-CD59 bound to human erythrocytes but to none of the borrelial proteins. The mean values from three separate experiments are shown, and error bars show SD. Download