

ABSTRACT
SurA is a component of the periplasmic chaperone network that plays a central role in biogenesis of integral outer membrane β-barrel proteins (OMPs) in Escherichia coli. Although SurA contains two well-conserved proline isomerase (PPIase) domains, the contribution of these domains to SurA function is unclear. In the present work, we show that defects in OMP assembly caused by mutation of the β-barrel assembly factors BamA or BamB can be corrected by gain-of-function mutations in SurA that map to the first PPIase domain. These mutations apparently bypass the requirement for a stable interaction between SurA and the Bam complex and enhance SurA chaperone activity in vivo despite destabilization of the protein in vitro. Our findings suggest an autoinhibitory mechanism for regulation of SurA chaperone activity through interdomain interactions involving a PPIase domain. We propose a model in which SurA activity is modulated by an interaction between SurA and the Bam complex that alters the substrate specificity of the chaperone.
IMPORTANCE
The dominant surA mutations described here alter amino acid residues that are highly conserved in eukaryotic homologs of SurA, including Pin1, the human proline isomerase (PPIase) implicated in Alzheimer’s disease and certain cancers. Consequently, a mechanistic description of SurA function may enhance our understanding of clinically important PPIases and their role(s) in disease. In addition, the virulence of Gram-negative bacterial pathogens, such as Salmonella, Shigella, and Escherichia coli O157:H7, is largely dependent on SurA, making this PPIase/chaperone an attractive antibiotic target. Investigating the function of SurA in outer membrane (OM) biogenesis will be useful in the development of novel therapeutic strategies for the disruption of the OM or the processes that are essential for its assembly.
Introduction
The outer membrane (OM) of Gram-negative bacteria, which primarily serves as a robust diffusion barrier, is distinguishable from the inner membrane (IM) in its relative asymmetry and unique constitution. Specifically, the OM harbors a distinct species of integral membrane protein that adopts a β-barrel fold and is specific to this extracytoplasmic compartment. These integral OM proteins (OMPs) are highly insoluble, aggregation-prone polypeptides that achieve native structure only in the cell membrane; consequently, molecular chaperones are required to maintain nascent OMPs in a folding-competent state as they exit the IM translocase (Sec) and traverse the aqueous periplasmic compartment in transit to the OM-localized OMP assembly complex known as Bam (1).
The periplasmic protein SurA has long been known to participate in assembly of integral outer membrane β-barrel proteins (2–4). The significant reduction in the levels of folded OMPs and the accumulation of unfolded periplasmic OMP species observed upon disruption of surA (4) provided early evidence for the contribution of SurA to OMP biogenesis. SurA plays a vital role in the maintenance of OM integrity in Escherichia coli, as surA mutants possess a defective OM that leaks periplasmic contents and permits the influx of detergents and toxic small molecules (2–4). SurA has been regarded as a periplasmic OMP chaperone since the time of its discovery, but a clear understanding of the mechanism by which SurA promotes OMP biogenesis is lacking (5).
Although the folding and assembly of many OMPs are impacted by deletion of surA (2, 4), only a small number of OMPs are strongly SurA dependent; a notable example is LptD, the essential lipopolysaccharide (LPS) insertase (6). The viability of surA mutants reflects the existence of an auxiliary OMP transport pathway comprising the chaperone Skp and the protease/chaperone DegP (7, 8). Yet, given the indirect but critical role for SurA in LPS assembly, the accumulation of misassembled OMPs observed in its absence, and the fact that disruption of surA is poorly tolerated (especially in combination with other mutations affecting OM biogenesis) (8–11), SurA is considered to be the primary OMP chaperone in the E. coli periplasm (6, 7, 12).
The architecture of SurA raises interesting questions about its activity. Structural analysis of SurA (13, 14) revealed a core module, which is formed from the N and C termini, and two proline isomerase (PPIase) domains of the parvulin type (referred to here as P1 and P2). The P1 domain associates extensively with the core module, whereas the peripheral P2 domain is tethered to the core by an extended linker. In vivo and in vitro analysis of SurA derivatives lacking one or more of the various domains described above led to the conclusion that the chaperone activity of SurA is contained within the core module; deletion of one or both parvulin domains has little impact on SurA chaperone activity, and the core module alone is largely sufficient to complement the function(s) of SurA in vivo (15). Only the second PPIase domain (P2) actually exhibits significant PPIase activity (4, 15), and this activity seems to be dispensable for SurA function (15). However, the strong conservation of PPIase domains among SurA homologs and the presence of multiple proline isomerases in the periplasm suggest that these domains of SurA have some physiological relevance (16).
SurA interacts with the periplasmic domain of the central OMP assembly factor, BamA, in a manner that can be stabilized by cross-linking agents (7, 17), and formation of this complex does not require the accessory lipoprotein BamB (18). The fact that deletion of BamB has no effect on the binding of SurA to Bam is somewhat surprising, as SurA and BamB have been proposed to function in the same folding pathway for several OMPs (6, 18–21). The molecular determinants of the interaction between SurA and BamA are not yet clear, although recent evidence suggests that the N-terminal POTRA domain of BamA (POTRA 1) is involved in this physical interaction (17, 22). Although the BamA periplasmic domain likely influences various aspects of the OMP assembly reaction, the importance of the POTRA domains must, at least in part, reflect their role in chaperone docking and substrate transfer.
We report here the isolation of a BamA mutant that impacts the functional and physical connection between SurA and the Bam complex. We show that the OMP misassembly that occurs upon disruption of the BamB pathway can be reversed by compensatory mutations within surA. Strikingly, these surA mutations are found within the first parvulin domain of SurA (P1). This finding represents the first direct evidence that implicates a PPIase domain of SurA in OMP biogenesis. Furthermore, the identification of these parvulin domain mutations in SurA provides compelling genetic evidence for the existence of two separable SurA-dependent OMP assembly pathways: one that involves BamB, and one that does not. The mutants reported here inform an integrated model for OMP targeting in the periplasm that offers insight into the role of Bam and a proline isomerase domain in the regulation of SurA activity.
RESULTS
Bam function is compromised in a bamA616 background.
To generate a library of partial loss-of-function bamA mutants, we conducted a screen for bamA mutations that decrease resistance to the detergent SDS, a phenotype that indicates impaired OM biogenesis (23). We describe here bamA616, a detergent-hypersensitive mutant isolated in this screen. This allele was found to encode three missense mutations (R91H, R162L, Y317H), but through construction of single and double mutants of every combination, we determined that the R91H and R162L mutations together are necessary and sufficient to cause the bamA616 phenotype. The pertinent substitutions that define bamA616 involve two residues lying at the interface between the first and second POTRA domains of BamA (see Fig. S1 in the supplemental material).
In addition to SDS, bamA616 strains exhibit increased susceptibility to antibiotic compounds with various physicochemical properties (see Table S1a in the supplemental material). To determine whether this increased drug sensitivity reflects a defect in OMP assembly, we determined the steady-state levels of various OMPs in this mutant strain. We observed reduced levels of abundant OMPs (particularly LamB and OmpF) in this background (Fig. 1a), an effect attributable to misassembly, degradation, and reduced synthesis of major OMPs (6, 24, 25). In addition, we observed a marked increase in the levels of the periplasmic protease DegP (Fig. 1a) and a 4-fold increase in expression from the rpoHP3 promoter (Fig. 1b); both of these findings indicate activation of the σE extracytoplasmic stress response through a signaling cascade induced by unfolded periplasmic OMPs (26). Together, these findings indicate that OMP biogenesis is impacted by the bamA616 mutations. This effect is limited to a subset of OMPs, as the levels of LptD, BamA, and TolC are unchanged in the mutant background (Fig. 1a).
FIG 1 .

bamA616 impacts biogenesis of major OMPs. (A) Relative OMP levels were determined in whole-cell extracts of bamA+ and bamA616 strains that were subjected to SDS-PAGE and immunoblotting for the proteins indicated. (B) σE activity was assayed by measuring β-galactosidase activity in bamA+ and bamA616 strains containing a chromosomal copy of the σE-dependent rpoHP3-lacZ reporter fusion. (C) Whole-cell extracts of bamA+, bamA616, and bamB8 strains were prepared, and envelope proteins were detected as described for panel A.
We note that bamA616 strains bear a striking phenotypic resemblance to those that carry bamB8, a previously characterized null mutation that abrogates the function of the accessory lipoprotein BamB (9). bamA616 and bamB8 have similar antibiotic and small molecule sensitivity profiles, and the levels of affected OMPs are similarly reduced in each mutant background (Fig. 1c). In addition, rather than observing a global decrease in OMP levels in bamA616 and bamB8 strains (as occurs upon BamA or BamD depletion), each mutation results in a decrease in levels of major OMPs (such as LamB and OmpF) but not LptD or BamA (6). Finally, we are unable to construct a strain containing both bamA616 and imp4213 by P1 transduction (data not shown); this is striking considering the reported synthetic lethality of bamB8 imp4213 double mutants (23, 27). While bamA616 strains seem to exhibit a bamB-null-like phenotype, we do not observe an obvious change in binding of BamB to BamA616 compared with that of BamA (see Text in the supplemental material and Fig. 2), suggesting that the bamA616 phenotype is not simply caused by the physical dissociation of BamA and BamB.
FIG 2 .

The interaction between SurA and BamA is compromised by bamA616. DSP cross-linking and affinity purification were performed in cells expressing His6-tagged BamA+ (lanes 1, 2, 5, and 6) or BamA616 (lanes 3, 4, 7, and 8). Cells were incubated in the presence of DSP (even lanes) or DMSO (odd lanes) prior to lysis. Whole-cell extracts (lanes 1 to 4) and Ni-nitrilotriacetic acid (NTA)-enriched eluate fractions (lanes 5 to 8) were subjected to SDS-PAGE and immunoblotting for the indicated envelope proteins.
Binding of SurA to BamA is impaired in a bamA616 background.
Several lines of evidence suggest that some OM β-barrels are targeted to and assembled by BamA via a folding pathway that includes both BamB and the periplasmic chaperone SurA (6, 18–20). Given the bamB-null-like phenotype of the bamA616 mutant and the apparent functional relationship between SurA and BamB, we considered the possibility that SurA function is somehow compromised in a bamA616 background.
The Bam proteins form a stable complex that is readily purified intact, and no other OMPs or envelope proteins are known to stably associate with Bam in E. coli (see Fig. S2 in the supplemental material) (28, 29). However, SurA is known to interact with the Bam complex via a physical interaction that can be trapped with chemical cross-linking agents (7, 17, 18). As such, we first sought to determine whether the bamA616 mutations influences this interaction. Copurification experiments using wild-type His-Bam (His-BamAWT) or His-BamA616 as described above were repeated in the presence of the OM-permeable cross-linking agent DSP [dithiobis(succinimidyl propionate)]. Upon purification, we observe that both His-BamAWT and His-BamA616 form detectable interactions with SurA. However, there is an apparent 50% decrease in the amount of SurA that copurifies with His-BamA616 compared to that with His-BamAWT (Fig. 2, top). This decrease suggests that the physical interaction between BamA and SurA is impacted by the bamA616 mutations.
Mutations in surA suppress bamA616.
In order to better understand the physical and functional interaction(s) between SurA, BamB, and the N-terminal POTRA domains of BamA, we exploited the detergent sensitivity of bamA616 mutant to select for mutations that reverse the OM biogenesis defects observed in a bamA616 background. By selecting spontaneous SDS-EDTAr revertants at 37°C, we identified two extragenic suppressor mutations that confer wild-type SDS-EDTA resistance to a bamA616 strain but differ in their effect on colony size: one suppressor strain forms normal colonies on rich media, whereas the other forms abnormally small colonies. Despite the difference in colony morphology, we found that both of these suppressor strains carry mutations in the surA gene. We refer to the suppressor alleles here as surA10 and surA13, which give rise to normal and small colonies, respectively, in a bamA616 background. The surA10 mutation results in a Ser-to-Ala substitution in SurA at residue 220 (SurAS220A). surA13 encodes a deletion of Leu215 and Ala216 (SurA∆LA).
In addition to restoring SDS-EDTA resistance, both surA mutations reduce the sensitivity of a bamA616 strain to other antibiotics, albeit to different degrees (see Table S1a in the supplemental material). The increase in resistance to multiple antibiotic compounds shows that these suppressors are not acting to restrict the influx of SDS exclusively. This suggests that the surA10 and surA13 mutations influence the composition of the OM, perhaps by correcting the defect in OMP maturation that is observed in a bamA616 background. To test this, we measured steady-state OMP levels and quantified σE stress response activation in the suppressor strains. In the bamA616 surA10 double mutant, we observe a substantial increase in the steady-state levels of LamB, OmpF, and OmpA, such that the cellular amounts of these OMPs approximated the levels observed in wild-type cells (Fig. 3a). In addition, transcriptional activity at the rpoHP3 promoter (a direct σE target) is reduced to baseline levels in the double mutant (Fig. 3b). Taken together, these data show that the surA10 mutation restores efficient OMP biogenesis in a bamA616 background.
FIG 3 .

The surA10 and surA13 suppressors restore detergent resistance but differentially influence OMP levels. (A) Whole-cell extracts of strains indicated were prepared by boiling after harvesting cells at an OD600 of 1. Major OMPs were detected by SDS-PAGE and immunoblotting. Detergent sensitivity of each strain is shown below the corresponding lane (SDSR, detergent resistant; SDSS, detergent sensitive). (B) β-Galactosidase activity was measured in the indicated strains containing a transcriptional fusion with the σE-dependent rpoHP3 promoter driving expression of lacZ. (C) The strains indicated were processed as described for panel A, and the indicated envelope proteins were detected by immunoblotting.
Surprisingly, LamB levels in the bamA616 surA13 (small colony) suppressor strain are decreased in comparison to levels in the bamA616 strain (Fig. 3c). Additionally, we do not observe a significant change in transcriptional activity at the rpoHP3 promoter compared to that of the bamA616 single mutant (Fig. 3b). Thus, the bamA616 surA13 strain, although quite similar to the wild type with respect to OM permeability, has an OMP profile and colony morphology distinct from that of a bamA616 surA10 strain, its surA+ parent, and the wild type. We have found that this apparent incongruity reflects activation of the Cpx stress response in the bamA616 surA13 double mutant specifically; because of the pleiotropic effects caused by the surA13 allele, we here concentrate our analysis on the surA10 suppressor (see Text and Fig. S3a to c in the supplemental material).
surA10 does not restore the SurA-BamA616 interaction.
To determine whether the surA10 mutation restores binding of SurA to BamA616, we performed copurification experiments in surA10 strains expressing either bamA+ or bamA616. We find that SurA+ and SurAS220A can be cross-linked to wild-type BamA in roughly equivalent amounts. However, the amount of SurAS220A that copurifies with BamA616 is even less than is observed for wild-type SurA (Fig. 4). The S220A mutation does not impair the ability of SurA to interact with BamA+, nor does it restore the physical interaction between SurA and BamA616.
FIG 4 .

The surA10 mutation does not restore the SurA-BamA616 interaction. DSP cross-linking and affinity purification were performed using His6-tagged BamA+ (lanes 1, 4, and 5) or BamA616 (lanes 2 and 3). All strains indicated were incubated with the cross-linker DSP prior to lysis. Whole-cell extracts (left) and Ni-NTA-enriched eluate fractions (right) were subjected to SDS-PAGE and immunoblotting for BamA and SurA.
Mutations in SurA P1 suppress the loss of BamB.
If the phenotypic similarity between the bamA616 and the bamB-null mutants reflects a common defect in an OMP assembly pathway, surA10 might also be expected to suppress the bamB-null mutant. Intriguingly, we found that the surA10 mutation indeed suppresses the detergent sensitivity and OMP assembly defects observed in a bamB mutant background (Fig. 5), suggesting that the variant overcomes a defect that arises upon deletion of bamB or substitution of BamA residues R91 and R162.
FIG 5 .
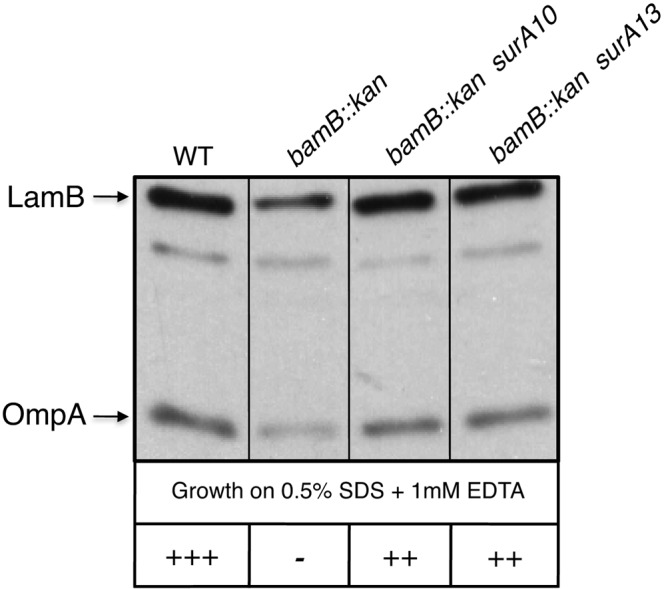
bamB-null phenotypes are suppressed by surA10 and surA13. Whole-cell extracts of bamA+ and bamB::kan strains (containing wild-type or suppressor alleles of surA) were analyzed by Western blotting using antisera that recognize LamB and OmpA. Detergent sensitivity phenotypes are shown below each lane; the qualitative growth score on agar plates containing SDS-EDTA is indicated by the number of “+” symbols present. −, no growth; ++, intermediate growth; +++, wild-type growth.
The observation that surA10 is neither gene specific nor capable of restoring the SurA-BamA616 interaction is consistent with the notion that surA10 is a gain-of-function allele. Several additional observations support this hypothesis (see the text in the supplemental material): (i) surA10 is dominant to surA+ in diploid analysis (see Table S1b in the supplemental material), (ii) surA10 is sufficient to fully complement ∆surA (Fig. 6a), (iii) SurAS220A exhibits increased activity relative to SurA+ (Fig. 6b), and (iv) SurA overexpression is not sufficient to suppress bamA616 (see Table S1b).
FIG 6 .
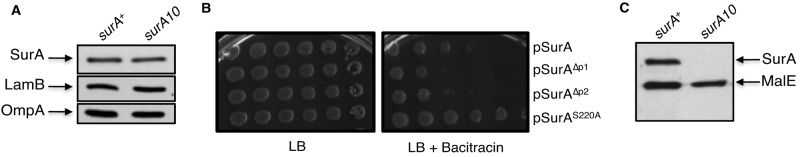
SurAS220A is hyperactive in vivo and unstable in vitro. (A) Western blot analysis was performed with wild-type and surA10 strains to determine relative levels of SurA, LamB, and OmpA. (B) Serial 10-fold dilutions of stationary-phase cultures of ∆surA strains containing plasmids expressing SurA+, SurAS220A, SurA∆P1, or SurA∆P2 were spotted onto LB with or without bacitracin and incubated at 37°C. (C) Periplasmic contents in cultures of wild-type and surA10 strains were released by chloroform shock, and samples were incubated at 37°C prior to SDS-PAGE. SurA and the periplasmic protein MalE were detected by Western blotting using the appropriate antisera.
The S220A mutation destabilizes SurA in vitro.
All residues affected by the surA10 and surA13 mutations (Leu215, Ala216, and Ser220) are highly conserved in both eukaryotic and bacterial parvulins (see Fig. S4a in the supplemental material) and map to the α2 helix of the P1 domain; this helix lies along one edge of the antiparallel β-sheet within P1 and forms one side of the peptide binding pocket (see Fig. S4b), although none of the residues described above interact directly with peptide substrates that bind P1 (14). In the full-length SurA protomer, the P1 α2 helix also forms a major interface between the P1 domain and the core chaperone domain of SurA (see Fig. S4c). We therefore speculated that the surA10 mutation might influence the stability of the P1-core interaction, potentially impacting global stability of the protein. In order to test this, we released periplasmic proteins from cells expressing surA+ or surA10 by chloroform shock and determined the fate of SurA and SurAS220A following release. We found that SurA+ can be observed in protein extracts following incubation at 37°C. However, SurAS220A is undetectable under identical conditions, suggesting that the protein is destabilized and degraded after periplasmic release (Fig. 6c). As a control, we show that the periplasmic protein MalE remains resistant to degradation in either a surA+ or surA10 background.
To confirm this finding by an independent method, we prepared cell lysates of strains expressing surA+ or surA10 under conditions in which cellular proteins are solubilized but not denatured. When these strains are lysed gently in a mild detergent solution, brief incubation of cell extracts at room temperature prior to boiling in SDS-PAGE sample buffer leads to the complete degradation of SurAS220A but not SurA+ (see Fig. S5a). Because this degradation can be partially stabilized by the addition of protease inhibitors (data not shown), and because levels of SurAS220A in whole cells are comparable to those observed for SurA+ (Fig. 6a), we interpret this finding to mean that SurAS220A is much more susceptible to degradation by cellular proteases following lysis than is SurA+.
DISCUSSION
The mutations that cause the bamA616 phenotype affect residues at the interface of POTRA 1 and POTRA 2 near the periplasmic N terminus of BamA. POTRA 1 and 2 form a rigid body that is maintained by extensive interdomain contacts (33, 42, 43), and it is possible that this POTRA pair operates as a single module (32). POTRA 1 and 2 have previously been implicated in binding of SurA (17, 22) and BamB (33), and it has been suggested that POTRA 1 is involved in the assembly of a subset of OMPs that includes the archetypal porins OmpF/OmpC and the major OMPs LamB and OmpA (17). Consistent with this hypothesis, we find that bamA616 specifically affects these abundant OMPs without compromising the assembly of LptD, TolC, or BamA itself.
BamA616 interacts normally with BamB, so the bamB-null-like phenotype of a bamA616 strain cannot be attributed to the physical dissociation of these two components. We argue instead that POTRA 1/2 and BamB are each involved in a common function that is unmasked in a bamA616 background or in the absence of BamB. Because binding of SurA to BamA is reduced by the bamA616 mutations and because both bamA616 and bamB mutant strains exhibit OMP biogenesis defects that are suppressed by mutations in SurA, we conclude that the primary defect in each of these mutant strains likely relates to the activity of SurA (or lack thereof). Our observations implicate the N-terminal POTRA module and BamB in the regulation of SurA function. We suggest that BamAB modulates SurA chaperone activity and that the SurAS220A variant mimics the BamAB-induced activity state of SurA.
SurA activity is potentiated by the surA10 mutation in a manner that cannot be recreated by simply increasing its concentration. Unexpectedly, the S220A (surA10) and ∆215-216 (surA13) mutations are found not in the chaperone domain of SurA but in the first PPIase domain (P1). surA10 is a “gain-of-function” mutation in that it increases SurA chaperone activity. Yet, a comparative analysis of parvulins suggests that S220A and ∆215-216 should represent loss-of-function mutations, since substitutions at those highly conserved residues abrogate function in homologs of SurA (34, 35). These observations can be reconciled by proposing that the P1 domain inhibits SurA chaperone activity and that the surA10 and surA13 mutations disinhibit the chaperone domain by crippling the inhibitory P1 domain (antirepression). In this way, loss-of-function mutations in an autoinhibitory domain lead to the increased activity of a covalently linked chaperone domain. This model presumes that the activity of full-length SurA is normally at least partially inhibited. Consistent with this proposal, the in vitro chaperone activity of SurA is significantly increased upon the deletion of one or both parvulin domains (15). Since the parvulin domains have no intrinsic chaperone activity, they likely act by modulating the activity of the chaperone domain.
Our model further implies that the relief of this autoinhibition involves the dissociation of the P1 and chaperone domains of SurA and that the S220A and ∆215-216 mutations somehow destabilize the P1-core complex. In support of this, we find that SurAS220A is particularly protease susceptible, a possible indication that the disruption of interdomain contacts destabilizes SurA. This is corroborated by the reported instability of a SurA variant lacking both parvulin domains (14) and suggests that the P1 domain stabilizes and inhibits the core chaperone domain; a similar regulatory function has been proposed for the autoinhibited signaling adaptor protein Crk (36). Structural analysis has shown that the P1 domain can dissociate completely from the chaperone core while bound to a model peptide (14), suggesting that SurA may normally undergo conformational changes that involve the transient uncoupling of the P1 and core domains. The α2 helix of SurA, which contains the residues that are mutated in both surA10 and surA13, forms multiple contacts with residues in the chaperone domain of SurA (13). It is possible, then, that mutations in P1 destabilize the P1-core complex, thereby eliminating an interaction that occludes both the P1 active site and a large surface in the core chaperone domain. It is tempting to suggest that BamAB catalyzes the displacement of P1 from the core domain.
We favor a model in which SurA, by default, primarily assists the folding of high-priority substrates (such as LptD, which is principally dependent on SurA for assembly) (6) but can be induced by POTRA 1/2 and BamB to promote the assembly of abundant OMPs through modulation of its chaperone activity via the P1 domain. We suggest that the role of SurA in LptD biogenesis can be separated from its role as a general OMP chaperone and that BamA and BamB regulate these activities by adjusting the equilibrium between the autoinhibited and activated SurA conformations (Fig. 7).
FIG 7 .
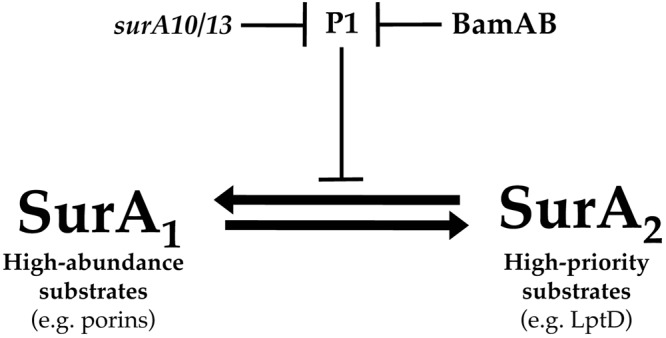
Model for regulation of SurA activity by BamAB and the P1 domain of SurA. SurA exists in equilibrium between two conformations, one (SurA1) that is optimized for the targeting of major, abundant OMPs (such as OmpF, OmpC, and LamB), and another (SurA2) that promotes the prioritized assembly of critical, lower-abundance substrates (such as LptD). A shift in conformational equilibrium toward the SurA1 state involves relief of P1-mediated inhibition of SurA chaperone activity through the combined action of BamB and the BamA N terminus. The surA10 and surA13 mutations bypass the requirement for BamAB in regulation of SurA activity, potentially by destabilizing the interaction between P1 and the core chaperone domain.
An obvious advantage of this mode of regulation is that it permits selectivity by maintaining dedicated and semi-independent OMP delivery pathways. This model provides a mechanism for both efficient assembly of highly abundant OMPs—several of which are present in numbers approaching 105 molecules/cell (37)—and the prioritized delivery of essential minor OMPs, such as LptD. This enables both the concentration-driven assembly of major OM proteins and the selective assembly of low-abundance OMP species that are critical for cell growth.
Using this model as a framework, we suggest that the SurA variants described in this report effectively bypass a regulatory requirement for POTRA 1/2 and BamB by artificially relieving the P1-mediated autoinhibition of SurA chaperone activity (Fig. 7). Although this model provides a satisfying explanation for the genetic and biochemical data presented here, it does not explain why the P1 domain is required for suppression of bamA616 (see Table S1a in the supplemental material). One possibility is that P1 performs a second role in addition to SurA autoinhibition; such a role is not without precedent, as cooperativity between proline isomerase and chaperone domains has been observed in several other bacterial PPIase-chaperone hybrid proteins, such as SlyD (30), trigger factor (31), and PrsA (38). If cooperative catalysis of protein folding occurs between P1 and the disinhibited SurA chaperone domain, this may account for the additional requirement for P1 in suppression of bamA616 by surA10 and surA13.
In addition to unmasking a regulatory function for a SurA PPIase domain, the model presented here has implications for the interplay between the factors comprising the E. coli periplasmic chaperone network. Recent work in our lab revealed an unexpected role for the secondary OMP chaperone Skp in assembly of LptD (39). According to the model diagrammed in Fig. 7, SurA conformational equilibrium should be shifted toward the form that favors assembly of LptD (SurA2) in a bamB mutant background. If this is the case, the profound synthetic LptD biogenesis defect that is observed upon simultaneous deletion of Skp and BamB (39) may in fact indicate a critical role for Skp in the prioritized assembly of LptD by SurA2 in particular, or it may even suggest that Skp is somehow involved in shifting the equilibrium toward the SurA2 conformation.
MATERIALS & METHODS
Bacterial strains and plasmids.
Strains and plasmids used in this study are described in Table S2 in the supplemental material. All strains were constructed using standard microbiological techniques. When necessary, media were supplemented with 125 µg/ml ampicillin, 25 µg/ml kanamycin, or 25 µg/ml tetracycline. All bacterial cultures were grown under aerobic conditions at 37°C unless otherwise noted.
Random and site-directed mutagenesis.
For a detailed description of mutagenesis procedures, see Materials and Methods in the supplemental material. Oligonucleotide primer sequences are provided in Table S3 in the supplemental material.
Quantification of small molecule and antibiotic sensitivity.
Sensitivities to bacitracin, erythromycin, novobiocin, and rifampin were measured using the BBL Sensi-Disc antimicrobial susceptibility test discs (BD) as described (9). Susceptibility was quantified by measuring the diameter of the zones of inhibition around the discs. For efficiency of plating (EOP) assays, serial dilutions of stationary-phase cultures were spotted onto LB agar with or without 625 µg/ml bacitracin and incubated overnight at 37°C.
Genetic selection.
Spontaneous detergent-resistant suppressors of bamA616 were obtained by plating 1 ml of an overnight culture at 37°C on LB agar containing SDS-EDTA (0.5%:1.0 mM). Suppressors that arose following overnight incubation were purified on selective media at 37°C. Mutations were mapped by P1 transduction and identified by DNA sequencing.
Western blot analysis.
For a detailed description of Western blot procedures, see Materials and Methods in the supplemental material.
Quantification of β-galactosidase activity.
Cells were grown in LB liquid medium with aeration at 37°C to mid-exponential phase. Aliquots of 1 ml were withdrawn from each culture, and cells were then pelleted and permeabilized by treatment with chloroform and SDS. β-Galactosidase activity from the chromosomal σE-dependent rpoHP3::lacZ reporter (26) was spectrophotometrically assayed in triplicate using the Miller assay (40) as previously described.
Affinity purification and in vivo cross-linking.
For a detailed description of affinity purification and in vivo cross-linking procedures, see Materials and Methods in the supplemental material.
Release of periplasmic proteins.
Periplasmic contents were released by chloroform shock as previously described (41). As an alternative method, cells were grown to an optical density at 600 nm (OD600) of 1, harvested by centrifugation, washed in 20 mM potassium phosphate (pH 7.2) and 150 mM NaCl, resuspended in BugBuster solution (Novagen) containing lysozyme (5 µg/ml), DNase I (50 µg/ml), RNase I (50 µg/ml), and allowed to lyse gently by rocking for 15 min at room temperature. Following lysis, the lysates were centrifuged at 10,000 × g for 10 min to remove debris. SDS-PAGE buffer was added to the samples, which were then boiled for 10 min prior to Western blot analysis as described above.
SUPPLEMENTAL MATERIAL
Supplemental text. Download
Supplemental materials and methods. Download
The bamA616 phenotype is caused by substitutions at two Arg residues in POTRA 1 to 2. A model of full-length BamA [R. Albrecht and K. Zeth, J. Biol. Chem. 286(31):27792–803, 2011] is shown in the left panel in the context of a schematically depicted outer membrane (OM). The model was generated by superimposing POTRA 4 and 5 of BamA (PDB code 3OG5) onto the N-terminal POTRA 1 and 2 domains of the related transporter FhaC in the full-length crystal structure (PDB code 2QDZ). The complete structural model was then obtained by superimposing POTRA 4 and 5 with the crystal structure of POTRA 1 to 4 of BamA (PDB code 3EFC) at POTRA 4. The N-terminal POTRA pair (POTRA 1 and 2) is shown in the right panel with R91 and R162 highlighted in red on the structure and the corresponding mutations in bamA616 indicated in red text. Download
The envelope proteins SurA and LamB do not stably interact with the Bam complex. Affinity purification was performed in cells expressing C-terminally His6-tagged BamD+ (BamD-His) in the absence of a chemical cross-linker. A whole-cell extract (left) and Ni-NTA-enriched eluate fraction (right) were subjected to SDS-PAGE and immunoblotting for SurA, LamB, and the indicated Bam proteins. Download
The downregulation of envelope proteins in a bamA616 surA13 double mutant is a consequence of Cpx stress response induction. Whole-cell extracts of the strains shown were prepared after harvesting cells at late exponential phase. Samples were subjected to SDS-PAGE, and antibodies were used to specifically detect MalE (A) or LamB (B, C). Download
The surA10 and surA13 mutations affect α2 helix residues conserved across parvulins. (A) An alignment of parvulin-like PPIase domain sequences from highly diverse species was generated using ClustalW. The surA10 and surA13 mutation sites are shaded in purple and orange (respectively) and are indicated by arrows. (B) The structure of SurA-P1 (cyan) is shown bound to a model helical peptide (pink) to highlight the positions of Leu215, Ala216, and Ser220 (indicated by arrows and colored as described above) relative to the binding pocket of P1 (PDB code 2PV3). (C) Left, space-filling model of full-length SurA (PDB code 1M5Y) with the PPIase domains labeled and colored in cyan and the core chaperone domain colored in orange. The dashed arrow indicates the perspective shown in the right panel. Right, ribbon representation of the structure shown on the left, rotated 90° and amplified for clarity. The α2 helix (which contains the residues affected in surA10 and sur13 mutants) is indicated by a black line, and Leu215, Ala216, and Ser220 are shaded in red. For simplicity, the P2 domain is hidden in the right panel. Download
SurAS220A is unstable in vitro. Late-exponential-phase cultures (OD600 = 1) of the indicated strains were harvested and gently lysed at room temperature in a mild detergent buffer (see Materials and Methods). The cleared lysates were boiled and subjected to SDS-PAGE, and SurA was detected immunologically. The asterisk (*) indicates an unknown protein that cross-reacts with the anti-SurA antiserum. Download
bamA616 exhibits increased OM permeability that is suppressed by surA mutations in a dominant fashion. (A) Antimicrobial test discs soaked in bacitracin (Bac), novobiocin (Nov), erythromycin (Ery), and rifampin (Rif) were added to LB agar containing a lawn of the strains indicated. The data are expressed as the diameter of the zone of clearing surrounding each disc in millimeters. Numbers in parentheses indicate a hazy zone of clearing. (B) The strains listed were grown on LB plates containing 0.5% SDS-1.0 mM EDTA either in the presence (+ara) or absence (−ara) of arabinose, which induces expression of plasmid-borne surA and its derivatives. R, resistant to SDS-EDTA; S, sensitive to SDS-EDTA.
Strains and plasmids used in this study.
Mutagenic oligonucleotides used for site-directed mutagenesis.
ACKNOWLEDGMENTS
We thank R. Misra for generously providing anti-TolC antisera.
This work was supported by National Institute of General Medical Sciences grant GM34821.
Footnotes
Citation Ricci DP, Schwalm J, Gonzales-Cope M, Silhavy TJ. 2013. The activity and specificity of the outer membrane protein chaperone SurA are modulated by a proline isomerase domain. mBio 4(4):e00540-13. doi:10.1128/mBio.00540-13.
REFERENCES
- 1. Ricci DP, Silhavy TJ. 2012. The bam machine: a molecular cooper. Biochim. Biophys. Acta 1818:1067–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lazar SW, Kolter R. 1996. SurA assists the folding of Escherichia coli outer membrane proteins. J. Bacteriol. 178:1770–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Missiakas D, Betton JM, Raina S. 1996. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol. 21:871–884 [DOI] [PubMed] [Google Scholar]
- 4. Rouvière PE, Gross CA. 1996. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10:3170–3182 [DOI] [PubMed] [Google Scholar]
- 5. Behrens-Kneip S. 2010. The role of SurA factor in outer membrane protein transport and virulence. Int. J. Med. Microbiol. 300:421–428 [DOI] [PubMed] [Google Scholar]
- 6. Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet JF. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21:2473–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizzitello AE, Harper JR, Silhavy TJ. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183:6794–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruiz N, Falcone B, Kahne D, Silhavy TJ. 2005. Chemical conditionality: a genetic strategy to probe organelle assembly. Cell 121:307–317 [DOI] [PubMed] [Google Scholar]
- 10. Onufryk C, Crouch ML, Fang FC, Gross CA. 2005. Characterization of six lipoproteins in the sigmaE regulon. J. Bacteriol. 187:4552–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, Braberg H, Yamamoto N, Takeuchi R, Wanner BL, Mori H, Weissman JS, Krogan NJ, Gross CA. 2008. High-throughput, quantitative analyses of genetic interactions in E. coli. Nat. Methods 5:781–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denoncin K, Schwalm J, Vertommen D, Silhavy TJ, Collet JF. 2012. Dissecting the Escherichia coli periplasmic chaperone network using differential proteomics. Proteomics 12:1391–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bitto E, McKay DB. 2002. Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10:1489–1498 [DOI] [PubMed] [Google Scholar]
- 14. Xu X, Wang S, Hu YX, McKay DB. 2007. The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J. Mol. Biol. 373:367–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Behrens S, Maier R, de Cock H, Schmid FX, Gross CA. 2001. The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J. 20:285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merdanovic M, Clausen T, Kaiser M, Huber R, Ehrmann M. 2011. Protein quality control in the bacterial periplasm. Annu. Rev. Microbiol. 65:149–168 [DOI] [PubMed] [Google Scholar]
- 17. Bennion D, Charlson ES, Coon E, Misra R. 2010. Dissection of β-barrel outer membrane protein assembly pathways through characterizing BamA POTRA 1 mutants of Escherichia coli. Mol. Microbiol. 77:1153–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vuong P, Bennion D, Mantei J, Frost D, Misra R. 2008. Analysis of YfgL and YaeT interactions through bioinformatics, mutagenesis, and biochemistry. J. Bacteriol. 190:1507–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ureta AR, Endres RG, Wingreen NS, Silhavy TJ. 2007. Kinetic analysis of the assembly of the outer membrane protein LamB in Escherichia coli mutants each lacking a secretion or targeting factor in a different cellular compartment. J. Bacteriol. 189:446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hagan CL, Kim S, Kahne D. 2010. Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palomino C, Marín E, Fernández LA. 2011. The fimbrial usher FimD follows the SurA-BamB pathway for its assembly in the outer membrane of Escherichia coli. J. Bacteriol. 193:5222–5230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Workman P, Heide K, Giuliano N, Lee N, Mar J, Vuong P, Bennion D, Misra R. 2012. Genetic, biochemical, and molecular characterization of the polypeptide transport-associated domain of Escherichia coli BamA. J. Bacteriol. 194:3512–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruiz N, Wu T, Kahne D, Silhavy TJ. 2006. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem. Biol. 1:385–395 [DOI] [PubMed] [Google Scholar]
- 24. Charlson ES, Werner JN, Misra R. 2006. Differential effects of yfgL mutation on Escherichia coli outer membrane proteins and lipopolysaccharide. J. Bacteriol. 188:7186–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol. 4:e2. 10.1371/journal.pbio.0040002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mecsas J, Rouviere PE, Erickson JW, Donohue TJ, Gross CA. 1993. The activity of sigma E, an Escherichia coli heat-inducible sigma-factor, is modulated by expression of outer membrane proteins. Genes Dev. 7:2618–2628 [DOI] [PubMed] [Google Scholar]
- 27. Ruiz N, Silhavy TJ. 2005. Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr. Opin. Microbiol. 8:122–126 [DOI] [PubMed] [Google Scholar]
- 28. Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ. 2007. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 104:6400–6405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245 [DOI] [PubMed] [Google Scholar]
- 30. Zoldák G, Schmid FX. 2011. Cooperation of the prolyl isomerase and chaperone activities of the protein folding catalyst SlyD. J. Mol. Biol. 406:176–194 [DOI] [PubMed] [Google Scholar]
- 31. Liu CP, Zhou QM, Fan DJ, Zhou JM. 2010. PPIase domain of trigger factor acts as auxiliary chaperone site to assist the folding of protein substrates bound to the crevice of trigger factor. Int. J. Biochem. Cell Biol. 42:890–901 [DOI] [PubMed] [Google Scholar]
- 32. Jacob-Dubuisson F, Villeret V, Clantin B, Delattre AS, Saint N. 2009. First structural insights into the TpsB/Omp85 superfamily. Biol. Chem. 390:675–684 [DOI] [PubMed] [Google Scholar]
- 33. Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D. 2007. Structure and function of an essential component of the outer membrane protein assembly machine. Science 317:961–964 [DOI] [PubMed] [Google Scholar]
- 34. Wu X, Wilcox CB, Devasahayam G, Hackett RL, Arévalo-Rodríguez M, Cardenas ME, Heitman J, Hanes SD. 2000. The Ess1 prolyl isomerase is linked to chromatin remodeling complexes and the general transcription machinery. EMBO J. 19:3727–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Behrsin CD, Bailey ML, Bateman KS, Hamilton KS, Wahl LM, Brandl CJ, Shilton BH, Litchfield DW. 2007. Functionally important residues in the peptidyl-prolyl isomerase Pin1 revealed by unigenic evolution. J. Mol. Biol. 365:1143–1162 [DOI] [PubMed] [Google Scholar]
- 36. Cho JH, Muralidharan V, Vila-Perello M, Raleigh DP, Muir TW, Palmer AG. 2011. Tuning protein autoinhibition by domain destabilization. Nat. Struct. Mol. Biol. 18:550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nikaido H. 1996. Outer membrane, p 29–47 In Neidhardt F, Curtiss R, Ingraham J, Lin EC, Low K, Magasanik B, Reznikoff W, Riley M, Schaechter M, Umbarger HE, Escherichia coli and Salmonella typhimurium: cellular and molecular biology, 2nd ed. ASM Press, Washington, DC [Google Scholar]
- 38. Vitikainen M, Lappalainen I, Seppala R, Antelmann H, Boer H, Taira S, Savilahti H, Hecker M, Vihinen M, Sarvas M, Kontinen VP. 2004. Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J. Biol. Chem. 279:19302–19314 [DOI] [PubMed] [Google Scholar]
- 39. Schwalm J, Mahoney TF, Soltes GR, Silhavy TJ. 14 June 2013. A role for Skp in LptD assembly in Escherichia coli. J. Bacteriol.[Epub ahead of print.] 10.1128/JB.00431-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 41. Ames GF, Prody C, Kustu S. 1984. Simple, rapid, and quantitative release of periplasmic proteins by chloroform. J. Bacteriol. 160:1181–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ward R, Zoltner M, Beer L, El Mkami H, Henderson IR, Palmer T, Norman DG. 2009. The orientation of a tandem POTRA domain pair, of the beta-barrel assembly protein BamA, determined by PELDOR spectroscopy. Structure 17:1187–1194 [DOI] [PubMed] [Google Scholar]
- 43. Gatzeva-Topalova PZ, Warner LR, Pardi A, Sousa MC. 2010. Structure and flexibility of the complete periplasmic domain of BamA: the protein insertion machine of the outer membrane. Structure 18:1492–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental text. Download
Supplemental materials and methods. Download
The bamA616 phenotype is caused by substitutions at two Arg residues in POTRA 1 to 2. A model of full-length BamA [R. Albrecht and K. Zeth, J. Biol. Chem. 286(31):27792–803, 2011] is shown in the left panel in the context of a schematically depicted outer membrane (OM). The model was generated by superimposing POTRA 4 and 5 of BamA (PDB code 3OG5) onto the N-terminal POTRA 1 and 2 domains of the related transporter FhaC in the full-length crystal structure (PDB code 2QDZ). The complete structural model was then obtained by superimposing POTRA 4 and 5 with the crystal structure of POTRA 1 to 4 of BamA (PDB code 3EFC) at POTRA 4. The N-terminal POTRA pair (POTRA 1 and 2) is shown in the right panel with R91 and R162 highlighted in red on the structure and the corresponding mutations in bamA616 indicated in red text. Download
The envelope proteins SurA and LamB do not stably interact with the Bam complex. Affinity purification was performed in cells expressing C-terminally His6-tagged BamD+ (BamD-His) in the absence of a chemical cross-linker. A whole-cell extract (left) and Ni-NTA-enriched eluate fraction (right) were subjected to SDS-PAGE and immunoblotting for SurA, LamB, and the indicated Bam proteins. Download
The downregulation of envelope proteins in a bamA616 surA13 double mutant is a consequence of Cpx stress response induction. Whole-cell extracts of the strains shown were prepared after harvesting cells at late exponential phase. Samples were subjected to SDS-PAGE, and antibodies were used to specifically detect MalE (A) or LamB (B, C). Download
The surA10 and surA13 mutations affect α2 helix residues conserved across parvulins. (A) An alignment of parvulin-like PPIase domain sequences from highly diverse species was generated using ClustalW. The surA10 and surA13 mutation sites are shaded in purple and orange (respectively) and are indicated by arrows. (B) The structure of SurA-P1 (cyan) is shown bound to a model helical peptide (pink) to highlight the positions of Leu215, Ala216, and Ser220 (indicated by arrows and colored as described above) relative to the binding pocket of P1 (PDB code 2PV3). (C) Left, space-filling model of full-length SurA (PDB code 1M5Y) with the PPIase domains labeled and colored in cyan and the core chaperone domain colored in orange. The dashed arrow indicates the perspective shown in the right panel. Right, ribbon representation of the structure shown on the left, rotated 90° and amplified for clarity. The α2 helix (which contains the residues affected in surA10 and sur13 mutants) is indicated by a black line, and Leu215, Ala216, and Ser220 are shaded in red. For simplicity, the P2 domain is hidden in the right panel. Download
SurAS220A is unstable in vitro. Late-exponential-phase cultures (OD600 = 1) of the indicated strains were harvested and gently lysed at room temperature in a mild detergent buffer (see Materials and Methods). The cleared lysates were boiled and subjected to SDS-PAGE, and SurA was detected immunologically. The asterisk (*) indicates an unknown protein that cross-reacts with the anti-SurA antiserum. Download
bamA616 exhibits increased OM permeability that is suppressed by surA mutations in a dominant fashion. (A) Antimicrobial test discs soaked in bacitracin (Bac), novobiocin (Nov), erythromycin (Ery), and rifampin (Rif) were added to LB agar containing a lawn of the strains indicated. The data are expressed as the diameter of the zone of clearing surrounding each disc in millimeters. Numbers in parentheses indicate a hazy zone of clearing. (B) The strains listed were grown on LB plates containing 0.5% SDS-1.0 mM EDTA either in the presence (+ara) or absence (−ara) of arabinose, which induces expression of plasmid-borne surA and its derivatives. R, resistant to SDS-EDTA; S, sensitive to SDS-EDTA.
Strains and plasmids used in this study.
Mutagenic oligonucleotides used for site-directed mutagenesis.