

Abstract
Melanoma is the most aggressive form of skin cancer, responsible for the majority of skin cancer related deaths. Thus, the search for natural molecules which can effectively destroy tumors while promoting immune activation is essential for designing novel therapies against metastatic melanoma. Here, we report for the first time that a natural triterpenoid, Ganoderic Acid DM (GA-DM), induces an orchestrated autophagic and apoptotic cell death, as well as enhanced immunological responses via increased HLA class II presentation in melanoma cells. Annexin V staining and flow cytometry showed that GA-DM treatment induced apoptosis of melanoma cells, which was supported by a detection of increased Bax proteins, co-localization and elevation of Apaf-1 and cytochrome c, and a subsequent cleavage of caspases 9 and 3. Furthermore, GA-DM treatment initiated a possible cross-talk between autophagy and apoptosis as evidenced by increased levels of Beclin-1 and LC3 proteins, and their timely interplay with apoptotic and/or anti-apoptotic molecules in melanoma cells. Despite GA-DM's moderate cytotoxicity, viable cells expressed high levels of HLA class II proteins with improved antigen presentation and CD4+ T cell recognition. The antitumor efficacy of GA-DM was also investigated in vivo in murine B16 melanoma model, where GA-DM treatment slowed tumor formation with a significant reduction in tumor volume. Taken together, these findings demonstrate the potential of GA-DM as a natural chemo-immunotherapeutic capable of inducing a possible cross-talk between autophagy and apoptosis, as well as improved immune recognition for sustained melanoma tumor clearance.
Keywords: Melanoma, Ganoderic acid DM, Apoptosis, Autophagy, HLA class II, CD4+ T cells
Introduction
Melanoma is the most aggressive form of skin cancer with an ever-increasing prevalence in the Western world [1, 2]. Among human tumors, melanoma has one of the poorest prognosis, with mean survival <1 year in patients with unresectable distant metastases [3]. In these patients with metastatic melanoma, 5 year survival is <10% with a mean of 4-6 months [4, 5]. The lessinvasive treatment options for melanoma include: radiation, chemotherapy, and immunotherapy, but these have proved rather ineffective in treating the disease [6, 7]. Standard therapies have also failed to show any meaningful impact on survival, and in those patients who do show a positive initial response, a rapid deterioration often ensues with the exception of the recently discovered monoclonal antibody, ipilimumab, which has shown a significant survival benefitin metastatic melanoma [3, 8]. Over the past three decades, the incidence of melanoma has increased more rapidly than all other solid malignancies, and it is now the sixth most commonly diagnosed malignancy in the United States [4]. The steady increase in the incidence of melanoma has resulted in a dire need for relatively low-cost therapies with greater efficacy.
Despite toxicities, chemotherapeutic drugs have become a general mainstay of cancer treatment, destroying cancer cells by inducing apoptosis [9], which effectively reduces the size of the tumor and prevents further tumor growth. Another less well-defined survival pathway of drug-induced cell death is autophagy, a process within the cell utilizing lysosomal machinery to sequester and degrade its own molecules [10]. Autophagy is crucial for the recycling of cellular components to generate nutrients and enable survival in times of cellular stress. However, excessive autophagy may lead to cell death following the hallmarks typical of apoptotic events [11]. Apoptosis and autophagy are distinct cellular processes, with distinguished molecular mediators. Nevertheless, there could be cross-talk between the two pathways, which may regulate cell death/survival pathway under particular cellular conditions. Understanding this delicate balance might help investigators design new anticancer therapeutics, which modulate both of these cellular processes to reduce tumor burden.
Considering the facts that immune evasion is essential for tumor development and standard chemotherapies have the drawback of treatment-associated toxicities, it has become crucial to search for natural therapeutics that are less cytotoxic to normal cells and can enhance the immune response. One attractive source of antitumor products is the Ganoderma lucidum mushroom, which has been used for centuries as an herbal medicine for the prevention and treatment of a variety of inflammatory and malignant diseases [12-14]. While crude extracts of Ganoderma lucidum mushroom have been shown to cause cytotoxicity in some tumor cell lines, a triterpenoid extract of Ganoderma, Ganoderic Acid DM (GA-DM), has never been tested for anti-tumor activity in human melanoma cells.
The majority of human melanoma cells express HLA class II molecules which can bind peptides generated from degradation of exogenous or endogenous antigens, and present class II-peptide complexes to CD4+ T cells for their immune recognition [15-18]. Intracellular proteins or tumor fragments may get access to vacuolar compartments by sequestration in double-membrane organelles or autophagosomes, which can then fuse with vesicles of the endolysosomal compartments where antigen processing and peptide loading onto HLA class II proteins take place [19-21]. Recent studies suggest that cellular autophagic activity could give rise to HLA class II ligands for CD4+ T cell recognition [19, 22, 23]. Thus, our current study has also investigated whether this natural product GA-DM could influence intracellular machinery of melanoma cells for HLA class II processing and presentation to T cells for immune recognition. We report that GA-DM treatment induces autophagic and apoptotic events as well as immune activation in human melanoma cells, which may be important in the regulation of cancer development and progression, and in determining the response of tumor cells to anticancer therapy. We also show that GA-DM treatment reduces tumor burden in vivo in a B16 mouse melanoma model. Our current findings highlight the potential of GA-DM as a natural chemo-immunotherapeutic capable of inducing tumor destruction and also activating a possible immune response imperative to sustain melanoma tumor remission.
Materials and methods
Cell lines
Human melanoma cell lines HT-144, 1359-mel, and DM-331 were maintained in complete RPMI-1640 (Invitrogen, Grand Island, NY) medium supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT), 50 U/ml penicillin, 50 μg/ml streptomycin (Mediatech Inc., Manassas, VA), and 1% L-glutamine (Mediatech) [16, 24]. HT-144, 1359-mel, and DM-331 cells constitutively express HLA-DR4 molecules on their cell surface [16, 24]. The human melanoma cell line J3 was maintained in complete IMDM with 10% heat-inactivated bovine growth serum (BGS) (HyClone), 50 U/ml penicillin, 50 μg/ml streptomycin, and 1% L-glutamine[16, 25, 26]. J3 cells were transduced using retroviral vectors for constitutive expression of HLA-DR4 (DRB1-0401) with linked drug selection markers for hygromycin and histidinol resistance [25, 26]. Expression of surface HLA-DR4 complexes on cells was confirmed by flow cytometric analysis using the DR4-specific mAb, 359-F10 [16, 27]. The B16 mouse melanoma cell line was a gift from Dr. Mark Rubinstein (Medical University of South Carolina, Charlerston), and was cultured in complete RPMI supplemented with HEPES and nonessential amino acids (Invitrogen) as described above.
Antigens, peptides and antibodies
Human serum albumin (HSA) was purchased from Sigma-Aldrich (St. Louis, MO). The human HSA64-76K peptide (VKLVNEVTEFAKTK) was produced using Fmoc technology and an Applied Biosystems Synthesizer as described [16, 28]. Peptide purity (>99%) and sequence were analyzed by reverse phase HPLC purification and mass spectroscopy. Peptides were dissolved in DMSO and stored at -20°C until used [25]. The primary antibodies used were human caspase 3 (31A1067) (Alexis Biochemicals, Plymouth Meeting, PA); caspase 9 (ICE-LAP6, Mch6), cytochrome c (136F3) (Cell Signaling technologies, Danvers, MA); Beclin-1 (G-11), LC3β (N-20), Apaf-1 (2E10), Bcl-2 (C-2), Bax (B-9), survivin, HLA-DM, HLA-DR, LAMP-2, and CD3-ε (M-20) (Santa Cruz Biotechnology Inc., Santa Cruz, CA); and β-actin (clone AC-15) (Sigma, St. Louis, MO); HLA class II (L243), and invariant chain Ii (Pin 1.1) antibody was obtained from Dr. Janice Blum (Indiana University School of Medicine, Indianapolis). The secondary antibodies used were horseradish peroxidase conjugated anti-mouse (Pierce, Rockford, IL, USA), anti-rabbit or anti-goat IgG (Santa Cruz).
Ganoderic acid DM and cellular cytotoxicity assay
Ganoderic Acid DM (GA-DM), originally isolated from the Ganoderma lucidum mushroom, was purchased from Planta Analytica, LLC (Danbury, CT) (Cat# G-032). The purity of GA-DM was determined by the vendor as 99.9% using LC/MS analysis. GA-DM was dissolved in DMSO (Sigma) to make a 10 mM stock solution for use in the cytotoxicity assay. For all GA-DM treatments, DMSO final concentration was ≤1%. Melanoma cell lines J3, HT-144, 1359-mel, and DM-331 were seeded at 1×104 cells/well in 100 μl of appropriate culture medium in a flat-bottom 96-well plate. GA-DM was then added to appropriate wells for final concentrations of 0, 10, 20, 30, 40, 50, 60 and 80 μM for 24 h at 37°C. Cells (J3 and HT-144) were also treated with 40 μM of GA-DM for 3, 6, 12, and 24 h, in the presence or absence of an autophagy inhibitor [3-methyladenine (3-MA, 5mM)] (Sigma) or a pan caspase inhibitor (Z-VAD-FMK, 50 μM) (R&D systems #FMK001, Minneapolis, MN) for overnight. Following treatment, cell viability was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS; Promega, Madison, WI) [28]. Twenty μl of MTS reagent was added to each well, and the plate was incubated for 2 h at 37°C. After incubation, absorbance was read at 490 nm. Cells treated with vehicle alone were used as controls. The percent cell death induced by GA-DM was calculated using the equation: [ (Absorbance control - Absorbance treated)/Absorbance control] ×100. Experiments were repeated at least three times and the data were expressed as percent cytotoxicity ± S.D. of triplicate wells.
Hoechst staining and DNA fragmentation assay
J3 cells (4×105/ml) were treated with vehicle (DMSO) alone or 40 μM of GA-DM in 6-well tissue culture plate (Corning #3506) for 24 h. Cells were then incubated with Hoechst stain (1X, Sigma Aldrich) for 10 minutes at 37°C, and examined under fluorescence microscope with an Axiovert 200 ultraviolet filter (Carl Zeiss, Jena, Germany) at 200× magnification. For DNA fragmentation assay, J3 cells (2×106) were treated with vehicle alone or GA-DM (40 μM) for 24 h. Cell samples treated with a known apoptosis inducer (staurosporine, 1 μM) for 24h, was used as a positive control. DNA fragments were extracted using a DNA Gentra Puregene Cell Kit (D5500A; QIAGEN) as described in the manufacturer's protocol. DNA samples containing 5μg of DNA each were mixedwith 2 μL of loading dye (Sigma), and analyzed by 1.8% agarose gel (Sigma) electrophoresis pre-stained with 1 mmol/L of ethidium bromide (Sigma)[29]. Each gel electrophoresis included a DNA marker (100-2000 bp).
Isolation of cytoplasmic fraction
Melanoma cells (J3.DR4 and HT-144) were treated with vehicle alone or 40 μM of GA-DM for 24 h, washed in Dulbecco's phosphate-buffered saline (D-PBS), and were fractionated as described previously [16, 30]. Briefly, cells (1×107) were resuspended in 200 μl of ice cold buffer A (10 mM HEPES, pH 7.9, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol, protease inhibitor cocktail) by gentle pipetting, and kept on ice for 15 min, followed by the addition of 15 μl of 10% NP-40 and vigorous vortexing for 10 seconds. The samples were first spun at 7,000 rpm for 1 min. Then the supernatants were collected and further spun at 10,000 rpm for 10 min. The supernatant obtained was used as a cytoplasmic fraction and subjected to western blot analysis for the detection of Apaf-1 and cytochrome c proteins.
Western blot analysis
J3.DR4 and HT-144 cells were cultured for 24 h in the presence of vehicle alone or 40 μM of GA-DM. Following treatment, cells were washed and cell lysate was obtained using a standard lysis buffer (10 mM Trizma base, 150 mm NaCl, 1% Triton-X 100) [16, 28]. Equal protein concentrations from designated cell lysates were separated on a 4-12% Bis/Tris NuPage gel (Invitrogen, Grand Island, NY). Proteins were transferred onto a nitrocellulose membrane (Pierce, Rockford IL), and probed with antibodies for the expression of caspase 3 and 9, cytochrome c, Apaf-1, survivin, Bcl-2, Bax, Beclin-1, LC3, HLA class II (HLA-DR), HLA-DM, invariant chain (Ii), and LAMP-2 proteins [31, 32]. The secondary antibodies used were horseradish peroxidase conjugated anti-mouse (Pierce), anti-rabbit or anti-goat IgG (Santa Cruz Biotechnology). Monoclonal antibody for β-actin was used as a protein loading control.
Caspase inhibition assays
To analyze caspase activity, melanoma cells were treated with GA-DM (0, 20, and 40 μM) in the presence or absence of the pan-caspase inhibitor Z-VAD-FMK (R&D systems #FMK001, Minneapolis, MN) at a 50 μM final concentration. Cells were then incubated at 37°C for 24 h, and cell viability was measured using the MTS assay as described above [28].
Flow cytometry
J3.DR4 cells were cultured for 24 h in the presence of vehicle alone or 40 μM of GA-DM. Following treatment, cells were washed with staining buffer (PBS+1% heat-inactivated BGS) (HyClone), and resuspended in a binding buffer (cat. no. 556454, BD Bioscience: 0.1 M HEPES (pH 7.4), 1.4 M NaCl2, and 25 mM CaCl2). Cells were stained with Annexin V-FITC (cat. no. 556420, 556419, BD Bioscience), followed by the addition of propidium iodide (PI, cat# 556463, BD Bioscience). Samples were then analyzed on a FACScan using CellQuest software (BD Bioscience, Mountain View, CA) for apoptotic cells. To examine mitochondrial dysfunction, cells treated with vehicle alone or GA-DM (40 μM) were also stained with TMRE (tetramethylrhodamine ethyl ester, 200 nM, Sigma), and analyzed by flow cytometry according to the manufacturer's protocol.
Confocal microscopy
J3.DR4 cells were cultured on glass coverslips (cat#12-545-80, Fisher Scientific Co) in the presence of vehicle alone or 40 μM of GA-DM for 24 h. Cells were washed, fixed with (1:1) acetone/methanol mixture for 10 min at room temperature. Non-adherent cells were collected in a 24 well plate containing coverslips, centrifuged at 50×g for 5 min, and fixed identically as the adherent cells. Cells were then permeabilized with 0.1% Triton X-100 for 15 min, blocked with 5% normal serum for 10 min, and incubated with the Apaf-1 and cytochrome c antibodies at 37° C for 1 h. Following incubation, cells were washed twice with 1% BSA (cat. no. 2930, OmniPur, EMD, Cincinnati, OH) in PBS, and incubated with Alexa 488-FITC conjugated donkey anti-goat Ig (Santa Cruz) and Alexa 543-rhodamine conjugated goat anti-mouse Ig (Santa Cruz) for 1 h. Cells were also counter-stained with DAPI (4′-6-Diamidino-2-phenylindole) for nuclear localization. The slides were mounted in fluorescent mounting medium G (South Biotechnology, Inc), observed with a 63× N.A.1.4 oil immersion objective lens, and analyzed by a Leica TCS SP5 confocal laser scanning microscope using Las-AF software (Leica Lasertechnik) [24].
HLA class II antigen presentation assays
J3.DR4, HT-144, and 1359-mel cells (5×105 cells/ml) were treated with either vehicle alone or 20 μM of GA-DM for 24 h at 37° C in culture media in 96-well plate (cat# 3595, Corning Incorporated). The HSA64–76K peptide (10 μM) was added to the appropriate wells for the last 4-6 h of incubation as described [16, 28]. J3.DR4 and HT-144 cells were also treated with an autophagy inhibitor(3-methyladenine (3-MA)) or a pan caspase inhibitor (Z-VAD-FMK) for 3 h, followed by the addition of HSA64-76K peptide (10 μM) for overnight. Cells were then washed and cocultured with the HSA64-76K peptide specific T cell hybridoma (17.9) for 24 h. Following incubation, the plates were stored at -80oC until tested for IL-2 production. T cell production of IL-2 was measured by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA) [16, 28]. Anti-IL-2 was purchased from Sigma-Aldrich. All assays were repeated at least three times.
In vivo tumor induction and evaluation
Tumors were induced by the subcutaneous injection of B16 cells (1×105 cells, >90% viability by Trypan blue exclusion) into the right flanks of C57BL/6 mice [11]. Treatment was initiated by i.p. injection of GA-DM (50 mg/kg) or control vehicle (DMSO) at day 7, and day 10 after B16tumor implantation. Following treatment, tumor volumes were determined at periodic intervals using a digital caliper by measuring the longest diameter (a) and the next longest diameter (b) perpendicular to (a). Using these measurements, the tumor volume was calculated by the formula: V=ab2 × π/6 [33]. The mice were kept under observation until tumor volume was determined to be 1400 mm3 at which point they were requisitely euthanized owing to the size of the tumor. In some experiments, tumor tissues obtained from GA-DM (50 mg/kg) or vehicle-only treated mice at days 14, 17, and 19 after B16 melanoma implantation, were analyzed by immunohistochemistry for T cell infiltration. Tumor samples were first fixed in 10% formalin, embedded in paraffin and stained with CD3-ε antibody at our Hollings Cancer Center immunohistochemistry facility. Treatment or control groups consisted of 6 mice (n=6), and the data shown were representative of at least three separate experiments. All work with mice was approved by the Medical University of South Carolina Animal Protocols Review Board and was performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
Statistical analysis
Data from each experimental group were subjected to statistical analysis. ANOVA with post-hoc tests, Repeated Measures ANOVA with post-hoc tests or Student's t-tests were used, as appropriate. Two-sided tests were used in all cases and P values <0.05 were considered statistically significant.
Results
GA-DM induces anti-proliferative/apoptotic effects in human melanoma cells
Initial screening of GA-DM anti-cancer effects was carried out on four human melanoma cell lines, J3, HT-144, 1359-mel, and DM-331. Cells were simultaneously treated with vehicle alone (DMSO) or serial doses of GA-DM (10-80 μM), and the detrimental effects were measured by MTS assay as described in the Methods. Results showed that GA-DM reduced the viability of all tested melanoma cells in a dose-dependent manner with a mean IC50 of 25.5±3.4μM (Fig. 1A). A time course study (3, 6, 12, and 24 h treatment) was also performed using 20 and 40 μM of GA-DM and two melanoma cell lines J3 and HT-144 (Supplemental Fig. 1). Data showed that at early time points (3-6 h), both 20 and 40 μM of GA-DM concentrations were not effective in killing melanoma cells (Supplemental Fig. 1A and B). At later time points (12-24 h), 20 μM of GA-DM induced detectable cell death; but, 40 μM of GA-DM was very effective in inducing cytotoxicity in both cell lines (Supplemental Fig. 1B). To detect apoptotic changes induced by GA-DM, J3 melanoma cells were incubated (10 min) with Hoechst dye, which is commonly used to stain genomic DNA. Hoechst staining of J3 cells showed that GA-DM treatment induced apoptotic events characteristic of chromatin condensation. Microscopic observation in Fig. 1B demonstrates typical morphology of apoptotic nuclei stained with Hoechst, in which chromatin was condensed and aggregated at the nuclear membrane as indicated by a bright fluorescence at the periphery. Also, DNA fragmentation assay showed that GA-DM (40 μM) treatment induced internucleosomal DNA fragmentation in J3 cells (Fig 1C). A known apoptosis inducer, staurosporine (1 μM), was used as a positive control which also showed internucleosomal DNA fragmentation in the same gel [29]. Thus, the chromatin condensation detected in Fig 1B, may be a consequence of DNA cleavage within the dying J3 cells. Further analysis of cell cycle progression suggested that GA-DM treatment caused G1 arrest in J3 melanoma cells (Supplemental Fig. 2). GA-DM treatment also induced dose-dependent anti-proliferative activities by inducing apoptotic events in mouse B16 melanoma cells (Supplemental Fig. 3, and data not shown).
Figure 1.
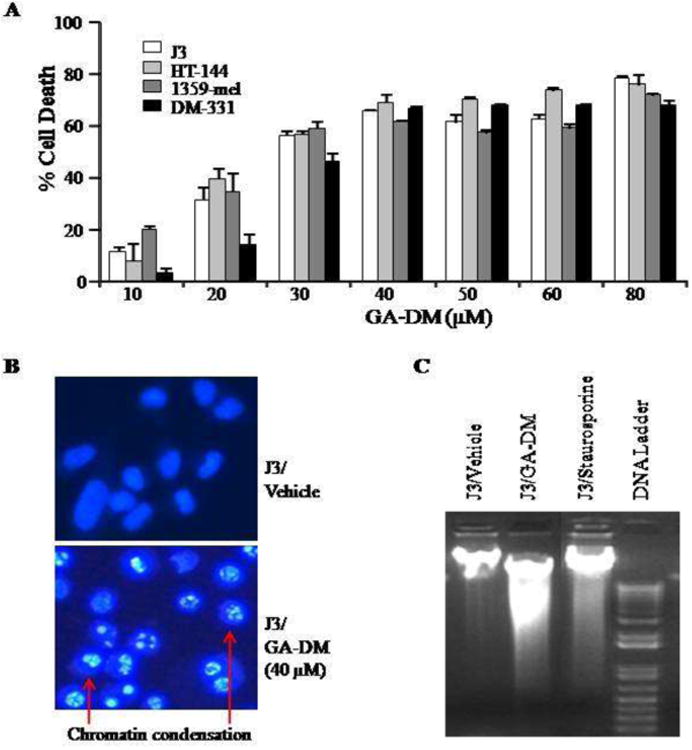
Anti-proliferative and apoptotic activity of GA-DM on human melanoma cells. (A) Cells were treated with vehicle (DMSO) alone or GA-DM (10-80μM) for 24h at 37°C, followed by the MTS viability assay as described in the Materials and Methods. Control cells treated with vehicle alone were utilized to calculate the percent cell death induced by GA-DM as indicated. The data shown are results of at least three separate experiments that were performed in triplicate wells. Error bars represent mean ± S.D. (B) Hoechst staining of J3 melanoma cells treated with vehicle alone or 40 μM of GA-DM. Cells treated with GA-DM showed typical morphology of the apoptotic nuclei stained with Hoechst, in which chromatin was condensed and aggregated at the nuclear membrane as indicated by bright fluorescence at the periphery (arrows). (C) Agarose gel electrophoresis of DNA extract showing internucleosomal DNA fragmentation after treatment of J3 cells with GA-DM (40μM) for 24 h. Cells treated with staurosporine (1 μM) was used as positive control.
GA-DM induces caspase-dependent apoptosis in melanoma
We further investigated the GA-DM-induced apoptotic pathway in melanoma cells and examined the underlying molecular mechanism of its antitumor effects. Western blot analysis of whole cell lysates showed that GA-DM treatment induced a concurrent surge of active forms of caspases 3 and 9 proteins in J3 and HT-144 cells, suggesting the role of caspases in executing melanoma cell death. Caspase activation was indicated by the cleavage of pro-caspases 3 and 9, to a number of catalytic subunits (Fig. 2A). In order to test whether in vitro caspase inhibition could reverse cytotoxicity, we used a pan-caspase inhibitor Z-VAD-FMK known to bind to the catalytic sites of active caspases rendering them inactive. Briefly, J3 and HT-144 cells were treated with GA-DM (40 μM) in the presence or absence of Z-VAD-FMK (50 μM) for 3-24 h (Fig. 2B and 2C). Data showed that inhibition of caspases by Z-VAD-FMK at early time points (3-6 h) did not alter GA-DM-induced cell death. However, at later time points (12-24 h), inhibition of caspases significantly reversed cell death in both J3 and HT-144 cell lines. These data suggest that GA-DM treatment may initiate a time-dependent processing and activation of cell survival and apoptotic proteins in melanoma cells.
Figure 2.
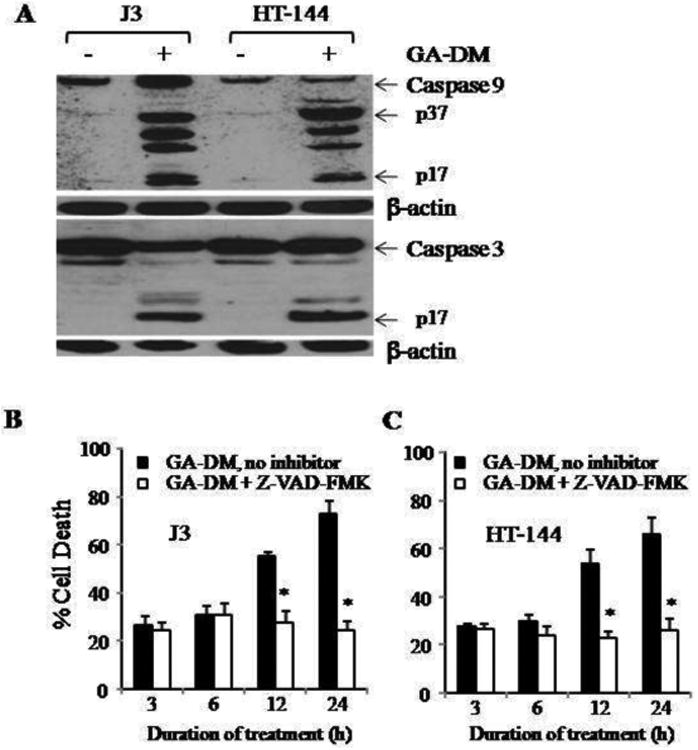
GA-DM induces caspase-dependent apoptosis in melanoma cells. (A) Western blot analysis shows protein expression and cleavage of caspases 9 and 3 in melanoma cells treated with vehicle alone (control) versus 40μM of GA-DM for 24 h. β-actin was utilized as a loading control. (B, C) Inhibition of caspases by a pan caspase inhibitor, Z-VAD-FMK, decreased GA-DM-induced cell death in J3 and HT-144 melanoma cells. Cells were treated with vehicle alone or GA-DM (40μM) for 3, 6, 12, and 24 h in the presence or absence of Z-VAD-FMK at 37°C, followed by the MTS viability assay as described. Experiments were repeated at least three times, and data were expressed as mean ± S.D. Significant differences were calculated by student's t test; *p <0.001.
To determine the molecular mechanism underlying the regulation of GA-DM initiated apoptosis, we analyzed cellular expression of Bcl-2 family proteins Bcl-2 and Bax, and an inhibitor of apoptosis protein survivin. Western blot analysis showed that GA-DM treatment reduced survivin protein expression, but up-regulated the expression levels of Bax proteins in whole cell lysates of J3 and HT-144 cells (Fig. 3A, B). No significant changes in Bcl-2 protein expression were detected in both J3 and HT-144 cells (Fig. 3A and B). Meanwhile, GA-DM treatment caused a significant increase of apoptotic cytochrome c in the cytosolic fraction of J3 cells, along with the induction of Apaf-1 (Fig. 3C). The disturbance of the Bcl-2/Bax ratio further suggests the role of GA-DM in inducing mitochondrial dysfunction and the increase of cytosolic cytochrome c in melanoma cells. Experiments using TMRE staining of J3 melanoma cells confirmed that GA-DM treatment caused significant loss of mitochondrial membrane potential preceding cytochrome c release (Fig. 3D). The release of cytochrome c may also subsequently promote Apaf-1 oligomerization, which in the presence of caspase 9 could form a large multimeric apoptosome, activating the downstream caspase cascades [34]. Immunofluorescence studies confirmed a remarkable upregulation and co-localization of cytochrome c (green) and Apaf-1 (red) proteins in the cytoplasm of GA-DM-treated J3 melanoma cells, implying apoptosome formation can trigger cellular apoptosis (Fig.4A). Furthermore, flow cytometric analysis of annexin V and propidium iodide-stained melanoma cells showed measurable increased levels of apoptotic events developed in GA-DM-treated cells compared to those treated with vehicle alone (Fig.4B). Apparently, both early and late apoptotic events were most prominent in J3 cells treated with GA-DM for 24 h (gated in R2: 1.8% in control and 19% in treated cells). Taken together, these data indicate that GA-DM treatment can activate the intrinsic pathway of apoptosis where caspase processing plays a key role in melanoma cell death.
Figure 3.
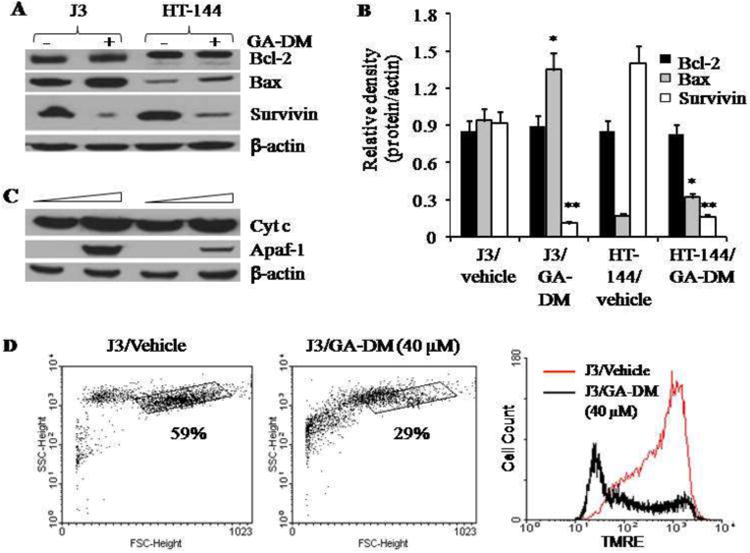
GA-DM treatment alters apoptosis regulatory proteins in melanoma cells. (A) Western blot analysis of whole cell lysates shows the expression of Bcl-2, Bax, and survivin proteins. Cells were treated with vehicle alone or 40 μM of GA-DM for 24 h at 37°C, followed by western blotting as described. β-actin was used as a loading control. (B) Densitometric analysis of protein bands detected in Fig. 3A. Although there was no detectable change in Bcl-2 proteins, upregulation of pro-apoptotic Bax was consistent with a sharp decline in anti-apoptotic survivin. Data represent average ± S.D. Significant differences from controls were calculated by student's t test; *p<0.05, **p<0.01. (C) Western blot analysis shows upregulation of cytochrome c and Apaf-1 in cytoplasmic fractions of melanoma cells. β-actin was used as a loading control. The figures shown are representative of three independent experiments. (D) J3 cells were treated with vehicle alone or GA-DM (40 μM) for overnight, followed by staining with TMRE as described. Cells were then analyzed by flow cytometry for depolarized mitochondria. Data are representative of at least three separate experiments.
Figure 4.
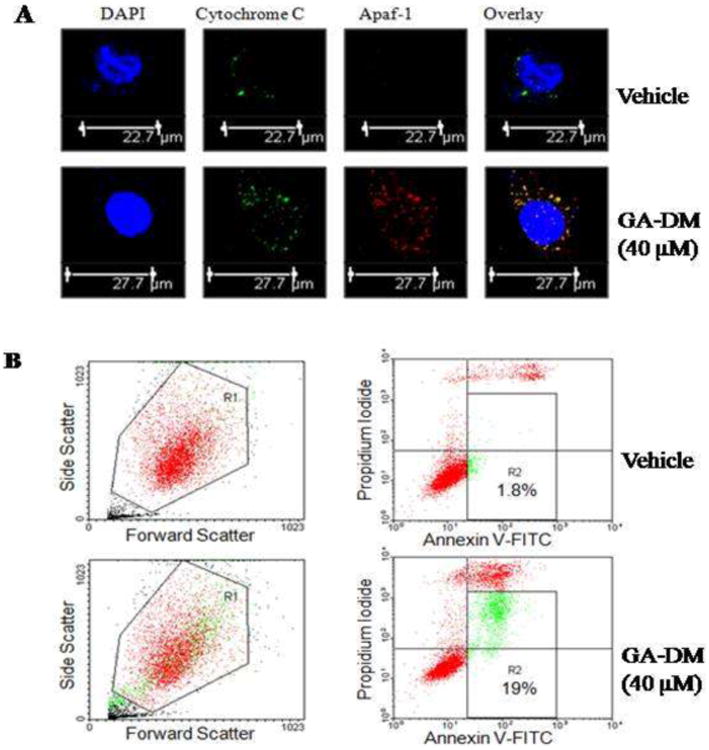
GA-DM treatment upregulates cytochrome c and Apaf-1 proteins in melanoma cells. (A) Cells treated with vehicle alone or GA-DM (40 μM) were simultaneously stained with primary antibodies against cytochrome c and Apaf-1, followed by fluorophore-conjugated secondary antibodies as described in the Materials and Methods. Representative confocal microscopy images of J3 melanoma cells indicate increased levels of cytochrome c (green) and Apaf-1 proteins (red) and their co-localization (overlay, yellow) following GA-DM treatment. DAPI (blue) was used to stain nuclei of J3 cells. (B) Dot plot depicting forward-(FSC) versus side-scatter (SSC) profile of J3 cells (at least 20,000 events/sample) after incubation with GA-DM (40 μM) or vehicle for 24 h at 37°C. The bulk of J3 cells (in the gate R1) distinguishable from smaller cellular debris (low FSC) and larger cell clumps (high FSC), were analyzed for annexin V and PI double staining (right panels). The quadrants (R2) represent the annexin V-positive apoptotic cells. Data are representatives of at least three independent experiments with similar patterns.
GA-DM treatment induces cross-talk between autophagy and apoptosis in melanoma cells
Depending on the type of the stimulus, autophagy can function as a cell survival or a cell death mechanism [11, 35, 36]. To evaluate the role of GA-DM in the induction autophagy, we conducted a post-treatment timeline western blotting to analyze changes in autophagic protein Beclin-1 that has long been identified as a Bcl-2-interacting partner [35, 37, 38]. Results showed a significant increase in Beclin-1 protein expression at 3-6 h after GA-DM treatment, which was drastically declined after 12 h of treatment (Figure 5A, B). These data indicate a possible binding of Beclin-1 to the survival protein Bcl-2, triggering apoptotic events characterized by the activation of caspase 3. We also monitored GA-DM-treated cells for another autophagic protein LC3, which is a marker of autophagosome formation [38]. Our results showed that GA-DM treatment induced a time-dependent activation of LC3 protein that reached a peak at about 6 h post-treatment, suggesting that treated melanoma cells underwent autophagy as a possible survival mechanism (Fig. 5A, B). Concurrent timeline cytototoxic assay in the presence or absence of an autophagy inhibitor, 3-MA, showed that blocking autophagic events at relatively earlier time points (3-6 h) induced cell death (Fig. 5C). By contrast, remarkable cell execution occurred beyond 12 h of treatment regardless of 3-MA treatment (Fig. 5C), indicating that excessive autophagy may have triggered a subsequent caspase dependent apoptotic pathway. Taken together, these data suggest the potential of autophagy and apoptosis cross-talk as a possible mechanism of action for effective antitumor responses, which can be exploited for melanoma therapies.
Figure 5.
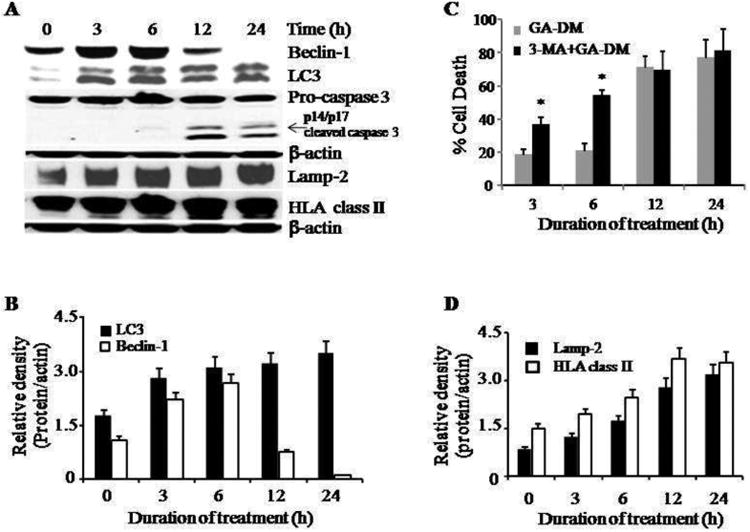
GA-DM treatment induces cross-talk between autophagy and apoptosis in melanoma cells, and concurrent activation of HLA class II and Lamp-2 proteins. (A) J3 melanoma cells were treated with GA-DM (40 μM) for 3-24 h. Western blot analysis shows that GA-DM treatment induced an earlier time-dependent upregulation of autophagic proteins Beclin-1 and LC3 (3-6 h), and a later time-dependent activation and processing of effector caspase 3 (12-24 h) in J3 cells. Treatment of melanoma cells with GA-DM (40 μM) also induced a time-dependent increase in HLA class II and Lamp-2 proteins. (B, D) Protein bands detected in Fig. 5(A) were analyzed by densitometric analysis, which showed time-dependent expression of Beclin-1, LC3, HLA class II, and Lamp-2 molecules in GA-DM-treated cells, β-actin was used as a reference band to normalize the original protein loading and to quantitate the expression of these proteins. (C) Data showing that GA-DM induces autophagy (cell survival) at earlier time points (3-6 h), and apoptosis (cell death) at later time points (12-24h) in GA-DM treated cells. Blocking autophagy with 3-MA treatment (5 mM) induced melanoma cell death at early time points (3-6 h). Apoptotic death of melanoma cells might have occurred by overwhelmed autophagy in the first 6 h of GA-DM treatment. Data represent mean ± S.D of triplicate wells. Significant differences were calculated by student's t test; *p<0.01.
GA-DM treatment activates immune components in melanoma cells and promotes CD4+ T cell recognition of tumors
Studies suggest that autophagic activity facilitates antigen processing and presentation via HLA molecules, and is critical for the development of immunity against malignant tumors. In order to evaluate whether GA-DM-induced autophagy in melanoma cells influenced intracellular HLA class II proteins and their presentation to CD4+ T cells, J3.DR4 and HT-144 cells were subjected to western blotting and functional antigen presentation assays. Data showed a significant upregulation of HLA class II molecules as well as lysosomal LAMP-2 proteins in GA-DM-treated melanoma cells (Fig. 5A, D), which correlated with early autophagic events as evidenced by Beclin-1 expression in those cells. Further analysis of HLA class II components by western blotting showed a significant increase in HLA-DR and HLA-DM proteins with a differential Ii expression (Fig. 6A,B), indicating the activation of HLA class II antigen processing machinery in GA-DM-treated melanoma cells. However, no significant changes in heat shock proteins (both Hsp70 and Hsp90) in GA-DM-treated cells were observed as analyzed by western blotting (data not shown), indicating that chaperone-mediated autophagy may not be involved in cellular stress, but rather macroautophagy played a role in HLA class II activation[18-20]. Cell-mediated immune activation and recognition of GA-DM-treated melanoma cells were also examined in vitro by using whole HSA protein or HSA64-76K peptide as a model for the study of antigen processing and presentation to specific CD4+T cells. Results showed that GA-DM (20 μM) treatment increased HLA class II antigen presentation and CD4+ T cell recognition of melanoma tumor cells (Fig. 6C). It was also observed that 20 μM of GA-DM enhanced HSA processing and presentation via HLA class II proteins, although a decline in antigen presentation was observed in treatment with higher doses (40 μM and above) of GA-DM (data not shown). This is not unexpected as higher doses of GA-DM induced significant cytotoxicity and cell death. However, the remaining viable tumor cells remained capable of inducing T cell responses.
Figure 6.
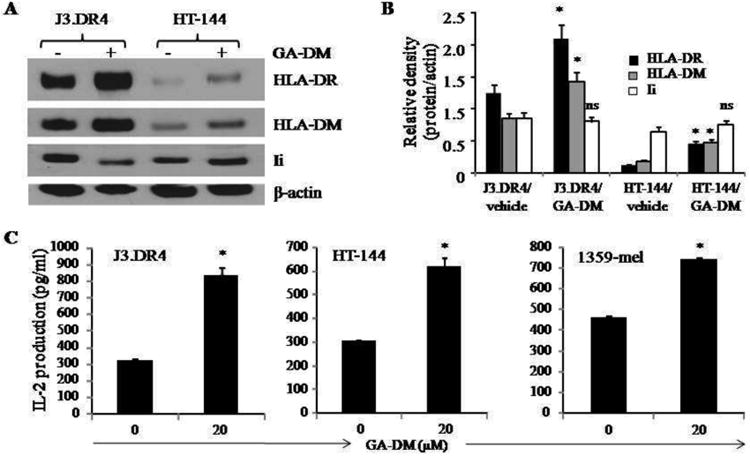
GA-DM treatment alters the components of the HLA class II pathway, and increases antigen presentation and CD4+ T cell recognition of melanoma cells. (A) Western blot analysis shows an increased expression of HLA-DR and HLA-DM molecules, and a slight change in Ii protein expression in J3.DR4 and HT-144 cells treated with GA-DM (20μM) at 37°C for 24 h. (B) Densitometric analysis of protein bands detected in Western blotting in Fig. 6(A). β-actin was used as a reference to quantitate the relative expression of proteins in both control and GA-DM treated cells. Significant differences to controls were calculated by student's t test; *p<0.01, ns = not significant. (C) Antigen presentation assay shows an improved antigen presentation and CD4+ T cell recognition of three different melanoma cells treated with GA-DM. J3 cells were transduced with HLA-DR4 molecules as described. HT-144 and 1359-mel cells innately express cell surface HLA-DR4 proteins. These cells were treated with vehicle alone or 20μM of GA-DM for 24 h in 96-well plates, followed by the addition of HSA64-76K peptide for another 4 h. Cells were then washed, and co-cultured with the peptide specific CD4+ T cell hybridoma for 24 h. The production of IL-2 was measured by ELISA and expressed as pg/ml ± SD of triplicate wells of at least three independent experiments. *p<0.01
To study whether blocking autophagy alters CD4+ T cell recognition of melanoma cells, J3.DR4 and HT-144 cells were treated with either vehicle alone or GA-DM (20 μM) and an autophagy inhibitor, 3-MA. Data showed that blocking autophagy reduced CD4+ T cell responses to antigens presented by GA-DM-treated J3.DR4 and HT-144 cells (Supplemental Fig. 4A). Our results also showed that the pan caspase inhibitor Z-VAD-FMK treatment restored the expression of Beclin-1 (Supplemental Fig. 4B), as well as antigen presentation and CD4+ T cell recognition of GA-DM (40 μM)-treated J3.DR4 and HT-144 cells (Supplemental Fig. 4C).
GA-DM treatment reduces tumor burden in vivo in B16 mouse melanoma model
In order to assess in vivo antitumor efficacy of GA-DM, we used a murine B16 melanoma model as described previously [33]. Mice were injected i.p. with GA-DM (50 mg/kg) or control vehicle at days 7 and 10 following the subcutaneous implantation of B16 melanoma tumor cells as described. Data showed that GA-DM treatment reduced the normal growth of B16 tumor (Fig. 7A), which resulted in a significant diminution in tumor volume of treated mice that lasted up to three weeks after tumor implantation (Fig. 7B). After day 21, treated mice still possessed a reduced tumor volume at least until day 23, as compared to untreated mice (Supplemental Fig. 5). Our data also showed that all the mice developed tumors, and the GA-DM-treated mice develop smaller tumors overall. Immunohistochemical analysis with anti-CD3 staining confirmed host T cell infiltration at a greater extent in tumor tissues of treated mice than those treated with the vehicle alone (Fig. 7 C). These findings suggest that GA-DM may serve as a potential chemo- and immunotherapeutic natural product for attenuation of melanoma growth in vivo.
Figure 7.
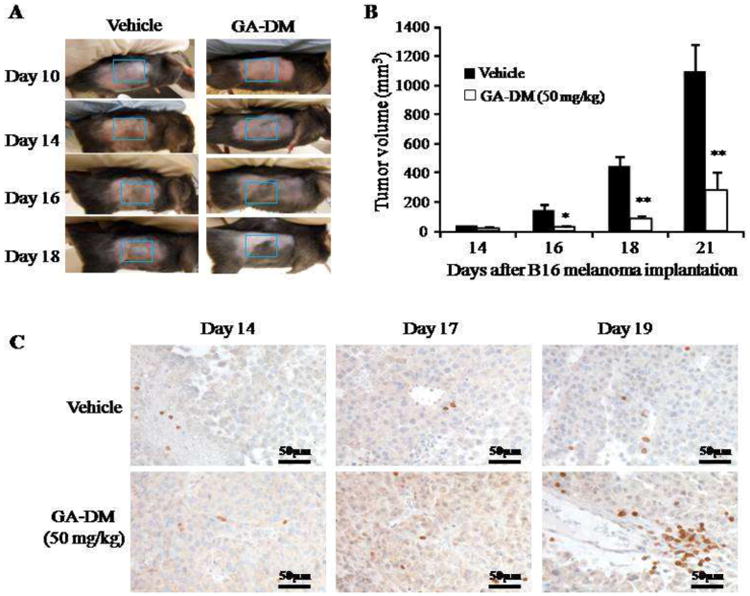
GA-DM treatment reduces melanoma growth in vivo in murine B16 melanoma model. Mice were injected with two doses (50mg/kg) of GA-DM intraperitoneally (i.p.) on days 7 and 10 after subcutaneous (s.c.) injection of B16 melanoma cells as described. (A) Photographs showing a slower tumor growth in GA-DM-treated mouse as compared to the control group. (B) Analysis of tumor growth shows a significant decline in tumor volume in GA-DM-treated mice (n=6). Data represent average tumor volume ± S.D. (C) Immunohistochemistry of tumor tissue with anti-CD3 showing T cell infiltration at day 14, 17 and 19 after B16 tumor implantation. Significant differences were calculated by ANOVA tests as described in the methods. *p<0.05, **p<0.01.
Discussion
A defining feature of conventional anticancer therapies is their capacity to evoke cellular death by inducing cell apoptosis. However, inherent or acquired resistance of cancer cells to chemotherapy has been a major obstacle to complete treatment and sufficiently prevent tumor reoccurrence. Therefore, the search for new natural drugs that sensitize cancer cells to chemotherapy-mediated apoptosis, while also restoring antitumor immunity has increasingly become a focus of investigation [9, 39]. More recently, exploiting other mechanisms, such as autophagy, has emerged as a potential strategy to induce tumor cell death [11, 40]. In this study, we have shown that GA-DM, a natural product of the medicinal mushroom Ganoderma lucidum, not only inhibited proliferation and killed human metastatic melanoma in low micromolar concentrations, but also significantly restored immune recognition of tumor cells. We also provide evidence that GA-DM induces an orchestrated molecular cross-talk with an early autophagy that may contribute to apoptotic cell death.
Our data indicate that a 24 h GA-DM treatment of melanoma cells results in typical apoptotic events such as degradation of DNA distinguished by a subsequent cleavage of chromatin DNA into internucleosomal fragments as observed by Hoechst staining and DNA fragmentation assay of treated melanoma. Apoptotic cell death was carried out through upregulation and cleavage of initiator caspase 9 and executioner caspase 3. Biochemical analysis showed that caspase protein expression was correlated with a surge of cellular caspase catalytic activity, which was markedly diminished by the pan caspase inhibitor Z-VAD-FMK, indicating a key role for caspases in executing GA-DM-treated melanoma. Further mechanistic investigations of melanoma cell death revealed that GA-DM treatment induced the expression of pro-apoptotic Bax with a marked suppression of pro-survival protein survivin, and destabilization of mitochondrial membrane potential. Analysis of cytoplasmic fractions of treated cells detected remarkable elevation of apoptotic protein Apaf-1, which also co-localized with cytochrome c in melanoma cells, suggesting the involvement of the intrinsic apoptotic pathway where cellular insult starts by the migration of Bax to the surface of the mitochondria that subsequently inhibit the protective effects of Bcl-2, and mediates the release of cytochrome c as suggested previously [41]. Cytochrome c then complexes with Apaf-1, forming the apoptosome, to activate caspase 9, triggering the effector caspase cascade and leading to an induction of apoptosis in melanoma cells as described [42]. Annexin V staining of treated melanoma cells supported this notion because a significant portion of GA-DM-treated cells were undergoing apoptosis, which included both early and late apoptotic events.
Autophagy functions as both a cell survival and a cell death mechanism depending on the context and the stimuli, which are likely exploitable for cancer therapy [11, 40]. Time-dependent cell death of treated melanoma suggests an apparent role of GA-DM in promoting a toxic form of autophagy that possibly contributes to an activation of the intrinsic apoptosis pathway. Protein analysis of melanoma cells showed early events (3 to 6 h, following GA-DM treatment) featuring significant upregulation of autophagy regulatory proteins Beclin-1 and LC-3. Yet, autophagic markers, especially Beclin-1, are drastically reduced at a later time point (12-24 h), suggesting a shift to apoptosis. Autophagy protein Beclin-1 has long been identified as a Bcl-2-interacting partner [37, 43, 44]. Thus, our data suggest that an early increase of Beclin-1 in treated melanoma cells may contribute to the interplay of apoptotic and survival proteins, deactivating mitochondrial protection and triggering intrinsic apoptotic events.
Although, current chemotherapy for melanoma has shown a high success rate at early stage disease, it has failed to relieve those suffering from late stage metastatic disease [6, 45, 46]. Thus, treatment strategies have been adopted to combine chemotherapy with immunotherapy, to destroy the cancer cells as well as elevate antitumor immunity to prevent further re-occurrence [4]. Unfortunately, in many clinical trials, immunotherapy did not prove advantageous over current chemotherapy strategies, suggesting prominent defects in tumor cell-expressed immune components, a pre-requisite for T cell stimulation via the cell-mediated immune pathway. Thus, an agent that could enhance tumor immunogenicity while reducing tumor burden would be an ideal chemo-immunotherapeutic to overcome major constraints bound to melanoma treatment. A key aspect of this immune response to tumor cells was observed in amplification of HLA class II-mediated antigen presentation to T cells [16, 28, 47]. Our data clearly showed that GA-DM treatment is capable of augmenting the HLA-class II pathway of immune recognition. An increase level of Lamp-2 by GA-DM may also complement CD4+ T cell recognition via the endolysosomal class II pathway. Blocking autophagy may reduce immune recognition of tumor cells, while suboptimal killing of tumors may enhance immune recognition via the HLA class II pathway. Despite the fact that doses of GA-DM (40 μM or higher) induce >50% cytotoxicity in J3 melanoma cells after 12 h of treatment, the few remaining viable cells are able to present antigen and stimulate CD4+ T cells. Evidently, lower concentrations of GA-DM showed a much stronger effect in inducing immune response, where surviving cells were capable of processing whole antigens for presentation to T cells. The antitumor effects of GA-DM such as slower growth and host T cell infiltration in tumor mass were also observed in vivo in the B16 mouse melanoma model, suggesting the potential of GA-DM as a complementary chemotherapeutic against malignant tumors. Future studies are underway to determine the molecular mechanisms of the in vivo therapeutic effects of GA-DM treatment, but our current findings suggest an enormous potential of GA-DM as a chemo-immunotherapeutic agent for treating malignant diseases.
Supplementary Material
Supplemental Figure 1. Time course of anti-proliferative activity of GA-DM in human melanoma cells. J3.DR4 and HT-144 cells were treated with either 20 μM of GA-DM (A) or 40 μM GA-DM (B) for 3, 6, 12, and 24 h. After incubation, MTS viability assay was performed and percent cell death was calculated as described in the methods. Experiments were repeated at least three times, and data are representative of mean ± S.D. of triplicate wells.
Supplemental Figure 2. Representative DNA histograms show the GA-DM-induced alteration of normal distribution of J3 cells during cell cycle progression. Cells were treated with vehicle alone or 20 and 40 μM of GA-DM for 24h, stained with propidium iodide (PI) in the presence of RNAse (1 mg/ml), and analyzed by flow cytometry. Appropriate gating was used to select the easily distinguished single population. Twenty thousand events per sample were acquired, and determinations were made at least in triplicate to assure the distribution of each cell cycle. The table presented below shows percent distribution and G1 arrest of GA-DM-treated melanoma cells in each cycle.
Supplemental Figure 3. Anti-proliferative and apoptotic activity of GA-DM on mouse melanoma cells. (A) B16 cells were treated with vehicle alone or 20, 40 and 80 μM of GA-DM for 24 h. Following incubation, MTS viability assay was performed and percent cell death was calculated as described in the Materials and Methods. Data are representative of at least three separate experiments and expressed as percent cell death ± S.D. (B) B16 cells were also subjected to Hoechst staining, where chromatin was condensed and aggregated at the nuclear membrane as indicated by bright fluorescence at the periphery. (C) GA-DM treatment induces caspase-dependent apoptosis in mouse B16 melanoma cells. Western blot analysis shows active caspase3 proteins inB16 cells treated with 80 μM of GA-DM for 24 h. β-actin was used as a loading control.
Supplemental Figure 4. Antigen presentation assays showing the effects of autophagy and apoptosis on CD4+ T cell recognition of J3.DR4 and HT-144 melanoma cells. (A) Cells were treated with an autophagy inhibitor 3-MA (0 and 5 mM) and GA-DM (0 and 20 μM) for 3 h, followed by the addition of HSA664-76K peptide for another 4 h. Cells were then washed, and co-cultured with the peptide specific CD4+ T cell hybridoma for 24 h. The production of IL-2 was measured by ELISA and expressed as pg/ml ± SD of triplicate wells of at least three independent experiments. (B) J3.DR4 and HT-144 cells were treated with either vehicle alone, GA-DM (40 μM) or GA-DM plus a pan-caspase inhibitor Z-VAD-FMK (50μM) for 24 h. Cells were washed and cell lysates were subjected to western blotting for detecting beclin-1 proteins as described. β-actin was used as a loading control. (C) J3.DR4 cells were also treated with vehicle alone or GA-DM (40 μM) in the presence or absence of Z-VAD-FMK (50 μM) for 20 h, followed by the addition of HSA64-76K peptide for another 4 h. Cells were then washed, and co-cultured with the peptide specific CD4+ T cell hybridoma for 24 h as described. The production of IL-2 was measured by ELISA and expressed as pg/ml ± SD. Significant differences to controls were calculated; *p<0.05, #p<0.01, ns = not significant.
Supplemental Figure 5. GA-DM treatment attenuates melanoma growth in vivo. C57BL/6 mice were injected with two doses (50 mg/kg) of GA-DM i.p. on days 7 and 10 after s.c. injection of B16 melanoma cells as described in the methods. The graph shows tumor growth in individual mice with a significant decline of tumor volume in GA-DM-treated mice (n=6).
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA129560 and CA129560-S1 to A. Haque). The research presented in this article was also supported in part by the Tissue Biorepository and Flow Cytometry Shared Resource as part of the Hollings Cancer Center at the Medical University of South Carolina which is funded by a Cancer Center Support Grant P30 CA138313. We thank Drs. Janice Blum (Indiana University, Indianapolis), Craig Slingluff Jr. (University of Virginia, Charlottesville) for melanoma cell lines; Drs. L. Xiang, Christina Johnson, and Carola Neumann (MUSC) for antibodies; Dr. Sakamuri Reddy (Dept. of Pediatrics, MUSC) for his critical reading of the manuscript. We also gratefully acknowledge Dr. Chenthamarakshan Vasu (MUSC) for his Fow Cytometry Facility.
Footnotes
Conflict of Interest: No conflicts of interest were disclosed by the authors.
References
- 1.Rigel DS, Russak J, Friedman R. The evolution of melanoma diagnosis: 25 years beyond the ABCDs. CA Cancer J Clin. 2010;60:301–316. doi: 10.3322/caac.20074. [DOI] [PubMed] [Google Scholar]
- 2.Reed KB, Brewer JD, Lohse CM, Bringe KE, Pruitt CN, Gibson LE. Increasing incidence of melanoma among young adults: an epidemiological study in Olmsted County, Minnesota. Mayo Clin Proc. 2012;87:328–334. doi: 10.1016/j.mayocp.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blank CU, Hooijkaas AI, Haanen JB, Schumacher TN. Combination of targeted therapy and immunotherapy in melanoma. Cancer Immunol Immunother. 2011;60:1359–1371. doi: 10.1007/s00262-011-1079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle GM. Therapy for metastatic melanoma: an overview and update. Expert Rev Anticancer Ther. 2011;11:725–737. doi: 10.1586/era.11.25. [DOI] [PubMed] [Google Scholar]
- 5.Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology (Williston Park) 2009;23:488–496. [PMC free article] [PubMed] [Google Scholar]
- 7.Alexandrescu DT, Ichim TE, Riordan NH, et al. Immunotherapy for Melanoma: Current Status and Perspectives. J Immunother. 2010 doi: 10.1097/CJI.0b013e3181e032e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boasberg P, Hamid O, O'Day S. Ipilimumab: unleashing the power of the immune system through CTLA-4 blockade. Semin Oncol. 2010;37:440–449. doi: 10.1053/j.seminoncol.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 12.Boh B, Berovic M, Zhang J, Zhi-Bin L. Ganoderma lucidum and its pharmaceutically active compounds. Biotechnol Annu Rev. 2007;13:265–301. doi: 10.1016/S1387-2656(07)13010-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhu M, Chang Q, Wong LK, Chong FS, Li RC. Triterpene antioxidants from ganoderma lucidum. Phytother Res. 1999;13:529–531. doi: 10.1002/(sici)1099-1573(199909)13:6<529::aid-ptr481>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 14.Miyamoto I, Liu J, Shimizu K, et al. Regulation of osteoclastogenesis by ganoderic acid DM isolated from Ganoderma lucidum. Eur J Pharmacol. 2009;602:1–7. doi: 10.1016/j.ejphar.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Norton DL, Haque A. Insights into the Role of GILT in HLA Class II Antigen Processing and Presentation by Melanoma. J Oncol. 2009;2009:142959. doi: 10.1155/2009/142959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao D, Amria S, Hossain A, et al. Enhancement of HLA class II-restricted CD4+ T cell recognition of human melanoma cells following treatment with bryostatin-1. Cell Immunol. 2011;271:392–400. doi: 10.1016/j.cellimm.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li P, Gregg JL, Wang N, et al. Compartmentalization of class II antigen presentation: contribution of cytoplasmic and endosomal processing. Immunol Rev. 2005;207:206–217. doi: 10.1111/j.0105-2896.2005.00297.x. [DOI] [PubMed] [Google Scholar]
- 18.Nimmerjahn F, Milosevic S, Behrends U, et al. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33:1250–1259. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- 19.Gannage M, Munz C. Autophagy in MHC class II presentation of endogenous antigens. Curr Top Microbiol Immunol. 2009;335:123–140. doi: 10.1007/978-3-642-00302-8_6. [DOI] [PubMed] [Google Scholar]
- 20.Strawbridge AB, Blum JS. Autophagy in MHC class II antigen processing. Curr Opin Immunol. 2007;19:87–92. doi: 10.1016/j.coi.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 21.Paludan C, Schmid D, Landthaler M, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 22.Dengjel J, Schoor O, Fischer R, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A. 2005;102:7922–7927. doi: 10.1073/pnas.0501190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumamoto Y, Iwasaki A. MHC class II-presentation of antigen by autophagy. Tanpakushitsu Kakusan Koso. 2008;53:2286–2291. [PubMed] [Google Scholar]
- 24.Goldstein OG, Hajiaghamohseni LM, Amria S, Sundaram K, Reddy SV, Haque A. Gamma-IFN-inducible-lysosomal thiol reductase modulates acidic proteases and HLA class II antigen processing in melanoma. Cancer Immunol Immunother. 2008;57:1461–1470. doi: 10.1007/s00262-008-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haque MA, Hawes JW, Blum JS. Cysteinylation of MHC class II ligands: peptide endocytosis and reduction within APC influences T cell recognition. J Immunol. 2001;166:4543–4551. doi: 10.4049/jimmunol.166.7.4543. [DOI] [PubMed] [Google Scholar]
- 26.Haque MA, Li P, Jackson SK, et al. Absence of gamma-interferon-inducible lysosomal thiol reductase in melanomas disrupts T cell recognition of select immunodominant epitopes. J Exp Med. 2002;195:1267–1277. doi: 10.1084/jem.20011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amria S, Hajiaghamohseni LM, Harbeson C, et al. HLA-DM negatively regulates HLA-DR4-restricted collagen pathogenic peptide presentation and T cell recognition. Eur J Immunol. 2008;38:1961–1970. doi: 10.1002/eji.200738100. [DOI] [PubMed] [Google Scholar]
- 28.Radwan FF, Zhang L, Hossain A, Doonan BP, God JM, Haque A. Mechanisms regulating enhanced human leukocyte antigen class II-mediated CD4 + T cell recognition of human B-cell lymphoma by resveratrol. Leuk Lymphoma. 2011 doi: 10.3109/10428194.2011.615423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu GS, Lu JJ, Guo JJ, et al. Ganoderic acid DM, a natural triterpenoid, induces DNA damage, G1 cell cycle arrest and apoptosis in human breast cancer cells. Fitoterapia. 2012;83:408–414. doi: 10.1016/j.fitote.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 30.Surjit M, Kumar R, Mishra RN, Reddy MK, Chow VT, Lal SK. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J Virol. 2005;79:11476–11486. doi: 10.1128/JVI.79.17.11476-11486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haque A, Das A, Hajiaghamohseni LM, Younger A, Banik NL, Ray SK. Induction of apoptosis and immune response by all-trans retinoic acid plus interferon-gamma in human malignant glioblastoma T98G and U87MG cells. Cancer Immunol Immunother. 2007;56:615–625. doi: 10.1007/s00262-006-0219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Younger AR, Amria S, Jeffrey WA, et al. HLA class II antigen presentation by prostate cancer cells. Prostate Cancer Prostatic Dis. 2008;11:334–341. doi: 10.1038/sj.pcan.4501021. [DOI] [PubMed] [Google Scholar]
- 33.Ugen KE, Kutzler MA, Marrero B, et al. Regression of subcutaneous B16 melanoma tumors after intratumoral delivery of an IL-15-expressing plasmid followed by in vivo electroporation. Cancer Gene Ther. 2006;13:969–974. doi: 10.1038/sj.cgt.7700973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 35.Raftopoulou M. Mitochondrial wrinkles: the first signs of ageing? Nat Cell Biol. 2005;7:853. doi: 10.1038/ncb0905-853. [DOI] [PubMed] [Google Scholar]
- 36.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 37.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Kadowaki M, Karim MR. Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 2009;452:199–213. doi: 10.1016/S0076-6879(08)03613-6. [DOI] [PubMed] [Google Scholar]
- 39.Kerr JF, Winterford CM, Harmon BV. Apoptosis. Its significance in cancer and cancer therapy. Cancer. 1994;73:2013–2026. doi: 10.1002/1097-0142(19940415)73:8<2013::aid-cncr2820730802>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 40.Mujumdar N, Saluja AK. Autophagy in pancreatic cancer: an emerging mechanism of cell death. Autophagy. 2010;6:997–998. doi: 10.4161/auto.6.7.13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999;18:3586–3595. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reubold TF, Wohlgemuth S, Eschenburg S. A new model for the transition of APAF-1 from inactive monomer to caspase-activating apoptosome. J Biol Chem. 2009;284:32717–32724. doi: 10.1074/jbc.M109.014027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene. 2009;28:2128–2141. doi: 10.1038/onc.2009.60. [DOI] [PubMed] [Google Scholar]
- 44.Vazquez CL, Colombo MI. Beclin 1 modulates the anti-apoptotic activity of Bcl-2: insights from a pathogen infection system. Autophagy. 2010;6:177–178. doi: 10.4161/auto.6.1.10743. [DOI] [PubMed] [Google Scholar]
- 45.Slipicevic A, Herlyn M. Narrowing the knowledge gaps for melanoma. Ups J Med Sci. 2012;117:237–243. doi: 10.3109/03009734.2012.658977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nikolaou VA, Stratigos AJ, Flaherty KT, Tsao H. Melanoma: new insights and new therapies. J Invest Dermatol. 2012;132:854–863. doi: 10.1038/jid.2011.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haque A, Blum JS. New insights in antigen processing and epitope selection: development of novel immunotherapeutic strategies for cancer, autoimmunity and infectious diseases. J Biol Regul Homeost Agents. 2005;19:93–104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Time course of anti-proliferative activity of GA-DM in human melanoma cells. J3.DR4 and HT-144 cells were treated with either 20 μM of GA-DM (A) or 40 μM GA-DM (B) for 3, 6, 12, and 24 h. After incubation, MTS viability assay was performed and percent cell death was calculated as described in the methods. Experiments were repeated at least three times, and data are representative of mean ± S.D. of triplicate wells.
Supplemental Figure 2. Representative DNA histograms show the GA-DM-induced alteration of normal distribution of J3 cells during cell cycle progression. Cells were treated with vehicle alone or 20 and 40 μM of GA-DM for 24h, stained with propidium iodide (PI) in the presence of RNAse (1 mg/ml), and analyzed by flow cytometry. Appropriate gating was used to select the easily distinguished single population. Twenty thousand events per sample were acquired, and determinations were made at least in triplicate to assure the distribution of each cell cycle. The table presented below shows percent distribution and G1 arrest of GA-DM-treated melanoma cells in each cycle.
Supplemental Figure 3. Anti-proliferative and apoptotic activity of GA-DM on mouse melanoma cells. (A) B16 cells were treated with vehicle alone or 20, 40 and 80 μM of GA-DM for 24 h. Following incubation, MTS viability assay was performed and percent cell death was calculated as described in the Materials and Methods. Data are representative of at least three separate experiments and expressed as percent cell death ± S.D. (B) B16 cells were also subjected to Hoechst staining, where chromatin was condensed and aggregated at the nuclear membrane as indicated by bright fluorescence at the periphery. (C) GA-DM treatment induces caspase-dependent apoptosis in mouse B16 melanoma cells. Western blot analysis shows active caspase3 proteins inB16 cells treated with 80 μM of GA-DM for 24 h. β-actin was used as a loading control.
Supplemental Figure 4. Antigen presentation assays showing the effects of autophagy and apoptosis on CD4+ T cell recognition of J3.DR4 and HT-144 melanoma cells. (A) Cells were treated with an autophagy inhibitor 3-MA (0 and 5 mM) and GA-DM (0 and 20 μM) for 3 h, followed by the addition of HSA664-76K peptide for another 4 h. Cells were then washed, and co-cultured with the peptide specific CD4+ T cell hybridoma for 24 h. The production of IL-2 was measured by ELISA and expressed as pg/ml ± SD of triplicate wells of at least three independent experiments. (B) J3.DR4 and HT-144 cells were treated with either vehicle alone, GA-DM (40 μM) or GA-DM plus a pan-caspase inhibitor Z-VAD-FMK (50μM) for 24 h. Cells were washed and cell lysates were subjected to western blotting for detecting beclin-1 proteins as described. β-actin was used as a loading control. (C) J3.DR4 cells were also treated with vehicle alone or GA-DM (40 μM) in the presence or absence of Z-VAD-FMK (50 μM) for 20 h, followed by the addition of HSA64-76K peptide for another 4 h. Cells were then washed, and co-cultured with the peptide specific CD4+ T cell hybridoma for 24 h as described. The production of IL-2 was measured by ELISA and expressed as pg/ml ± SD. Significant differences to controls were calculated; *p<0.05, #p<0.01, ns = not significant.
Supplemental Figure 5. GA-DM treatment attenuates melanoma growth in vivo. C57BL/6 mice were injected with two doses (50 mg/kg) of GA-DM i.p. on days 7 and 10 after s.c. injection of B16 melanoma cells as described in the methods. The graph shows tumor growth in individual mice with a significant decline of tumor volume in GA-DM-treated mice (n=6).