

Abstract
This article provides an overview of principles and barriers relevant to intracellular drug and gene transport, accumulation and retention (collectively called as drug delivery) by means of nanovehicles (NV). The aim is to deliver a cargo to a particular intracellular site, if possible, to exert a local action. Some of the principles discussed in this article apply to noncolloidal drugs that are not permeable to the plasma membrane or to the blood–brain barrier. NV are defined as a wide range of nanosized particles leading to colloidal objects which are capable of entering cells and tissues and delivering a cargo intracelullarly. Different localization and targeting means are discussed. Limited discussion on pharmacokinetics and pharmacodynamics is also presented. NVs are contrasted to micro-delivery and current nanotechnologies which are already in commercial use. Newer developments in NV technologies are outlined and future applications are stressed. We also briefly review the existing modeling tools and approaches to quantitatively describe the behavior of targeted NV within the vascular and tumor compartments, an area of particular importance. While we list “elementary” phenomena related to different level of complexity of delivery to cancer, we also stress importance of multi-scale modeling and bottom-up systems biology approach.
Keywords: distribution, pharmacokinetics/pharmacodynamics, gene delivery, liposomes, in silico modeling, controlled release/delivery, polymeric drug delivery, nanovehicle, nanoparticle, targeted drug delivery, polymeric drug carrier, cancer
INTRODUCTION
Delivery to Cells and Tissues of Agents—Controlled Release Versus Intracellular Delivery
Delivery systems have been used as clinical tools for rationalizing and executing different treatment modalities (dose escalations, administration sites, etc.). The design of delivery systems is an increasingly valuable discipline in pharmaceutical development, allowing rational manipulation of the pharmacological profiles of drugs and their concomitant therapeutic indices. Delivery systems are now used to modify potentially therapeutic agents toward: (a) creation of new pharmaceutical moieties (e.g., liposomal anthracyclines); (b) improvement in the effectiveness or reduction of the side-effects of an existing therapeutic by limiting the shortcomings of current cytotoxic drugs due to their dose-limiting toxicities; (c) extension of the patent lifetime for an already marketed drug1 and (d) tissue-specific targeting. This is of advantage for drugs and gene products which often exhibit a narrow therapeutic index, short half-time in the blood stream and a high overall clearance rate. The therapeutic index is defined as the ratio of the toxic to the therapeutic dose of a drug.
Therapeutic agent is defined as a chemical, biological, genetic, and radiological agent, an entity to be delivered to a disease site for the purpose of treatment or detection (imaging).
The development of delivery systems that are able to alter the biological profiles (biodistribution, tissue uptake, pharmacokinetics, and pharmacodynamics) of therapeutic agents is considered of utmost importance in biomedical research and pharmaceutical industry.2 Biodistribution of an agent is usually time-dependent. The tissue distribution of a particulate/macromolecular drug among different body locations is also highly dependent on the nonspecific effects of the reticuloendothelial system (RES). RES is the cellular system responsible for protection and clearance of “foreign” material. The RES primarily consists of macrophages or macrophage precursors, specialized endothelial cells lining the sinusoids of the liver (e.g., Kupffer cells–liver macrophages), spleen, and bone marrow and reticular cells of lymphatic tissue (macrophages) and bone marrow (fibroblasts) as well as circulating monocytes. RES is also called the mononuclear phagocytic system (MPS). RES represents a preferential drug distribution site, following the first pass.1
Bioavailability3 is one of the essential parameters in pharmacokinetics, as bioavailability must be considered when calculating dosages for nonintravenous routes of administration. Even the systemic administration does not guarantee that the drug is freely available because of plasma-protein binding. It is generally assumed that only free drug can cross membranes (some drugs bind surface receptors, however) and bind to the intended molecular target, and it is therefore important to estimate the fraction of drug bound to plasma proteins. Drugs can bind to a variety of particles in the blood, including red blood cells, leukocytes, and platelets, in addition to proteins such as albumin (HSA; particularly relevant to acidic drugs and negative zeta potential nanovehicles), glycoproteins, basic drugs including gene delivery vehicles, lipoproteins (neutral and basic drugs), erythrocytes, and globulins.4 The significance of HSA is expanded further in relation to RES system and nanovehicles (NV) colloidal stability.
Recent years we have seen an explosion in the field of novel microfabricated and nanofabricated devices for drug delivery. Such devices seek to develop a platform of well-controlled functions in the micro- or nano-level. The distinction is often made between micro- and nanoparticles on the basis of size although the justification of dimension is arbitrary. Drug encapsulation within microparticles (1–1000 μm) and nanoparticles (1–1000 nm) is typically achieved with biodegradable and biocompatible polymers. Microparticles are composed of synthetic or natural polymers that can be modified to speed up or slow down the degradation of the polymer reservoirs (and, therefore, modify drug release kinetics). The most commonly used polymers are polylactide (PLA) and poly (lactide-coglycolide) (PLGA). Drug diffusion rates through the polymer reservoirs can be altered as desired. Depending on these factors and others, degradation of the biodegradable polymers can occur over from months to years5 via enzymatic/hydrolytic scission mechanisms. For example, TCA cycle metabolism can result in the biotolerable metabolites of lactic acid and glycolic acid.
Controlled release in drug delivery can significantly enhance the therapeutic effect of a drug. Typically, controlled release is used to achieve sustained or pulsatile drug release. Sustained release is used to achieve a constant concentration of a drug over an extended period of time keeping the drug delivered within the optimum range for maximum therapeutic effect. The advantage of such a microdevice include very accurate dosing, the ability to have a certain release patterns, potential for local delivery, and possible biological drug stability enhancement. Microdevices act as an external depot of a drug which is then released into an interstitial space between the cells and tissues with potential long-lasting effect.6 Due to their size, microparticles, when injected into a variety of tissues or deposited directly tend to stay where they are placed (local delivery) while minimizing system toxicity.7a In contrast, NV are taken up, in most cases, very efficiently by cells, internalized, and sorted into different organelles or cytoplasm where they exert their function. This basic distinction dictates a separation between the macro-/micro-devices and NV and serves a basis of this article. A special case of microparticle delivery to cells is a delivery to phagocytic antigen-presenting cells, capable of taking up larger cargo (e.g., In Reference 7b).
NV are thus quasi-soluble, delivery systems for intracellular delivery as contrasted to microparticulate and other macroscopic vehicles (polymeric scaffolds, etc.) typically used for a slow-release of drugs. The macroscopic vehicles are, as a matter of distinction from NV, not taken up by cells and internalized and sorted into different organelles or cytoplasm where they exert their function. Many different nanovehicular technology platforms have been employed, each with different properties, strengths, and weaknesses. Most frequently discussed among these are polymer-based nanoparticulate platforms,8 dendrimers,9 liposomes,10,11 gold nano-shells,12 semiconductor nano-crystals,13 silicon- and silica-based nano-systems14, and superparamagnetic nanoparticulates,15,16 among others.
We introduce here a concept of NV as contrasted to nanoparticles, although not popular in the field. We propose that the term of NV comprises very different chemical and morphological categories, including liposomes (not often denoted as nano-particles), liquid-core nanocapsules (walled), quasi-soluble dendrimers or polymer–drug conjugates, nanoparticles as such (generated as a result of different processing modes), as well as nanosuspensions or polymeric films. Although stated above, not all (nano-targeted) NV can overcome the cell membrane barrier without a targeting or internalizing motif. In this respect, the cell-type is perhaps a controlling factor, as some cells are more susceptible to uptake of nonfunctionalized particles via their design (e.g., macrophages).
The fundamental opportunities for nanovehicular delivery are summarized in three, closely interrelated aspects: first, the recognition of target cells and tissues; second, the ability to reach the diseased sites where the target cells and tissues are located; and third, ability to deliver multiple therapeutic agents. The first two aspects comprise the notion of achieving preferred, substantially higher concentration of a therapeutic agent at site, a phenomenon that will be called “localization”, as opposed to the term “targeting” that is often used to identify drugs that provide specific action against a target biological pathway.17 It should be also noted that the term localization is more often employed to denote an intracellular, organelle-specific, site delivery.
The nanovehicular systems offer certain distinct advantages for drug delivery. Due to their subcellular and submicron size, NV can penetrate deep into tissues through fine capillaries, cross the fenestrations present in the epithelial lining (e.g., liver), and generally be taken up efficiently by the cells. This allows efficient delivery of therapeutic agents to sites in the body. Also, by modulating polymer characteristics, one can control the release of a therapeutic agent from NV to achieve desired therapeutic level in target tissue. NV can be delivered to specific sites by means of conjugation or adsorption of a biospecific ligand. Targeted delivery can improve the therapeutic index of drugs by minimizing the toxic effects to healthy (nondiseased) tissues/cells. The parameter “intracellular delivery index,” the ratio of intracellular delivery to the extracellularly delivered drug (on mass basis), provides a suitable measure for assessing the effectiveness of intracellular drug delivery. It largely depends on the extent of circulation time of NV in the central compartment (see below), release rate, and rate of uptake of NV (their internalization; see below).
The present article overviews the new potential therapeutic applications of NV based on their mechanism of action. The mechanism of their intracellular uptake, different pathways of their uptake, intracellular trafficking, and sorting into different intracellular compartments, and the mechanism of enhanced therapeutic efficacy of the NV-entrapped agent both in vitro and in vivo is elaborated more below.
INTRACELLULAR DELIVERY: PHARMACOKINETICS
Many of the following salient features of this discussion below were derived from Petrak.18 According to him, several elementary steps in pharmacokinetics are important to consider. They are summarized below (from (A) to (F)) and in Figure 1. It should be re-stated that the intracellular delivery may involve both the extracellular drug release at the interstitium (tissue site) followed by the intracellular delivery upon the NV internalization.
-
(A)
Removal from the circulation: It is essential that the NV, loaded with a drug or gene, is not cleared too quickly from the circulation. Rapid clearance may prevent the vehicle from reaching the required concentration at the site of localization. Many drugs will bind to plasma components (principally HSA) or within other compartments of the tissue. Binding can greatly influence the transport and elimination in individual organs and can influence the overall pharmacokinetics. The design and the production of the delivery system need to eliminate (or minimize) all nonspecific interactions occurring between the nanovehicular drug-carrier and the environment of the systemic compartment.19 The central compartment of the body (blood and lymph) is essentially an aqueous, polar medium, featuring many different types of noncovalent interactions. The most frequently employed approach is to use water- soluble, inert macromolecules as drug carriers, or to attach them (covalently or by adsorption) to the surface of drug-carrying particles. The function of the carrier is to mask all unwanted interactions between the drug and the environment until the drug is released from the carrier at the target site. The specifics of targeted drug delivery system are more discussed below.
-
(B)
Release of free payload at nontargeted sites: Depending on the amount of drug/gene vector, the release of drug/gene vector away from the target site could nullify any benefits that might potentially come from delivering the drug/gene vector to the target site. This could be because the amount of drug reaching sites of systemic toxicity might become too high or, second, the amount of free drug that reaches the target site after it has been released from the NV at nontarget sites might be greater than the amount of drug actually being delivered to the target using the delivery system.
-
(C)
Delivery of drug/gene vehicle to the target site: If the drug NV reaches the target site too slowly, the supply of free drug might never be sufficient to generate the concentration required to elicit the desired therapeutic effect at the site of action (delivery window). The total amount of drug delivered (i.e., the area under the curve in a drug concentration vs. time plot for the target site) is irrelevant if, at any time, the free-drug concentration at the target site does not reach its pharmacologically effective level. Delivery of the drug NV to the target organ might not guarantee that an adequate amount of the drug will be available at the actual target (intracellular targets).
-
(D)
Release of free payload at the target site: The capacity of the system selected for the release of payload from the NV should be considered at a rate that also ensures drug accumulation at the target site.
-
(E)
Removal of free payload from the target site: Agents that benefit most from target-selective delivery are those that are retained at the site while acting on their target of action. Certain drugs will need to be delivered into the cytoplasm; therefore, it would be preferential for the drug to be fully retained within the NV and delivered, intracellularly, to a proper place within the cells.
-
(F)
Elimination of the vehicle from the body: For optimal targeting, elimination of the payload vehicle should be minimal. NV and their payloads could be eliminated via hepatobiliary and renal excretions, following the pay-load release. The liver sinusoidal domains in the liver lack a basement membrane and possess pores of 100–1000 nm in size,20 thus allowing the NV to freely access hepatocytes and Kupffer cells from Disse space. Kupffer cells belong to the RES and are primarily located at the sinusoidal domains of the liver. At large, the liver, kidney, and lungs are organs specialized in the removal of leaking drug from the circulation. The rate of elimination of free drug from the systemic circulation should be rapid relative to its rate of transfer from the target site to the central compartment of the body. This way, the drug delivery system will at least achieve a decrease in the drug-associated toxicity. Most of water-soluble substances are eliminated from the body in urine via glomerular filtration and renal excretion. The liver is a major site for drug metabolism. This organ aids in elimination by converting lipid-soluble substances into more hydrophilic compounds which are more easily excreted by the kidney. Peripheral blood mediated elimination is mainly due to proteolytic enzymes, affecting a portion of peptides and protein drugs. Receptors for peptides and proteins can serve as potential source for elimination of these substances via receptor-mediated uptake and subsequent intracellular metabolism. In terms of NV, in the liver, endothelial filtration can remove NV up to 150 nm, whereas particles below 10 nm can leave the systemic circulation via the lymph nodes.21
Figure 1.

Systemic administration of a drug-nanovehicle into the central (blood-lymph) compartment. In pharmacokinetic models each organ could be composed if multiple compartments, which reflect the anatomy/morphology of the organ. Red arrow symbolizes the lymph drainage from each organ(s). Note that the tumor lymph drainage is often impaired. For peripheral compartment, most organs are drained.
Some useful details of the elementary steps and associated mathematical modeling tools which encompass the above considerations (from (A) to (F)) can be found in the reference by Boddy et al.22 It would be prudent to pay attention to these considerations at the start of any drug-carrier-system development. It might also be worth determining some of the key characteristics of the drug-delivery system and the drug to be delivered in vitro, before using them in vivo. For example, opsonizing protein–drug-carrier interactions should be determined in vitro.23 The opsonization of foreign particles, such as bacteria, with so-called opsonins (e.g., IgG antibody molecules, fibrinogen, and complement protein C3b) by phagocytes is a method of marking them for destruction. Phagocytes contain surface receptors that bind to these opsonins and the invading particle is engulfed, surrounded, and phagocytozed. To test the opsonization of putative drug-delivery particles with fibrinogen in vitro, measurement of the isothermal adsorption of 125I-fibrinogen onto the particles can be used. The lower the plateau adsorption of fibrinogen, the less likely the particles will be opsonized in vivo and the more likely it is that they will remain in the circulation.24 Likewise, an opsonization BSA test would be useful.25
Pharmacokinetics
Some basics of pharmacokinetic (PK) modeling will be discussed.26 The goal of pharmacokinetics is synthesis into a coherent model of physical and biological phenomena involved in drug distribution in the body, although a development of comprehensive (elementary) model may not be practical. The four components of PK are commonly referred to as absorption, distribution, metabolism, and excretion (ADME). Drug absorption (bioavailability) is normally determined from the drug concentration in plasma as a function of time, from plasma concentration-time curves. Integrated area under the curve (AUC) is obtained as a primary measure of the amount of drug in systemic circulation. Drug distribution is a drug concentration attained for the appropriate duration in the target tissue for the desired pharmacological effect. Hepatic metabolism and renal filtration are the main contributors to the drug clearance. The other part of clearance is that of excretion (hepatic biliary and renal). Clearance, together with the volume of distribution, defines the half-life and thus the dosing of a drug. The volume of distribution is a theoretical concept that connects the administered dose with the actual initial concentration in circulation.
Being empirical, the utility of compartmental models is limited, as they are not valid beyond their experimental domain. In contrast, detailed physiological models often contain parameters which are difficult or impossible to measure. A physiological pharmacokinetic model (physiologically-based PK model, PBPK model) may include a multiple compartment approach, whereas each organ is composed of multiple subcompartments, reflecting an anatomy/morphology of the organ. In a typical pharmacological model the target is directly accessible from the central compartment (whereas a delivery to a peripheral compartment is required in another case; e.g., in delivery to skin, muscle, peritoneum). The central compartment is defined as a blood/lymphatic circulation system. In the most favorable case, traditionally measured plasma drug concentration is connected with tissue distribution and pharmacokinetic measurements. Targeted drug delivery systems (see below) may substantially affect both drug disposition and pharmacological properties. The pharmacokinetic modeling may provide insights into appropriate dosing regimens (amount of drug, capacity, and dosing frequency), optimal binding affinity, and how receptor-mediated effects may be anticipated from natural or mimetic drug ligands.27 Simple scheme of systemic administration of a drug-vehicle into the central (blood-lymph) compartment is presented in Figure 1. In this scheme, a simplification is presented to obtain an easy understanding. Besides the central compartment, only liver, kidney, and tumor compartments are presented in some detail. Such compartmentalization for most part is neglected for other organs.
The key to our analysis is the target compartment, tumor/vascular tissue. This is due to the fact that two-thirds of clinical trials are currently conducted in the cancer area. The tumor compartment is presented as a one entity comprising of vascular subcompartment with cell surface receptors, interstitial and intracellular tumor space (the target tissue). Further breakdown within the intracellular space is presented below (in the Section “Role of Receptor-Ligand-Signaling and Clustering in Agent Delivery”). Well-perfused interstitial compartment is typical for liver and kidneys. Other tissue compartments are lumped into a common peripheral compartment with no details presented. That is, the organs or tissues that contain negligible quantities of the drug are eliminated from investigation.
The tumor/vascular compartment could be presented as a one entity comprising of vascular subcompartment with cell surface receptors, interstitial and intracellular tumor space (the target tissue). Further breakdown within the intracellular space is also warranted. Well-perfused interstitial compartment is typical for liver and kidneys. Other tissue compartments are typically lumped into a common peripheral compartment with no details presented. That is, the organs or tissues that contain negligible quantities of the drug are eliminated from investigation. It is important to realize a possible fate of a drug as delivered to the solid tumor, intravenously. First, NV passage through a leaky tumor blood endothelium occurs. The attachment of the NV to the endothelium is either nonspecific or facilitated by a specific targeting (active targeting) motif directing NV to the tumor endothelium. NVs accumulate in tumor tissue because of their extended circulation time in conjunction with preferential extravasation from tumor vessels (EPR effect). This passive targeting process facilitates tumor tissue binding, followed by uptake (internalization). Resulting is intracellular drug release for drug action and cell killing. In addition, NV which fail to bind to tumor cells will reside in the extracellular (interstitial) space, where they eventually become destabilized because of enzymatic and phagocytic attack. This results in extracellular drug release for eventual diffusion to nearby tumor cells and bystander cell. The EPR effect features tumor hypervasculature, defective vascular architecture, and deficient lymphatic drainage system. Targeted drug delivery systems may substantially affect both drug disposition and pharmacological properties.
The choice (ligand affinity and avidity) and optimization of ligand surface density may be necessary in order not to allow an excessive attachment to the outer vascular/cancer compartments to facilitate nanovehicular penetration within the tumor interior (“binding site barrier”)28,29 as the excessive binding can retard such penetration of the NV.
The lymphatic administration is a means of minimizing general systemic drug exposure to modify the drug biodistribution. The primary function of the lymphatics is to drain the capillary beds and return extracellular fluid to the systemic circulation. Unlike the blood flow, lymph flow is unidirectional, recovering fluid from the periphery and returning it to the vasculature. Drug transport through the lymph may be utilized to prolong the time course of drug delivery to the systemic circulation, bypassing liver and avoiding hepatic first-pass metabolism. Consequently, intestinal, subcutaneous, and intramuscular areas represent poorly perfused peripheral compartment. Nanovesicular drug transport, however, is different from that of proteins.30 This is also potentiated by inherent anatomical differences between the blood and lymph capillaries. In case of interstitial administration (subcutaneous, intraperitoneal, and intestinal) smaller particles leak back into the blood capillaries (thus exhibiting a long circulation time), whereas larger particles (up to about 100 nm) may enter the lymphatic capillaries and lymph nodes where they may be trapped for a long time. The size of administered NV should < 100 nm if good drainage from the injection site is achieved. In addition, charge of NV may influence their distribution. Positively charged NV enhance drainage from the interstitial injection site and their localization in the regional lymph nodes.31a
The lymphatic delivery (lymphotropic delivery system) is prone to the same problems associated with the intravenous administration; that is the deleterious effect of interaction with RES system. Interstitially injected NV are in contact with sera and interact with its proteins (opsonins); consequently, they are attacked by macrophages within the lymph nodes draining the injection site.
Under pathological conditions, both the flow pattern and cellular content of the lymphatic system may be altered dramatically. Targeting to lymph nodes for therapeutic purpose has been attempted with different NV. This targeting is of importance for delivery to lymphatic cancers. Subcutaneous (s.c.) delivery prevents a rapid systemic clearance, to some degree. The s.c. delivery of PEGylated IFN-β 1a resulted in 16–27-fold increase in area under the concentration (AUC)-time curve in monkeys31b following a single injection. Intramuscular delivery is somewhat impaired because of low level of regional lymphatics. Gut-associated lymphoid tissue (GALT) is another area to be more explored as it is suitable for delivery of NV for oral vaccine design. The overall uptake of the NV across the GI epithelium is, however, very small, normally amounting to few percent of the original, oral dose.32 Drug delivery to lymphatic tissue has been reviewed by Papisov and Weissleder33. For tumors, excellent model has been proposed by Ferl et al.34 which includes lymph drainage from almost every organ/tissue. Flessner35 outlined a schematic of peritoneal lymph/blood transport. A simplified schematic of intraperitoneal lymph delivery is presented in Figure 2. A subcutaneous lymph pharmacokinetic model has been suggested.36
Figure 2.
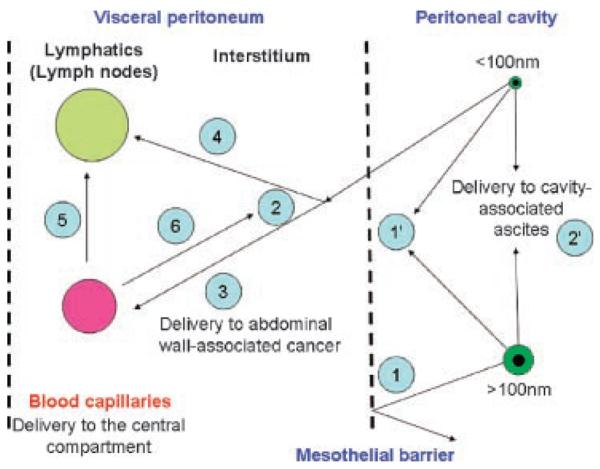
Major elements of the intraperitoneal drug delivery. Nanovehicles > 100 nm are rejected (1), but smaller NP can pass from the peritoneal cavity and through the mesothelium into the peritoneal interstitium (2). (1′) represents a delivery to mesotheliomas. (2′) denotes interstitial uptake (by abdominal tumors and ascites). The peritoneal membrane is an idealized partition barrier with heterogeneous sieving characteristics. By diffusion and convection, NP can enter discrete blood (3) and lymph capillaries (4) within the interstitium. Macromolecules and nanovehicles could also diffuse from the blood compartment to the lymphatic (5) or interstitial (6) compartments. Interstitium (peritoneal tissue) is dense network composed of collagenous, glycoprotein, and proteoglycan material. Adapted from Flessner.35
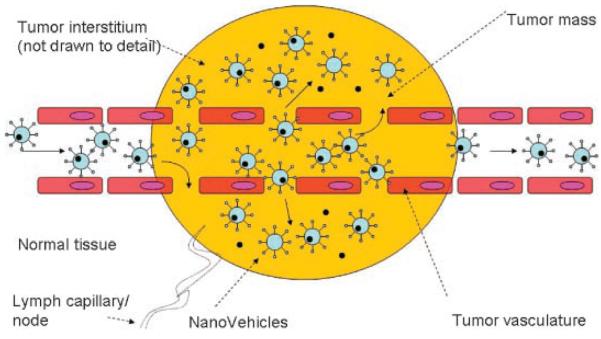
Figure 3 depicts possible fate of a drug as delivered to the solid tumor, intravenously. First, NV passage through a leaky tumor blood endothelium occurs. The attachment of the NV to the endothelium is either nonspecific or facilitated by a specific targeting (active targeting) motif directing NV to the tumor endothelium. NV accumulate in tumor tissue because of their extended circulation time in conjunction with preferential extravasation from tumor vessels (EPR effect)(Fig. 4). This passive targeting process facilitates tumor tissue binding, followed by uptake (internalization). Resulting is intracellular drug release for drug action and cell killing. In addition, NV which fail to bind to tumor cells will reside in the extracellular (interstitial) space, where they eventually become destabilized because of enzymatic and phagocytic attack. This results in extracellular drug release for eventual diffusion to nearby tumor cells and bystander cell. The EPR effect features tumor hypervasculature, defective vascular architecture, and deficient lymphatic drainage system.
Figure 3.

Schematic diagram of mechanism of targeted nanovehicle delivery of a therapeutic drug to vascular compartment and into solid tumors. Vascular targeting agents often exhibit an affinity to both endothelial and tumor cells. Targeted nanovehicles, endowed with a specific targeting ligand on their periphery, accumulate passively in tumor tissue because EPR (enhanced permeability and retention) effect and preferential extravasation (1) from tumor vessels. Endothelial cells are shed (partially) from the lining of tumor blood vessels, exposing underlying tumor cells. Consequently, increased vascular permeability of vascular tissue (leaky endothelium) enables nanovehicles to extravasate and reach the tumor interstitial fluid. This passive and nonspecific process of nanovehicle extravasation is statistically improved by the prolonged residence time of nanovehicles in circulation and repeated passages through the tumor microvascular bed. Nanovehicles with engineered (PEG and other technologies) long-circulating properties increase the number of passages through the tumor microvasculature. However, except for rare instances, tumor cells are not directly exposed to the blood stream. Therefore, for an intravascular targeting device to access the tumor cell, it must first cross the vascular endothelium and diffuse into the interstitial fluid, via extravasation (2). Extravasated nanovehicles then attach to cancer cells (3) and are taken up (internalized) by tumor tissue (4). Likewise, an attachment and internalization of nanovehicles may happen with endothelial cells because of specific vascular targeting agent. Subsequently to internalization, intracellular drug cytosolic release (5) occurs, followed by direct killing of tumor and endothelial cells (6). Once nanovehicles have penetrated the tumor interstitial fluid, binding of targeted ligand-endowed nanovehicles may occur vigorously, shifting the intratumor distribution from the extracellular compartment to the tumor cell intracellular compartment. This shift could be several times higher for targeted nanovehicles as compared to nontargeted ones. Also, the recirculation of nanovehicles within the blood compartment will be considerably reduced for nanovehicles with specific-binding affinity to tumor cell receptors. Because of limited diffusion capacity of nanovehicles within the interstitial space, binding is likely to be limited to the tumor cells in closest vicinity to blood vessels. In addition, the nanovehicles which fail to bind to tumor cells will reside in the extracellular (interstitial) space. Upon their destabilization, they slowly release (7) their drug content into the interstitial space which will eventually diffuse to nearby cancer cells and bystander cells (8) exerting a cytotoxic effect. Obviously, there will always be a combination of in situ release from an extracellular nanovehicle depot and intracellular release from internalized nanovehicles. Therefore, the theoretical advantages of targeted nanovehicles over the nontargeted are related to a shift of nanovehicle distribution to the tumor cell compartment, delivery of nanovehicular contents to an intracellular tumor compartment in nanovehicle-associated form, and, possibly, prolonged nanovehicle retention in the tumor (provided with a proper PEG chemistry). Adapted from Park et al.37 and Gabizon et al.38
Figure 4.
Schematic of EPR (enhanced permeability and retention) effect in solid tumors: nanovehicles passively target to vasculature and extravasate through fenestrated tumor vasculature. Nanovehicles accumulate in tumor tissue because of their extended circulation time in conjunction with preferential extravasation from tumor vessels (EPR effect) and lack of lymphatic drainage. This passive targeting process facilitates tumor tissue binding, followed by uptake (internalization). Resulting is intracellular drug release for drug action and cell killing. Nanovehicles which fail to bind to tumor cells will reside in the extracellular (interstitial) space, where they eventually become destabilized because of enzymatic and phagocytic attack. This results in extracellular drug release for eventual diffusion to nearby tumor cells and bystander cell. Normal blood vessels have a tight endothelium. Adapted from van Vlerken and Amiji.39
ROLE OF RECEPTOR-LIGAND-SIGNALING AND CLUSTERING IN AGENT DELIVERY
Cell surface receptors are complex transmembrane proteins which mediate highly specific interactions between cells and their extracellular milieu. Receptor binding is an area of potential importance to targeted drug delivery, including endocytosis, transcytosis, ligand–receptor interactions, and receptor regulation. Since biochemical and physiological properties of receptors vary depending on both receptor-type and cellular background, it is likely that some receptor systems may be more suitable than others for receptor-mediated drug delivery. Below, we review some basics of the cellular entry of macromolecules and particles.40
Receptor-Mediated Endocytosis and Signaling
An important function for receptors is to facilitate ligand internalization via receptor-mediated endocytosis. In this process, a ligand bound to its receptor is endocytosed from clathrin-coated pits on the plasma membrane, forming an endocytotic vesicle. This can occur in a constitutive manner in which the receptor is internalized at the same rate in the absence or in the presence of ligand or in a ligand-stimulated manner. In addition to being regulated by ligand, a number of other factors and agents (e.g., insulin, transferrin) can modulate receptor-mediated endocytosis via phosphorylation. There is a strong evidence that protein phosphorylation can regulate receptor endocytosis and intracellular trafficking.
The concept of targeting always exploits phenotypical differences between the disease target and normal tissues that are then translated into a dose differential (often very small but appreciable) between the target and off-target sites and therapeutic benefit. The pharmacokinetics between the specific and nonspecific sites favors the targeted delivery: there is often a predominance of transient occupation of nonspecific sites cleared at longer times as compared to more pronounced and stable accumulation at the target sites.41,42 The clearance from the occupied sites, both due to specific and nonspecific interactions, is of reversible nature, well-established concept in receptor–ligand interaction. In simple chemical reaction terms, it is denoted as mass transfer with reversible chemical reaction. As such, it is typically treated quantitatively (e.g., In Reference 34). This traditional view is being challenged, for certain drug category, by a nonequilibrium drug-binding action model which explains an amelioration of off-target-based drug toxicities (collateral binding) on the basis of their capacity to limit the concentration and duration of systemic exposure required for pharmacodynamic toxicity. The premise is that the dissociative half-life of a drug-target (receptor) binary complex, that is rather the period for which receptor is occupied by a ligand, is critical and could be extended to exhibit a prolong capacity to block the desired target site and not the ligand affinity as shown, for example, for the anticancer effects of Hsp90 inhibitors.43
Ligands for active targeting can be derived from endogenous physiological receptor–ligand combinations, such as transferrin (Trf)-TrfR and folate (Ft)-folate-R (FtR), but could also include other structures on target tissue (vascular, tumor) or in the extracellular environment where a ligand could be discovered, for example, such as antibodies, or peptide or synthetic libraries. Endogenous receptors, however, could be occupied and out competed by endogenous ligands. Such receptors are occupied to varying degrees at all times and thus compete with drugs for receptor binding. New ligands and antibodies, however, can bind with greater affinity than its receptors natural ligands.44
For targeting drug delivery vehicles, various endogenous ligands, such as peptides, proteins, lipoproteins, growth factors, vitamins, and carbohydrates can be used. The aim is to improve delivery by targeting receptors which initiate internalization by endocytosis. Following binding of the NV to target cells, delivery of the therapeutic to the cell occurs by one of two mechanisms, depending on whether the ligand is internalizing or noninternalizing.45 The potential advantage of targeted delivery may result from an altered intracellular distribution. After NV that is linked to a noninternalizing ligand binds to target cells, the drug is gradually released from the NV and is taken up by the cell as free drug, using standard uptake mechanisms. When the ligand is an internalizing one, the NV–drug is taken into the cell by receptor-mediated endocytosis and, assuming it is stable in the environment of the endosome, the drug is gradually released within the cell. The number of drug molecules that are delivered intracellularly are higher when an internalizing ligand is used as the diffusion and redistribution of the released drug seem to be higher for noninternalizing ligands, which leads to lower concentrations of drug being delivered to the target cells. It is probably for this reason that internalizing ligands have resulted in better therapeutic outcomes in animal models.46–48 For internalizing ligands, because not all of the NV–drug will immediately be internalized into target cells, the opportunity for a bystander effect exists, as a drug that is released extracellularly and it diffuses within the tumor to be taken up by receptor-negative cells45. Successful targeting has been achieved within the vascular compartment. Anti-CD19 Ab-targeted liposomes showed B-cell-specific killing in vivo in a B-cell lymphoma tumor model.49 For solid tumors, pharmacological improvements were reported to be only modest for long-circulating and targeted liposomes.50 For dendrimers (<5 nm in size), Kukowska-Latallo et al.46 reported quite high T/B ratios with folate targeting into solid tumors. These results could be explained on the basis of very small NV size and its penetration into the tumors as seen readily with small imaging motifs.
The internalization of a NV with its payload is a prerequisite for the induction of efficient cytotoxicity and therapy while also enabling to overcome drug resistance. While seeking the best ligands for surface antigens it should be noted that a considerable fraction of antibodies generated by immunization do not bind receptors in a manner that triggers internalization.51–53 It is necessary to screen various targeting ligands for their ability to elicit the desired effect because different targeting ligands and different receptors may traffic to very different intracellular pathways while others do not elicit any receptor activation at all. The same may apply for peptide or nonpeptide ligands. Enhanced efficacy in a cancer model was demonstrated to be due to anti-HER2 immunoliposomes, allowing an efficient internalization and intracellular delivery of doxorubicin (Park, 2002)37. On the other hand, Goren et al.54 have employed an IgG directed against HER2, coupled to immunoliposomes, with no in vivo effect because this ligand did not allow for internalization of NV. Hosokawa et al.55 employed successfully a GAH, human mAb against gastric cancer for their GAH-conjugated PEG-modified liposomal doxorubicin delivery in cancer model, while Sapra and Allen56a used liposomal targeting with CD19 B-cell antigen for lymphoma treatment. In both cases, ligands were allowed for efficient uptake and internalization. Receptor-mediated endocytosis is an essential first step for many targeted therapeutic interventions.
Receptors and Signaling
While the initial events in the endocytosis of the receptor–ligand complex are similar for most systems, the processing of the ligand can differ depending on both receptor and cell-type. In most instances, the role of receptor-mediated endocytosis is to internalize ligands for their subsequent proteolytic degradation in acidic pH of the endosomal compartment. The receptor is then recycled back to the plasma membrane, whereas the ligand is destined for lysosomal degradation. This proteolysis of ligand can serve to remove a signal or to clear an undesirable protein or delivery vehicle. Cells can also reduce the level of ligand stimulation both by degrading the ligand and reducing the number of receptors expressed on the plasma membrane (downregulation of receptors). The receptor internalization by endocytosis is one of the mechanisms for the termination of receptor activity and is typical for many adhesion molecules (which may serve as good acceptors of ligands for targeting).
In addition to their natural ligands, a number of proteins and peptides can bind the extracellular domain of receptors, including anti-receptor antibodies, and synthetic agonists and antagonists. The nonnatural receptor-binding peptides may overcome potential limitations of the natural ligand for receptor-mediated drug delivery.40 Targeting receptors via their natural ligand has several potential limitations, such as ligands could be rapidly degraded in the plasma or could bind to multiple receptor types or isoforms (below). Receptor antagonists block the binding of their natural ligand and thereby prevent receptor activation.
Further understanding of the biochemical and physiological characteristics of receptors is necessary to avoid undesirable effects of targeting these active cell surface proteins and to realize their full potential for receptor-mediated drug delivery. Receptors' primary functions are to mediate protein trafficking and to transduce signals across the cellular membrane. Studies of receptor structure have revealed specific domains and motifs which regulate receptor trafficking, kinase activity, and coupling with intracellular signaling networks. These findings can be utilized at the design of drug-delivery systems.
Receptor-Mediated Transcytosis
In cells that are polarized into apical and basolateral surfaces, such as endothelial and epithelial cells, certain receptors can mediate transcytosis. This involves the vectorial trafficking of the ligand, via endocytic vesicles, across the cellular layer. A number of ligands have been reported to be transcytosed in these cell types, including insulin and EGF.57–59 The role of transcytosis is to selectively facilitate the transport of ligands across diffusional barriers, such as the vascular endothelium. Receptor-mediated transcytosis can be utilized as a method for the delivery of drugs across cellular layers, including the blood–brain barrier.60 More details on transcytosis are presented below.
Receptor Clustering61
Transmembrane receptors are increasingly being found to be organized into higher-order structures in the cytoplasmic membrane. Clustering may be an evolved strategy to achieve greater effectiveness and/or higher strengthening of adhesion required by many biological functions other than uniform molecular distribution. Clustering renders high local densities of receptors and/or ligands that may not be independent of global or average densities, resulting in multiple bonds that do not obey existing criteria for receptor–ligand interaction. One way of visualizing this event is a mechanism where ligand binding induces rearrangement of a receptor oligomer and involves large-scale reorganizations of many membrane proteins into a highly structured and dynamic micrometer-scale region at the cell–cell interface, which has been termed as a “supramolecular activation cluster.” This mechanism in fact occurs through ligand-induced oligomerization of receptor subunits. The binding constants of bivalent interactions can be a factor of 1000 higher than monovalent binding, and for tri- and pentavalent interactions values up to 108 have been reported. Multiple receptor interactions of ligands interact with multiple receptors on the cell surface and may lead to increased affinity and avidity.62
A good example of the clustering comes from a carbohydrate–carbohydrate and protein–glycan interaction. Characteristic features of these interactions are their specificity, their strong dependency on divalent cations, and their extreme low affinity that has to be compensated by multivalent presentation of the ligands. Multivalent presentation (display) has been proved to be important in the study of carbohydrate–protein interaction.63
Receptor clustering after binding of multivalent ligands also occurs frequently in vivo. Such process is important for cell activation, signaling, and adhesion and participates in immune synapse. Pharmaceuticals which employ such phenomenon should have enhanced potency or enhanced binding. Polyvalent glycan presentation via NV is becoming more important at soliciting proper response.64 The exploration of multivalency is the key for enhancing the targeting effects.
INTRACELLULAR TRAFFICKING: NANOVEHICLE UPTAKE AND TRAFFICKING
The successful design of a delivery system requires a thorough understanding of the mechanisms involved in the interaction of the delivery systems with the target cells. We believe that an understanding of intracellular trafficking and targeting would help to design better and more efficient drug delivery systems. That is why we put so much emphasis in this article on the mechanism of intracellular events connected with drug/vehicle delivery. Intracellular targeting refers to the delivery of therapeutic agents to specific compartments or organelles within the cell. The therapeutic agent could be a small molecular weight drug or a macromolecule like protein or DNA (see below on payload). Targeted delivery could result in higher bioavailability of a therapeutic agent at its site of action, simultaneously reducing both the total dose and the side toxic effects associated with the drug. In many situations, however, it is not only important to deliver a therapeutic agent into a specific tissue, but also to deliver it within a specific cellular compartment. Poor permeability of the drug through cell membrane, low accessibility of the drug to its site of action within the cell, degradation of the drug in specific cell compartments, or toxicity due to exposure of the drug and/or the delivery system to different cellular organelles present challenges. Endocytosis (and some other forms of uptake, phagocytosis) is considered the most useful mechanism to describe multiple forms of internalization. Special devices, endowed with peptides that can enhance the cytosolic release of internalized molecules are essential for achieving successful delivery after endocytosis-mediated uptake (e.g., In References 65,66). To avoid the degradation problem associated with endocytosis, other nonendocytic strategies can be used to deliver drugs (genes) in a manner that circumvents endocytosis.
Nanovehicle Size
Our studies and those of others show that particle size significantly affects cellular and tissue uptake, and in some cell lines, only the submicron size particles are taken up efficiently but not the larger size microparticles (e.g., In References 67–71,72a). In Caco-2 cells, the uptake of nanoparticles (PLGA with PVA coating) of 100 nm in size had 2.3-fold greater uptake compared to that of 50 nm particles, 1.3-fold to that of 500 nm particles, about 1.8-fold that of 1000 nm particles. Thus, it is demonstrated that nanoparticles of 100–200 nm size acquire the best properties for cellular uptake. The better cellular uptake can result from an optimum size range.69 An optimal size for dendritic cells uptake in vitro is < 500 nm because they can phagocytose particles.72b The authors employed model polystyrene nanoparticles. A notable departure from these observations is that of lipoplex uptake, whereas large particles (size between 0.2 nm and 5000 nm) were preferable for efficient lipofection regardless of charge.73 The observed phenomena are related to lipoplex fusion directly with cytoplasmic membranes (lipid rafts), although the authors did not take enough precautions on the degree of external lipoplex adsorption while measuring the luciferase expression, employing only an extensive buffer washing.
Particle size controls the NV clearance and tumor penetrability. In general, smaller particles have slower clearance rates and higher penetration into solid tumor tissue as long as their size is above the exclusion limit for kidney filtratvion, 150 nm (size of fenestrae). Very small particles (1–20 nm) with long circulatory residence times slowly extravasate from the vasculature into the interstitial spaces to be transported by lymphatic vessels to lymph nodes.74 Larger particles, such as immunoliposomes (approximately 100–150 nm in diameter), can take 48 h or longer to reach peak levels in the tumor, and it is important not only that the particles circulate for sufficient time to allow for maximum tumor localization, but also that the particles retain their drug contents during this process. In vitro studies revealed that a viruslike particles (<100 nm) do lend themselves to be taken up by RES system. Likewise, for NV larger than 100 nm prevention of opsonization is a difficult task as the clearance of larger neutral and anionic liposomes increases progressively with increasing size.75 The size of fenestrae in certain inflammatory vessels as well as tumor capillaries can be up to 700 nm,76,77 depending, however, on animal/human model. Particles of less than 10 nm can leave the systemic circulation through the permeable vascular endothelium in lymph nodes.78 The sinus endothelium of bone marrow is also capable of removing small-sized particles via transcellular and intracellular pathways.79 The splenic filtration by the sinusoidal spleens at inter-endothelial cell slits (IES) can occur for size up to 200–500 nm, more often up to 250 nm, the larger size only for deformable particles. This sets the upper limit for an ideal NV from the circulation point of view as not exceeding 200 nm80,81 unless splenotropic targeting is sought.
The macrophages of the RES system have ability to remove unprotected NV from the circulation following their i.v. administration, via a process which involves opsonins. Immunoglobulins (Fc-receptor) and components of the complement system (C3, C4, and C5) as well as other serum proteins such as fibronectin, collagens, and laminin are involved in the process of opsonization of NV. Resulting is an adsorption of these components on the periphery of NV, their marking for elimination by macrophages, via a set of specialized receptors. Part of this step is complement activation, a size-dependent process: the larger the size, higher the activation rate.
For in vivo applications, it is required that NV are resistant to the effects of plasma and in part, size is the controlling factor. Larger particles (200 nm and above) are more efficient at activating the human complement system and are hence cleared faster from the circulation by Kupffer cells of liver than their smaller counterparts. Consequently, these NV can be administered into systemic circulation without the problems of particle aggregation or blockage of fine blood capillaries. The differences in opsonization may account for differences in clearance rates. This is due to the view that macrophages are heterogeneous with respect to phenotype and function, even within the same tissue. Problems related to PEC NV are discussed separately (in the Section “Gene Delivery”).
Nanovehicle Internalization
Many reports indicate that NV are internalized efficiently through an endocytic process and that uptake is concentration- and time-dependent as well as cell-type-dependent (for a different opinion see the section “Mechanism of NV Uptake and Efflux”). Endocytosis results in internalization of the cell's plasma membrane to form vesicles that capture macromolecules and particles present in the extracellular fluid and/or bound to membrane-associated receptors. These vesicles then undergo a complex series of fusion events directing the internalized cargo to an appropriate intracellular compartment (uptake of fluids, macromolecules, particles, and other ligands that sort to cell's processing pathways). Uptake of particulate systems could occur through various processes such as by phagocytosis, fluid phase pinocytosis, transport via clathrin-coated pits, caveolae-mediated transport, or nonendocytic pathway. Transcytosis and exocytosis are pathways communicating with the external environment82 (Fig. 5). The uptake is also cell-type-dependent. Following their uptake, NVs have been shown to be transported to primary endosomes and then to sorting endosomes. From sorting endosomes, a fraction of NV is sorted back to the cell exterior through recycling endosomes (efflux, exocytosis), while the remaining fraction is transported to secondary endosomes, which then fuse with lysosomes. The NV then escape the endo-lysosomes and enter the cytosolic compartment. Time-dependent uptake studies showed that NV escaped the endo-lysosomes within 10 min of incubation and entered the cytoplasmic compartment. Surface charge reversal of NV selectively in the acidic pH of endo-lysosomes is proposed as the mechanism responsible for the endo-lysosomal escape of these particles (see Fig. 11 in Reference 83). Surface charge reversal results from transfer of protons/hydronium ions from bulk solution to NV surface under acidic conditions. This is often due to typical cationic charge of NV periphery, leading to localized destabilization of the vesicle membrane and the escape of particles into cytoplasmic compartment. Polystyrene nanoparticles, which do not exhibit surface charge reversal with change in pH, were not seen escaping the endo-lysosomal compartment.84
Figure 5.
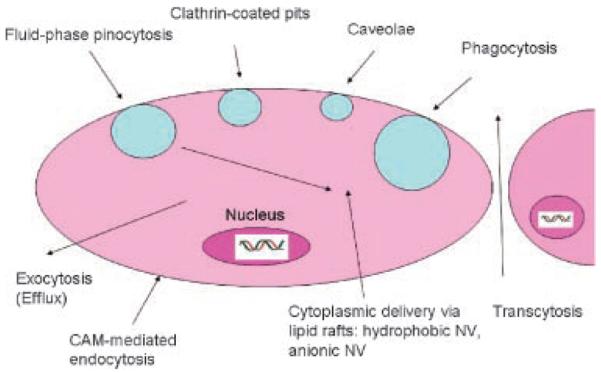
Schematic representation of uptake and exocytosis routes of nanovehicles. Redrawn from Panyam and Labhasetwar.82 Not drawn to scale.
Lysosomes had been recognized in several cell types and found to be involved in the digestion of extracellular material taken up by endocytosis and of intracellular material segregated by intracellular trafficking. Lysosomes are powerful digestion system, especially within the RES system macrophages (liver, spleen) and are the key clearance mechanism of NV elimination from the circulation. Constitutive polymers and macromolecules, making up the NV, are directed into the endocytic pathway and end up in lysosomes where they are degraded by late endosome and lysosome enzymes or inside the cytoplasm. Most of nonbiodegradable polymeric nanoparticles cannot be degraded by digestion in endosomal-lysomal compartment and depending on their size (molecular weight) they will either be removed by renal filtration or sequestered and stored in one of the RES organs. If the NV can undergo partial scission (chemically or enzymatically) individual components can be removed by renal system easily.85 Permanently stored (intracellularly) remnants of NV may represent a health hazard, not yet explored.
NV amounts inside the cell are maintained as long as NVs are present in the outside medium. Once the external concentration gradient is removed, exocytosis of intact NV begins and results in a substantial drop in a short time of minutes. However, at least some initial NV levels are maintained over several hours of incubation in vitro. Some researchers have observed that a NV exocytosis was inhibited in serum-free medium. Protein (albumin) present in the serum was found to be responsible for inducing NV exocytosis.86 Figure 6 A/B depicts NV adsorption/attachment, uptake, and efflux of NV.
Figure 6.
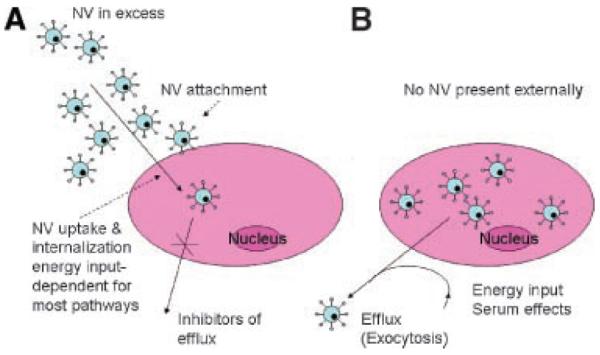
Kinetic considerations at nanovehicle uptake and exocytosis in vitro. The overall fate could be traced by flow cytometry (FACS) following the cell detachment via trypsin or EDTA (ethylenediamine tetraacetic acid). Nanovehicles must be present in medium in excess to be internalized. The efflux could be inhibited by specific inhibitors. The quantity of nanovehicles could be distinguished from total uptake by fluorescence quenching by Trypan Blue (A). When resuspended in fresh media without nanovehicles, they can efflux to the environment. This process requires energy and presence of serum (B).
The translation of the in vitro data to in vivo situation is not always straightforward. One can infer the internalization of NV from experiments on gene delivery in vivo as well as from those that follow the localization of NV in tissues and cells. This detection is possible due to recent developments in labeling and imaging. The intracellular localization of NV in tissues is not easily obtained in vivo.
Mechanism of NV Uptake and Efflux87
The plasma membrane is a dynamic structure that functions to segregate the chemically distinct intracellular milieu (the cytoplasm) from the extracellular environment by regulating and coordinating the entry and exit of small and large molecules. Essential small molecules, such as amino acids, sugars, and ions can traverse the plasma membrane through the action of integral membrane protein pumps or channels. Macromolecules must be carried into the cell in membrane-bound vesicles derived by the invagi-nation and pinching-off of pieces of the plasma membrane in a process termed endocytosis. Endocytosis occurs by multiple mechanisms that fall into two broad categories, “phagocytosis” or cell eating (the uptake of large particles) and “pinocytosis” or cell drinking (the uptake of fluid and solutes). Specialized efflux mechanisms serve to dispose the NV.
-
(1)
Phagocytosis (PhC) is typically restricted to specialized mammalian cells (dendritic, macrophages, monocytes, and neutrophils), whereas pinocytosis occurs in all cells by at least four basic morphological distinct mechanisms: macropinocytosis, clathrin-mediated endocytosis, caveolae-mediated endocytosis, and clathrin- and caveolae-independent endocytosis (Fig. 7A). These mechanistically diverse and highly regulated endocytic pathways function to control such complex physiological processes as hormone-mediated signal transduction, immune surveillance, antigen-presentation, and cellular and organismal homeostasis. The mechanistic complexities that govern endocytosis suggest that great evolutionary effort has been expanded to control entry into the cell and thereby to control cellular responses to the environment.
-
(2)
Macropinocytosis (MPC) or fluid-phase (FPE) endocytosis (Figs. 5 and 7A), can be measured by the intracellular accumulation of tracer molecules (for example, an enzyme or labeled compound) present in the medium. The degree of internalization of fluid-phase markers is directly proportional to their concentration in the medium and the volume encased by the transport vesicles. Greater efficiency of endocytosis is achieved by nonspecific binding of solutes to the cell membrane (adsorptive pinocytosis), but the most efficient uptake occurs when dilute solutes are captured by specific high-affinity receptors (receptor-mediated endocytosis), which are themselves concentrated into specialized endocytic transport vesicles. MPC accompanies the membrane ruffling that is induced in many cell types upon stimulation by growth factors or other signals. The signaling cascades that induce MPC involve Rho-family GTPases, which trigger the actin-driven formation of membrane protrusions. These protrusions then collapse onto and fuse with the plasma membrane to generate large endocytic vesicles called macropinosomes (>1 μm in size, up to 5 μm), which sample large volumes of the extracellular milieu. Although MPC accompanies seemingly chaotic membrane ruffling, it is likely to be a highly controlled and regulated process.
MPC fulfils diverse functions. It can be transiently induced in most cells and might have a role in the downregulation of activated signaling molecules (growth factors). Activation of antigen-presenting dendritic cells triggers extensive and prolonged macropinocytic activity, enabling these cellular sentries to sample large volumes of the extracellular milieu and to fulfill their role in immune surveillance. MPC is generally considered a nonreceptor-mediated process (nonspecific) where cells take up large volumes of extra-cellular fluids and solutes. It is constitutive in highly specialized cells (macrophages and dendritic cells) and in some tumors, and it can be induced by growth factors in epithelial cells. It is the principal endocytic pathway in endothelial cells (EC). MPC fulfills diverse functions, especially when massive fluid-phase endocytosis is necessary. Although the pH of macropinosomes decreases they do not fuse into lysosomes. MPC has been shown to be of importance for gene and drug delivery by means of CPP (see below). This pathway provides some advantageous aspects such as the increased uptake of particles and macromolecules, the avoidance of lysosomal degradation, and the ease of escape from macropinosomes because of their relatively leaky nature. This route may also be a significant pathway for polymer (carrier) elimination at the kidney site. Surprisingly, the majority of studies to date have ignored the biological fate of nonbiodegradable polymer-carrier used in nanovector delivery systems. When a targeting ligand is released from the particle surface a major assumption is that polymer excretion via the renal system occurs.21
-
(3)
Clathrin-mediated endocytosis (CME) occurs constitutively in all mammalian cells, and carries out the continuous uptake of essential nutrients, antigens, growth factors, and pathogens (Figs. 5 and 8A). The most common examples are internalization of cholesterol-laden low-density lipo-protein (LDL) particles that bind to the LDL receptor, and iron-laden transferrin (Tfn) that binds to Tfn receptors. CME is crucial for intercellular communication during tissue and organ development, it modulates signal transduction both by controlling the levels of surface signalling receptors, and by mediating the rapid clearance and down-regulation of activated signalling receptors. Clathrin-coated vesicles (CCV) of the CME pathway carry concentrated receptor–ligand complexes into the cells. They range in size from 100 nm to 150 nm in diameter. Molecules entering via this pathway will rapidly experience a drop in pH from neutral to pH 5.9–6.0 in the lumen of early endosomes, with a further reduction to pH 5 during progression from late endosomes to lysosomes.88 Ligands and receptors are then sorted to their appropriate cellular destinations, such as lysosomes, the Golgi apparatus, the nucleus, or the cell surface membrane. Some portion of receptor–ligand complex also recycles back, via what is called exocytosis (below). A receptor-rich region buds off to form a separate vesicle that recycles the receptors back to the cell membrane.89 CME is a highly regulated process; it is also an energy-dependent process. Perhaps, also, dependent on actin cytoskeleton as treatment of mammalian cells with actin-disrupting agents has a partial or no effect on CCV formation. CME can be targeted by using certain ligands, such as transferrin, which can specifically recognize certain receptors on the cell surface, is of significance for drug delivery.
-
(4)
Caveolar-mediate endocytosis (CavME). Caveolae are flask-shaped invaginations (50–60 nm is size) of the plasma membrane that are typically seen on the surface of endothelial cells, where they are extremely abundant (Figs. 5 and 7A). They were proposed to mediate the extensive transcellular shuttling of serum proteins from the bloodstream into tissues across the endothelial cell layer. Caveolae are derived from the Golgi complex and are known to be present on many specialized cells, and to demarcate cholesterol and sphingolipid-rich microdomains of the plasma membrane, in which many diverse signaling molecules and membrane transporters are concentrated.90 The shape and structural organization of caveolae are conferred by caveolin, a dimeric protein that binds cholesterol, inserts as a loop into the inner leaflet of the plasma membrane, and self-associates to form a striated caveolin coat on the surface of the membrane invaginations. Caveolae have been experimentally disrupted by depletion of plasma-membrane cholesterol (cyclodextrin). A major difference from CME is that the caveolar uptake is a nonacidic and nondigestive route of internalization, bypassing lysosomes,91 potentially a route of advantage for drug delivery. However, caveolae are slowly internalized and are small in size, and their fluid-phase volume is small; this results in low capacity uptake. Thus, it is unlikely that they contribute significantly to constitutive endocytosis, although the situation is different in endothelial cells in which caveolae constitute 10–20% of the cell surface.87 Some researchers consider caveolar up-take as a subclass of lipid-mediated rafts uptake92 (also see below).
-
(5)
Lipid rafts-mediated endocytosis (LRME). For anionic and neutral-lipid liposomes, solid-lipid nanoparticles, and hydrophobic nanoparticles (e.g., synthetic polystyrene nanoparticles) there is another potential pathway, endocytosis-independent uptake. Lipid rafts are small (40–60 nm in diameter), free cholesterol- and sphingolipid-rich planar microdomains characteristic for cell surfaces which lack caveolin and caveolae and clathrin, such as lymphocytes, many human cancer cells and rodent macrophages. A hydrophobic uptake via the lipid membrane fusion is likely to be involved. The likely mechanism is that of scavenger receptor, involved in the uptake of both lipophilic and anionic groups.93,94 As expected, anionically-charged liposomes (heparin-functionalized) exhibit an extended circulation time and provide better pharmacokinetics for encapsulated doxorubicin (Dox), following their intravenous injection in mice.95 The scavenger receptor class B (SRB1; CD36 superfamily of proteins) is expressed on mature macrophages is particularly promiscuous and ligands include proteins, polyribonucleo-tides, polysaccharides, and lipids for which the main common feature is that they are “polyanionic.” The involvement of SRB1 in the liposome uptake was demonstrated on the basis of polyinosinic acid (a strong polyanion) competitive abolishment of association of neutral phospholipids/cholesterol liposomes with hepatic cells.96 Other receptors, such as apolipoprotein E (apoE), LDL receptors, or some undefined receptors, have also been indicated in the clearance of neutral and negatively charged liposomes in hepatocytes, although differences have been noted between neutral and negatively charged liposomes.97 Fusogenic liposomes may also fall into this category. For example, liposomes prepared with the help of UV-inactivated Sendai virus enable membrane fusion with the liposomal bilayer and direct cytosolic delivery.98 Accessing this type of uptake pathway is one way of increasing the circulation time of NV, minimizing the RES, and delivering to cytosol. At this time, it is not really possible to label one particular uptake pathway and delineate the mechanism completely. It could be argued that the lipid raft uptake may be just a variant of caveolae-mediated uptake (or vice versa) because of their cholesterol association as caveolae and lipid rafts share many characteristics, that is, high cholesterol and glycosphingolipid contents. It appears that the lipid-raft-mediated uptake is facilitated by a transbilayer membrane potential that exists across cell membranes. This constitutes a major difference between the caveolae- and lipid raft-mediated endocytosis, the lack of energy input requirement for lipid raft-mediated uptake (see energy-independent flippase),99 a strong argument for this distinction, although not universal (see Tab. 4). The diffusion via the lipid raft would be rather slow process and would not provide very efficient way of transport (uptake). It may be operational for cholesterol efflux. Many investigators, however, consider raft/caveolin-dependent endocytosis as one integral system.
-
(6)
Clathrin- and caveolae-independent endocytosis (CCLIE) is a special endocytic pathway (Figs. 5 and 7A). Less well characterized, both clathrin- and caveolae-independent pathways may constitute a specialized high capacity constitutive endocytic pathway for lipids and fluid, as well as NV. Clathrin-independent pathways are now amenable to detailed analysis using the approaches that have proved so successful in studies of clathrin-coated pits. The ability to characterize clathrin-independent carriers by electron microscopy, follow their dynamics and precise protein–protein interactions in real-time by light microscopy, and manipulate their molecular machinery through large-scale downregulation of defined components should allow rapid insights to be gained into this poorly understood process.101
Recently discovered clathrin- and caveoli-independent pathway is of significance for ICAM-1-positive cells (CAM-mediated endocytosis).102–104a NV coated with anti-ICAM antibody induce ICAM-1 clustering by multivalent NV presentation. CAM-mediated endocytosis delivers material to lysosomes. CAM-1, a transmembrane glycoprotein from the Ig-like superfamily of adhesion molecules that is upregulated and functionally involved in inflammation and thrombosis, may provide a unique target for lysosomal disease (LSD): (i) it is expressed by cell-types affected in diverse forms of LSDs, including endothelial (EC), epithelial, glial, and Schwann cells; leukocytes; myocytes; and other cells; (ii) it is accessible to ligands administered via intravascular, intratracheal, or intracerebral routes; (iii) ICAM-1 levels are enhanced by pathological factors pertinent to LSDs, such as inflammation; and (iv) ICAM-1 targeting blocks its function as an anchor for leukocytes, thus providing anti-inflammatory benefits. However, ICAM-1 and another Ig superfamily cell adhesion molecule, platelet endothelial cell adhesion molecule-1 (PECAM-1) are not readily internalized by endothelial cells. Nevertheless, despite the inability of these cell adhesion molecules to act as receptors to mediate endocytosis of monomeric antibodies, endothelial cells internalize multimeric anti-PECAM-NV and anti-ICAMNV < 300 nm in diameter. Anti-CAM-NV uptake depends on signaling induced by CAM clustering and represents a unique actin-dependent process requiring activation of protein kinase C, Src kinase, and Rho kinase. Results from cell culture and in vivo studies showed that CAM-targeted systems, including anti-ICAM NV, deliver reporter, and model enzymes into target cells. Muro et al.103 found that anti-ICAM Ab greatly enhances binding, internalization, and lysosomal trafficking of recombinant human acid sphingomyelinase (hydrolase deficient in types A and B Nieman-Pick disease (NPD)). Most importantly, in situ functional activity of ICAM-targeted recombinant ASM alleviated cellular lipid storage, either induced by trafficking and fate of anti-CAM NV.
Besides the above five basic mechanisms, exocytosis and transcytosis are more specialized mechanisms (efflux).
-
(7) Exocytosis (EC). Exocytosis via exosomes is a constitutive mechanism of metabolite elimination for every cell (Figs. 5 and 7A). It may be also operational for NV. Limited studies are available at this point, however. The underlying observation is that once the cells are devoid of any external reservoir of NV for uptake, once internalized NV leave the intracellular sites and are expelled outside.86 This is probably because of high local concentration of NV in the vicinity of cell membranes result in the internalization along with extracellular liquid and solutes into the cell through endocytosis (pinocytosis). Recent observations testify on the role of targeting ligands associated with NV in exocytosis. Sahoo and Labhasetwar104b observed decreased exocytosis for Trf-conjugated PLGA nanoparticles as compared to nonfunctionalized ones. Salaün et al.105 argues that there is a constitutive exocytosis pathway operating in all cells, in addition to regulated one, facilitated by lipid rafts, cholesterol and sphingolipid-rich microdo-mains, enriched in the plasma membrane. Minimizing the exocytosis or its control would provide a special handle on retention of delivered drugs and cargo.
A special exocytic transport has been demonstrated in tumor cells causing an efflux of anti-cancer drugs out of the cancer compartment via molecular pump mechanism. P-glycoprotein (Pgp, ABCB1) is the archetypical mammalian ABC transport protein and its mechanism of action has received considerable attention. There is strong biochemical evidence that Pgp moves molecular cargo (apparently a broad range of structurally unrelated compounds) against a concentration gradient using the energy of ATP hydrolysis. However, the molecular details of how the energy of ATP hydrolysis is coupled to transport remain in dispute (Ambudkar et al.)106. The clinical importance of Pgp stems from the fact that this molecular pump is implicated in multidrug resistance (MDR), the phenomenon by which tumor cells simultaneously exhibit intrinsic or acquired cross-resistance to diverse chemotherapeutic agents, resulting in the failure of chemotherapy for many cancers.107 Overexpression of Pgp in cancer cells can undermine cancer chemotherapy. Transporters are involved in the intestinal absorption, hepatic excretion, and renal excretion of drugs. They are located in the plasma membrane, the nuclear membrane, the cytoplasm, and Golgi apparatus of many cell types. Multiple transporters are involved in the processes of drug absorption, distribution and excretion, particularly in excretion in the liver and kidney and intestine. The efflux system is effective in normal intestinal and colonic cells, and also at other epithelial sites.
For transcellular transport (Fig. 5), drugs need to cross two different membranes on the basal and apical sides. In the intestine, drugs are absorbed from the luminal side (brush border membrane) and excreted into the portal blood across the basolateral membrane. In the liver, drugs are taken up into hepatocytes across the sinusoidal membrane and excreted into the bile.
-
(8)
Transcytosis (TsC) reflects transport across the endothelial (and epithelial) cells to the subendothelial space (Figs. 5 and 7A). The endocytosis on the apical (luminal) surfaces is followed by transport through a series of intercellular compartments and delivery to the basolateral (ablumenal) plasma surface where the substances are secreted or diffusion via intercellular junctions (paracellular transport). In the kidney, drugs undergo secretion (urinary excretion) or reabsorption. The rate of transporter-mediated uptake and efflux determines the rate of renal and hepatobiliary elimination. Transporters are thus important as a determinant of the clearance in the body.108 Caveolar-mediated endocytosis serves as an entry point for transcytosis of any compounds to the subendothelial tissue. This process is highly inefficient as observed on passage through the gastrointestinal tract and blood–brain barrier (BBB), amounting to less than 1–2% of the luminal input.109
-
(9)
Markers for intracellular compartments and trafficking pathways. The use of the term “marker” is often misleading because it can be interpreted to imply an absolute specificity which does not reflect the dynamic nature of intracellular compartments.110 Use of multiple fluorescent probes facilitates the analysis of the trafficking of delivered vehicles/agents in live cells. Typically, localization of many of the markers is achieved through incorporation of a minimal targeting motif or molecule. Fluorescent transferrin and EGF are routinely used to functionally define endosomal compartments based on the time that it takes for the ligands to accumulate within them.111 Another mean to follow the fate of intracellularly delivered drugs is an employment of inhibitors (physical and chemical) of intracellular transport pathways. A widely used means for inhibiting transport is to apply a temperature block cell metabolism. For example, clathrin-dependent internalization is blocked at 4°C which facilitates the binding to cells of extracellular ligands before allowing endocytic uptake by raising the temperature. Pharmacological inhibitors of membrane traffic pathways are most commonly used. Tables 1 and 2 summarize typical inhibitors of uptake and trafficking, and Table 3 lists markers for specifying (visualizing) cellular compartments. Molecular targets of these inhibitors are discussed in References 100,103,110.
Figure 7.

Nanovehicle uptake and efflux routes. (A) Mechanistic view (pathways in red lettering indicate the preferred routes of uptake, bypassing endosomal trafficking, but not very efficient in terms of uptake rate). (B) Kinetic view (preferred routes). Adapted from Khalil et al.100
Figure 8.

Some possible disease intracellular targets.
Table 4.
Multiple Portals of Nanovehicle (NV) Entry as Differentiated by Cargo Chemistry and Size* and Cell Type
| Pinocytosis |
||||||
|---|---|---|---|---|---|---|
| Phagocytosis | Macropinocyto-sis | Clathrin-Mediated | Caveolae-Mediated Rafts | Lipid Rafts | Clathrin- and Caveolin-Independent | |
| Vehicle size | a1–10 μm | a1–5 μm | <150 nm | <60 nm | 40–60 nm | N/A |
| Actin involvement | Yes | Yes, to less extend | Yes | Yes | Yes | Yes, to less extend |
| Energy input | Yes | Yes | Yes | Yes | Yes/bNo | Yes |
| Cell types | Dendritic, macrophages, monocytes | Many cells, constitutive in some tumors | Many cells | Differentiated endothelial, adipocytes, epithelial and muscle cells | Lymphocytes, cancer cells, rodent macrophages | Specialized |
| Endo-lysosomal compartment | Yes, but early or late endosome arrest | By-pass or quick escape | Yes, main digestive route | By-pass or quick escape to allow for nonacidic and nondigestive route | By-pass | By-pass or quick escape; CAM-mediated uptake is strictly lysosomal |
| Receptor | Yes | Yes, rarely, in response to growth factors and mitogenic factors | Yes Uptake saturable | Yes; caveolae are internalised Upon activation of resident receptors Uptake saturable | Yes, rarely, nonspecific receptors (e.g., HSPG) uptake nonsaturable | Yes (e.g., IL-2 R) or nonspecific receptors (e.g., HSPG) |
| Adsorptive endocytosis | No | Yes | No | Yes | Yes | Yes |
| cFluid-phase endocytosis | +++++ | ++++ Uptake nonsaturable, nonspecific | ++ | ++ | ++ | ++ |
| dEfficiency of uptake | +++++ for specialized cells | ++ | +++++ | + Inefficient uptake may be offset by the lower proteolytic activity | + Rafts are internalized constitutively | ++++ |
Typically, cargo chemistry imposes itself a size range limit due to technological restrictions.
Phagocytosis and macropinocytosis, most likely, do not contribute to the nanovehicular uptake to a great extend.
Based on controversial literature entries demonstrating that the uptake of some cationic- and amphiphatic-peptide-modulated nanovehicles is energy-independent, besides a standard dependency (e.g., In Reference112). It is possible that a closer, more critical scrutiny will confirm energy-dependence of lipid-raft-mediated uptake.
The extend of fluid-phase endocytosis relates to the vehicle size; estimated on scale 1–5, 5 being the highest.
The efficiency of uptake may depend on NV type and chemistry. The proportion between different uptake mechanisms may vary; estimated on scale 1–5, 5 being the highest.
Table 1.
Inhibitors (Perturbations) of Nanovehicle Uptake
| Internalization Pathway | Inhibitor |
|---|---|
| Endocytosis (in general) | Low temperature, fluoride, azide |
| Macropinocytosis/phagocytosis | Cytochalasin D, wortmannin, phorbol ester (stimulator) |
| Clathrin-mediated | Low potassium in medium |
| Caveolae | Methyl-β-cyclodextrin, filipin |
Note that many inhibitors were tested for drug/macromolecular uptake; only some for nanovehicular uptake.
Table 2.
Some Inhibitors of Intracellular Trafficking
| Trafficking Pathway | Inhibitor |
|---|---|
| Receptor recycling | Chloropromazine |
| Lysosomal (and endosomal) degradation | Chloroquine |
| Endosome-lysosome trafficking | Nocodazole |
Table 3.
List of Some Markers of Cellular Compartments
| Compartment | Marker/Sources |
|---|---|
| Generic fluid phase marker | Lucifer yellow (IMP) |
| Clathrin coated pits | Clathrin-dsRed expression |
| Early endosomes | Fluorescent-transferrin (Molecular Probes) |
| Late endosomes | Fluorescent dextran, short pulse (Molecular Probes) |
| Lysosomes | Lamp 1 (Affinity BioReagents) fluorescent dextran, longer pulse (Molecular Probes) |
| Caveolae | GFP-caveolin-1 |
| Mitochondria | Mitotracker (Molecular Probes) |
| Nucleus | GFP-nls (Clontech) |
We summarize the above findings on portals of entry of nanovehicular drugs/vehicles in Table 4, Figs. 5 and 7. There is accumulating evidence that different endocytic pathways merge and are connected to the same early endosome that serves as a distribution site inside the cells. The cargo may be delivered to internal organelles (caveosomes) from noncaveolar uptake mechanisms (Sharma et al.)113. There are no definitive answers to all questions in terms of involvement of certain trafficking pathway in different cells. A considerable confusion exists because of many endocytosis studies relied on the use of pharmacological agents whose specificity remains a matter of debate. In addition, many artifacts have been generated and reported because of improper cell fixation prior to microscopic observation of the cells. Cryofixation method should be employed.114 The prevalent view is that the cargo (payload) also influences the type of uptake mechanism and trafficking.115a The problem with attempting to define specific endosomal and other secretory organelles and pathways is that they are highly dynamic, undergoing constant addition, removal, recycling, and maturation of components.115b
Kinetically (Fig. 7B), three modes of endocytosis can be defined: fluid-phase, adsorptive, and receptor-mediated endocytosis.116 Fluid-phase endocytosis refers to the bulk uptake of solutes in the exact proportion to their concentration in the extracellular fluid. This is a low-efficiency and nonspecific process. In contrast, in adsorptive and receptor-mediated endocytosis, macromolecules are bound to the cell surface and concentrated before internalization. In adsorptive endocytosis, molecules preferentially interact with generic complementary binding sites (e.g., by HSPG, lectin or charged, surface-bound macromolecules). Such distinction is of practical interest because of uncertainties of intimate uptake mechanism as discussed above. Fluid-phase endocytosis has a lower internalization capability compared to adsorptive endocytosis. It is also saturable process, meaning that the cellular uptake depends on the dose of NV. One more distinction is of practical aspect: both phagocytosis and MPC exhibit a nonsaturable linear profile with the NV dose, that is, their uptake is independent from concentration (uptake increases linearly with increase in NV dose) as opposite to often saturable uptake for the rest of mechanisms of endocytosis (e.g., in case of receptor-mediated endocytosis, uptake is saturated at higher doses because of saturating of finite number of receptors present on the cell surface117). This is often a neglected feature but of a great significance. Very few references are available to support this statement. Behrens et al.118 observed a strict liner uptake, related to the dose, for their chitosan-coated polystyrene nanoparticles of about 200 nm size for human intestinal cells (Caco-2). Likewise, Panyam and Labhasetwar86 described almost linear uptake for PLGA nanoparticles in vascular smooth muscle cells, a process partially obscured by a rapid exocytosis. Nonspecific, receptor-independent uptake of LDL particles by macrophages also shows a nonsaturable kinetics causing high levels of constitutive macrophage cholesterol accumulation,119 cytochalasin D-dependent. Similar nonsaturable LDL uptake was observed for cultured human fibroblasts,120 and of irreversibly glycated albumin-modified LDL lipoproteins.121 The above observations are in accord with the dose-independent elimination by RES system of PEG-PLGA nanoparticles in mice, while nonPEGylated nanoparticles followed nonlinear and dose-dependent pharmacokinetics.122 Flip-flop (lipid-raft)-mediated free fatty acid uptake at nonphysiologic concentrations123 is reminiscent of nonsaturable particle uptake.
STABILITY OF NANOVECTORS IN BUFFERS AND BIOLOGICAL FLUIDS: STERIC VERSUS ELECTROSTATIC STABILIZATION
To be effective for delivery, NV must remain in a colloidal form. This requires minimization of inter-particle interactions. The long-term stability of the NV under physiological conditions to avoid nonspecific aggregation presents itself as a serious problem.124 Steric stabilization is defined as reduction in particle interactions by a surface steric barrier. The presently prevailing view that repulsive forces result from the “compression” of tethered layers presented on the NV surface. In addition, the hydrophilic side chains extending outward from the particle surface provide stability to the particle suspension by a repulsion effect through a steric mechanism of stabilization involving both enthalpic and entropic contribution, while the dimension of the stabilizing chains exceed the range of Van der Waals force of attraction. Macrophages express surface receptors that do not recognize and internalize sterically protected NV.
Recently, Szleifer125 presented a general theoretical framework for studying the adsorption of proteins on surfaces with grafted polymers including PEG. It was postulated that the affinity of the proteins to the PEG chain is negligible. It could be calculated that the adsorption of the model protein lysozyme on a surface with PEG chains is independent of the PEG chain length if the molecules consist of more than 50 ethylene oxide units corresponding to a PEG Mw larger than 2200. If the PEG Mw exceeds this critical value the adsorption is only a function of the surface density of the PEG chains. In general, the theoretical value for maximum protein resistance of PEG-coated surfaces calculated by this author is well in agreement with the value experimentally determined in the present study for PEG-modified PLA particles. It has to be taken into account that the threshold in terms of PEG chain length which results in maximum protein resistance depends on the size of a particular protein. In addition, the coating by PEG layer should protect the nanoparticles not only against plasma protein adsorption, but also against recognition by phagocytic cells. For particles of the type PEG-PLA (45K) with different PEG Mw, a maximal reduction in protein adsorption was found for a PEG Mw of 5000. The existence of a threshold in terms of PEG (5K) surface density to avoid the uptake by human MPS cells, circulating cells endowed with a high phagocytic activity has been documented. The same surface density threshold was found to mask the surface charge and ensure efficient steric stabilization, and to avoid the uptake by MPS cells. These findings have a great consequence for the design of long-circulating NV.
A word of caution: one of the factors diminishing the success of specific targeting is shielding of the targeting functional groups by the protective polymer layer. There are a number of reports of a reduction of transfection efficiency of large PEG-covered NV carrying targeting groups.126,127 Also, recent experimental studies of phospholipid and liposome adhesion show low or no adhesion achieved when functional groups were attached either directly to the surface via a polymer spacer.128 Similar observations were made by Torchilin et al.129 in studying TAT-targeted liposome internalization by several cell lines: when targeting groups were attached directly to liposome surface or to a spacer shorter than stabilizing PEG chains, no cell uptake was registered. In all the above-mentioned cases shielding of targeting groups by the protective polymer layers is thought to be the reason for the targeting failure. Using a bidisperse protective polymer layer formed by short nonfunctional polymers and long functionalized polymers considerably increases the fraction of functional groups on the periphery of the layer.130
One interesting experimental observation on limited opsinization capacity of the organism (saturable elimination) should be listed. The macrophage clearance is mediated by a pool of blood opsonins. Therefore, it appears to be a limited pool of opsonic factors in the blood that can interact with PEG-bearing vehicles, a fact of significance at drug delivery. That is the blood clearance of PEG liposomes was shown to be dose-dependent and at liposome doses lower than certain level liposomes do not show the long-circulation property but instead are cleared relatively rapidly from the bloodstream.131 In addition, imperfect coverage with PEG molecules can cause an uptake of sterically-stabilized particles. Recent study demonstrated that 30% of the total population of PEG-coated polystyrene nanoparticles is prone to phagocytosis because of inadequate surface coverage by PEG molecules.21 Macrophages express surface receptors that can recognize and internalize sterically protected particles.
Besides PEG molecules, a special set of polymers is available for steric stabilization. They are lipid-based. 2-methacryloyloxyethyl phosphorylcholine (MPC) polymers have been synthesized as biomimetics of biomembrane structures.132 MPC polymers have a surface that resists nonspecific protein adsorption and cell adhesion, that is, “biofouling.”133 It has been shown that cells in contact with MPC polymers do not exhibit activation or an inflammatory response.134 It has been reported that nanoparticles coated with MPC polymer were not recognized by macrophages, and they could be used as long-term circulating drug carriers in the blood stream.135 The MPC polymers are available commercially (Purebright® MB series, MPC–MBA copolymer, MBA is n-butyl methacrylate, NOF, Tokyo, Japan and Biocompatibles, UK). No comparison has been carried out with PEG stabilization strategy.136 However, the large hydration shell afforded by the phospholipid headgroup may mean that it is a more effective protein-resistant technology than PEG-based systems, offering reduced opsonisation, extended plasma half-lives, and sustained efficacy.
In contrast to the kinetic stability achieved by electrically neutral polymers above, electrostatic stabilization employs repulsive, Coulombic interactions between the charged surfaces (Pincus, 1991)137. Note that most of NVs are endowed with a charge. Any ionizable groups in the hydrophilic domains presented at the NV periphery act as electrostatic stabilizers. This method is sensitive to dissolved salts, which suppress the screening effect. A coupled mechanism involving both electrostatic and steric mechanisms is denoted as electrosteric stabilization. A good example is an employment of anionic copolymers with suitably sized PEG molecules. Attached to gene delivery vectors via coating, such complexes readily prevent opsonization by serum proteins, maintain their size, and do not aggregate at near electroneutrality (low zeta potential).138
In general, the PEC stability in plasma could present a problem (see some coverage in the Section “Types of Payload”). PLL/DNA complex can undergo rapid clearance from the plasma following intravenous administration. The plasma proteins, particularly serum albumin, can loose the complexes and enlarge them. Dash et al.139 reported on a possibility of ternary complex between the PLL/DNA and albumin which can undergo a disruption by positively charged serum proteins at physiological pH. All forms of PEC complexes may require stabilization to improve their circulation times in vivo. Cationic polymers with short side chains were shown to form smaller complexes, resistant to destabilization by polyanions. The destabilization of cationic lipid/DNA complexes in vitro and in plasma by HSPG polymers and heparin facilitates DNA release.140a,140b The multicomponent PEC-based nanoparticles141,142 are more resistant to inter-polyelectrolyte complex exchange mechanism (competitive displacement of the components of the polyelectrolyte complex with other charged species, external polyelectrolytes, and small ion due to cooperativity of molecular interactions characteristic of polymer systems). The inter-polyelectrolyte exchange often leads to unpacking of the assembled particle.143 The PEC complex is considered to be a result of a reversible mono-molecular reaction, while there is a competition between complexation and counterion binding. The multicomponent system, results from, apparently, an interaction of two pairs of polyelectrolytes. Oupicky et al.144 demonstrated an increased resistance to polyelectrolyte exchange and prolonged plasma circulation time for polycation/DNA polyplexes modified with multivalent PHPMA N-(2-hydroxypropyl)methacrylamine] laterally-crosslinked polymer (of the surface of polyplex) as the only presence of PEG molecules has no beneficial effect on susceptibility to polyelectrolyte exchange reaction. This paper also provides an evidence for saturable elimination mechanism mentioned earlier.
LOCALIZATION AND TARGETING
Localization
The following concepts have been introduced by Ferrari.145
-
(1)
Localization by size and shape. Years of liposome research and clinical use have demonstrated that tailoring nanovector size to match the fenestrations of the cancer neovasculature yields preferential concentration at tumor sites—a phenomenon termed enhanced penetration and retention (EPR). Yet, the spherical shape of liposomes, and sizes of about 50–100 nm tend to keep them in the middle of blood flow146 as opposed to the vicinity of the endothelial wall, and therefore diminish the likelihood of convection through the fenestrations, and molecular targeting to the neovascular endothelium. Similar considerations apply to most of nanovectors presented in the literature, since they are all spherical or nearly spherical. Recent advances in nanofabrication technology, however, may open new way toward the development of more suitable nonregular geometries for injectable, silicon- and polymer-based delivery systems.147
-
(2)
Localization by physical properties. The surface charge of nanovectors influences tumor uptake. Differences between positively versus negatively charged nanovectors have been noted. Several investigators demonstrated that negatively charged NV could distribute away from the RES system,148–150 unlike the typical behavior of cationically charged nanovectors. Preadministration of protamine increases the accumulation of cationic liposomes in a solid tumor animal model because of an increased selectivity of cationic liposomes in targeting angiogenic microvessels151 by occupying negatively charged HSPG target sites. Albumin (BSA) pretreatment has similar effects. Likewise, Takagi et al.152 have used several cationic compounds to reduce nonspecific hepatic clearance of recombinant IL-11 protein. Such pretreatments could be denoted as pretargeting.
-
(3)
Localization by remote or environmental activation. Regardless of the details of the distribution of injected nanovectors in the body, exquisite localization of the effect may be attained if the cytotoxic action is released only at the intended target sites, by irradiation with an exogenous, and locally focused source of energy. Remote activation approaches that have been demonstrated to include the triggering of gene expression by X-ray irradiation,153 gold nanoshells by near-infrared radiation, leading to localized thermal ablation of tumor xenografts in animal models.154 Enhanced photodynamic therapy by targeted silica nanoparticles,155 neutron-capture therapy with gadolinium nanoparticles,156 and the magnetic field activation of cytolysis157 have been reported. Localized release may also be activated by environmental conditions, such as metabolite (ATP),158 two-step activation methodologies such as polymer-directed enzyme prodrug therapy (PDEPT) and polymer-enzyme liposome therapy (PELT).159 Other methods to control exogenous gene expression include glucocorticoids, estrogens, progesterone, androgens, and heavy metals.
An excellent example of remote activation is a TNFerade Biologic™, an adenovirus carrying the TNFα gene under the control of a radiation-inducible promoter.160 It has entered phase II trial where intratumoral injection is used in combination with radiotherapy. By this means, a control of different spatial and temporal levels is achieved.
Targeting
“Targeting” is a term commonly used to describe the above-mentioned aims when delivering therapeutics, whereby selective delivery and prolonged retention is desired via the control of the pharmacological parameters characteristic of the administered moieties. This can be achieved by designing delivery systems with the aim to either: (a) enhance the deposition or accumulation of the therapeutic in a particular tissue; (b) associate the therapeutic with a particular cell population; (c) associate the therapeutic with a specific intracellular component; (d) prolong the association of the therapeutic within a specific organ (e.g. brain, blood, etc.); or any combination of these.1
-
(1)
Active targeting refers to delivery systems designed to associate or interact with specific biological moieties, most commonly by attachment on their outer surface of ligands (peptides, antibodies, antibody fragments, and proteins) with an enhanced binding affinity for cellular receptors. The requirements of the targeting agents for delivery of molecules into the cell are that the targeting agent must bind to the cell surface and then be internalized. Besides a requirement for high-binding affinity, one should distinguish between the binding, enhanced vehicle internalization, and subsequent downstream biological activity. The ligand specificity is still another issue: some ligands may be promiscuous, binding to different receptors.161 On the other hand, antibodies can be exquisitely specific.
To achieve active drug targeting it is necessary to:162
-
(i)
Define valid targets as markers of disease and disease-associated cells,
-
(ii)
Develop and validate the necessary chemistry to bind drugs to cell-selective vectors, and
-
(iii)
Release the drugs at the right place and time from its vector by developing “intelligent linkers.”
In addition, the delivery systems of active therapeutic agents must:
-
(iv)
Resist hydrostatic, hydrophilic/hydrophobic, and biophysical/biochemical barriers,
-
(v)
Resist cellular resistance to treatment,
-
(vi)
Resist biotransformation, degradation, and clearance mechanisms.
Ideal characteristics of linkers are:163
-
(i)
Stable in circulation (physiological pH) and release drug at appropriate rate within the tumor.
-
(ii)
Drug should be released at appropriate site of action.
-
(iii)
Biodegradable (ideally).
-
(iv)
Stable during conjugate elimination to avoid normal tissue damage.
-
(v)
Toxic or immunogenic byproducts should not be generated.
Linkers or spacers are essential for appropriate presentation of ligands. Vyas et al.,164 Veronese and Caliceti,165 Nobs et al.166, and Khandare et al.167a list some frequent spacers, reagents with homobifunctional or heterobifunctional coupling potential. Among the heterobifunctional reagents, the most popular covalent conjugation reagent has terminal amine/carbonyl/sulphydryl reactive ends, electrophilic groups capable of reacting with these end-groups. Examples are N-hydro-xysuccinimide ester (NHS)-PEG, MA(maleimide)-PEG-NHS, and carbodiimide coupling reagent (1-ethyl-3-(3-dimethylaminopropyl))carbodiimide (EDC) (zero-length crosslinker). The latter may not be optimal for ligand presentation due to steric effects. Hartig et al.71a,71b clearly demonstrated that EDC-coupled ligand (peptide) to the nanoparticle corona doesn't represent efficient presentation as compared to PEG (linker) presentation, using FACS-based affinity assays.
Special degradable ligands or linkers to drugs (coupled to high-molecular-weight polymers through a cleavable linker) are used as an effective method for improving the therapeutic index of clinically established agents via targeting or prodrug presentation. For intracellular drug delivery, the design of drug–polymer conjugates focused on incorporating enzymatically cleavable bonds that allow the prodrug to be cleaved intracellularly after cellular uptake at the requested site.167b Such cleavage is useful, particularly, for avoiding lysosomal degradation of gene vectors. Similar approach has been applied for design of pH-sensitive liposomes.168
A special type of targeting is that termed “Externally stimulated targeting,” whereby localization of the therapeutic agent at a specific site is achieved by an externally applied stimulus such as temperature, light, ultrasound, ionizing radiation, or magnetic force. Many examples of specific biological applications do exist (below) whereby each of the delivery aims has been achieved by physical manipulation of a colloidal or interfacial property of the delivery system.
The benefits of targeted delivery are:163
-
(i)
Localised therapy is possible.
-
(ii)
Prolonged action as a result of the appropriate drug concentration at the target site.
-
(iii)
Reduced dosage as therapeutic efficiency is enhanced.
-
(iv)
Reduced systemic concentration because of direct action on target.
-
(v)
Reduced adverse effects and reduced toxicity as a result of selective enhancement.
-
(vi)
Localised delivery of multiple agents resulting in targeted combination therapy.
Active targeting may also affect the internalization mechanism. The utilization of receptor-mediated endocytosis offers means for controlling the intracellular localization and trafficking of the NV and their cargoes. The intracellular fate of particulate drug delivery systems is determined by the initial mode of internalization and the subsequent intracellular trafficking. Different cellular uptake mechanisms for internalization could be considered and strategies devised how to control their availability. For a targeted drug delivery, the mechanism of caveolae uptake seems to be the most promising since it does not transport internalized material to endosomes and lysosomes where internalized material might be degraded. The knowledge of the mode of internalization is therefore the first step for optimized drug targeting. As there is diversity in endocytotic mechanisms employed by each cell-type this offers the possibility to enhance the uptake via the desired route. For this purpose, the NV's surface could be modified by attaching specific linkers and ligands to either guide the drug to targets different from endosomal/lysosomal compartments by mediating vesicular escape, or to directly accomplish intracellular (cytoplasmic and nuclear) localization. Examples of successful intracellular and organelle-specific delivery of biologically active molecules, including DNA, are presented below and challenges are also discussed.
To date, there are very few references available which investigate the targeting within the scope of the mechanism of internalization. It appears logical that the targeting ligand might dramatically change the NV destination and its uptake mode. Some evidence is available that targeted NV behave very differently as compared to those naked ones (nontargeted). Often, a dramatic increase in the uptake of targeted NV is noted. Thus, Sahoo and Labhasetwar104b observed triple increase in uptake of PLGA nanoparticles endowed with Trf ligand as compared to non-conjugated ones in breast cancer cell line. Likewise, selective uptake of Trf-liposomes by brain capillary endothelial cells (13-fold vs. nontargeted) was noted by Soni et al.169 Kakudo et al.170 observed no internalization of Trf-free liposomes (only cell attachment noted) as compared to Trf-functionalized ones (similar data were obtained with Trf-PEG-liposomes).171 Likewise, galactosylated (Gal) PLA nanoparticles, targeted to ASGP (asialoglycoprotein) receptors, exhibited increased uptake in HepG2 cells,172 while Managit et al.173 observed Gal-liposomes to be preferentially (15-fold) taken up by hepatocytes and parenchymal cells. Adachi et al.174 only observed intracellular localization within hepatocytes of their synthetic (PVLA) nanoparticles bearing a high surface density of Gal ligands. Folate has been successfully applied for targeting of Ft-liposome-formulated oligodeoxynucleotide delivery (sixfold increase into cancer cells175 and Ft-conjugated iron oxide nanoparticles (sevenfold increase in internalization into KB cells176). Finally, RGD-4C cyclic peptide targeted to integrin was employed for targeting of lipid-protamine lipopolyplexes featuring 15-fold increase in uptake and 100-fold increase in transfection efficacy in KB cells.177 It is quite possible that the low basal uptake of nonfunctionalized NV in most above references is due to another functional endocytic uptake mechanism as multiple uptake mechanisms are often observed.
The differences in cellular entry pathway were also noted in relation to NV polymer chemistry. DOTAB DNA polyplexes were preferentially internalized by clarthrin-mediated endocytosis, while PEI polyplexes were internalized both by clathrin- and caveolae-mediated endocytosis, partially evading the lysosomal compartment in A549 and HeLa cells. This may be due to, possibly, easier DNA release from the lipocomplexes while PEI complex is destined for degradation.178
Dual targeting. It should be noted that the dual targeting should be limited to issues of employment of two ligands interacting with two different surface molecules. The only one literature reference is that of Saul et al.,179a who employed dual-targeted liposomes (folate and antibody), targeting overexpressed tumor folate and EGFR receptors. The experiments were designed to limit the off-target effects and demonstrate an increase in selectivity. Dual-targeting of a NV should be strictly distinguished from an application of two drugs with two different targets (e.g., In Reference 179b a combination of anti-EGFR monoclonal antibody and tyrosine kinase inhibitor, targeting extracellular and intracellular domains of the same receptor, respectively). A theoretical foundation of dual targeting has been described.180a
-
(1)
Passive targeting refers to all strategies attempting to achieve the defined delivery aim(s), without utilizing specific biological (ligand–receptor) interactions, accompanied by correlating the physicochemical and surface characteristics of the delivery systems with the pathophysiology and anatomy of the target sites (EPR effect). Passive targeting exploits the anatomical differences between normal and diseased cells (tissues). Illustrative examples of passive targeting include the extravasation of sterically stabilized nanoparticles from leaky tumor capillaries into the interstitium, and the extended blood circulation half-lives of polymer-coated moieties (proteins, drugs, and NV). The drug carrier accumulates in the interstitial fluid of the diseased tissue by exploiting the enhanced permeability and retention (EPR) effect.48 Extravasation is critical in this setting. In addition, release of the drug from the carrier is required for bioavailability.
Passive targeting is thus defined as a method whereby the physical and chemical properties of carrier systems increase the target/nontarget ratio of the quantity of drug delivered by adjusting these properties to the physiological and the histological characteristics of the target and nontarget tissues, organs, and cells. The EPR effect, initially described by Maeda to account for increased deposition of macromolecular drug carriers in tumors, also applies to liposomes and other NV. The mechanism underlying the EPR effect is related to the increased vascular permeability of tumor vessels characteristic of the tumor neoan-giogenic process. The vascular permeability of tumor tissues is enhanced by the actions of secreted factors resulting in loss of junction integrity between endothelial cells and loss of cell–cell and cell–matrix interactions.180c It is also heterogeneous with respect to tumor type and tumor microenvironment. As a result of this increased vascular permeability, macromolecules (and NV) selectively increase their transport from blood vessels to tumor tissues. In addition, the lack of functional lymphatic drainage prevents the outflow of the extravasated NV. Therefore, NVs are selectively retained for a prolonged time in the tumor interstitium and are not taken up by cells and internalized. These NVs will gradually release the entrapped drug in the vicinity of tumor cells (by-stander effect). Data obtained with lipid vesicles of different mean size in an experimental tumor model suggest that the threshold vesicle size above which major hindrance to extravasation occurs is around 400 nm.181 The occurrence of EPR in tumors coupled with the low permeability of most normal tissues to NV results in a high tumor/normal tissue ratio for liposomal drug concentration. It should be emphasized that in this scenario, the drug enters the tumor cells as free drug. The rate-limiting step of drug bioavailability is the release from liposomes or any other NV. Employing fluorescently labeled liposomes in the mouse skin-fold tumor chamber model, most liposomes appear to accumulate in the immediate perivascular area with little or no penetration into the tumor cell layers.182 Excellent examples of passive accumulation of NV are that of specific liposomal accumulation in tumor sites,183–185 particularly for highly vascularized tumors. In most cases, liposomes localize in the tumor interstitium and are not internalized up by tumor cells56a,186,187a no matter whether they are sterically stabilized (PEG) or not. For certain liposome chemistry (coformulated with N-octanoyl-glucosylceramide, C8-GlcCer-liposomes) some lipid-raft uptake is noted, beside standard passive targeting.185 This is in contrast to an interaction with the RES system, whereas liposomes are readily endocytosed and internalized by macrophages.
-
(3)
Indirect targeting (to tumors) involves generating a specific antitumor response by targeting the cellular or humoral arm of the immune system of cancer cells.187b Anticancer immunotherapy to innate immunity has been demonstrated by means of delivering an activating stimulus of the N-acetyl-glucosamine motif, bound to PAMAM dendrimer polymers.188 Delivering GM-CSF gene (or the cytokine itself) to express this cytokine in the inflammatory milieu of cancer environment provides a possibility of switch from Th-2 to Th-1 type of response, facilitating proper anticancer effects.187b This type of targeting is extremely useful for accessing secondary sites of tumors (metastases) via specific antibodies or specialized immune cells.
Targeting and Bio-Barriers
No level of targeting sophistication will produce substantial benefits in therapeutic index, unless the agents of the therapeutic actions can reach the intended lesions sites. Intrinsic to the body defense systems are several extremely effective obstacles (collectively termed “biobarriers”) that largely prevent injected chemicals, biomolecules, NV, and any other foreign agents of therapeutic action from reaching their intended destinations.145
-
(1)
Endothelial and epithelial barriers. The blood–brain barrier (BBB) possesses a formidable obstacle to penetration by therapeutic agents. A good example of nanotechnologies for overcoming epithelial barriers involves the colocalized delivery of a therapeutic biomolecule with a penetration enhancer, such as a zonula occlu-dens toxin (ZOT). This acts to open intracellular tight junctions in a short-term, reversible and localized fashion, thus affording transport of the therapeutic biomolecule into the vascular compartment.189
-
(2)
Sequestration by the reticulo-endothelial system (RES). The circulatory clearance half-time of NV has been upgraded by the attachment of polyethylene glycol (PEG) to their surfaces. PEG provides a shielding “stealth” effect, delaying recognition and sequestration by the resident macrophages of the RES. Unfortunately, this “immunos-tealthing” function is often concurrent with the annihilation of biomolecular targeting capabilities because the PEG molecules hide localizing antibodies conjugated on the liposomal surfaces. An attachment to the terminal PEG groups on the NV periphery represents one way to present the targeting molecules.37 Avoiding or minimizing the RES localization represents a major hurdle for drug delivery.
A word of caution of PEGylation of NV: recently, several pieces of evidence exist on the fate of PEGylated liposomal drug formulation administered in multiple injections for effective clinical applications. An accelerated blood clearance (ABC) of liposomes has been noted and investigated by several investigators, notable by Ishida et al.190,191 The authors reported on immune reaction in the spleen against the Dox-entrapped-PEGylated liposomes occurring during 2–3 days following the first administration, resulting in abundant IgM production. This phenomenon can be partially abrogated by a high dose of liposomes as the complement activation was observed to be inversely dependent of the lipid dose. Laverman et al.131 proposed that hepatos-plenic macrophages are responsible for induction of the high clearance rate phenomenon. Others (e.g., in Reference 192) reported on Dox-entrapped PEG-liposomes as having a capability to exert a toxic effect on liver macrophages (and spleen) indicating that such liposomes may be relatively safe for clinical use even if repeated injections are required. Spleens are transiently depleted of macrophages and may not cause an excessive induction of the ABC effect. Opposite to reports on rapid liposomal clearance, Park et al.37 did not observe different clearance rates for repeatedly injected anti-HER2-ligand-targeted, long-circulating (PEG) immunoliposomes versus a single-dose. Concluding, these considerations may pertain to all NV which employ PEGylated technology and should be more extensively studied.
-
(3)
Adverse osmotic pressure. As cancer lesions grow, they develop an increased internal (interstitial) hydrostatic pressure that counters convective extravasation from the vascular compartment into the tumor. This leaves diffusion as the only mechanism of transport of therapeutic agents into the tumor—a very unlikely route for large molecules or nanovectors.145
Intracellular Targeting
Nanovectors could be used as efficient delivery vehicles for intracellular targeting. Types of intracellular targeting include (a) endo-lysosomal; (b) cytoplasmic; (c) nuclear; (d) mitochondrial. An important feature of NV is that physical properties such as size, surface charge, hydrophobicity, and release characteristics can be easily varied by altering the composition of the formulation and/or the formulation method.
Lysosomal targeting, occurring in many forms of endocytic uptake has shown an application in therapy of lysosomal storage diseases (e.g., Tay-Sachs, Gaicher's disease, Lesh-Nyhan syndrome, or adenosine deaminase insufficiency), including exogenous delivery of enzymes (or genes) such as glucocerebrosidase, fucosidases, phenylalanine ammonia lyases, and others.193
The ability of NV to escape the endo-lysosomes depends on the surface charge of the nanoparticles.84 Nanoparticles which show transition in their surface charge from anionic (at pH 7) to cationic in the acidic endosomal pH (pH 4–5) were found to escape the endosomal compartment, whereas the nanoparticles which remain negatively charged at pH 4 were retained mostly in the endosomal compartment.86 Thus, by varying the surface charge, one could potentially direct the nanoparticles either to lysosomes or to cytoplasm (Fig. 5). CPPs (see Section “Types of Payload”) have been successfully used for the intracellular delivery of drugs by means of nanoparticles, micelles, and liposomes.
A special targeting to subsurface structures is that of targeting to cytoskeleton and caveolae. Many diseases have now been associated with abnormalities in cytoskeleton and nucleoskeletal proteins, including several blistering skin diseases.194 Very little has been proposed for such targeting. Targeting drugs to caveolae structures (as subcompartment of the plasma membrane) could potentially interfere with diseases such as muscular dystrophy, cancer, type II diabetes, atherosclerosis, inflammation, and Alzheimer's disease.195
It seems possible to localize NV to mitochondria by modifying the vehicle surface to obtain a net positive charge.196 Delivery to mitochondria has been reviewed.197,198 The membrane potential of mitochondria in vitro is in between 180 mV and 200 mV, which is the maximum a lipid bilayer can sustain while maintaining its integrity. Therefore, permanently positive motifs with delocalized cationic charge, like oligoguanidines, can target molecules and NV to mitochondria in response to the highly negative membrane potential.199 Positively charged NV or molecules are attracted by mitochondria in response to the highly negative membrane potential. The correction of a mitochondrial deficiency, for example, of neuromuscular disease or myopathy, has been demonstrated by this mechanism.200
Organelle-specific localization signals such as fusogenic endosomal membrane peptide or nuclear localization signal (NLS) can be attached to the NV surface, enabling particles to target the nucleus. Simoes et al.201 reported that liposomes associated with GALA fusogenic peptide (a mimic of viral fusion proteins, 30-residue synthetic amphipathic peptide) enhanced gene delivery to cytoplasm. Tachibana et al.202 attached NSL peptide from the SV40 large T-antigen to BSA labeled with a fluorescence marker and observed the nuclear trafficking of the NLS-BSA by confocal microscopy. BSA without the NSL peptide did not transport to nucleus.
Current studies on successful intracellular delivery and tumor targeting of small selective peptides and peptide-conjugated molecules have shown promise. A peptide should be both specific to target tissues (binding to cell surface receptors overexpressed in tumors) and it could have antineoplastic properties.203 Natural peptides have low bioavailability and short half-life in the mammalian circulation system, while synthetic peptides have potential cytotoxicities.204
Tissue Targeting
Targeted delivery of therapeutic agents to specific tissues has been made feasible due to a number of developments such as monoclonal antibodies, discovery of specific receptors that are either over-expressed or expressed only in specific tissues, and development of conjugation techniques to attach antibodies or ligands to drug delivery systems. Targeted delivery results in higher bioavailability of the therapeutic agent at its site of action and at the same time results in reduced side-effects. Several methodologies have been developed to enable tumor-specific drug delivery. An alternative approach to targeted delivery is to directly deliver the therapeutic agent locally to a peripheral or the disease tissues.205,206 This is a typical route for immunization, inhalation (pulmonary), ocular, diabetes (insulin—pulmonary), intratumoral, and skin applications. Macrophages (as well as monocytes and dendritic cells) as a target represent a special and valid pharmaceutical target, particularly for the development of new generation of vaccines and adjuvants. This target has been explored early at the NV development as their uptake is more pronounced and easier for larger sizes (including microparticles). As such, nanovehicular vaccine development is not very much covered in this review.
Targeting in Cancer Treatment
We now briefly review the importance of the active and passive strategies in cancer treatment. Tumor targeting with NV may use passive or active strategies. Passive targeting occurs as a result of extravasation of the NVs at the diseased site (tumor) where the microvasculature is hyperpermeable and leaky, a process aided by tumor-limited lymphatic drainage. Combined, these factors lead to the selective accumulation of NVs in tumor tissue (EPR). However, passive targeting would be limited to tumors in MPS organs (liver, spleen, and bone marrow). Addressing other tumor tissues does not seem feasible without active targeting strategies because of the short circulation times involved and the low concentration of NPs that is achieved in the tumor area (despite the EPR effect), leading to drug concentrations below the therapeutic level. Active targeting is based on the over or exclusive expression of different epitopes or receptors in tumor cells, and on specific physical characteristics. Thus, vectors sensitive to physical stimuli (e.g., temperature, pH, electric charge, light, sound, magnetism) have been developed and conjugated to drugs. Alternatively, active targeting may be based on overexpressed species such as low molecular weight ligands (folic acid, thiamine, monosacharides), peptides (RGD, LHRD), proteins (transferrin, antibodies, lectins), polysaccharides (hyaluronic acid), polyunsaturated fatty acids, peptides, or DNA.207–210
Targeting to Tumor Vascular Compartment
Targeting to tumor vascular compartment provides an improved cancer therapy. Tumor vasculature, blood flow, and lymphatic drainage as contrasted to normal vasculature are reviewed in Jain211,212 and Jang et al.213 Advantages of targeting tumor vasculature are numerous:214,215
-
(a)
Better selectivity of the treatment against proliferative tumor-derived endothelial cells and minimal toxicity because angiogenesis in the adult is limited to wound healing, ovulation, and pregnancy.
-
(b)
Easy access for drugs from the blood to tumor vascular endothelial cells (compared with drugs that have to penetrate large bulky tumor masses).
-
(c)
Low mutation rate (high genetic stability of endothelial and stromal cells) within the vasculature and thus treatment is independent of tumor-cell resistance mechanisms.
-
(d)
Broad applicability (both solid tumors, and leukemia are dependent on angiogenesis for their survival) and high efficacy because each tumor capillary supplies hundreds of tumor cells.
However, there are some concerns on the genetic stability and development of resistance. Some experimental and clinical data associated with anti-angiogenic drugs has led to the recent identification of various forms of intrinsic and acquired resistance. To overcome these different modes of resistance, broad-spectrum antiangiogenic modalities should be used or, alternatively, a combination of antiangiogenic agents or a combination of an antiangiogenic agent with drugs that decrease the survival threshold of the tumor vascular compartment could be considered (e.g., doxorubicin). Besides, an administration strategy of a chemotherapeutic agent could be modified. An optimized administration via increasing the frequency of administration and decreasing the doses of the chemotherapeutic drug is possible. This strategy has been termed as antiangiogenic or metronomic chemotherapy.216
A special comment should be given to the importance of embolization (chemoembolization) in cancer treatment. Although mostly achieved with microparticles, NV may reach very small blood vessels and cause their necrotic death due to a cut-off of the nutrient supply.217,218
Targeting Issues and Examples
The development of nanovehicular delivery systems for targeted delivery has been reviewed recently by Moghimi et al.80 Active targeting of a therapeutic agent or a carrier is achieved by conjugating the therapeutic agent or the carrier to a tissue- or cell-specific ligand.219,220 Passive targeting is achieved by coupling the therapeutic agent to a macromolecule or carrier that passively reaches the target organ.221 NV can be passively targeted to the RES system and circulating macrophages. Similarly, antitumor drugs coupled to macromolecules such as high molecular weight polymers passively target the tumor tissue through the EPR effect.222 Accumulated macromolecules and lipids in tumors are, unfortunately, not readily removed by the lymphatic system, as they are in inflammated tissues; this EPR effect provides an opportunity for tumor-selective treatment. According to Senger's findings,223 polymeric drugs accumulate in tumor tissue at 5–10 times greater concentrations in the 24 h after intravenous injection. Ideally, the nanovector size should not exceed 300 nm for optimum transport to tumor sites.224 Such sizes above a critical 300 nm are also most vulnerable to macrophage phagocytosis.
Because the number of receptors on the cell's periphery is limited, their binding and the related drug uptake (internalization) can usually be saturated within therapeutic concentrations, at relatively low molar ratios between the ligand and receptor. This fact represents a major source of nonlinear pharmacokinetic behavior, especially for peptides and protein drugs, resulting in a lack of dose proportionality.225 It should be also mentioned that the occupancy of peptide receptors occurs at an early period of intravenous drug delivery, followed by a re-distribution (dissociation) of drug onto nonspecific targets.226 In this respect, the pretargeting could affect the uptake and retention of the delivery vehicle at the disease site. Although coined for the optimal retention of targeted radiolabel for tissue imaging,227a the two initial steps of this procedure are applicable to therapeutic applications. In cancer therapy scenario, typically, targeted therapeutic vehicle is injected and allowed to distribute within the body to bind to its appropriate surface receptors of the tumor vasculature/tissue and to clear substantially from nontumor tissues and circulation. The optimization of dosing of such initial bolus is often required. Second, a clearing step (chasing) is often applied to enhance the tumor/background tissue uptake ratio (T/B), usually many folds. An antibody that reacts against the targeting agent could be applied, or agents reactive with the targeted receptor (mild concentrations of the targeted free ligand) in order to compete with the specific binding at the equilibrium state. The retention of the targeted therapeutic vehicle is due to internalization of or strong affinity to the receptor molecule. (Third, at the radioimaging/radioimmunoimaging, only then the bolus (or a continuous infusion) of the radioligand is injected to maximize its contrast; e.g., in References 227,228). Recently, two-step immunotargeting has been applied for PLA nanoparticle delivery by means of avidinbiotin technology.229 Obviously, any suitable high-affinity receptor–ligand pair could be used to enable the second step of this two-step targeted delivery technology.
Below we list several successful examples of targeting accompanied by details on mechanism. With regulatory approvals of drugs such as Herceptin (trastuzumab) for the treatment of metastatic breast cancer and Gleevec (imatinib mesylate) for the treatment of chronic myelogenous leukemia, a new class of therapeutics, based on selective drug targeting to pathologic components, has been clinically validated. Within this growing class of targeted pharmaceuticals, folate–drug conjugates constitute a well-studied example of a distinct subclass of receptor-targeted therapeutics. Folic acid selectively binds to and delivers attached drugs into any cell that expresses a cell surface folate receptor (FR). Because FR is expressed and accessible primarily on pathologic cells, folate conjugation allows delivery of nonspecific drugs selectively into pathologic cells. As a consequence, normal tissues lacking FR are spared the toxicity that commonly limits nontargeted therapies.230 In this case, quite intimate knowledge of the mediated mechanism is available. Receptor-mediated uptake of folate–drug conjugates proceeds through a series of distinct steps, beginning with conjugate binding to a cell surface FR and culminating with release of at least part of the therapeutic cargo into the cytoplasm. After membrane invagination and internalization to form an endocytic vesicle, acidification of the endosomal compartment to pH ~5 results in release of some (but not all) folate conjugates from their receptor.231 Trafficking of the acidic endosome to a recycling center then allows separation of membrane-bound FR from released conjugates/free drug. Released folate conjugates are seen to escape the endosome through an unknown mechanism, resulting in drug deposition in the cytoplasm. In contrast, membrane bound FR largely recycle back to the cell surface, allowing for delivery of additional folate-linked drugs into the cell. In cancer FR-positive cells, the recycling rate may be quite fast. Importantly, it has been observed that relatively few folate conjugates enter lysosomes for destruction232 enabling the delivery of hydrolytically sensitive materials such as drugs, genes, and ribozymes into cells.46
Polymer–peptide conjugates have recently been shown to target, very successfully, to tumor angiogenesis sites. Line et al.233 have conjugated doubly cyclized RGD motif KACDCRGDCFCG (RGD4C) to water-soluble N-(2-hydroxypropyl)-methacrylamide (HPMA) to yield 30.2 kDa molecular weight conjugate, and found its enhanced relative localization at sites of angiogenesis, that is the ratio of tumor uptake to normal tissue background (T/B) in two tumor animal models (DU145 and PC-3) of about 18 at 24 h scintigraphic imaging while the nonspecific localization still persisted. The same group234 employed 43.3 kDa conjugate to deliver a 99mTc and 90Y chelation radiotherapy with help of the same targeting peptide and found an effective tumor volume reduction in DU145 in SCID mice.
Targeting with help of transferrin represents another example. Iron-transporting protein (Trf) is upregulated in tumor cells. Plasmid-encoding tumor necrosis factor alpha was systemically delivered (with help of Trf-polycation/NV system) to fibroblastoma model, leading to delayed tumor growth and cure rate 60%.60,235 The authors reported that gene transfer after intratumoral application was 10–100-fold more efficient with Trf-PEI/DNA as compared to naked DNA. DNA/lipid-based liposomes, coated with anti-TrfR single-chain antibody fragment and PEG lead to an efficient delivery to tumors, inhibiting the first pass clearance observed with nonPEG containing liposomes.236 Other targeting strategies to tumor tissues are reviewed in Reference 237.
A note of caution: while considering endothelium as a target, we should differentiate between the endothelium as a portal for delivery versus destruction of tumor vasculature. While attempting the latter, one should point out that avascularity of tumor (or of its regions) often translates into limited drug delivery to tumors238 as poorly vascularized portions of tumor will accumulate fewer drugs. The widely held view is that these antiangiogenic therapies should destroy the tumor vasculature, thereby depriving the tumor of oxygen and nutrients. Jain239 has put forward emerging evidence supporting an alternative hypothesis that certain antiangiogenic agents can also transiently “normalize” the abnormal structure and function of tumor vasculature to make it more efficient for oxygen and drug delivery. Drugs that induce vascular normalization can alleviate hypoxia and increase the efficacy of conventional therapies if both are carefully scheduled.
DRUG LOADING
Nanovector cores serve as a nano-reservoir for loading, releasing, and delivering of drugs. The small internal cavity or extravehicular surface does not allow a high drug loading. Some nanovectors due to their small size (e.g., dendrimer-based) have a poor capacity to incorporate active compounds. Thus, the application is limited to very potent drugs (biological modifiers), such as peptides, oligonucleotides, antibody-fragments, cytokines, etc. The capacity of a NV is primarily determined by the chemistry of the vehicle, both of the core or surface. Polymeric vehicles are promising drug carriers, of which the critical parameters such as the size, drug loading, and release can be controlled by engineering the constituent polymers. As a rule, the core chemistry controls the drug loading (and release). Hydrophilic core is amenable to charged drugs and noncharged entities. Unfortunately, no systematic research is available to assess the drug loading as related to polymeric carrier chemistry.
TYPES OF PAYLOAD
A number of pharmacological agents can be delivered directly to cells because they can readily cross the plasma membrane. Many weekly charged compounds will accumulate in cellular organelles. From the delivery perspective, they are considered a challenge because of their small molecular weight. As such, in most cases, can be delivered when attached to a larger carrier (e.g., see polymeric drug in the Subsection “Functionalized Polymer–Drug Conjugates” of the Section “Advanced Drug Delivery (ADD) Systems”), and subsequently entrapped in a particulate vehicle141,142 Macromolecular entities cannot diffuse through the cellular membrane but can be internalized inside the cells through endocytosis. For most part, this chapter considers only drugs which enter the cells via endocytosis or related uptake.
Typical payload-agents include: polymeric drug conjugates, macromolecules: proteins, peptides, gene vectors, therapeutic oligonucleotides: anti-sense, ribozymes, DNAzymes, aptamers, small interfering RNAs—siRNAs and imaging agents for diagnostic, radiotherapeutic, and imaging purposes.
Therapeutic Agents
Standard therapeutic agents include those of injectables, oral, transdermal, and other topical delivery technologies. Specifically, proteins (cytokines, lymphokines, growth factors, immunotherapy products—antibody drugs, and pegylated proteins), hormones (peptides—e.g., insulin; nonpetide hormones), small molecule drugs (both nonprotein and protein drugs—antineoplastic and immunomodulating agents), anti-infectives (e.g., antibiotics), RNA/DNA agents (see below) and vaccines have been attempted to be delivered via nanovehicular approaches (listing just few in References 239–248). Successful applications are listed with the different nanovehicular classes (see Section“Advanced Drug Delivery (ADD) Systems” below).
A novel class of payload is represented by therapeutic oligonucleotides. Selective silencing of genes was initially accomplished with short antisense oligonucleotides, pieces of DNA which hybridize with the target mRNA and inhibit their translation into the protein. A clinical trial is ongoing targeting certain genes involved in oncogenesis.249 Specifically, in this case, oligonucleotides, sensitive to enzymatic breakdown, were replaced by derivatives with the phosphorothioate backbone in place of original phosphoester backbone. Ribozymes are naturally occurring enzymes consisting of RNA, resulting in cleaving of RNA. A downregulation of oncogenes resulting in tumor suppression has been reported.250 Deoxyribozymes or DNAzymes (enzymes made of DNA) are artificial molecules and are not found in nature. Silverman251 has isolated a multitude of DNAzymes that catalyze RNA ligation. RNA-cleaving DNAzymes, DNAzymes able to catalyze RNA hydrolysis in the absence of divalent metal ions have been discovered. Aptamers are synthetic RNA molecules capable of capturing ligands with a high-binding selectivities and affinities for a specific target. They have proven to be valuable therapeutic agents with enhanced properties relative to antibodies.252,253 Short interfering RNAs serve for a specific knockdown of genes. Sumimoto et al.254 demonstrated an in vivo melanoma inhibition with lentiviral vector encoding for iRNA directed against the mutated oncogene BRAF. While plasmid DNA needs to enter nucleus, siRNA and other carriers mentioned above need only to reach cytoplasm since they act by mRNA degradation.
Molecular Diagnostics, Radiotherapeutics, and Imaging Drugs and Agents
These include quantum dots (QDs) and contrast agents, imaging and radionuclide agents (e.g., gadolinium contrast agent, Gd). Low-molecular weight gadolinium chelates are clinically used in MRI since they rapidly diffuse into the tissue and interstial space. Long-circulating agents do not extravasate as easily and have longer circulation times. Gd-PEG-liposomes (e.g., In Reference 255) or Gd-PEG NV exhibit longer circulation properties and can be further coupled with targeting ligands such as Ab to selectively target contrast agent to tumors, angiogenesis, atherosclerotic plaque, etc. Gd-QDs are currently considered as special delivery vehicles and as such are treated below. Recent innovations in imaging have allowed to employ intense and photostable quantum dot-based tumor imaging, enabling multi-color detection of cell receptors with a single optical excitation source. Advantages of NV-based imaging and targeting by use of gold nanoshells and QDs are presently being explored. Nanoshells are promising new candidates for global tumor treatment therapy which combine optimum size for traversing endothelium to reach tumor sites, with externally triggered release by IR-induced thermal ablation.
Recent advances have improved detection in vivo by Semiconductor QDs are bright, photostable CdSe/ZnS fluorescent nanocrystal and as such are promising nanoscale visualization tools for biological applications, due to their facile surface chemistries, which promote biocompatibility and heteroconjugation, and their long fluorescence lifetimes with broadband absorption and narrow emission. Narrow bandwidth emission for multiple color imaging, broad absorption spectrum for single excitation sources, and superior photostability, demonstrate key advantages over conventional dyes. As QDs exhibit tunable emission properties for a wide range of color possibilities, they seem to be ready to be implemented in diagnosis and medical imaging. The unique properties of QD have been applied toward multiplexed imaging of cellular cancer targets,256in vivo multiphoton fluorescence for deep tissue visualization,257 and FRET-based sensing.258 Kim et al.259 recently used near-infrared-emitting oligomeric phosphine-coated QD for sentinel lymph node (SLN) mapping at 1 cm tissue depth. It has been also shown that QDs can be rendered noncytotoxic by suitable polymeric coatings260. QDs are also brighter than most conventional fluorescent dyes by ~10-fold and have been significantly easier to detect than green fluorescent protein (GFP) among background autofluorescence in vivo. Furthermore, QDs are far less susceptible to photobleaching, fluorescing more than 20 times longer than conventional fluorescent dyes.
The emergence of molecular imaging,261–263 as a result of unprecedented advances in molecular and cell biology and the availability of a cohort of molecular probes that are highly target specific, as well as the successful development of small-animal imaging instrumentation, allows noninvasive visualization of molecular events within living subjects. On the molecular side, tremendous development occurred in the area of targeted bioluminescence probes, fluorescent proteins, and exogenous fluorescent probes. NIR fluorescence imaging probes that emit at longer wavelength, with considerably lower autofluorescence, lower tissue scattering, and more photon penetration into living tissue is one example.264 It is expected that an NIR dye with longer excitation and emission wavelengths and an appropriate filter set will reduce the interference from the background and, thus, improve the tumor contrast and attain deeper penetration for visualization of deep-lying tumors and organs. The use of an optical-coupling medium and/or tomographic imaging265 may also be helpful to minimize the effects of skin scattering. The very sensitive, cryogenically cooled CCD camera allows for acquiring a fluorescence image superimposed on a gray-scale photographic image of the small animal with overlay and imaging analysis software. Dynamic imaging has also been introduced.226 According to Hirsch et al.,266a near-infrared-absorbing 65-nm gold nanoshells were capable of heating solid tumors to a temperature sufficient to cause irreversible tissue damage at 37.4°C at up to only 1 mm depth of skin. Recently, imaging and quantification of fluorochrome reporters in deep structures (>1 mm) has resulted in considerable improvements. Ntziachristos et al.265 described an in vivo implementation of fluorescence molecular tomography (FMT), a novel, volumetric imaging technique that accounts for the diffusive propagation of photons in tissue. The technique is capable of resolving and quantifying pico- to femtomole quantities of fluorochromes in whole animals and can achieve several centimeters of tissue penetration. They demonstrated the molecular specificity and three-dimensional (3D) imaging capacity in small animals. Recent measurements and modeling266b,267 show that NIR fluorescent signals from tumorlike structures can propagate for more than 12 cm in breast or lung tissue and more than 5 cm in the adult brain.
Several small molecular weight compounds have been recently developed for imaging at tumor targeting. Citrin et al.269 employed endostatin-Cy5.5 probe for in vivo LLC tumor imaging in mice in NIR regime. As endostatin (20-kDa C-terminal fragment of collagen XVIII) localizes to PECAM-1 endothelial surface marker, upregulated in tumor tissues, a high NIR signal (not quantified) was noted at 3 h following the i.v. injection and persisting to 72 h. The same compound will also localize to tumors from distant i.p. sites. Cheng et al.269 employed tetrametic-RGD-Cy5.5 as a contrast agent in vivo in s.c. U87MG glioblastoma xenografts and obtained 3–3.6 T/B ratio with NIR imaging at 0.5–4 h. Ye et al.270 used terameric-RGD-cypate to monitor ABIR-positive tumor cells A549 in vivo and observed nonexistent blood NIR fluorescence at 24 h postinjection (r.o.) while a high tumor affinity (and kidneys and liver) still persisted. These successes induced a quest for nanovehicular formulations (below).
Kelly et al.271 and Montet et al.272 developed nanosized imaging agents for imaging of vascular targets. Kelly used 30 nm magnetofluorescent nanoparticles endowed with VINP28, phage-display-selected peptide for targeting to VCAM-1 endothelial vascular adhesion molecule and obtained 54-fold T/B ratios in atherosclerotic lesions in mice. Montet employed the same vehicle (but of 10–100 nm size) decorated with linear RGD-Cy5.5 peptide for targeting to integrins in BT-20 tumors but observed a 6.5 increase in the T/B ratio. A general observation for above imaging studies is that T/B ratios always show a continuous and significant increase for all the organs analyzed over time, indicating prolonged retention in the tumor and clearance from background tissues. These results were concurrently translated into the therapeutic area with dendimers and liposomes.46,47
Gadolinium contrast agents have been used for some time. Good example of recent development is a series of dendrimer-based macromolecular MRI agents which have a spherical shape and possess similar surface charges.262,273,274 Changes in molecular size alter the route of excretion and pharmacokinetics. Smaller sized contrast agents, of less than 60 kD molecular weight, are excreted through the kidney resulting in these agents being potentially suitable as functional renal contrast agents. Less hydrophilic and larger sized contrast agents were found better suited for use as blood pool (systemic) contrast agents. Hydrophobic variants of polypropylenimine diaminobutane dendrimer cores quickly accumulate in the liver and can function as liver contrast agents. Larger hydrophilic agents are also useful for lymphatic imaging. Finally, contrast agents conjugated with either monoclonal antibodies or with avidin are able to function as tumor-specific contrast agents and might also be employed as therapeutic drugs for either gadolinium neutron capture therapy or in conjunction with radioimmunotherapy. Thus, a special care must be paid to the selection of agents and their macromolecular properties.
Cowpea mosaic virus (CPMV) particles of 25–31 nm size can be fluorescently labeled to high densities with no measurable quenching, allowing a high-resolution intravital imaging of vascular endothelium for period of 72 h to visualize vasculature to a depth of up to 500 μm, finding of significance for tumor vasculature.275
It should be mentioned that successful imaging often requires an internalization of imaging agent at the process of targeted delivery in order to achieve a high therapeutic/imaging ratio, for example, T/B for tumors. There is an ongoing discussion on the importance of imaging agent internalization; it is certainly justified for imaging of intracellular targets.276,277
Much of imaging research today is aimed at the development of biomarkers in order to derive information on specific biological processes (even molecules) or responses to treatment. In cancer, imaging-based biomarkers of tumor vascular properties may be used to predict early stages of a disease progression, via the molecular imaging tools.278,279 Imaging of the status of tumor angiogenesis, gene expression, receptor imaging, cell death and apoptosis, drug resistance, and clinical drug development are areas which are receiving a great deal of attention.280 Several NV-based imaging agents are on market. Endorem® (Guerbet LLC, Bloomington, IN) uses supermagnetic iron oxide nanoparticles to facilitate MRI imaging.
ADVANCED DRUG DELIVERY (ADD) SYSTEMS
The ideal nanovehicular vector would achieve long circulation time, low immunogenicity, good biocompatibility, selective targeting, efficient penetration of physiological barriers such as vascular endothelium and the blood–brain barrier, external activation or self-regulating drug release, and have no clinical side-effects. Universal tools such as PEGylation and antibody decoration become requisite features to improve circulation time and enable selective targeting. Metal-cored dendrimers may provide simultaneous contrast imaging and drug release, nano-shells enable NIR imaging and ablation therapy, and QD-virus hybrids provide fluorescence-based imaging and oncolytic activity.261 Only liposome-based delivery has made its clinical debut. A NV based on paclitaxel crystals embedded into an albumin matrix (Abraxane, ABI-007) represents another successful application.281 ABI-007 is an albumin-bound, 130-nm particle formulation of paclitaxel, which is devoid of any solvents; its lyophilized formulation comprises of albumin and paclitaxel, reconstituted in 0.9% NaCl, and forms a colloidal suspension which internalizes tumor cells via the specific receptor gp60 and activation of caveolin-1.282 The human serum albumin (HSA)-stabilized paclitaxel particles have an average particle size of 130 nm. This very first “nano” medicine on the market is also internalized by cells and tissues (via endocytosis). Other possible intracellular targets are schematically depicted in Figure 8.
As there are many types and approaches, with many modifications and modalities, we will be only able to list some few examples of concrete chemistry. Particularly, we may discern some surface modifications which enable to engineer a stealth character or to provide a targeting capability. The following ADD types of vehicles will be discussed below: nanosuspensions, nanoparticles, micelles, multi-layered polyelectrolyte films, dendrimers, liposomes, funtionalized polymers–drug conjugates, cell penetrating peptides (CPP), and special systems: QDs, nanotubes, calcium phosphate, and silica NPs; as well as gene delivery vehicles. Typical size limits of different vehicle types are depicted in Figure 10.
Figure 10.
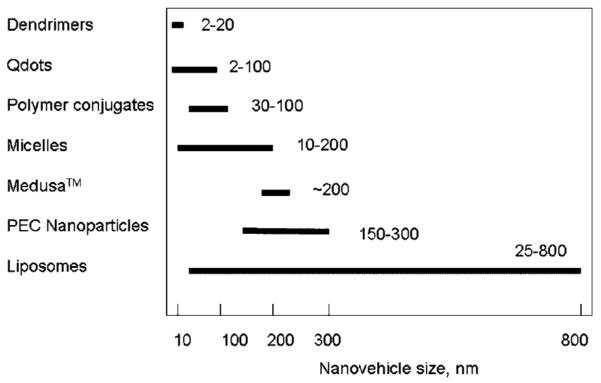
Typical size range of representative examples of nanovehicles. Note a lower limit on liposomes (vehicle stability), nanoparticles (condensation limit) and upper limit on dendrimers, Qdots, and polymer conjugates (due to their chemistry/structure). Larger sizes may not be optimal for accessing/delivery of certain cells and tissues.
Nanosuspensions
Nanosuspensions is a commercial technology for generating solid crystal-size particle of insoluble drugs by a continuous, large scale, micronization (wet milling, high-pressure homogenization, emulsification, precipitation from an antisolvent, rapid expansion from a liquefied gas-solution, freeze drying, and evaporative precipitation) from larger insoluble aggregates.283 Similarly to other NV, nanosuspensions undergo rapid cellular uptake.284 The chemistry of such vehicles does not provide easy possibility for functionalization or further modification.
NanoCrystal® Technology (Elan, Dublin, Ireland). For poorly water-soluble compounds, Elan's proprietary NanoCrystal technology can enable formulation and improve compound activity and final product characteristics. The NanoCrystal technology can be incorporated into all dosage forms both parenteral and oral, including solid, liquid, fast-melt, pulsed release, and controlled release dosage forms. Poor water solubility correlates with slow dissolution rate, and decreasing particle size increases the surface area, which leads to an increase in dissolution rate. NanoCrystal particles are small particles of drug substance, typically less than 1000 nanometers (nm) in diameter, which are produced by milling the drug substance using a proprietary, wet-milling technique. The particles of the drug are stabilized against agglomeration by surface adsorption of selected GRAS (FDA approved) stabilizers. The result is an aqueous dispersion of the drug substance that behaves like a solution—a NanoCrystal colloidal dispersion, which can be processed into finished dosage forms for all routes of administration. Since 2000, several products have been approved for use on the US market. Products on the market are Rapamune® (Wyeth, Madison, NJ) and Emend® (Merck, Whitehouse Station, NJ) based on the pearlmilling technology licensed by Elan Nanosystems. Baxter Healthcare Corporation (Round Lake, IL) has developed a similar technology (NanoEdge).
Nanoparticles
-
(1)
PLGA nanoparticles: Aliphatic poly (α-hydoxy ester)s, such as poly(l-lactic acid) [PLA], poly(glycolic acid) [PGA], and their copolymers [PLGA], belong to the most widely utilized class of polymers approved for human clinical use. They were among the first synthetic biodegradable polymers to be used clinically285 and show compatibility with living tissue. These polymers are commercially available with different molecular weights and copolymer compositions. The degradation time of these polymers can span from several months to several years, depending on the molecular weight and copolymer ratio.286,287 The first PLA nanoparticles were reported by Bazile et al.288 and the first PLGA nanoparticles by Venier-Julienne and Benoit.289 A recent review on PLGA nanoparticles290 covers their potential use as carriers for several classes of drugs such as anticancer agents, antihypertensive agents, immunomodulators, and hormones; and macromolecules such as nucleic acids, proteins, peptides, and antibodies. The various methods used for preparation of nanoparticles with their advantages and limitations have been discussed. The recognized problems include nanoparticle stability after preparation; a freeze-drying using different classes of cryoprotectants has been developed. PLGA nanoparticles were also designed for the site-specific delivery of drugs (targeting) and evasion of RES system.
-
(2)
PACA nanoparticles: Polyalkylcyanoacrylate nanoparticles seem to be an interesting drug carrier owing to their size, structure, degradability, and drug sorptive properties. They were first prepared by Couvreur et al.291 Nanospheres preparation methods based on the polymerization of monomers generally consist in introducing a monomer into an aqueous phase or in dissolving the monomer in a nonsolvent of the polymer. The polymerization reaction in these systems generally occurs in two steps: a nucleation phase followed by a growth phase. Poly(alkylcyanoacrylates) have been used for several years as surgical glues and are bioerodible292 which is the most significant advantage of alkylcyanoacrylates over other acrylic derivatives previously used. In contrast to other acrylic derivatives, requiring an energy input for the polymerization, alkylcyanoacrylates can be polymerized easily without such a contribution which is another advantage regarding the stability of the associated drug. These nanospheres are prepared by emulsion polymerization of cyanoacrylic monomers dispersed in an acidic aqueous phase. The size of the nanospheres obtained is approximately 200 nm, but it can be reduced to 30–40 nm using a nonionic surfactant in the polymerization medium. A drug can be incorporated by adsorption or is added to the reaction mixture before the polymerization. An extensive review of the developments and applications of PACAs as nanoparticles for the delivery of drugs is then given. Many different polymeric chemistries and modifications of this technology, including PEGylation, exist. The degradation, in vivo distribution, toxicity, and cytotoxicity of the nanoparticles are also included, as well as different therapeutic applications.293 Recent work demonstrated a low toxicity and cytotoxicity of PACA nanoparticles.294 A phase I clinical trial has been conducted in 2002.295a Past and ongoing clinical trials are summarized on a website (Anonymous296) from the company conducting these trials (BioAlliance Pharma, Paris, France). The trade name of the nanoparticles is Transdrug.
-
(3)
Polyelectrolyte complex nanoparticles (PEC): Nanoparticles can be made by a nonstoichiometric mixing of polyelectrolytes of opposite charge (by ionotropic gelation) by a spontaneous association. Only several approaches will be presented here. Prokop et al.141,142 reported on multicomponent polymeric complexes that are candidates for delivery vehicles of biological molecules such as proteins and drugs. Biocompatible and mostly natural polymers can be fabricated into thermodynamically stable nanoparticles insoluble in water and buffered media, in the absence of organic solvents, featuring core-shell morphology. A careful choice of construction materials and the superposition of several interacting principles during their production allow for the customization of the physicochemical properties of the structures. In fact, two overlapping polyeletrolyte pairs were used to assemble nanoparticles. Several chemistries of particles are available. A recent development involves low-molecular weight polymers. Typically, nanoparticles are made by mixing of polyanionic core polymers, such as alginate, chondroitin sulfate, and the corona polycations, such as spermine hydrochloride, and poly(methylene-co-guanidine) hydrochloride (PMCG). PMCG is a synthetic oligomer which mimics peptide structure. It contains highly charged cationic guanidine groups.
A multipolymeric nanoparticulate system was shown to be very effective for in vitro gene transfer,296b particularly in cell systems that are normally refractory to gene transfer, such as pancreatic islets and antigen-presenting cells. The findings suggest a nonspecific uptake system that permits adenoviral particle release within the transfected cells. A comparison with literature data revealed that this system is efficient at much lower levels (at least three orders of magnitude) of infectious viral particles. Earlier, we have shown that this PEC provides an efficient antigen delivery under different delivery modes (subcutaneous, oral).296c Recent data revealed that such nanoparticles are taken up by a nonspecific adsorptive endocytosis and/or by fluid-phase endocytosis.
Alonso297,298 introduced chitosan-based nanoparticles. Chitosan (CS) nanoparticles are obtained by the process of ionotropic gelation based on the interaction between the negative groups of the pentasodium tripolyphosphate (TPP) and the positively charged amino groups of CS. This process has been used to prepare nanoparticles for the delivery of peptides and proteins, insulin, and cyclosporine. Blank nanoparticles were obtained upon the addition of a TPP aqueous solution into a (CS) solution stirred at room temperature. The formation of nanoparticles is a result of the interaction between the negative groups of TPP and the positively charged amino groups of chitosan. The problem associated with this kind of delivery vehicles is a rather limited stability in ionic environments (buffers, media, and sera) and some degree of swelling of these nanoparticles. The instability results from the ion exchange (followed by dissolution) between the polyions incorporated into the nanoparticle and small ions present in the above environments. However, this system offers an interesting potential for transmucosal delivery.
PEC stability is covered in the Section “Gene Delivery”. Other nanovehicular chemistries are also available.299
Polymeric Micelles
This technology has been pioneered and developed by K. Kataoka in Japan (e.g., In Reference 300). Polymeric micelles, core-shell-typed colloidal carriers for drug and gene targeting, prepared from the self-assemblies of block copolymers, are one of the most refined and promising modalities of DDSs, since the critical parameters such as size, drug loading, and release can be controlled by engineering the constituent block copolymers. It has been demonstrated that polymeric micelles show unique disposition characteristics in the body suitable for drug targeting (e.g., prolonged blood circulation and significant tumor accumulation). Micelles exhibit very high thermodynamic stability due to entropy driven self-assembly of block copolymers followed by a hydrophobic or electrostatic interaction in the core. This stability is characterized by low critical micelle concentration (CMC). A slow dissociation/dissolution rate in vivo helps to retain their integrity and long circulation time. This is accentuated by the fact that micelles that are typically < 200 nm, between 10 nm and 50 nm in size. While small size may lead to extravascular and cellular uptake it may also restrict the space for drug entrapment and limit the ability for release and sufficient local drug delivery. No details are available on the scale-up strategy for micelle production.
Novel approaches for the formation of functionalized poly(ethylene glycol) (PEG) layers as hydrophilic outer shell were developed to attain receptor-mediated drug and gene delivery through PEG-conjugated ligands with a minimal nonspecific interaction with other proteins. Surface organization of block copolymer micelles with cross-linking core was also described from a standpoint of the preparation of a new functional surface-coating with a unique macromolecular architecture.301 Also, recent advances in polymer synthesis have allowed the development of polymeric micelles with integrated smart functions such as environmental sensitivity and targetability.302 As the assembly of micelles is posed to happen during a dialysis step, it is rather difficult scale-up strategy. Apparently, a corporate partner has been able to design a new scaleable process, which is currently in a commercial exploration. Previously, a preclinical study with incorporated doxorubicin demonstrated a strong EPR effect and is posed for cancer therapeutic applications, especially for intractable cancers with limited vasculature.303,304 Cisplatin- and paclitaxel-based formulations were successfully tested in animal studies and currently are in clinical trials.305,306 Other micellar chemistries are also available.307
Multilayered Polyelectrolyte Films
These films can be manufactured by deposition of polyelectrolyte solutions layer-by-layer. If assembled on a planar substrate, thin films results. If applied on a suitable globular matrix a hollow vesicle can be formed following the dissolution of the matrix. In the latter case, often, microcapsules, loaded with a suitable drug, could be produced.308 Nanocapsules with an affinity to cancer cells in vitro have also been reported.309 Obviously, such vehicles are uptaken by cells. In either case, a controlled release could be accomplished310 as a result of a diffusion or erosion of the capsule (film) itself,311,312 permitting a broad range of spatial and temporal control over the release of therapeutic agents from the surfaces. Polypetide-based multilayers have also been reported.313
Dendrimers
Dendrimers are attractive macromolecular systems for drug delivery because of their nanometer size range (5–20 nm), ease of preparation and functionalization, and their ability to display multiple copies of surface groups for biological recognition processes314 and are used for the systemic delivery of water-insoluble drugs. Dendrimer molecules are monodisperse symmetric macromolecules built around a small molecule or in a linear polymer core using connectors and branching units. Interaction of dendrimer macromolecules with the molecular environment is predominantly controlled by their terminal groups. By modifying their termini, the interior of a dendrimer may be made hydrophilic, while its exterior surface is hydrophobic or vice versa. Dendrimers can be synthesized starting from the central core and working out toward the periphery (divergent synthesis) or in a top-down approach starting from the outermost residues (convergent synthesis). Commonly used and commercially available polyamidoamine dendrimers are (PAMAM) form spherical polymers with good aqueous solubility because of their highly charged exposed surface groups with abundant primary amines for convenient conjugation.315 PEGylation of amino-terminated PAMAM dendrimers has reduced immunogenicity, improving circulation lifetime.316 Glycodendrimers can be grown to a controlled size, at low polydispersity, with functional groups exposed on the exterior rather than folded inside, like those of glycopolymers. They are already used commercially as target-specific contrast agents for MRI.317 In a system to deliver DNA using PEGylated PAMAM dendrimer, high transfection efficiency was observed,318 but with significant cytotoxicity.319 There is, therefore, an optimum dendrimer composition and preferable cell target for minimum toxicity and efficient delivery. Majoros et al.320 described multifunctional dendrimers for targeted delivery of chemotherapeutics and imaging agents to specific cancer cells.
Phase I clinical trials of Starpharma's dendrimer-based microbicide321 (Viva-Gel) is also the first human dendrimer pharmaceutical clinical trial. Cytotoxicity issues, drug-release kinetics, and uptake of the next generation of polyvalent dendrimers are the issues that must be addressed.
Liposomes and Solid-Lipid nanoparticles
Liposomes are small artificial vesicles of spherical shape that can be produced from natural nontoxic phospholipids and cholesterol. Because of their size, hydrophobic and hydrophilic character, as well as biocompatibility, liposomes are promising systems for drug delivery. Drug molecules can be either entrapped in the aqueous space or intercalated into the lipid bilayer of liposomes, depending on the physicochemical characteristics of the drug. Liposome properties vary substantially with lipid composition, size, surface charge, and the method of preparation. They are therefore classified into three classes based on their size and number of bilayers. Small unilamellar vesicles (SUV) are surrounded by a single lipid layer and are 25–50 nm in diameter. Large unilamellar vesicles (LUV) are a heterogeneous group of vesicles similar to SUVs and are surrounded by a single lipid layer. Multilamellar vesicles (MLV), however, consist of several lipid layers separated from one another by a layer of aqueous solution. Although SUV's can carry soluble substances in their lipid bilayers structure, MLV's can also carry membrane bound lipophilic drugs for sustained release. A combination with a cationic lipids results in a cationically charged liposomes (lipoplex). The incorporation of neutral lipids leads to neutral liposomes (at physiological conditions).
Early liposomes, designed as carriers inside their hydrophilic core or within their hydrophobic phospholipid bilayer coat, have shown promise in improving the solubility of many amphiphilic drugs, enhancing the transfer of therapeutics into cells and tissues, avoiding particular organs (e.g. brain, heart, kidneys), and enabling sustained release to reduce drug toxicity. Shielding from the RES has been achieved by PEGylation (addition of PEG).322–324 Liposomes have excellent biocompatibility and, after rupture, are readily integrated into cells, while the PEG is readily excreted via exocytosis. Antibody-conjugated liposomes were developed as an extension of earlier prototypes. Ishida et al.325 reported that 100–200 nm PEG-liposomes had the longest circulation time and greatest tumor accumulation in mice. Because of their ability to target immune-system proteins, liposomes can also amplify macrophage response during infection;326 they have, therefore, been used in vaccination327 and adjuvant therapy328 during cancer treatment to boost the immune system.
Numerous studies have investigated the reasons behind the observed prolonged blood residence times of sterically stabilized liposomes. The most prevalent mechanism is thought to be the liposome surface protection offered by the hydrophilic groups against protein adsorption and opsonisation. Other mechanisms contributing to rapid clearance, leading to reduced circulation half-lives of delivery systems include: complement activation, filtration, and blockade through anatomical barriers in specific tissues such as lung capillaries, spleen sinusoids, or entrapment in blood-clotted regions in the circulation.1 Although PEGylated liposomes improve the colloidal stability of cationic lipid/DNA complexes and their biodistribution, these liposomes interfere with the intracellular trafficking of the complexes resulting in reduced transgene expression.329
Inex Pharmaceuticals (Burnaby, BC, Canada) successfully employs SPLP, “stabilized plasmid lipid particles” of approximately 70 nm diameter composed of 1,2-dioleoyl-3-phosphatidyl-etahnolamine (DOPE), the cationic lipid N,N-dioleoyl-N,N-dimethylammonium chloride (DODAC) and poly(ethylene glycol) conjugated to ceramide (PEG-Cer), with a potential for systemic drug (e.g., siRNA) and gene delivery with very suitable pharmacokinetics and the biodistribution. Circulation lifetimes observed for SPLPs were approximately 10 h. In specific case, the SPLPs are stable while circulating in the blood and the encapsulated DNA is fully protected from degradation by serum nucleases. Significant accumulation (approximately 10% of injected dose) of the long circulating SPLPs into a distal tumor (Lewis lung tumor) was observed following i.v. application versus delivery of intact plasmid to tumor tissue at approximately 6% injected dose/g tissue.330
Although liposomes can undergo endothelial transcytosis in cell cultures, they likely leave the vasculature via paracellular pathway in vivo.331In vivo, liposomes, because of their amphiphilic structure, may suffer from physical instability in solution. Once destabilized, liposomes can aggregate. This is main reason for their limited use in clinical practice. Some commercial aspects of liposome technology, currently pursued in clinical trials, including a number of liposome-based formulations, are reviewed in Goyal et al.332 Major progress is expected from developments of many different modalities of liposome preparation, including those of pH-sensitive status, cytosolic delivery, delivery of cytotoxic agents and toxins, antigen delivery, a combination of viral and peptide delivery systems, and from functionalized liposomes.333 The resistance of these systems to polyanion-mediated disassembly in plasma, particularly pronounced in case of complexes, may significantly enhance the liposome applications.334,335
Neutral liposomes neither appear readily capable of internalization (fusion with membranes of cells) nor they internalize in most tumor cells. The uptake of such liposomes was inhibited by polyanions which are the competing ligands for scavenger receptors. Based on such in vitro experiments, there appears to be different patterns of uptake and the competition with various polyanions. The differences observed in the uptake rate of liposomes with different lipid compositions seemed to be primarily due to the differences in the binding between liposomes and cell membrane components. The in vitro interaction of various liposomes with these cell lines shows significant similarities to the in vivo clearance rates of the liposomes.336b The inhibition of neutral liposomes uptake by polyanions testify on the role of scavenger receptor in this process. Anionic liposomes are probably taken up via this route.
Some liposomal formulations have been applied in clinical use or development. DaunoXome® (Gilead Sciences Ltd., Granta Park, Great Abington, Cambridge, UK), Doxil®/Caelyx™ (Johnson & Johnson, Bridgewater, NJ) and Myocet® (Sopherion Therapeutics, Princeton, NJ), loaded with doxorubicin, have all been used in cancer treatment, while Amphotech® (Amphotech, Beverly, MA), loaded with amphotericin B has an application in fungal infection area. Cytotoxic drugs entrapped in new types of liposomes, endowed with targeting and long-circulatory properties to confer favorable in vivo pharmacokinetics and biodistribution patterns, are becoming established component of our tools for treatment of cancer, after suffering a number of setbacks over the last a quarter of century. It has been accepted that such new preparations alter the efficacy and toxicity profiles of the parent compounds substantially, although still in clinical tests.
Solid-lipid nanoparticles (SLN) belong to the liposome category by virtue of their chemical matrix composition. These are submicron range particles made up of biocompatible and biodegradable material capable of incorporating lipophilic and hydrophilic drugs. They are usually made of lipid matrix material, emulsifiers, coemulsifiers, and water (and liquid or solid drug) by means of high-pressure homogenization (hot and cold technique), a microemulsion technology, emulsification by sonication, and other methods. Resulting is an aggregate produced from lipids and an enclosed surfactant. Drug could be incorporated directly into the lipid core or associated with the surface. Both passive and active targeting (particularly suitable for active targeting to gastrointestinal tract) can be accomplished. The cellular entry is believed to be via transcellular or paracellular routes.336,337 Paracellular route explores intercellular junctions between cells of epithelium and endothelium. One application of SLN is topical dermal application.338 A first product has recently been introduced to the Polish market (Nanobase™, Yamanouchi) as a topical application moisturizer.
Another lipid-based delivery is that of lipoprotein-based delivery system that deserves an attention. Rensen et al.339 described recombinant chylomicron, and low and high density lipoproteins to facilitate delivery of different substances, including lipophilic in nature, as well as hydrophilic and oligonucleotides cargo. Lipoproteins are biodegradable and do not trigger immune reactions and are not recognized by the RES, resulting in efficient delivery to hepatocytes and tumor cells in animal models.
Functionalized Polymer–Drug Conjugates (Polymer Therapeutics)
Polymer-based nanomedicines constitute water-soluble hybrid constructs, designed for intravenous administration. They fall into two main categories: polymer–protein conjugates or polymer–drug conjugates. Polymer–drug conjugates typically include a minimum of three components: a natural or synthetic polymer, a biodegradable polymer–drug linkage (spacer), and a bioactive drug (e.g., antitumor agent). Conjugates can also be synthesized to contain ligands to promote receptor-mediated targeting (antibody, peptide, glycan). The conjugates with peptidyl polymer–drug linkers are amenable to cleavage by lysosomal thiol-dependent proteases.340 In this case, prodrug activation occurs intracellularly. In contrast, conjugates that contain an ester link between drug and polymer can release its cargo by chemical hydrolysis or esterase degradation extracellularly. They provide a prolonged circulation times. Because of higher molecular weights of more than 40 kDa, a renal excretion is overcome. For the same reason, such drugs are not uptaken into the cells by a simple diffusion and require some form of endocytosis, typically into the endosomes. This method of uptake, serves to circumvent the problem of the P-glycoprotein-dependent drug efflux in MDR.341,342 This resistance to MDR, through a bypassing, is believed to be mediated by better selectivity (receptor-mediated uptake) and efficiency of delivery to cancer cells, including the EPR effect.343
The notable members of efflux proteins include P-glycoprotein, multi-drug resistance protein, and breast cancer resistance protein. These efflux pumps play a pivotal role in extruding drugs. The role of efflux pumps in drug delivery and the importance of their tissue distribution have been recently discussed as a means for improving pharmacokinetic parameters of drugs.344 One general approach to overcome this problem has been to inhibit specific resistance mechanisms, such as P-glycoprotein (PGP)-mediated drug efflux, using small molecule agents or other therapeutic strategies.345 Several inputs are available on employment of nanovehicular drug delivery vehicles to overcome MDR.346–348 It has been proposed that a local drug (e.g., nanovehicular-bound doxorubicin) delivery in high concentration close to the cell membrane would affect the local microconcentration of the drug which subsequently saturates the MDR efflux pumps.
Polymer–drug conjugation promotes tumor targeting through the EPR effect and, at the cellular level following endocytic capture, allows lysosomotropic drug delivery.8 Poly(ethyleneglycol) (PEG), N-(2-hydroxypropylmethacrylamide) (HPMA) copolymers and poly(glutamic acid) (PGA) are the most widely tested in a clinical setting. Although many HPMA copolymer conjugates have been designed to contain glycans, antibodies, proteins, and peptides as targeting ligands, only one conjugate has progressed into clinical trial: HPMA copolymer–doxorubicin–galactosamine (PK2, FCE28069). In this case, the overhanging galactosamine moieties localize to the asialoglycoprotein receptor in hepatocytes. Hepatocellular carcinoma cells have been used to assess the potential of this product as a possible treatment for liver cancer in a clinical study.349 The first polymeric antiangiogenic conjugate, HPMA copolymer–TNP-470, has been recently described.350 Due to the EPR effect, macromolecular drug concentration in tumor tissue far exceeded that of the blood plasma, usually by 5–10-fold. A recent review on diverse polymer chemistries that can be used in drug delivery, covering linear, graft, branched, cross-linked, block, star-shaped, and dendrimer topology (architecture) is available.351
Cell Penetrating Peptides (CPP)
CPP are up to 30 amino acid amphiphilic peptides, which can be internalized by cells by mechanisms that require no energy and are receptor mediated or not. Protein-derived CPPs usually consist of the minimal effective partial sequence of the parent translocation protein, and are known also as protein transduction domains or membrane translocation sequences.352 CPPs from different classes do not share common amino acid sequence motifs. The only two common features of all CPPs appear to be a positive charge and amphipathicity. All known CPPs are net positively charged at physiological pH, and incorporate from approximately 17% to 100% (polyarginines) of positively charged amino acids. Most CPPs have only one negative charge—at the C-terminus—or even none, when the C-terminus is amidated. The CPPs are able to promote translocation of various types of useful cargo, ranging from small molecules to proteins and large NV, with great efficiency.
Protein transduction occurs by a rapid, lipid raft-mediated MPC mechanism that is independent of receptors and transporters that all cells perform. A class of cationic peptides, such as the TAT, penetratin, and VP22 peptides may have the ability to be taken up by cells without endocytosis events.353,354 It was initially suggested that these peptides directly penetrate cell membranes by an energy-independent route.353 Although many controversial data were published, there is a consensus that the fluid-phase and adsorptive endocytosis may contribute to the nonspecific uptake of such extracellularly applied substances.114 This is based on observations with an inhibition of this uptake by negatively charged GAG molecules (glycosaminoglycans, HSPG), dextran sulfate, and heparin. This is supported by observations on inhibition of CPP uptake in GAG-deficient cells.355 Positively charged CPP molecules, CPP-conjugated drugs (e.g., oligonucleotides), and CPP-conjugated NV have been shown to bypass endosomes (endosomal escape) and deliver the cargo to cytoplasm.355–358
A mechanistic hypothesis has been presented for how water-soluble guanidinium-rich transporters (polyarginines) attached to small cargoes (MW ca. < 3000) can migrate across the nonpolar lipid membrane of a cell and enter the cytosol. Positively charged and water-soluble, arginine oligomers can associate with negatively charged (HSPG), bidentate hydrogen bond acceptor groups of endogenous membrane constituents, leading to the formation of membrane-soluble ion pair complexes. The resultant less polar, ion pair complexes partition into the lipid bilayer and migrate in a direction, and with a rate, influenced by the membrane potential. The complex dissociates on the inner leaf of the membrane and the transporter conjugate enters the cytosol. This mechanism could also be involved in the translocation of guanidinium-rich molecules that are endocytosed due to their size or the conditions of the assay, across the endosomal membrane.199 Polyelectrolyte complex nanoparticles,141,142,206b rich in arginine motifs on the outer surface of nanoparticles (due to constitutive polymers of spermine and poly-methylene-co-guanidine copolymer, PMCG), probably share the same pathway of internalization. Oligoguanidinium vectors have been shown to localize to mitochondria of cytoplasm.359 This finding could be of significance for targeting of apoptosis. Another supporting hypothesis for uptake of cationically charged NV is that they are drawn into cells by “piggybacking” onto the normal internalization pathway of HSPG, which occurs with a half-life of 5–20 h,360 still employing the above explanation.
CPPs do not belong to NV. They are listed here because such motifs are often used to functionalize NV in order to enable their uptake and quick penetration. It is implied that the mechanism of uptake of such vehicles may be similar to those of plain CPPs. Such NV functionalization is expected to play a significant role in future.
Special Systems
Besides imaging, CDs have been shown to have a potential for controlled release of drugs and magnetically guided thermal responsive matrices, in which drug release is triggered by the heating as well as for vectors of nucleic acids for vaccination and gene transfer.361 Nanogold and QDs are usually treated together as metallic vehicles.
Nanoshells (nanogold) are powerful nonselective multifunctional nanoparticles which rely on tumor extravasation (EPR) for targeting combined with external activation for localized thermal ablation therapy. In addition to providing NIR-absorbing properties, the gold shell acts as a biocompatible barrier and enables conjugation of stabilizing moieties for attachment of expression systems. Loo et al.362 recently reported use of immunolabeled and PEGylated gold nanoshells for both NIR imaging and therapy in cancer cells.
Several inorganic systems have been reported for drug and gene delivery. These include calcium phosphate, nanogold, carbon material, silicon oxide, and iron oxide.363–367 Carbon nanotubes are one-dimensional tubular objects of carbon which can be either multiwalled or single-walled. In addition, they can be also functionalized permitting an attachment of ligands and drugs.368
A biomedical application has been reported on biodistribution of labeled carbon single-wall tubes in mice369 and has been reported to be uptaken by endocytosis.370 This probably holds of the rest of these special case delivery vehicles. Carbon tubes are in early stages of development and no clinical use has been suggested. Bianco371 summarized their therapeutic prospects.
A new class of NV is based on assembly of amphiphilic polymers. Flamel Technologies (Venissieux, France) have designed a mimetic system which is based on the self-assembly of hydrophobic segments coupled to alternating hydrophilic segments of the diblock polymer poly(l-leucine-block-l-glutamate). They are synthesized by acid hydrolysis of the ester poly(l-leucine-block-l-methyl glutamate). During the hydrolysis reaction, the leucine block precipitates from the reaction mixture forming nanosized particulate structures, which upon purification results in stable, colloidal dispersions (200 nm in diameter). The core of the nanoparticles is composed of crystalline, helical leucine segments, the corona is made from the poly(l-glutamate) polyelectrolyte brush extending out, allowing for particle stability. At pH 7.4, the nanoparticles spontaneously adsorb proteins, such as insulin, directly from solution. Partial desorption of the protein in its native configuration can be induced by simple dilution. This system is being explored commercially as a new delivery vehicle.372 This technology features an ability of a polymer, with suitable repeating segments, to self-assemble into NV. Apparently, the α-helix conformation of the hydrophobic segments was shown to be a driving force for assembly. The stability and resistance of this product to the polyelectrolyte exchange reaction is, however, questioned in physiologic media.373 The cellular uptake mechanism has not yet been determined.
GENE DELIVERY
These vehicles comprise nonviral and viral systems. Nonviral gene delivery refers to the use of naked DNA, biolistic delivery, cationic lipids formulated into liposomes and complexed with DNA (lipoplexes), cationic polymers complexed with DNA (polyplexes), polymeric vesicles complexed with DNA (cholesterol vesicles), polymeric nanoparticles or a combination of both cationic lipids and cationic polymers complexed with DNA (lipopolyplexes). Many different chemistries and modifications have been introduced and we cannot cover them in detail.
Nonviral Systems
Since DNA molecules are large and water-soluble they do not permeate through lipid bilayers of the cell membrane. Small DNA fragments (oligonucleotides or plasmids) enter the nucleus by passive diffusion, while larger fragments are transported through the nuclear pore complex in an energy-dependent manner.374 Nuclear pore transport can potentially be improved through the attachment of nuclear localization signal peptides which redirect intracellular vehicle transport to the nucleus,375 or the inclusion in the plasmid of nucleotide sequences with affinity for cellular proteins such as transcription factors; these then mediate the actual nuclear transport.376 Enhancing DNA delivery is obtained via complexing this molecule with suitable agents that form complexes and facilitate the membrane transport. The instability of the delivered DNA is also an issue, as its half-life is only 60–90 min in the cytoplasm.377 Gaining access to the cytoplasm is only the first step in DNA delivery. The DNA molecule must traverse the cytoplasm and gain access into the nucleus. Although polymeric nonviral vectors still display a very poor transfection efficiency compared to viruses,377 improving the design of pH-responsive endosomolytic polymers gives hope that a synthetic viral mimetic will eventually become reality.159
Several polymer-, lipid-, and peptide-based nonviral vehicles are currently under investigation for gene delivery.378 Commonly used polymers include polyelethyleneimine (PEI), Poly-l-lysine (PLL), chitosans, and dendrimers. Nonviral vectors are receiving increasing attention due to compound stability and potential easy of chemical modification, low cost and consistent standard of production, higher biosafety, and flexibility.379 The cationic polymers usually bear protonable amines enabling their complexation with nucleic acids and securing their transfection efficiency. Due to electrostatic interactions between the positively charged amino groups and negatively charged phosphate groups of plasmid DNA, polycations spontaneously form complexes with a display of rapid condensation. The ratio of polycation/DNA then allows for a charge control. Aggregation of originally colloidal structures (although of different submicroscopic shape) is one of the problems. Similarly, in vivo, biological fluids often cause an aggregation when complexes are introduced. That is why in vivo gene delivery is rather difficult and inefficient. Low incorporation into tissues and induction of thrombosis provide additional obstacles. Recent data suggest that coating the polycation/DNA complexes with polyanions can diminish particle aggregation (increasing colloidal stability).380 Some targeting of gene complexes with suitable ligands has been achieved. A new synthesis method can produce polyethylenimine derivatives with terminally galactose-grafted poly(ethylene glycol) for specific gene targeting to the liver.381 Several cationic lipids have been proposed for nonviral gene delivery to lungs, brain, tumors, and the skin. They face still serious limitations including low transfection efficiency and high toxicity. PEG–PEI copolymers have also been introduced.382
Bioplex technology combines advantages of nonviral vector with a functional entity allowing for targeting. To enable this, as peptide-nucleic acid complex with functional properties is hybridized to DNA plasmid, providing better formulation stability and minimal inactivation.383 The functional element can include localization or targeting signals.
Other polymers have been suggested for condensing large genes into smaller structures and to mask negative DNA charges. These include neutral polyvinyl alcohol, polyvinylpyrrolidone, and PEG copolymers. Many different polymeric approaches were summarized by Piskin et al.384 Cholesterol-based vesicles have been described by Brown et al.385
Various NV formulations have been shown to transfer genes in a significantly higher portion as compared to naked DNA386–389 but much less than the viral systems. A high in vivo activity, using a luciferase reporter, has been demonstrated with dendrimers.390 Carlesso et al.206b have demonstrated highly efficient, nanoparticle-based gene delivery in vitro and in vivo.
Biolistic method uses the hand-held gene gun with a pulse of helium to fire small gold particles coated with DNA at target cells. This method of biolistic transfection is becoming increasingly popular as an effective means of rapid gene delivery into mammalian tissue. It is restricted to local expression in the dermis, muscle, or mucosal tissue. Current methods of microcarrier preparation, however, are slow (up to 2 days) and can result in variations in transfection efficiency due to a number of problems including shearing of DNA, agglomeration, and adhesion of gold particles. An improved, more rapid method of microcarrier preparation has been reported.391 Accell® (PowderJect Vaccines, Inc.) and the Helios™ (Bio-Rad) gene gun designs available on the market. One of the major applications of gene gun is genetic vaccination in form of intramuscular or cutaneous injection of DNA.
Viral Systems
A survey of the distinct advantages and disadvantages of these systems is available.392 Having evolved to deliver their genes to target cells, viruses are currently the most effective means of gene delivery and can be manipulated to express therapeutic genes or to replicate specifically in certain cells. Gene therapy with attenuated viruses is being developed for a range of diseases including inherited monogenic disorders and cardiovascular disease, but it is in the treatment of cancer that this approach has been most evident, resulting in the recent licensing of a gene therapy for the routine treatment of head and neck cancer in China. A variety of viral vectors have been employed to deliver genes to cells to provide either transient (e.g., adenovirus, vaccinia virus) or permanent (e.g., retrovirus, adeno-associated virus) transgene expression and each approach has its own advantages and disadvantages.
Adenoviruses (AdV) are the most commonly used vitro-therapeutic models, where mechanisms for initial fiber protein binding with CAR cell surface receptors (followed by endocytosis) have been actively investigated for tissue and organ targeting in human gene therapy.393 The AdV vectors remain in an extrachromosomal location and are replication-defective, resulting in transient expression. AdV vector has an outstanding capability of escaping from endosome to cytoplasm. Since it also has a nuclear transfer function, a high gene expression can be obtained in nucleus. Despite extensive investigation of dozens of viral vectors for cancer treatment, and promising results in clinical trials, the FDA has not approved any virus-based therapeutics because of toxicities and immunogenicities widely reported in gene therapy.394 For example, potential complications with retrovirus-based vectors include activation of proto-oncogenes and inactivation of tumor-suppressor genes by insertional mutagenesis. Some of the immunological responses of viral vectors maybe circumvented by encapsulation and masking of virions within nanoparticles.
To circumvent the nuclear barrier a cytoplasmic expression system has been developed, in which the transgene is administered with T7 RNA polymerase and incorporates a T7 promoter sequence.395
Other Nanosized Vehicles (Polymeric Nanoscaffolds) For Gene Delivery
Luu et al.244 reported on use of nanoscaffolds manufactured by electrospinning to incorporate DNA for therapeutic application in gene delivery for tissue engineering. The scaffolds are nonwoven, nano-fibered, membranous structures composed predominantly of PLGA random copolymer and a PLA-PEG block copolymer. Release of plasmid DNA from the scaffolds was sustained over a 20-day period, with maximum release occurring at approximately 2 h. This could be applied in control of temporal and spatial sequence in tissue regenerative processes.
Comparison of Viral and Nonviral Methods
Usually, researchers do not normalize the gene expression data per protein or DNA and thus such data are difficult to compare. We refer to such presentation as activity or amount of gene product per mass of protein for in vitro studies or per gram of tissue in vivo. By large, the nonviral gene transfer methods do not reach the expression levels of the viral-based methods. It is the safety consideration which will dictate the final selection and choice of polymer/method. Although only three DNA-based pharmaceuticals (an antisense oligonucleotide inhibitor of CMV formulated in bicarbonate buffer, Vitravene, ISIS Pharmaceuticals Inc., USA, 1998; an adenoviral gene therapy treatment, Gendicine, virus particles carrying p53 gene, injected intratumorally once a week; China, 2003; and Rexin-G, Epeius Biotechnologies Corporation, Glendale, CA) have received approval from regulatory agencies, numerous candidates are in advanced stages of human clinical trials.396 Rexin-G has received orphan drug status to assist in the development of this new cancer treatment, since the first patients receiving multiple intravenous infusions of the targeted adenoviral constructs have all responded favorably. None of them are nanovector-formulated. Albumin-formulated phosphodiester oligonucleotide analog to formivirsen–Vitragene has been reported.397
Safety Considerations
Although at present the in vivo expression levels of synthetic molecular gene vectors are lower than for viral vectors and gene expression is transient, these vehicles are likely to present several advantages including safety, low-immunogenicity, and capacity to deliver large genes and large-scale production at low-cost. Interestingly, no major clinical toxicities have been reported with the nonviral delivery systems. Acute toxicity and induction of innate immunity may be two leading concerns.398,399 The two leading classes of synthetic gene delivery systems that have been mostly investigated are cationic lipids and cationic polymers. Liposomes are generally nonimmunogenic owing to lack of proteinaceous components. Viral vectors typically offer higher transduction efficiency and long-term gene expression, but may be associated with toxicity, immunogenicity, restricted target cell specificity, and high cost. Nonviral methods have become widespread because of their relative safety, capacity to transfer large genes, site-specificity and their noninflammatory, nontoxic, and noninfectious properties. However, the clinical usefulness of nonviral methods is limited by their low transfection efficiency and relatively poor transgene expression.
As with any emerging new biomedical discipline, gene therapy has also faced a number of setbacks, and there have been concerns regarding the safety of some gene delivery approaches, however, these hurdles are not insurmountable. Gene transfer technologies are improving rapidly and have led to the development of new and more efficacious gene delivery approaches with fewer side-effects. The success of gene therapy is still highly dependent upon the continuous development of improved gene delivery technologies, the progress of which should hopefully and ultimately cure diseases that are refractory to current treatment paradigms. No systematic discussion of these issues is available. A partial coverage is presented in Verma and Somia400 and in Wiethoff and Middaugh.401
EMPLOYING QUANTITATIVE SYSTEMS APPROACH TOOLS TO GUIDE DESIGN AND DEVELOPMENT OF TARGETED NANOVEHICULAR DRUG DELIVERY SYSTEMS (IN SILICO MODELING)
Discrepancy between drug potency between the laboratory and clinic is a recurrent problem in pharmaceutical science and is one of the major stumbling blocks in effective drug design. There is growing recognition in both academia and industry that the prevailing trial an error design of drug delivery techniques is a serious limiting factor and mathematical modeling has been suggested as an important tool in the design of drug delivery protocols. Issues include the rational design of appropriate agents, strategies for their optimal application, and technologies for the spatial and temporal control of their delivery to desired sites of action for a given disease model. Systems biology provides the methods, computational capabilities, and inter-disciplinary expertise to facilitate such development.
We very briefly review the existing modeling tools and approaches to quantitatively describe the behavior of targeted NV within the vascular and tumor compartments and how systems approach can help to speed-up the drug development. While we list “elementary” phenomena related to different level of complexity of delivery to cancer, we also stress importance of multi-scale modeling and bottom-up systems biology approach, a new paradigm which is going to shape the future of medicine. We reveal some quantitative tools for multi-scale modeling available in literature, including the drug delivery issues. We have chosen this special topic because of impressive number of references and work done in this area. The goal is to develop a therapeutic cancer systems model, or at least show where we stand and what else should be done in order to get there. Although some companies claim to have such quantitative tools, only the open literature provides unhindered access to such scenario. As we will see, while most of elementary descriptions are available, the systems approach designed for bottom-up is not available. In a strict sense, elementary steps are defined as unidirectional reactions (each enzyme-substrate may have two, for each reversible direction and any possible combination of E-S complexes, including inhibitors, activators, etc.), based on mass action model. Such approach is useful for description of metabolic and signaling pathways. Elementary events (phenomena) in the context of this article are defined as the simplest physical or chemical phenomena (reactions) relevant to each level of hierarchy that describes the whole organism behavior.
Systems Biology is the field that emerged from the need to address the shockwave of genetic and proteomic information in an integrative, quantitative manner. We define Systems Biology as “quantitative, postgenomic, postproteomic, dynamic, multi-scale physiology.” Computer models are central to the field, but the greatest challenge of Systems Biology is that these models may eventually have 106 dynamic variables with complex nonlinear interactions.402 Historically, biologists have been able to focus on one component of a biological system at a time (e.g., a gene or a protein), with the expectation that knowledge of the individual components will eventually enable an understanding of the entire system. As a result, individual data are often divorced from the context of the entire system—the functioning organism. Systems Biology attempts to define relevant global properties, relations, and functions of biological systems.403,404 Others have used different terms, including organismic system,405,406 emergent characteristics, emergent (systems) properties407a,407b, or systemic variables.404 Modularizing systems networks, that is, defining structure and revealing function of complex biological systems are topics of significant current interest.408 By making systematic perturbations (using inhibitors, activators, changes in external signals, etc.) and measuring global responses only, one can discover a network “interaction map” that can be expressed in terms of module-to-module connection strengths.409 The global network response to a signal or experimental perturbation can be predicted and expressed in terms of the individual (local) responses by using a “map” of network connections.410 The key is to obtain both the structural (modular, topological) and functional information. The same reasoning applies to cancer which could be considered as another systems biology problem.411,412 In the following, we will briefly review available elementary steps in terms of availability of quantitative tools and emerging properties relevant to cancer biology and its treatment.
For determining emerging properties, a concept of time characteristics (or time constants) is usually useful. It is defined as a relaxation time of individual subsystems, which is considered to be the time for a change from one state to another (due to reaction, diffusion, change in the adjoining levels, etc.) and are conveniently defined as the time to reach 90% of changed variable. We can easily demonstrate that the time constants change with size of structures. In fact, complex biological systems span multiple characteristic time and length scales from hours and meters to femtoseconds and tenths of nanometers. Usually, responses of higher levels of the system to disturbances coming from a higher level are slower than responses at the next lower level. Thus, the relaxation times of systemic elements are longer than those of individual elementary events. The determination of systemic variables and the neglect of unimportant ones is the key question of transition from a certain lower to a higher level and include system decomposition (reduction) into hierarchical levels (often by means of determining time constants). Such an approach is important for defining more complex (but not all inclusive) models having certain complexity which is much higher than the “minimal” models of biological systems. Such multi-scale models are useful for further development of comprehensive cancer models. Such models are preferred to be of linear-type as such treatment is often used in describing network and signaling pathway models. The most common nonlinearities (type of Michaelis–Menten) are only introduced when concentration-dependent clearance and binding/dissociation (equilibrium) are considered. The linear system is easier to simulate because of easier parameter identification and process simulation.
Table 5 presents rather short and incomplete outline of cancer-related quantitative tools, listing only few examples which deem to be of importance for the bottom-up scheme. Besides listing some important “elementary steps” the key is, via mathematical modeling, to identify emergent, intrinsic properties relevant to each level of hierarchy, validate process model and employ them for design of drug design, delivery schedule, and control strategy for cancer treatment. The recently developed GBN model provides a common framework for the simulation of the hybrid models (automatons), allowing to compute within the deterministic and stochastic domain and a wide range of time constants.486a In future, as comprehensive cancer treatment protocols are eventually developed and model parameters and initial states become individualized through patient-specific laboratory measurements, treatment course schedules will become individualized as a direct consequence of using model-based control system designs. We will briefly address the following hierarchical levels (Tab. 6):
-
(a)
Subcellular models of metabolism, signaling, NV uptake, and trafficking.
-
(b)
Interactions at cellular level: nutrients and drug transport, models of combination therapy, tumor invasion/metastasis, cell growth versus apoptosis, NV adsorption.
-
(c)
Multi-scale pharmacokinetic models: interaction with RES system, immune, vascular (EPR effect), hematopoietic and lymphatic cells, blood flow hydrodynamics, physiologically based pharmacokinetic models, structured according to physiological connectivity.
-
(d)
Hierarchical description toward a therapeutic cancer systems model: solving large-scale, multi-scale metabolic models.
Table 5.
List of Hierarchical Levels and “Elementary” Steps (Modular Units) Relevant to Drug Delivery and Cancer Therapy with Corresponding Quantitative Models
| Hierarchy | “Elementary” Phenomena and Models | Description and Reference(s) |
|---|---|---|
| Drug and polymer | Molecular level properties of drugs (small molecule species, macromolecular drugs, gene vectors, imaging agents): structure, solubility in water and lipid environments, adsorption | In References 413–416 |
| Molecular level properties of constitutive delivery polymers | In Reference 417 | |
| Modeling of associative (self-assembling) properties of drugs and polymers | In Reference 418 | |
| Transport properties of drugs via lipid structures | In Reference 419 | |
| Transport (controlled-release) properties of polymeric-drug superstructures, including hydrogel constructs | In Reference 420 | |
| Molecular modeling of in vitro receptor–ligand interaction | In Reference 421 | |
| Subcellular | Genetic control model | In References 422,423 |
| “Elementary” model of cancer metabolism | In References 424–429; cancer stem cells430–431a | |
| Signaling pathway models | In References 411,431b,432–437 | |
| Models of nanovehicle uptake, trafficking, degradation, and efflux | Analytical model of nanovehicle diffusion, adsorption, desorption, and endocytosis;438 ligand-induced internalization439,440 | |
| Cellular | Nutrient and oxygen effects | Compartmental (subcellular) analysis of nutrient influx and efflux441 |
| Radiation response | In References 442,443 | |
| Response to chemotherapy | In References 444–446 | |
| Models of combination therapy | In References 447–449 | |
| Models of cell cycle | In Reference 450 | |
| Models of tumor invasion and metastasis | In References 451,452 | |
| Models of hematopoiesis | In Reference 453 | |
| Capillary network growth | In References 454,455 | |
| Models of cell growth, quiescence, and apoptosis | In References 456–458a | |
| Models of nanovehicle/cell interaction; ligand-mediated targeting models | In;458b magnetic nanovehicle transport and capture;459 Folate targeting of liposomes;460 optimal tumor targeting by antibodies461 | |
| Multicellular/Tissue | Nutrient and vehicle/drug transport; convective interstitial transport | Tumor blood perfusion and oxygen transport;462 vascular transport—permeable versus nonpermeable capillaries;463 tumor spheroid penetration by antibody;464 hypoxia model;465 interstitial transport466 |
| Interaction with RES | In Reference 467 | |
| Interaction with immune system | In Reference 468 | |
| Interaction within the vascular system (EPR effect) | In Reference 469 | |
| Interaction with hematopoietic system | In Reference 453 | |
| Interaction with lymphatics | In Reference 471 | |
| Physiologically-based pharmacokinetic models: compartmental analysis and biodistribution | Tumor uptake of antibodies: compartmental analysis;471,472 first-pass model;473–476 pharmacokinetic cancer mode133 | |
| Systems model | Solving large-scale, multi-scale metabolic and signaling models coupled with upper system boundary conditions | Dynamic cancer network inference model476–478; network model479 |
| Cancer as a systems disease | The most comprehensive models yet available, still very far from ideal situation412,480,481 | |
| Cancer systems diagnostics | In Reference 482 | |
| Cancer systems epidemiology | In Reference 483 | |
| Bottlenecks in big Pharma and Biotech industries: discovery and development | Systems biology in drug discovery484 |
The information flow is mostly linear from lower levels to the upper ones, although a forward feedback may occur between the “Drug and polymer” and “Multicellular/Tissue” levels. Note, only very few entries (references) were selected to demonstrate tools are available.
Table 6.
Identification of Possible Emergent Phenomena For Comprehensive, Quantitative Cancer Treatment Model/Drug Delivery
| Level of Hierarchy | Emergent Phenomena |
|---|---|
| Subcellular | Elementary cancer metabolic and signaling quantitative model |
| Elementary model of nanovehicular uptake, targeting, internalization and trafficking | |
| Cellular | Model of tumor invasion and metastasis |
| Model of capillary network growth | |
| Tissue | Comprehensive pharmacokinetic model |
| Organism/Systems | Comprehensive model of cancer as a systems disease |
Possible emergent properties, only based on intuition at this time, are listed in Table 6. They can serve as a basis for identification of a minimal cancer model as a starting point for more comprehensive, all inclusive model which would include all levels of complexity of events involved in cancer initiation, progression, and treatment. Computational network multi-scale modeling can make predictions that challenge assumptions and motivate further experimental efforts. The cycle of model building and hypotheses testing will lead to a deeper understanding of metabolic/disease state. The inclusion of multivariate dependencies among molecules of complex network can potentially be used to identify combinatorial targets for therapeutic interventions and drug delivery. The challenge is to integrate all of the relevant knowledge and data in a systematic way to devise the best therapeutic and diagnostic strategies. The promise of systems biology is its capability to deconstruct (decompose) complex phenomena to allow for better understanding of the role of each level of hierarchy (elementary phenomena) in an integrated system. The accurate models will require comprehensive experimental data at multiple levels of complexity. Particularly demanding will be collecting the dose-response and pharmacodynamic profile in vivo from experiments encompassing perturbations for uncovering disease mechanisms allowing for hypothesis testing and verification, including high resolution imaging and noninvasive experimental data obtained on targeted drug delivery on different organs485b already demonstrated on targeted imaging agents by means of NIR and quantitative PET imaging.246,268,269 A term of quantitative systems biology or computational systems biology has been suggested for this type of simulation.486,487 Even term of modular systems biology would be adequate.
EPILOGUE
NV loaded with an appropriate drug offer improved spatio-temporal control (window) over drug kinetics and distribution. The amount of drug released from NV within the blood stream relative to the amount taken up intracellularly is governed by the circulatory properties of vehicles in the central compartment, by the release rate from NV (most NV have this capability, except polymeric conjugates) in this compartment and by the efficiency of intracellular uptake, independednt of the mechanism of uptake. This ratio is defined as the nanovehicular ratio of intracellular to systemic localization (I/S ratio). In order to optimize this ratio one should maximize the circulation time and release and minimize the uptake. NV then serve as a micro-reservoir in the circulation. In other instances, the intracellular delivery is desirable. Thus, there is a prospect for safer and mode-specific treatment therapies.
There is no single list of procedures or guidance available to choose a suitable drug delivery vehicle, that is, a unifying strategy. It will largely depend on the drug therapeutic niche and its final clinical outcome. It seems likely that the nanovector optimization will have to be performed for each application. We now present a scheme of idealized NV (of a particular charge), which can be fitted to different goals and applications (Fig. 9). This platform features a multifunctional character which is possible for most of nanovector systems discussed above. However, there is not an ideal NV suited for just for any application. One idealized NV for intravenous application would be < 100 nm in diameter, negatively charged, and capable of avoiding RES system. This is our generalization, based on critical evaluation of the situation in nanovehicular delivery and transport presented in this review.
Figure 9.
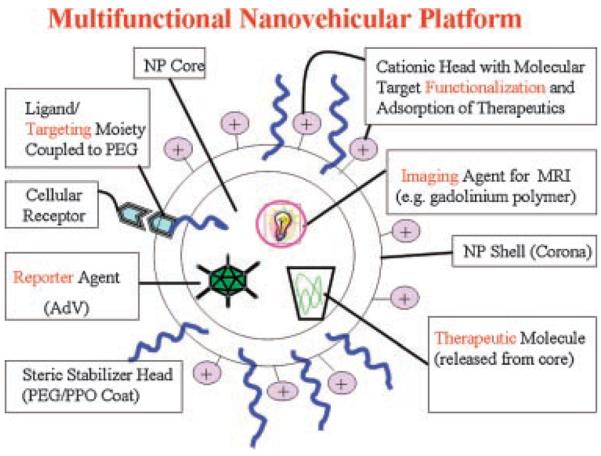
Multifunctional nanovehicle platform. NV (nanovehicle) core contains a therapeutic molecule, which can be released from NP (or delivered intracellularly, following NV internalization); it could also possess a molecular imaging agent—for example, a magnetic resonance imaging (MRI) agent, and reporter of efficacy—luminescence/imaging reporter agent (adV). The corona is typically cationically charged, with primary amino groups which serve as a electrostatic stabilizer, and could be used to functionalize the NV surface, for example, with a targeting moiety (e.g., a peptide with an affinity to HSPG molecules); a therapeutic (peptide, protein) could be adsorbed onto the NP surface via the electrostatic interactions to provide a suitable solid-phase signal; a targeting ligand could also be coupled to a terminal end-group of PEG molecule (Pluronic F-68), serving, in the first place, as a steric stabilizer.
In the coming years, we expect a vigorous progress on the cell and molecular biology sides which will further enhance our understanding of basic phenomena involved in drug/vehicle uptake. Internalization of the delivery systems results in poorly defined trafficking process (Tab. 7). Elucidation of both of endocytic and nonendocytic pathway(s) mechanism may help to rationally designed vehicles. The uptake of NV is a rather understudied area even through a lot of similarities and ideas come from similar area of uptake of macromolecules. Endosomal localization, if not escaped, often leads to degradation of the payload, an area which needs an attention. Cytosomal (mitochondrial-apoptosis) and nuclear localization will receive more concentrated effort.497 Recent efforts employing confocal fluorescence microscopy and fluorescence-activated cell sorter (FACS), coupled with proper discrimination between cell-bound and internalized nanovectors revealed that there exist diverse sets of processes for the uptake of nanovectors and that different cell types may utilize various types in parallel. Proper quantitation of different uptake pathways for internalization and in particular their relative contribution to the uptake is still in very early stages of understanding. There may not be a single specific uptake pathway responsible for intracellular delivery, and not every pathway may be equally effective. A series of inhibiting agents with preferably different modes of inhibition should be applied and combined with colocalization studies. Besides, it is not clear how macromolecular properties of nanovectors, particle size, sterical stabilization and specific homing devices, as well as the cargo chemistry influence the mechanism of uptake. Such diversity in NV uptake mechanisms employed by each cell-type then offers a possibility to manipulate and enhance the uptake via the desired route, via releasing the cargo into the cytosol or bypassing the lysosomal system.498
Table 7.
Summary on Nanovehicles: Likely Uptake Mechanism
| Type of Vehicle | Possible Mechanism/Explanation |
|---|---|
| Nanosuspension | Macropinocytosis384 or lipid-raft mediated due to hydrophobic character |
| PLGA | Depending on chemical modification of the nanovehicle periphery, adsorptive endocytosis and clathrin-coated pathway84 |
| PACA | Endocytosis (not specified)346 |
| PEC | Nonspecific adsorptive endocytosis or fluid-phase endocytosis71b |
| Micelles | Endocytosis-dependent, clathrin mediated488 |
| Dendrimers | Lipid raft-mediated or combination of lipid-raft and caveolar endocytosis for PAMAM dendrimers;489,490 surface modified with ligand—HER2 mAb via receptor-mediate endocytosis491 |
| Liposome & SLN | Endocytosis-independent (lipid rafts)492 |
| Polymeric drug conjugates | Adsorptive endocytosis or receptor mediated endocytosis for targeted drugs493 |
| CPP | Various endocytic mechanisms plus lipid rafts494 |
| Amphiphilic polymers (micelles) | Macropinocytosis or lipid-raft mediated (speculated, mechanism not yet determined) |
| Cationically charged gene vehicles | Nonspecific adsorptive endocytosis or fluid-phase endocytosis495 |
| Lipid-based gene vehicles | Speculated to be macropinocytosis or lipid-raft mediated unless functionalized (endocytosis, receptor-mediated)496 |
| Adenoviral particle delivery | Endocytosis (receptor-mediated)438 |
PLGA—polylactate/glycolate; PACA—polyalkylcyanoacrylate); PEC—polyelectrolyte complex; SLN—solid lipid nanoparticles; CPP—cell permeating peptides; PAMA—poly amidoamine); HER2 mAb—hercepin monoclonal antibody.
Trafficking of vesicles and organelles is mediated by molecular motors,499,500 with help of cytoskeleton proteins. Rab proteins (small GTP-binding proteins), which function as key regulators of intracellular trafficking, vesicular transport, endocytosis, endosome fusion and exocytosis have now been shown to recruit specific motors to organelle membranes.501 In addition, Rab-independent recruitment of motors by adaptor or scaffolding (sorting) proteins is also a key mechanism. Once recruited to vesicles and organelles, these motors can then drive directed transport; this directed transport could in turn affect the efficiency of trafficking events.502 It appears that the cargo takes control of endocytosis, through cargo-specific adaptors, although proofs are very rare.503
The most important aspects of future development of NV are that of their decoration by means of internalizing ligands to achieve an effective localizing effect. The knowledge of NV uptake and intracellular trafficking will have to be further delineated and augmented, particularly in terms of NV uptake routes, their proportions, and control over desirable trafficking route to enable their rational design. It appears that the shift in the tissue biodistribution of NV does not have to be absolute; a marginal improvement might be enough to achieve a dramatic change in the drug efficacy at the site of action.
On a practical side, further progress will be dictated by economic reasons. Approaches such as macroimplants, microdevices (such as microelectomechanical systems—MEMS—employing microfabrication of polymeric substrates), gelled structures, NanoCrystal® Technology (Elan, Corporation, Plc, Ireland), spheronization, nanosuspensions and solid-lipid nanoparticles, the areas of vigorous present growth, would still have higher success rate because of their simplicity and easy to implement (e.g., In Reference 504). Targeted NV, however, bring in novel features which cannot be substituted by the above-mentioned means. The NV will be in the center of attention, particularly in the area of development of new therapies against cancer. A convenient summary listing examples of application and of marketed nanovehiclular products is reported in Tables 8 and 9. In general, this field is in early phase of development and number of years away from maturity.
Table 8.
Examples of Nanovehicle Applications/Products
| Type of Product/Generic Vehicle | References |
|---|---|
| Nanosuspension/different lipophilic drugs | In Reference 505 |
| PLGA nanoparticle/lipophilic drugs | In Reference 289 |
| PACA nanoparticle/lipophilic drugs | In Reference 293 |
| PEC nanoparticle/proteins | In Reference 298 |
| Polymeric micelle/lipidic drugs | In Reference 302 |
| Dendrimer/different drugs | In Reference 316 |
| Liposome/lipophilic drugs | In Reference 324 |
| CPP peptide/different cargo | In Reference 356 |
| Polyplex (PEI)/gene | In Reference 506 |
| Lipoplex-liposome/lipophilic drugs | In Reference 507 |
| Molecular diagnostic | In Reference 257 |
| Radiotherapeutic | In Reference 508 |
| Imaging agent | In Reference 262 |
| Gold nanoparticle biolistic gene delivery | In Reference 391 |
| Antisense nucleotide | In Reference 249 |
| Ribozyme | In Reference 250 |
| DNAzyme | In Reference 251 |
| Aptamer | In Reference 252 |
| siRNA | In Reference 254 |
PLGA—polylactate/glycolate; PACA—polyalkylcyanoacrylate; PEC—polyeletrolyte complex; CPP—cell permeating peptides; PEI—polyethylenemine.
Table 9.
Examples of Marketed or Tested Nanovehicular Products
| Product Designation | Application | Company/References |
|---|---|---|
| Endorem® | Supermagnetic iron oxide MRI imaging agent | Guebert CCL, Bloomington, IN |
| ABI-007, Abraxane™ | Paclitaxel crystals in albumin matrix/cancer | American BioScience, Inc., Santa Monica, CA |
| NanoCrystal® Technology | Generic | Elan, Dublin, Ireland |
| Rapamune® (Sirolimus) | Immunosuppressant rapamycin | Elan/Wyeth, Madison, NJ |
| Emend® | Antinausea drug | Elan/Merck & Co, Whitehouse Station, NJ |
| NanoEdge | Generic | Baxter Health Corp., Round Lake, IL |
| Transdrug | Phase I clinical/doxorubicin/cancer | BioalliancePharma, Paris, France |
| Polymer micelle | Cisplatin in clinical trial/cancer | In Reference 305 |
| PAMAM-dendrimer | MRI contrast agent | In Reference 317 |
| Viva-gel | Phase I/microbicide | In Reference 321 |
| Nanobase™ | Solid-lipid nanoparticles/cosmetic | Yamanouchi, Japan on Polish market |
| DaunoXome® | Doxorubicin/cancer | Gilead Sciences, Ltd, Cambridge, UK |
| Doxil®/Caelyx™ | Doxorubicin/cancer | Johnson & Johnson, Bridgewater, NJ |
| Myocet® | Doxorubicin/cancer | Sepherion Therapeutics, Princeton, NJ |
| Amphotec® | Amphotericin B/fungal | Amphotec, Beverly, MA |
| PK2/FCE28069 | HPMA-copolymer-doxorubicin-galactosamine/cancer | In Reference 349 |
| TNP-470, ciplostatin | Antiangiogenic TNP-470-HPMA conjugate | In Reference 350 |
| Medusa™ | Amphiphilic polymer, generic technology/water-soluble drugs | Flamel Technologies, Venissieux, France |
| Vitravene | Antisense oligonucleotide for AIDS-related CMV retinitis | ISIS Pharmaceuticals Inc, Carlsbad, CA |
| Gendicine | Injectable AdV particles carrying p53 gene | Approved in China |
| Rexin-G | Injectable retroviral vector carrying a mutant form of the cyclin G1 gene | Epeius Biotechnologies Corp., Glendale, CA |
| Accell® | Gene gun/generic | PowderJet Vaccines, Inc., Middleton, WI |
| Helios™ | Gene gun/generic | Bio-Rad Laboratories, Hercules CA |
PAMA—polyamidoamine; MRI—magnetic resonance imaging; HPMA—hydroxypropyl metacrylate; CMV—cytomegalovirus.
Biomimetic NV will also gain future attention. Synthetic materials with biomimetic properties are attracting growing attention as possible new dosage formulations and the potential applications of these increasingly sophisticated polymers in cell-specific drug targeting and in gene therapy. A design of structured material based on peptide mimetics (peptide-amphiphile) has been suggested.509 Delivery nanovector design based on amphiphilic polypeptide has been documented.372 NanoCarrier Co, Ltd, Tokyo, Japan, is developing a micellar nanovehicular system, composed of copolymers of polyamino acids of the hydrophilic and hydrophobic type (MediCelle™ cisplatin anticancer formulation, suitable for other water-soluble drugs). Mimicking nature is an approach for developing delivery of therapeutics using active or “smart” polymers.510,511
The adjustable size of NV to a rather narrow window of size determined by the size of blood vessels (both by the upper and lower limits of normal and pathological vasculature) and epithelial barrier openings in case of oral applications and an opportunity of targeting precludes other delivery vehicles to gain a strong footing in future applications. In all cases, the safety considerations will control an ultimate application and its success. Because of rather steep cost of preclinical toxicology testing of novel delivery vehicles, the pace of implementation of new delivery modes will not be that high. Since NV have new and unique biologic properties because they are not the same as those of the bulk material of the same chemistry, they may represent a potential risk. Physical and chemical properties of materials can significantly improve or radically change as their size is scaled down to small clusters of atoms. Greater surface are per mass renders NV more active biologically than larger-sized particles of the same chemistry particle surface area appear to be better predictor for inflammatory and oxidative responses (to be tested on one-to-one basis).512 On the other hand, large surface area-to-volume ratio of NV (and chemical groups on their periphery) offers potential for designing multifunctional nanosystems.
Other considerations, such as patent expiry, may help to drive the introduction of new or re-formulated drug entities and place them on the market. Besides, large-scale manufacturing (scale-up) is most critical. Also, the batch-to-batch uniformity of NV preparation must be assured. The material requirements for Phase II clinical trials may define the first scale-up target, followed up by market demand. Residual solvents in case solvent-based methods are used to manufacture NV must be minimized via a complete evaporation in the final preparation. The quantity of the residual solvents should be below the prescribed limits. Issues relating to NV clearance and tolerance do need to be investigated.
Nanoscale-based delivery strategies are beginning to make a significant impact on global pharmaceutical planning and marketing (life cycle management).513–515 Such strategies have solid commercial prospects, but the process of converting basic discoveries into marketable products will be long and hard. Chavanpatti et al.516 state that NV have shown “enormous potential for sustained cellular drug and gene delivery. This is because of their ability to increase the cellular uptake of encapsulated therapeutic agents.” We will conclude with rather ambivalent comments. “After early attempts,517 Petrak518 points out that “four decades of research have not yet produced an effective, generally applicable, site-targeted drug delivery system.” He also states that “very little progress has been made in developing targeted drug-delivery systems for effective and realistic human therapy.” “Many recently proposed drug-delivery systems, such as liposomes and nanoparticles, have been linked to the promise of new advanced therapies…However, none have been able to offer a practical useful solution.” “So much is expected from the material, and particular macromolecular science to help to realize this dream.”…“It will need to progress in full recognition of all the requirements biology places on the acceptability of exogenous materials.” We can only agree with the latter statement.
ACKNOWLEDGMENTS
We acknowledge support of National Institutes of Health Grant 1R01EB002825-01 (J.M.D. and A.P.) and support from the Department of Veterans Affairs (J.M.D.). Authors also appreciate input and valuable suggestions from two anonymous reviewers.
REFERENCES
- 1.Kostarelos K. Rational design and engineering of delivery systems for therapeutics: Biomedical exercises in colloid and surface science. Adv Colloid Interface Sci. 2003;106:147–168. doi: 10.1016/s0001-8686(03)00109-x. [DOI] [PubMed] [Google Scholar]
- 2.Moses MA, Brem H, Langer R. Advancing the field of drug delivery: Taking aim at cancer. Cancer Cell. 2003;4:337–341. doi: 10.1016/s1535-6108(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 3.Shargel L, Yu AB. Applied biopharmaceutics & pharmacokinetics. 4th edition McGraw-Hill; New York: 1999. [Google Scholar]
- 4.van de Waterbeemd H, Gifford E. ADMET in silico modelling: Towards prediction paradise? Nat Rev Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- 5.Okada H, Toguchi H. Biodegradable microspheres in drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12:1–99. doi: 10.1615/critrevtherdrugcarriersyst.v12.i1.10. [DOI] [PubMed] [Google Scholar]
- 6.Varde NK, Pack DW. Microspheres for controlled release drug delivery. Expert Opin Biol Ther. 2004;4:35–51. doi: 10.1517/14712598.4.1.35. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kohane DS. Microparticles and nanoparticles for drug delivery. Biotechnol Bioeng. 2006;96:203–209. doi: 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]; (b) Kasturi SP, Sachaphibulkij K, Roy K. Covalent conjugation of polyethyleneimine on bio-degradable microparticles for delivery of plasmid DNA vaccines. Biomaterials. 2005;26:6375–6385. doi: 10.1016/j.biomaterials.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 8.Vicent MJ, Duncan R. Polymer conjugates: Nanosized medicines for treating cancer. Trends Biotechnol. 2006;24:39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Lee CC, MacKay JA, Frechet JM, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 10.Sato T, Sunamoto J. Recent aspects in the use of liposomes in biotechnology and medicine. Prog Lipid Res. 1992;31:345–372. doi: 10.1016/0163-7827(92)90001-y. [DOI] [PubMed] [Google Scholar]
- 11.Bendas G. Immunoliposomes: A promising approach to targeting cancer therapy. BioDrugs. 2001;15:215–224. doi: 10.2165/00063030-200115040-00002. [DOI] [PubMed] [Google Scholar]
- 12.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Colloidal gold: A novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004;11:169–183. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 13.Akerman ME, Chan WC, Laakkonen P, Bhatia SN, Ruoslahti E. Nanocrystal targeting in vivo. Proc Natl Acad Sci USA. 2002;99:12617–12621. doi: 10.1073/pnas.152463399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen JF, Ding HM, Wang JX, Shao L. Preparation and characterization of porous hollow silica nanoparticles for drug delivery application. Biomaterials. 2004;25:723–727. doi: 10.1016/s0142-9612(03)00566-0. [DOI] [PubMed] [Google Scholar]
- 15.Ito A, Shinkai M, Honda H, Kobayashi T. Medical application of functionalized magnetic nanoparticles. J Biosci Bioeng. 2005;100:1–11. doi: 10.1263/jbb.100.1. [DOI] [PubMed] [Google Scholar]
- 16.Arias JL, López-Viota M, Ruiz MA, López-Viota J, Delgado AV. Development of carbonyl iron/ethylcellulose core/shell nanoparticles for biomedical applications. Int J Pharmaceut. 2007;339:237–245. doi: 10.1016/j.ijpharm.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari M. Cancer nanotechnology: Opportunities and challenges. Nat Rev Cancer. 2005a;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 18.Petrak K. Essential properties of drug-targeting delivery systems. Drug Discov Today. 2005;10:1667–1673. doi: 10.1016/S1359-6446(05)03698-6. [DOI] [PubMed] [Google Scholar]
- 19.Opanasopit P, Nishikawa M, Hashida M. Factors affecting drug and gene delivery: Effects of interaction with blood components. Crit Rev Ther Drug Carrier Syst. 2002;19:191–233. doi: 10.1615/critrevtherdrugcarriersyst.v19.i3.10. [DOI] [PubMed] [Google Scholar]
- 20.Gabriele S-G. Liver contrast media for magnetic resonance imaging: Interactions between pharmacokinetics and imaging. Invest Radiol. 1993;28:753–761. doi: 10.1097/00004424-199308000-00018. [DOI] [PubMed] [Google Scholar]
- 21.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 22.Boddy A, Aarons L, Petrak K. Efficiency of drug targeting: Steady-state considerations using a three-compartment model. Pharm Res. 1989;6:367–372. doi: 10.1023/a:1015971113161. [DOI] [PubMed] [Google Scholar]
- 23.Morgan PJ, Harding SE, Petrak K. Interactions of a model block copolymer drug delivery system with two serum proteins and myoglobin. Biochem Soc Trans. 1990;18:1021–1022. doi: 10.1042/bst0181021. [DOI] [PubMed] [Google Scholar]
- 24.O'Mullane JE, Davison CJ, Petrak K, Tomlinson E. Adsorption of fibrinogen on to polystyrene latex coated with the non-ionic surfactant, poloxamer 338. Biomaterials. 1988;9:203–204. doi: 10.1016/0142-9612(88)90124-x. [DOI] [PubMed] [Google Scholar]
- 25.Ayhan H, Tuncel A, Bor N, Piskin E. Phagocytosis of monosize polystyrene-based microspheres having different size and surface properties. J Biomater Sci Polym Ed. 1995;7:329–342. doi: 10.1163/156856295x00355. [DOI] [PubMed] [Google Scholar]
- 26.Saltzman WM. Drug delivery: Engineering principles for drug therapy. Oxford University Press; New York: 2001. [Google Scholar]
- 27.Mager DE. Target-mediated drug disposition and dynamics. Biochem Pharmacol. 2006;72:1–10. doi: 10.1016/j.bcp.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 28.Saga T, Neumann RD, Heya T, Sato J, Kinuya S, Le N, Paik CH, Weinstein JN. Targeting cancer micrometastases with monoclonal antibodies: A binding-site barrier. Proc Natl Acad Sci USA. 1995;92:8999–9003. doi: 10.1073/pnas.92.19.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kostarelos K, Emfietzoglou D, Papakostas A, Yang WH, Ballangrud A, Sgouros G. Binding and interstitial penetration of liposomes within avascular tumor spheroids. Int J Cancer. 2004;112:713–721. doi: 10.1002/ijc.20457. [DOI] [PubMed] [Google Scholar]
- 30.Hawley AE, Davis SS, Illum L. Targeting of colloids to lymph nodes: Influence of lymphatic physiology and colloidal characteristics. Advan Drug Del Rev. 1995;17:129–148. [Google Scholar]
- 31.(a) Moghimi SM, Rajabi-Siahboomi R. Advanced colloid-based systems for efficient delivery of drugs and diagnostic agents to the lymphatic tissues. Prog Biophys Mol Biol. 1996;65:221–249. doi: 10.1016/s0079-6107(96)00012-0. [DOI] [PubMed] [Google Scholar]; (b) Mager DE, Neuteboom B, Jusko WJ. Pharmacokinetics and pharmacodynamics of PEGylated IFN-beta 1a following subcutaneous administration in monkeys. Pharm Res. 2005;22:58–61. doi: 10.1007/s11095-004-9009-z. [DOI] [PubMed] [Google Scholar]
- 32.Galindo-Rodriguez SA, Allemann E, Fessi H, Doelker E. Polymeric nanoparticles for oral delivery of drugs and vaccines: A critical evaluation of in vivo studies. Crit Rev Ther Drug Carrier Syst. 2005;22:419–464. doi: 10.1615/critrevtherdrugcarriersyst.v22.i5.10. [DOI] [PubMed] [Google Scholar]
- 33.Papisov M, Weissleder R. Drug delivery to lymphatic tissue. Crit Rev Ther Drug Carrier Syst. 1996;13:57–84. [PubMed] [Google Scholar]
- 34.Ferl GZ, Kenanova V, Wu AM, DiStefano JJ., III A two-tiered physiologically based model for dually labeled single-chain Fv-Fc antibody fragments. Mol Cancer Ther. 2006;5:1550–1558. doi: 10.1158/1535-7163.MCT-06-0072. [DOI] [PubMed] [Google Scholar]
- 35.Flessner MF. Peritoneal transport physiology: Insights from basic research. J Am Soc Nephrol. 1991;2:122–135. doi: 10.1681/ASN.V22122. [DOI] [PubMed] [Google Scholar]
- 36.McLennan DN, Porter CJ, Edwards GA, Martin SW, Heatherington AC, Charman SA. Lymphatic absorption is the primary contributor to the systemic availability of epoetin Alfa following subcutaneous administration to sheep. J Pharmacol Exp Ther. 2005;313:345–351. doi: 10.1124/jpet.104.078790. [DOI] [PubMed] [Google Scholar]
- 37.Park YS. Tumor-directed targeting of liposomes. Biosci Rep. 2002;22:267–281. doi: 10.1023/a:1020190606757. [DOI] [PubMed] [Google Scholar]
- 38.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56:1177–1192. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 39.van Vlerken LE, Amiji MM. Multi-functional polymeric nanoparticles for tumour-targeted drug delivery. Expert Opin Drug Deliv. 2006;3:205–216. doi: 10.1517/17425247.3.2.205. [DOI] [PubMed] [Google Scholar]
- 40.King GL, Feener EP. The biochemical and physiological characteristics of receptors. Adv Drug Deliv Rev. 1998;29:197–213. doi: 10.1016/s0169-409x(97)00079-3. [DOI] [PubMed] [Google Scholar]
- 41.Erdogan S, Roby A, Torchilin VP. Enhanced tumor visualization by gamma-scintigraphy with 111In-labeled polychelating-polymer-containing immunoliposomes. Mol Pharm. 2006;3:525–530. doi: 10.1021/mp060055t. [DOI] [PubMed] [Google Scholar]
- 42.Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res. 2006;16:175–183. doi: 10.1080/08982100600848769. [DOI] [PubMed] [Google Scholar]
- 43.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 44.Colburn WA, Lee JW. Biomarkers, validation and pharmacokinetic-pharmacodynamic modelling. Clin Pharmacokinet. 2003;42:997–1022. doi: 10.2165/00003088-200342120-00001. [DOI] [PubMed] [Google Scholar]
- 45.Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 46.Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR., Jr. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 47.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66:6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 48.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 49.Lopes de Menezes DE, Pilarski LM, Allen TM. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res. 1998;58:3320–3330. [PubMed] [Google Scholar]
- 50.Tardi P, Ickenstein L, Bally M, Mayer L. The development of liposomes for enhanced delivery of chemotherapeutics to tumors. In: Page M, editor. Cancer drug discovery and development: Tumor targeting in cancer therapy. Humana Press; Totowa, NJ: 2002. pp. 119–135. [Google Scholar]
- 51.Klapper LN, Vaisman N, Hurwitz E, Pinkas-Kramarski R, Yarden Y, Sela M. A subclass of tumor-inhibitory monoclonal antibodies to ErbB-2/HER2 blocks crosstalk with growth factor receptors. Oncogene. 1997;14:2099–2109. doi: 10.1038/sj.onc.1201029. [DOI] [PubMed] [Google Scholar]
- 52.Hurwitz E, Stancovski I, Sela M, Yarden Y. Suppression and promotion of tumor growth by monoclonal antibodies to ErbB-2 differentially correlate with cellular uptake. Proc Natl Acad Sci USA. 1995;92:3353–3357. doi: 10.1073/pnas.92.8.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nielsen UB, Kirpotin DB, Pickering EM, Drummond DC, Marks JD. A novel assay for monitoring internalization of nanocarrier coupled antibodies. BMC Immunol. 2006;7:24. doi: 10.1186/1471-2172-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: In vitro and in vivo studies. Br J Cancer. 1996;74:1749–1756. doi: 10.1038/bjc.1996.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hosokawa S, Tagawa T, Niki H, Hirakawa Y, Nohga K, Nagaike K. Efficacy of immunoliposomes on cancer models in a cell-surface-antigen-density-dependent manner. Br J Cancer. 2003;89:1545–1551. doi: 10.1038/sj.bjc.6601341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.(a) Sapra P, Allen TM. Improved outcome when B-cell lymphoma is treated with combinations of immunoliposomal anticancer drugs targeted to both the CD19 and CD20 epitopes. Clin Cancer Res. 2004;10:2530–2537. doi: 10.1158/1078-0432.ccr-03-0376. Erratum in: Clin Cancer Res 10:4893. [DOI] [PubMed] [Google Scholar]; (b) Sapra P, Allen TM. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002;62:7190–7194. [PubMed] [Google Scholar]
- 57.Kurihara A, Pardridge WM. Imaging brain tumors by targeting peptide radiopharmaceuticals through the blood-brain barrier. Cancer Res. 1999;59:6159–6163. [PubMed] [Google Scholar]
- 58.Brandli AW, Adamson ED, Simons K. Transcytosis of epidermal growth factor. The epidermal growth factor receptor mediates uptake but not transcytosis. J Biol Chem. 1991;266:8560–8566. [PubMed] [Google Scholar]
- 59.Shah D, Shen WC. Transcellular delivery of an insulin-transferrin conjugate in enterocyte-like Caco-2 cells. J Pharm Sci. 1996;85:1306–1311. doi: 10.1021/js9601400. [DOI] [PubMed] [Google Scholar]
- 60.Olivier JC. Drug transport to brain with targeted nanoparticles. NeuroRx. 2005;2:108–119. doi: 10.1602/neurorx.2.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochim Biophys Acta. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 62.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Hitting multiple targets with multimeric ligands. Expert Opin Ther Targets. 2004;8:565–586. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 63.de la Fuente JM, Penades S. Understanding carbohydrate-carbohydrate interactions by means of glyconanotechnology. Glycoconj J. 2004;21:149–163. doi: 10.1023/B:GLYC.0000044846.80014.cb. [DOI] [PubMed] [Google Scholar]
- 64.Shaunak S, Thomas S, Gianasi E, Godwin A, Jones E, Teo I, Mireskandari K, Luthert P, Duncan R, Patterson S, Khaw P, Brocchini S. Polyvalent dendrimer glucosamine conjugates prevent scar tissue formation. Nat Biotechnol. 2004;2:977–984. doi: 10.1038/nbt995. [DOI] [PubMed] [Google Scholar]
- 65.Plank C, Oberhauser B, Mechtler K, Koch C, Wagner E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J Biol Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- 66.Dokka S, Rojanasakul Y. Novel non-endocytic delivery of antisense oligonucleotides. Adv Drug Deliv Rev. 2000;44:35–49. doi: 10.1016/s0169-409x(00)00082-x. [DOI] [PubMed] [Google Scholar]
- 67.Zauner W, Farrow NA, Haines AM. In vitro uptake of polystyrene microspheres: Effect of particle size, cell line, and cell density. J Control Release. 2001;71:39–51. doi: 10.1016/s0168-3659(00)00358-8. [DOI] [PubMed] [Google Scholar]
- 68.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:2713–2722. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 70.Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: Effect of particle size. Pharm Res. 1996;13:1838–1845. doi: 10.1023/a:1016085108889. [DOI] [PubMed] [Google Scholar]
- 71.(a) Hartig S, Carlesso G, Davidson JM, Prokop A. Improved polyelectrolyte complex physico-chemistry by non-stoichiometric mixing of polyions with similar molecular weights. Biomacromol. 2006;8:265–272. doi: 10.1021/bm0604754. [DOI] [PubMed] [Google Scholar]; (b) Hartig SM, Greene R, Carlesso GL, Higginbotham JN, Khan WN, Prokop A, Davidson JM. Kinetic analysis of nanoparticulate polyelectrolyte complex interactions with endothelial cells by flow cytometry. Biomaterials. 2007;28:3843–3855. doi: 10.1016/j.biomaterials.2007.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.(a) Carlesso GL, Hartig SM, Higginbotham JN, Kozlov E, Roberts DD, Prokop A, Davidson JM. Enhancement of binding and gene delivery to endothelial cells by targeted nanoparticulate polyelectrolyte complexes. Mol Ther (Submitted for publication) 2007 [Google Scholar]; (b) Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 73.Almofti MR, Harashima H, Shinohara Y, Almofti A, Li W, Kiwada H. Lipoplex size determines lipofection efficiency with or without serum. Mol Membr Biol. 2003;20:35–43. doi: 10.1080/09687680210035104. [DOI] [PubMed] [Google Scholar]
- 74.Winter PM, Caruthers SD, Kassner A, Harris TD, Chinen LK, Allen JS, Lacy EK, Zhang H, Robertson JD, Wickline SA, Lanza GM. Molecular imaging of angiogenesis in nascent Vx-2 rabbit tumors using a novel alpha(nu)beta3-targeted nanoparticle and 1.5 tesla magnetic resonance imaging. Cancer Res. 2003;63:5838–5843. [PubMed] [Google Scholar]
- 75.Senior J, Crawley JC, Gregoriadis G. Tissue distribution of liposomes exhibiting long half-lives in the circulation after intravenous injection. Biochim Biophys Acta. 1985;839:1–8. doi: 10.1016/0304-4165(85)90174-6. [DOI] [PubMed] [Google Scholar]
- 76.Dams ET, Laverman P, Oyen WJ, Storm G, Scherphof GL, van Der Meer JW, Corstens FH, Boerman OC. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. 2000;292:1071–1079. [PubMed] [Google Scholar]
- 77.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. [PubMed] [Google Scholar]
- 78.Moghimi SM, Bonnemain B. Subcutaneous and intravenous delivery of diagnostic agents to the lymphatic system: Applications in lymphoscintigraphy and indirect lymphography. Adv Drug Deliv Rev. 1999;37:295–312. doi: 10.1016/s0169-409x(98)00099-4. [DOI] [PubMed] [Google Scholar]
- 79.Moghimi SM. Exploiting bone marrow microvascular structure for drug delivery and future therapies. Advan Drug Del Rev. 1995;17:61–73. [Google Scholar]
- 80.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: Current status and future prospects. FASEB J. 2005;19:311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 81.Awasthi VD, Garcia D, Goins BA, Phillips WT. Circulation and biodistribution profiles of long-circulating PEG-liposomes of various sizes in rabbits. Int J Pharm. 2003;253:121–132. doi: 10.1016/s0378-5173(02)00703-2. [DOI] [PubMed] [Google Scholar]
- 82.Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly(D,L-lactide-coglycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003a;20:212–220. doi: 10.1023/a:1022219003551. [DOI] [PubMed] [Google Scholar]
- 83.Hartig SM, Greene R, Dikov MM, Prokop A, Davidson JM. Multifunctional nanoparticulate polyelectrolyte complexes. Pharm Res. 2007;24(12):2353–2369. doi: 10.1007/s11095-007-9459-1. Epub 2007 Oct 12. [DOI] [PubMed] [Google Scholar]
- 84.Panyam J, Labhasetwar V. Targeting intracellular targets. Curr Drug Deliv. 2004;1:235–247. doi: 10.2174/1567201043334768. [DOI] [PubMed] [Google Scholar]
- 85.Owens DE, III, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 86.Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetwar V. Fluorescence and electron microscopy probes for cellular and tissue uptake of poly(D,L-lactide-co-glycolide) nanoparticles. Int J Pharm. 2003b;262:1–11. doi: 10.1016/s0378-5173(03)00295-3. [DOI] [PubMed] [Google Scholar]
- 87.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2002;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 88.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 89.Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. Receptor-mediated endocytosis: Concepts emerging from the LDL receptor system. Annu Rev Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 90.Sangiorgio V, Pitto M, Palestini P, Masserini M. GPI-anchored proteins and lipid rafts. Ital J Biochem. 2004;53:98–111. [PubMed] [Google Scholar]
- 91.Ferrari A, Pellegrini V, Arcangeli C, Fittipaldi A, Giacca M, Beltram F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol Ther. 2003;8:284–294. doi: 10.1016/s1525-0016(03)00122-9. [DOI] [PubMed] [Google Scholar]
- 92.Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- 93.Urban S, Zieseniss S, Werder M, Hauser H, Budzinski R, Engelmann B. Scavenger receptor BI transfers major lipoprotein-associated phospholipids into the cells. J Biol Chem. 2000;275:33409–33415. doi: 10.1074/jbc.M004031200. [DOI] [PubMed] [Google Scholar]
- 94.Fabriek BO, Dijkstra CD, van den Berg TK. The macrophage scavenger receptor CD163. Immunobiology. 2005;210:153–160. doi: 10.1016/j.imbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 95.Han HD, Lee A, Song CK, Hwang T, Seong H, Lee CO, Shin BC. In vivo distribution and antitumor activity of heparin-stabilized doxorubicin-loaded liposomes. Int J Pharm. 2006;313:181–188. doi: 10.1016/j.ijpharm.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 96.Adrian JE, Poelstra K, Scherphof GL, Molema G, Meijer DK, Reker-Smit C, Morselt HW, Kamps JA. Interaction of targeted liposomes with primary cultured hepatic stellate cells: Involvement of multiple receptor systems. J Hepatol. 2006;44:560–567. doi: 10.1016/j.jhep.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 97.Yan X, Kuipers F, Havekes LM, Havinga R, Dontje B, Poelstra K, Scherphof GL, Kamps JA. The role of apolipoprotein E in the elimination of liposomes from blood by hepatocytes in the mouse. Biochem Biophys Res Commun. 2005;328:57–62. doi: 10.1016/j.bbrc.2004.12.137. Erratum in: Biochem Biophys Res Commun (2005), 330: 1320. [DOI] [PubMed] [Google Scholar]
- 98.Kunisawa J, Masuda T, Katayama K, Yoshikawa T, Tsutsumi Y, Akashi M, Mayumi T, Nakagawa S. Fusogenic liposome delivers encapsulated nanoparticles for cytosolic controlled gene release. J Control Release. 2005;105:344–3453. doi: 10.1016/j.jconrel.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 99.Holthuis JC, Levine TP. Lipid traffic: Floppy drives and a superhighway. Nat Rev Mol Cell Biol. 2005;6:209–220. doi: 10.1038/nrm1591. [DOI] [PubMed] [Google Scholar]
- 100.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in oncoviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8. [DOI] [PubMed] [Google Scholar]
- 101.Kirkham M, Parton RG. Clathrin-independent endocytosis: New insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1746:349–363. doi: 10.1016/j.bbamcr.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 102.Muro S, Cui X, Gajewski C, Murciano JC, Muzykantov VR, Koval M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am J Physiol Cell. 2003;285:C1339–C1347. doi: 10.1152/ajpcell.00099.2003. [DOI] [PubMed] [Google Scholar]
- 103.Muro S, Koval M, Muzykantov V. Endothelial endocytic pathways: Gates for vascular drug delivery. Curr Vasc Pharmacol. 2004;2:281–299. doi: 10.2174/1570161043385736. [DOI] [PubMed] [Google Scholar]
- 104.(a) Ding BS, Dziubla T, Shuvaev VV, Muro S, Muzykantov VR. Advanced drug delivery systems that target the vascular endothelium. Mol Interv. 2006;6:98–112. doi: 10.1124/mi.6.2.7. [DOI] [PubMed] [Google Scholar]; (b) Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm. 2005;2:373–383. doi: 10.1021/mp050032z. [DOI] [PubMed] [Google Scholar]
- 105.Salaun C, James DJ, Chamberlain LH. Lipid rafts and the regulation of exocytosis. Traffic. 2004;5:255–264. doi: 10.1111/j.1600-0854.2004.0162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ambudkar SV, Kim I-V, Sauna ZE. The power of the pump: Mechanisms of action of P-glycoprotein (ABCB1) Europ J Pharmaceut Sci. 2006;27:392–400. doi: 10.1016/j.ejps.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 107.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multi-drug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 108.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Europ J Pharmaceut Sci. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 109.Florence AT, Hussain N. Transcytosis of nanoparticle and dendrimer delivery systems: Evolving vistas. Adv Drug Deliv Rev. 2001;50:S69–89. doi: 10.1016/s0169-409x(01)00184-3. [DOI] [PubMed] [Google Scholar]
- 110.Watson P, Jones AT, Stephens DJ. Intracellular trafficking pathways and drug delivery: Fluorescence imaging of living and fixed cells. Adv Drug Deliv Rev. 2005;57:43–61. doi: 10.1016/j.addr.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 111.Zeng Y, Tao N, Chung KN, Heuser JE, Lublin DM. Endocytosis of oxidized low density lipoprotein through scavenger receptor CD36 utilizes a lipid raft pathway that does not require caveolin-1. J Biol Chem. 2003;278:45931–45936. doi: 10.1074/jbc.M307722200. [DOI] [PubMed] [Google Scholar]
- 112.Levchenko TS, Rammohan R, Volodina N, Torchilin VP. Tat peptide-mediated intracellular delivery of liposomes. Method Enzymol. 2003;372:339–349. doi: 10.1016/s0076-6879(03)72019-9. [DOI] [PubMed] [Google Scholar]
- 113.Sharma P, Sabharanjak S, Mayor S. Endocytosis of lipid rafts: An identity crisis. Semin Cell Dev Biol. 2002;13:205–214. doi: 10.1016/s1084-9521(02)00049-6. [DOI] [PubMed] [Google Scholar]
- 114.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 115.(a) Johannes L, Lamaze C. Clathrin-dependent or not: Is it still the question? Traffic. 2002;3:443–451. doi: 10.1034/j.1600-0854.2002.30701.x. [DOI] [PubMed] [Google Scholar]; (b) Mousavi SA, Malerod L, Berg T, Kjeken R. Clathrin-dependent endocytosis. Biochem J. 2004;377:1–16. doi: 10.1042/BJ20031000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Amyere M, Mettlen M, Van Der Smissen P, Platek A, Payrastre B, Veithen A, Courtoy PJ. Origin, originality, functions, subversions and molecular signalling of macropinocytosis. Int J Med Microbiol. 2002;291:487–494. doi: 10.1078/1438-4221-00157. [DOI] [PubMed] [Google Scholar]
- 117.Catizone A, Medolago Albani L, Reola F, Alescio T. A quantitative assessment of non specific pinocytosis by human endothelial cells surviving in vitro. Cell Mol Biol (Noisy-le-grand) 1993;39:155–169. [PubMed] [Google Scholar]
- 118.Behrens I, Pena AI, Alonso MJ, Kissel T. Comparative uptake studies of bioadhesive and non-bioadhesive nanoparticles in human intestinal cell lines and rats: The effect of mucus on particle adsorption and transport. Pharm Res. 2002;19:1185–1193. doi: 10.1023/a:1019854327540. [DOI] [PubMed] [Google Scholar]
- 119.Zhao B, Li Y, Buono C, Waldo SW, Jones NL, Mori M, Kruth HS. Constitutive receptor-independent low density lipoprotein uptake and cholesterol accumulation by macrophages differentiated from human monocytes with macrophage-colony-stimulating factor (M-CSF) J Biol Chem. 2006;281:15757–15762. doi: 10.1074/jbc.M510714200. [DOI] [PubMed] [Google Scholar]
- 120.Brown MS, Goldstein JL. Receptor-mediated control of cholesterol metabolism. Science. 1976;191:150–154. doi: 10.1126/science.174194. [DOI] [PubMed] [Google Scholar]
- 121.Dobrian A, Lazar V, Tirziu D, Simionescu M. Increased macrophage uptake of irreversibly glycated albumin modified-low density lipoproteins of normal and diabetic subjects is mediated by nonsaturable mechanisms. Biochim Biophys Acta. 1996;1317:5–14. doi: 10.1016/0925-4439(96)00017-8. [DOI] [PubMed] [Google Scholar]
- 122.Panagi Z, Beletsi A, Evangelatos G, Livaniou E, Ithakissios DS, Avgoustakis K. Effect of dose on the biodistribution and pharmacokinetics of PLGA and PLGA-mPEG nanoparticles. Int J Pharm. 2001;221:143–152. doi: 10.1016/s0378-5173(01)00676-7. [DOI] [PubMed] [Google Scholar]
- 123.Berk PD, Stump DD. Mechanisms of cellular uptake of long chain free fatty acids. Mol Cell Biochem. 1999;192:17–31. [PubMed] [Google Scholar]
- 124.Szleifer I. Protein adsorption on surfaces with grafted polymers: A theoretical approach. Biophys J. 1997;72:595–612. doi: 10.1016/s0006-3495(97)78698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moghimi SM. Liposome recognition by resident and newly recruited murine liver macrophages. J Liposome Res. 2002;12:67–70. doi: 10.1081/lpr-120004778. [DOI] [PubMed] [Google Scholar]
- 126.Kunath K, Merdan T, Hegener O, Haberlein H, Kissel T. Integrin targeting using RGD-PEI conjugates for in vitro gene transfer. J Gene Med. 2003;5:588–599. doi: 10.1002/jgm.382. [DOI] [PubMed] [Google Scholar]
- 127.Dauty E, Remy J-S, Zuber G, Behr J-P. Intracellular delivery of nanometric DNA particles via the folate receptor. Bioconjugate Chem. 2002;13:831–839. doi: 10.1021/bc0255182. [DOI] [PubMed] [Google Scholar]
- 128.Lin JJ, Silas JA, Bermudez H, Milam VT, Bates FS, Hammer DA. The effect of polymer chain length and surface density on the adhesiveness of functionalized polymersomes. Langmuir. 2004b;20:5493–5500. doi: 10.1021/la036417a. [DOI] [PubMed] [Google Scholar]
- 129.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci USA. 2001;98:8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen CC, Dormidontova EE. Architectural and structural optimization of the protective polymer layer for enhanced targeting. Langmuir. 2005;21:5605–5615. doi: 10.1021/la047109v. [DOI] [PubMed] [Google Scholar]
- 131.Laverman P, Boerman OC, Oyen WJG, Corstens FHM, Storm G. In vivo applications of PEG liposomes: Unexpected observations. Crit Rev Ther Drug Carrier Syst. 2001;18:551–566. [PubMed] [Google Scholar]
- 132.Ho SP, Nakabayashi N, Iwasaki Y, Boland T, LaBerge M. Frictional properties of poly (MPC-co-BMA) phospholipid polymer for catheter applications. Biomaterials. 2003;24:5121–5129. doi: 10.1016/s0142-9612(03)00450-2. [DOI] [PubMed] [Google Scholar]
- 133.Iwasaki Y, Shibata N, Ninomiya M, Kurita K, Nakabayash N, Ishihara K. Importance of a biofouling-resistant phospholipid polymer to create a heparinized blood-compatible surface. J Biomater Sci Polym Ed. 2002a;13:323–335. doi: 10.1163/156856202320176556. [DOI] [PubMed] [Google Scholar]
- 134.Iwasaki Y, Sawada S, Ishihara K, Khang G, Lee HB. Reduction of surface-induced inflammatory reaction on PLGA/MPC polymer blend. Biomaterials. 2002b;23:3897–3903. doi: 10.1016/s0142-9612(02)00135-7. [DOI] [PubMed] [Google Scholar]
- 135.Konno T, Kurita K, Iwasaki Y, Nakabayashi N, Ishihara K. Preparation of nanoparticles composed with bioinspired 2-methacryloyloxyethyl phosphorylcholine polymer. Biomaterials. 2001;22:1883–1889. doi: 10.1016/s0142-9612(00)00373-2. [DOI] [PubMed] [Google Scholar]
- 136.Lewis AL. PC Technology as a platform for drug delivery: from combination to conjugation. Expert Opin Drug Deliv. 2006;3:289–298. doi: 10.1517/17425247.3.2.289. [DOI] [PubMed] [Google Scholar]
- 137.Pincus P. Colloid stabilization with grafted polyelectrolytes. Macromol. 1991;24:2912–2919. [Google Scholar]
- 138.Finsinger D, Remy JS, Erbacher P, Koch C, Plank C. Protective copolymers for nonviral gene vectors: Synthesis, vector characterization and application in gene delivery. Gene Ther. 2000;7:1183–1192. doi: 10.1038/sj.gt.3301227. [DOI] [PubMed] [Google Scholar]
- 139.Dash PR, Read ML, Barrett LB, Wolfert MA, Seymour LW. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther. 1999;6:643–650. doi: 10.1038/sj.gt.3300843. [DOI] [PubMed] [Google Scholar]
- 140.(a) Wiethoff CM, Smith JG, Koe GS, Middaugh CR. The potential role of proteoglycans in cationic lipid-mediated gene delivery. Studies of the interacttion of cationic lipid-DNA complexes with model glycosaminoglycans. J Biol Chem. 2001;276:32806–32813. doi: 10.1074/jbc.M007940200. [DOI] [PubMed] [Google Scholar]; (b) Chittimalla C, Zammut-Italiano L, Zuber G, Behr JP. Monomolecular DNA nanoparticles for intravenous delivery of genes. J Am Chem Soc. 2005;127:11436–11441. doi: 10.1021/ja0522332. [DOI] [PubMed] [Google Scholar]
- 141.Prokop A, Holland CA, Kozlov E, Moore B, Tanner RD. Water-based nanoparticulate polymeric system for protein delivery. Biotechnol Bioeng. 2002a;75:228–232. doi: 10.1002/bit.10025. [DOI] [PubMed] [Google Scholar]
- 142.Prokop A, Kozlov E, Carlesso G, Davidson JM. Hydrogel-based colloidal polymeric system for protein and drug delivery: Physical and chemical characterization, permeability control and applications. Advan Polymer Sci. 2002b;160:119–173. [Google Scholar]
- 143.Zelikin AN, Trukhanova ES, Putnam D, Izumrudov VA, Litmanovich AA. Competitive reactions in solutions of poly-L-histidine, calf thymus DNA, and synthetic polyanions: Determining the binding constants of polyelectrolytes. J Am Chem Soc. 2003;125:13693–13699. doi: 10.1021/ja036387y. [DOI] [PubMed] [Google Scholar]
- 144.Oupicky D, Ogris M, Howard KA, Dash PR, Ulbrich K, Seymour LW. Importance of lateral and steric stabilization of polyelectrolyte gene delivery vectors for extended systemic circulation. Mol Ther. 2002;5:463–472. doi: 10.1006/mthe.2002.0568. [DOI] [PubMed] [Google Scholar]
- 145.Ferrari M. Nanovector therapeutics. Curr Opin Chem Biol. 2005b;9:343–346. doi: 10.1016/j.cbpa.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 146.Decuzzi P, Lee S, Bhushan B, Ferrari M. A theoretical model for the margination of particles within blood vessels. Ann Biomed Eng. 2005;33:179–190. doi: 10.1007/s10439-005-8976-5. [DOI] [PubMed] [Google Scholar]
- 147.Zeng QH, Yu AB, Lu GQ, Paul DR. Clay-based polymer nanocomposites: Research and commercial development. J Nanosci Nanotechnol. 2005;5:1574–1592. doi: 10.1166/jnn.2005.411. [DOI] [PubMed] [Google Scholar]
- 148.Roser M, Fischer D, Kissel T. Surface-modified biodegradable albumin nano- and microspheres. II: Effect of surface charges on in vitro phagocytosis and biodistribution in rats. Eur J Pharm Biopharm. 1998;46:255–263. doi: 10.1016/s0939-6411(98)00038-1. [DOI] [PubMed] [Google Scholar]
- 149.Yang SC, Zhu JB. Preparation and characterization of camptothecin solid lipid nanoparticles. Drug Dev Ind Pharm. 2002;28:265–274. doi: 10.1081/ddc-120002842. [DOI] [PubMed] [Google Scholar]
- 150.Furumoto K, Nagayama S, Ogawara K, Takakura Y, Hashida M, Higaki K, Kimura T. Hepatic uptake of negatively charged particles in rats: Possible involvement of serum proteins in recognition by scavenger receptor. J Control Release. 2004;97:133–141. doi: 10.1016/j.jconrel.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 151.Eichhorn ME, Strieth S, Krasnici S, Sauer B, Teifel M, Michaelis U, Naujoks K, Dellian M. Protamine enhances uptake of cationic liposomes in angiogenic microvessels. Angiogenesis. 2004;7:133–141. doi: 10.1007/s10456-004-1428-2. [DOI] [PubMed] [Google Scholar]
- 152.Takagi A, Yabe Y, Oka Y, Takakura Y, Hashida M. Effect of co-administration of cationic macromolecules on the in vivo disposition and in situ renal disposition characteristics of rhIL-11. Drug Metab Pharmacokinet. 2002;17:136–141. doi: 10.2133/dmpk.17.136. [DOI] [PubMed] [Google Scholar]
- 153.Stacy DR, Lu B, Hallahan DE. Radiation-guided drug delivery systems. Expert Rev Anticancer Ther. 2004;4:283–288. doi: 10.1586/14737140.4.2.283. [DOI] [PubMed] [Google Scholar]
- 154.O'Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 155.Roy I, Ohulchanskyy TY, Pudavar HE, Bergey EJ, Oseroff AR, Morgan J, Dougherty TJ, Prasad PN. Ceramic-based nanoparticles entrapping water-insoluble photosensitizing anticancer drugs: A novel drug-carrier system for photodynamic therapy. J Am Chem Soc. 2003;125:7860–7865. doi: 10.1021/ja0343095. [DOI] [PubMed] [Google Scholar]
- 156.Oyewumi MO, Mumper RJ. Engineering tumor-targeted gadolinium hexanedione nanoparticles for potential application in neutron capture therapy. Bioconjug Chem. 2002;13:1328–1335. doi: 10.1021/bc025560x. [DOI] [PubMed] [Google Scholar]
- 157.Bergey EJ, Prasad PN. DC magnetic filed induced magnetocytolysis of cancer cells targeted by LH-RH magnetic nanoparticles in vitro. Biomed Microdev. 2002;4:293–299. [Google Scholar]
- 158.Ishii D, Kinbara K, Ishida Y, Ishii N, Okochi M, Yohda M, Aida T. Chaperonin-mediated stabilization and ATP-triggered release of semiconductor nanoparticles. Nature. 2003;423:628–632. doi: 10.1038/nature01663. [DOI] [PubMed] [Google Scholar]
- 159.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 160.McLoughlin JM, McCarty TM, Cunningham C, Clark V, Senzer N, Nemunaitis J, Kuhn JA. TNFerade, an adenovector carrying the transgene for human tumor necrosis factor alpha, for patients with advanced solid tumors: Surgical experience and long-term follow-up. Ann Surg Oncol. 2005;12:825–830. doi: 10.1245/ASO.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 161.Triggle DJ. Hijacked receptors. Pharm Acta Helv. 2000;74:287–290. doi: 10.1016/s0031-6865(99)00054-0. [DOI] [PubMed] [Google Scholar]
- 162.Juillerat-Jeanneret L, Schmitt F. Chemical modification of therapeutic drugs or drug vector systems to achieve targeted therapy: Looking for the grail. Med Res Rev. 2006;27:574–590. doi: 10.1002/med.20086. [DOI] [PubMed] [Google Scholar]
- 163.Parveen S, Sahoo SK. Nanomedicine: Clinical applications of polyethylene glycol conjugated proteins and drugs. Clin Pharmacokinet. 2006;45:965–988. doi: 10.2165/00003088-200645100-00002. [DOI] [PubMed] [Google Scholar]
- 164.Vyas SP, Singh A, Sihorkar V. Ligand-receptor-mediated drug delivery: An emerging paradigm in cellular drug targeting. Crit Rev Ther Drug Carrier Syst. 2001;18:1–76. [PubMed] [Google Scholar]
- 165.Veronese FM, Caliceti P. Bioconjugation and biodistribution. In: Page M, editor. Cancer drug discovery and development: Tumor targeting in cancer therapy. Humana Press; Totowa, NJ: 2002. pp. 411–429. [Google Scholar]
- 166.Nobs L, Buchegger F, Gurny R, Allemann E. Current methods for attaching targeting ligands to liposomes and nanoparticles. J Pharm Sci. 2004;93:1980–1992. doi: 10.1002/jps.20098. [DOI] [PubMed] [Google Scholar]
- 167.(a) Khandare J, Minko T. Polymer-drug conjugates: Progress in polymeric prodrugs. Progr Polymer Sci. 2006;31:359–397. [Google Scholar]; (b) Haag R, Kratz F. Polymer therapeutics: Concepts and applications. Angew Chem Int Ed Engl. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 168.Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 169.Soni V, Kohli DV, Jain SK. Transferrin coupled liposomes as drug delivery carriers for brain targeting of 5-florouracil. J Drug Target. 2005;13:245–250. doi: 10.1080/10611860500107401. [DOI] [PubMed] [Google Scholar]
- 170.Kakudo T, Chaki S, Futaki S, Nakase I, Akaji K, Kawakami T, Maruyama K, Kamiya H, Harashima H. Transferrin-modified liposomes equipped with a pH-sensitive fusogenic peptide: An artificial viral-like delivery system. Biochem. 2004;43:5618–5628. doi: 10.1021/bi035802w. [DOI] [PubMed] [Google Scholar]
- 171.Hatakeyama H, Akita H, Maruyama K, Suhara T, Harashima H. Factors governing the in vivo tissue uptake of transferrin-coupled polyethylene glycol liposomes in vivo. Int J Pharm. 2004;281:25–33. doi: 10.1016/j.ijpharm.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 172.Liang HF, Yang TF, Huang CT, Chen MC, Sung HW. Preparation of nanoparticles composed of poly(gamma-glutamic acid)-poly(lactide) block copolymers and evaluation of their uptake by HepG2 cells. J Control Release. 2005;105:213–225. doi: 10.1016/j.jconrel.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 173.Managit C, Kawakami S, Yamashita F, Hashida M. Effect of galactose density on asialoglycoprotein receptor-mediated uptake of galactosylated liposomes. J Pharm Sci. 2005;94:2266–2275. doi: 10.1002/jps.20443. [DOI] [PubMed] [Google Scholar]
- 174.Adachi N, Miyaike M, Kato S, Kanamaru R, Koyama H, Kikuchi A. Cellular distribution of mammalian DNA topoisomerase II is determined by its catalytically dispensable C-terminal domain. Nucleic Acids Res. 1997;25:3135–3142. doi: 10.1093/nar/25.15.3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chiu SJ, Marcucci G, Lee RJ. Efficient delivery of an antisense oligodeoxyribonucleotide formulated in folate receptor-targeted liposomes. Anticancer Res. 2006;26:1049–1056. [PubMed] [Google Scholar]
- 176.Sonvico F, Mornet S, Vasseur S, Dubernet C, Jaillard D, Degrouard J, Hoebeke J, Duguet E, Colombo P, Couvreur P. Folate-conjugated iron oxide nanoparticles for solid tumor targeting as potential specific magnetic hyperthermia mediators: Synthesis, physicochemical characterization, and in vitro experiments. Bioconjug Chem. 2005;16:1181–1188. doi: 10.1021/bc050050z. [DOI] [PubMed] [Google Scholar]
- 177.Harvie P, Dutzar B, Galbraith T, Cudmore S, O'Mahony D, Anklesaria P, Paul R. Targeting of lipid-protamine-DNA (LPD) lipopolyplexes using RGD motifs. J Liposome Res. 2003;13:231–247. doi: 10.1081/lpr-120026389. [DOI] [PubMed] [Google Scholar]
- 178.Rejman J, Conese M, Hoekstra D. Gene transfer by means of lipo- and polyplexes: Role of clathrin and caveolae-mediated endocytosis. J Liposome Res. 2006;16:237–247. doi: 10.1080/08982100600848819. [DOI] [PubMed] [Google Scholar]
- 179.(a) Saul JM, Annapragada AV, Bellamkonda RV. A dual-ligand approach for enhancing targeting selectivity of therapeutic nanocarriers. J Control Release. 2006;114:277–287. doi: 10.1016/j.jconrel.2006.05.028. [DOI] [PubMed] [Google Scholar]; (b) Huang S, Armstrong EA, Benavente S, Chinnaiyan P, Harari PM. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): Combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res. 2004;64:5355–5362. doi: 10.1158/0008-5472.CAN-04-0562. [DOI] [PubMed] [Google Scholar]
- 180.(a) Caplan MR, Rosca EV. Targeting drugs to combinations of receptors: A modeling analysis of potential specificity. Ann Biomed Eng. 2005;33:1113–1124. doi: 10.1007/s10439-005-5779-1. [DOI] [PubMed] [Google Scholar]; (b) van Nieuw Amerongen GP, van Hinsbergh VW. Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul Pharmacol. 2002;39:257–272. doi: 10.1016/s1537-1891(03)00014-4. [DOI] [PubMed] [Google Scholar]
- 181.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 182.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]
- 183.Koukourakis MI, Koukouraki S, Giatromanolaki A, Archimandritis SC, Skarlatos J, Beroukas K, Bizakis JG, Retalis G, Karkavitsas N, Helidonis ES. Liposomal doxorubicin and conventionally fractionated radiotherapy in the treatment of locally advanced non-small-cell lung cancer and head and neck cancer. J Clin Oncol. 1999;17:3512–3521. doi: 10.1200/JCO.1999.17.11.3512. [DOI] [PubMed] [Google Scholar]
- 184.Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG, Stewart JS. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin Cancer Res. 2001;7:243–254. [PubMed] [Google Scholar]
- 185.Veldman RJ, Koning GA, van Hell A, Zerp S, Vink SR, Storm G, Verheij M, van Blitterswijk WJ. Coformulated N-octanoyl-glucosylceramide improves cellular delivery and cytotoxicity of liposomal doxorubicin. J Pharmacol Exp Ther. 2005;315:704–710. doi: 10.1124/jpet.105.087486. [DOI] [PubMed] [Google Scholar]
- 186.Gabizon AA. Selective tumor localization and improved therapeutic index of anthracyclines encapsulated in long-circulating liposomes. Cancer Res. 2002;52:891–896. [PubMed] [Google Scholar]
- 187.(a) Moase EH, Qi W, Ishida T, Gabos Z, Longenecker BM, Zimmermann GL, Ding L, Krantz M, Allen TM. Anti-MUC-1 immunoliposomal doxorubicin in the treatment of murine models of metastatic breast cancer. Biochim Biophys Acta. 2001;1510:43–55. doi: 10.1016/s0005-2736(00)00334-5. [DOI] [PubMed] [Google Scholar]; (b) Minko T, Dharap SS, Pakunlu RI, Wang Y. Molecular targeting of drug delivery systems to cancer. Curr Drug Targets. 2004;5:389–406. doi: 10.2174/1389450043345443. [DOI] [PubMed] [Google Scholar]; (c) Tanaka K, Towata S, Nakao K, Mizuguchi H, Hayakawa T, Niwa M, Ishii N, Nagayama Y. Thyroid cancer immuno-therapy with retroviral and adenoviral vectors expressing granulocyte macrophage colony stimulating factor and inter-leukin-12 in a rat model. Clin Endocrinol (Oxf) 2003;59:734–742. doi: 10.1046/j.1365-2265.2003.01915.x. [DOI] [PubMed] [Google Scholar]
- 188.Vannucci L, Fiserova A, Sadalapure K, Lindhorst TK, Kuldova M, Rossmann P, Horvath O, Kren V, Krist P, Bezouska K, Luptovcova M, Mosca F, Pospisil M. Effects of N-acetyl-glucosamine-coated glycodendrimers as biological modulators in the B16F10 melanoma model in vivo. Int J Oncol. 2003;23:285–296. [PubMed] [Google Scholar]
- 189.Salama NN, Eddington ND, Fasano A. Tight junction modulation and its relationship to drug delivery. Adv Drug Deliv Rev. 2006;58:15–28. doi: 10.1016/j.addr.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 190.Ishida T, Atobe K, Wang X, Kiwada H. Accelerated blood clearance of PEGylated liposomes upon repeated injections: Effect of doxorubicin-encapsulation and high-dose first injection. J Control Release. 2006a;115:251–258. doi: 10.1016/j.jconrel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 191.Ishida T, Ichihara M, Wang X, Kiwada H. Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J Control Release. 2006b;115:243–250. doi: 10.1016/j.jconrel.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 192.O'Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C, CAELYX Breast Cancer Study Group Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 193.Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 194.Ramaekers FC, Bosman FT. The cytoskeleton and disease. J Pathol. 2004;204:351–354. doi: 10.1002/path.1665. [DOI] [PubMed] [Google Scholar]
- 195.Campbell L, Gumbleton M, Ritchie K. Caveolae and the caveolins in human disease. Adv Drug Deliv Rev. 2001;49:325–335. doi: 10.1016/s0169-409x(01)00145-4. [DOI] [PubMed] [Google Scholar]
- 196.Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002;16:1217–1226. doi: 10.1096/fj.02-0088com. [DOI] [PubMed] [Google Scholar]
- 197.Murphy MP, Smith RA. Drug delivery to mitochondria: The key to mitochondrial medicine. Adv Drug Deliv Rev. 2000;41:235–250. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 198.Holcik M. Targeting endogenous inhibitors of apoptosis for treatment of cancer, stroke and multiple sclerosis. Expert Opin Ther Targets. 2004;8:241–253. doi: 10.1517/14728222.8.3.241. [DOI] [PubMed] [Google Scholar]
- 199.Rothbard JB, Jessop TC, Wender PA. Adaptive translocation: The role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 200.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 201.Simoes S, Slepushkin V, Gaspar R, de Lima MC, Duzgunes N. Gene delivery by negatively charged ternary complexes of DNA, cationic liposomes and transferrin or fusigenic peptides. Gene Ther. 1998;5:955–964. doi: 10.1038/sj.gt.3300674. [DOI] [PubMed] [Google Scholar]
- 202.Tachibana R, Harashima H, Shono M, Azumano M, Niwa M, Futaki S, Kiwada H. Intracellular regulation of macromolecules using pH-sensitive liposomes and nuclear localization signal: qualitative and quantitative evaluation of intra-cellular trafficking. Biochem Biophys Res Commun. 1998;251:538–544. doi: 10.1006/bbrc.1998.9460. [DOI] [PubMed] [Google Scholar]
- 203.Mendoza FJ, Espino PS, Cann KL, Bristow N, McCrea K, Los M. Anti-tumor chemotherapy utilizing peptide-based approaches-apoptotic pathways, kinases, and proteasome as targets. Arch Immunol Ther Exp (Warsz) 2005;53:47–60. [PubMed] [Google Scholar]
- 204.Shadidi M, Sioud M. Selective targeting of cancer cells using synthetic peptides. Drug Resist Updat. 2003;6:363–371. doi: 10.1016/j.drup.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 205.Pawar R, Shikanov A, Vaisman B, Domb AJ. Intravenous and regional paclitaxel formulations. Curr Med Chem. 2004;11:397–402. doi: 10.2174/0929867043455981. [DOI] [PubMed] [Google Scholar]
- 206.Goldberg EP, Hadba AR, Almond BA, Marotta JS. Intratumoral cancer chemotherapy and immunotherapy: Opportunities for nonsystemic preoperative drug delivery. J Pharm Pharmacol. 2002;54:159–180. doi: 10.1211/0022357021778268. [DOI] [PubMed] [Google Scholar]
- 207.Bogdanov A, Wright SC, Marecos EM, Bogdanova A, Martin C, Petherick P, Weissleder R. A long-circulating co-polymer in “passive targeting” to solid tumors. J Drug Target. 1997;4:321–330. doi: 10.3109/10611869708995848. [DOI] [PubMed] [Google Scholar]
- 208.Brigger I, Dubernet C, Couvreur P. Nano-particles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 209.Némati F, Dubernet C, Fessi H, de Verdière AC, Poupon MF, Puisieux F, Couvreur P. Reversion of multidrug resistance using nanoparticles in vitro: Influence of the nature of the polymer. Int J Pharm. 1996;138:237–246. [Google Scholar]
- 210.Vauthier C, Dubernet C, Chauvierre C, Brigger I, Couvreur P. Drug delivery to resistant tumors: The potential of poly(alkyl cyanoacrylate) nanoparticles. J Control Release. 2003;93:151–160. doi: 10.1016/j.jconrel.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 211.Jain RK. Delivery of novel therapeutic agents in tumors: Physiological barriers and strategies. J Natl Cancer Inst. 1989;81:570–576. doi: 10.1093/jnci/81.8.570. [DOI] [PubMed] [Google Scholar]
- 212.Jain RK. Delivery of molecular medicine to solid tumors. Science. 1996;271:1079–1080. doi: 10.1126/science.271.5252.1079. [DOI] [PubMed] [Google Scholar]
- 213.Jang SH, Wientjes MG, Lu D, Au JL. Drug delivery and transport to solid tumors. Pharm Res. 2003;20:1337–1350. doi: 10.1023/a:1025785505977. [DOI] [PubMed] [Google Scholar]
- 214.Feron O. Targeting the tumor vascular compartment to improve conventional cancer therapy. Trends Pharmacol Sci. 2004;25:536–542. doi: 10.1016/j.tips.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 215.Alessi P, Ebbinghaus C, Neri D. Molecular targeting of angiogenesis. Biochim Biophys Acta. 2004;1654:39–49. doi: 10.1016/j.bbcan.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 216.Bocci G, Francia G, Man S, Lawler J, Kerbel RS. Thrombospondin 1, a mediator of the anti-angiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci USA. 2003;100:12917–12922. doi: 10.1073/pnas.2135406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sun ZG, Chen XP, Huang ZY, Yang GH, Guan J, Hu DY, Mu LD, Xia XG, Li GP, Zhang WG, Li Z. The effects of lipiodol-hydroxyapatite nanoparticle on apoptosis, proliferation, and angiogenesis in hepatic tumor: Experiment with rabbits. Zhonghua Yi Xue Za Zhi. 2007;87:409–413. in Chinese. [PubMed] [Google Scholar]
- 218.Stampfl S, Stampfl U, Rehnitz C, Schnabel P, Satzl S, Christoph P, Henn C, Thomas F, Richter GM. Experimental evaluation of early and long-term effects of microparticle embolization in two different mini-pig models. Part II: Liver. Cardiovasc Intervent Radiol. 2007;30:462–468. doi: 10.1007/s00270-005-0350-3. [DOI] [PubMed] [Google Scholar]
- 219.Cegnar M, Kristl J, Kos J. Nanoscale polymer carriers to deliver chemotherapeutic agents to tumours. Expert Opin Biol Ther. 2005;5:1557–1569. doi: 10.1517/14712598.5.12.1557. [DOI] [PubMed] [Google Scholar]
- 220.Marcucci F, Lefoulon F. Active targeting with particulate drug carriers in tumor therapy: Fundamentals and recent progress. Drug Discov Today. 2004;9:219–228. doi: 10.1016/S1359-6446(03)02988-X. [DOI] [PubMed] [Google Scholar]
- 221.Nori A, Kopecek J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv Drug Deliv Rev. 2005;57:609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 222.Seymour LW. Passive tumor targeting of soluble macromolecules and drug conjugates. Crit Rev Ther Drug Carrier Syst. 1992;9:135–187. [PubMed] [Google Scholar]
- 223.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 224.Green TR, Fisher J, Stone M, Wroblewski BM, Ingham E. Polyethylene particles of a `critical size' are necessary for the induction of cytokines by macrophages in vitro. Biomaterials. 1998;19:2297–2302. doi: 10.1016/s0142-9612(98)00140-9. [DOI] [PubMed] [Google Scholar]
- 225.Ayrton A, Morgan P. Role of transport proteins in drug absorption, distribution and excretion. Xenobiotica. 2001;31:469–497. doi: 10.1080/00498250110060969. [DOI] [PubMed] [Google Scholar]
- 226.Kwon S, Ke S, Houston JP, Wang W, Wu Q, Li C, Sevick-Muraca EM. Imaging dose-dependent pharmacokinetics of an RGD-fluorescent dye conjugate targeted to alpha v beta 3 receptor expressed in Kaposi's sarcoma. Mol Imaging. 2005;4:75–87. doi: 10.1162/15353500200505103. [DOI] [PubMed] [Google Scholar]
- 227.(a) Stoldt HS, Aftab F, Chinol M, Paganelli G, Luca F, Testori A, Geraghty JG. Pretargeting strategies for radio-immunoguided tumour localisation and therapy. Eur J Cancer. 1997;33:186–192. doi: 10.1016/s0959-8049(96)00477-7. [DOI] [PubMed] [Google Scholar]; (b) Sharkey RM, Cardillo TM, Rossi EA, Chang CH, Karacay H, McBride WJ, Hansen HJ, Horak ID, Goldenberg DM. Signal amplification in molecular imaging by pretargeting a multivalent, bispecific antibody. Nat Med. 2005;11:1250–1255. doi: 10.1038/nm1322. [DOI] [PubMed] [Google Scholar]
- 228.Meredith RF, Buchsbaum DJ. Pretargeted radioimmunotherapy. Int J Radiat Oncol Biol Phys. 2006;66:S57–S59. doi: 10.1016/j.ijrobp.2006.04.058. [DOI] [PubMed] [Google Scholar]
- 229.Nobs L, Buchegger F, Gurny R, Allemann E. Biodegradable nanoparticles for direct or two-step tumor immunotargeting. Bioconjug Chem. 2006;17:139–145. doi: 10.1021/bc050137k. [DOI] [PubMed] [Google Scholar]
- 230.Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: From therapeutics to diagnostics. J Pharm Sci. 2005;94:2135–2146. doi: 10.1002/jps.20457. [DOI] [PubMed] [Google Scholar]
- 231.Lee RJ, Wang S, Low PS. Measurement of endosome pH following folate receptor-mediated endocytosis. Biochim Biophys Acta. 1996;1312:237–242. doi: 10.1016/0167-4889(96)00041-9. [DOI] [PubMed] [Google Scholar]
- 232.Turek JJ, Leamon CP, Low PS. Endocytosis of folate-protein conjugates: Ultrastructural localization in KB cells. J Cell Sci. 1993;106:423–430. doi: 10.1242/jcs.106.1.423. [DOI] [PubMed] [Google Scholar]
- 233.Line BR, Mitra A, Nan A, Ghandehari H. Targeting tumor angiogenesis: Comparison of peptide and polymer-peptide conjugates. J Nucl Med. 2005;46:1552–1560. [PubMed] [Google Scholar]
- 234.Mitra A, Nan A, Papadimitriou JC, Ghandehari H, Line BR. Polymer-peptide conjugates for angiogenesis targeted tumor radiotherapy. Nucl Med Biol. 2006;33:43–52. doi: 10.1016/j.nucmedbio.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 235.Kircheis R, Ostermann E, Wolschek MF, Lichtenberger C, Magin-Lachmann C, Wightman L, Kursa M, Wagner E. Tumor-targeted gene delivery of tumor necrosis factor-alpha induces tumor necrosis and tumor regression without systemic toxicity. Cancer Gene Ther. 2002;9:673–680. doi: 10.1038/sj.cgt.7700487. [DOI] [PubMed] [Google Scholar]
- 236.Yu W, Pirollo KF, Rait A, Yu B, Xiang LM, Huang WQ, Zhou Q, Ertem G, Chang EH. A sterically stabilized immunolipoplex for systemic administration of a therapeutic gene. Gene Ther. 2004;11:1434–1440. doi: 10.1038/sj.gt.3302304. [DOI] [PubMed] [Google Scholar]
- 237.Gunther M, Wagner E, Ogris M. Specific targets in tumor tissue for the delivery of therapeutic genes. Curr Med Chem Anticancer Agents. 2005;5:157–171. doi: 10.2174/1568011053174855. [DOI] [PubMed] [Google Scholar]
- 238.Baish JW, Gazit Y, Berk DA, Nozue M, Baxter LT, Jain RK. Role of tumor vascular architecture in nutrient and drug delivery: An invasion percolation-based network model. Microvasc Res. 1996;51:327–346. doi: 10.1006/mvre.1996.0031. [DOI] [PubMed] [Google Scholar]
- 239.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 240.Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, Fujii T, Komuro A, Kiyono K, Kaminishi M, Hirakawa K, Ouchi Y, Nishiyama N, Kataoka K, Miyazono K. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci USA. 2007;104:3460–3465. doi: 10.1073/pnas.0611660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lutsiak ME, Kwon GS, Samuel J. Biodegradable nanoparticle delivery of a Th2-biased peptide for induction of Th1 immune responses. J Pharm Pharmacol. 2006;58:739–747. doi: 10.1211/jpp.58.6.0004. [DOI] [PubMed] [Google Scholar]
- 242.Kocbek P, Obermajer N, Cegnar M, Kos J, Kristl J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release. 2007;120:18–26. doi: 10.1016/j.jconrel.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 243.Huang M, Vitharana SN, Peek LJ, Coop T, Berkland C. Polyelectrolyte complexes stabilize and controllably release vascular endothelial growth factor. Biomacromol. 2007;8:1607–1614. doi: 10.1021/bm061211k. [DOI] [PubMed] [Google Scholar]
- 244.Luu YK, Kim K, Hsiao BS, Chu B, Hadjiargyrou M. Development of a nanostructured DNA delivery scaffold via electrospinning of PLGA and PLA-PEG block copolymers. J Control Release. 2003;89:341–353. doi: 10.1016/s0168-3659(03)00097-x. [DOI] [PubMed] [Google Scholar]
- 245.Yu JJ, Lee HA, Kim JH, Kong WH, Kim Y, Cui ZY, Park KG, Kim WS, Lee HG, Seo SW. Biodistribution and anti-tumor efficacy of PEG/PLA nano particles loaded doxorubicin. J Drug Target. 2007;15:279–284. doi: 10.1080/10611860701357235. [DOI] [PubMed] [Google Scholar]
- 246.Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, Chen X. Quantitative PET imaging of tumor integrin alphavbeta3 expression with 18F-FR GD2. J Nucl Med. 2006b;47:113–121. [PMC free article] [PubMed] [Google Scholar]
- 247.Nafee N, Taetz S, Schneider M, Schaefer UF, Lehr CM. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomed. 2007;3:173–183. doi: 10.1016/j.nano.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 248.(a) Ojeda R, de Paz JL, Barrientos AG, Martin-Lomas M, Penades S. Preparation of multifunctional glyconanoparticles as a platform for potential carbohydrate-based anticancer vaccines. Carbohydr Res. 2007;342:448–459. doi: 10.1016/j.carres.2006.11.018. [DOI] [PubMed] [Google Scholar]; (b) Lecaroz C, Gamazo C, Blanco-Prieto MJ. Nanocarriers with gentamicin to treat intracellular pathogens. J Nanosci Nanotechnol. 2006;6:3296–3302. doi: 10.1166/jnn.2006.478. [DOI] [PubMed] [Google Scholar]
- 249.Stephens AC, Rivers RP. Antisense oligonucleotide therapy in cancer. Curr Opin Mol Ther. 2003;5:118–122. [PubMed] [Google Scholar]
- 250.Ala-aho R, Ahonen M, George SJ, Heikkila J, Grenman R, Kallajoki M, Kahari VM. Targeted inhibition of human collagenase-3 (MMP-13) expression inhibits squamous cell carcinoma growth in vivo. Oncogene. 2004;23:5111–5123. doi: 10.1038/sj.onc.1207678. [DOI] [PubMed] [Google Scholar]
- 251.Silverman SK. In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res. 2005;33:6151–6163. doi: 10.1093/nar/gki930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fang X, Cao Z, Beck T, Tan W. Molecular aptamer for real-time oncoprotein platelet-derived growth factor monitoring by fluorescence anisotropy. Anal Chem. 2001;73:5752–5757. doi: 10.1021/ac010703e. [DOI] [PubMed] [Google Scholar]
- 253.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: An emerging class of therapeutics. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 254.Sumimoto H, Miyagishi M, Miyoshi H, Yamagata S, Shimizu A, Taira K, Kawakami Y. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;23:6031–6039. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 255.Ayyagari AL, Zhang X, Ghaghada KB, Annapragada A, Hu X, Bellamkonda RV. Long-circulating liposomal contrast agents for magnetic resonance imaging. Magn Reson Med. 2006;55:1023–1029. doi: 10.1002/mrm.20846. [DOI] [PubMed] [Google Scholar]
- 256.Wu X, Liu H, Liu J, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP. Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat Biotechnol. 2003;21:41–46. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 257.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science. 2003;300:1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 258.Kim JH, Morikis D, Ozkan M. Adaptation of inorganic quantum dots for stable molecular beacons. Sens Actuators B Chem. 2004a;102:315–319. [Google Scholar]
- 259.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Dor DM, Cohn LH, Bawendi MG, Frangioni JV. Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat Biotechnol. 2004b;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Derfus AM, Chan WC, Bhatia S. Probing the cytotoxicity of semiconductor quantum dots. Nano Lett. 2004;4:11–18. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Portney NG, Ozkan M. Nano-oncology: Drug delivery, imaging, and sensing. Anal Bioanal Chem. 2006;384:620–630. doi: 10.1007/s00216-005-0247-7. [DOI] [PubMed] [Google Scholar]
- 262.Kobayashi H, Brechbiel MW. Nano-sized MRI contrast agents with dendrimer cores. Adv Drug Deliv Rev 14. 2005;57:2271–2286. doi: 10.1016/j.addr.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 263.Kawata S, Inouye Y, Ichimura T. Near-field optics and spectroscopy for molecular nano-imaging. Sci Prog. 2004;87:25–49. doi: 10.3184/003685004783238580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Quaresima V, Matcher SJ, Ferrari M. Identification and quantification of intrinsic optical contrast for near-infrared mammography. Photo-chem Photobiol. 1998;67:4–14. [PubMed] [Google Scholar]
- 265.Ntziachristos V, Bremer C, Graves EE, Ripoll J, Weissleder R. In vivo tomographic imaging of near-infrared fluorescent probes. Mol Imaging. 2002a;1:82–88. doi: 10.1162/15353500200201121. [DOI] [PubMed] [Google Scholar]
- 266.(a) Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci USA. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ntziachristos V, Ripoll J, Weissleder R. Would near-infrared fluorescence signals propagate through large human oragns for clinical studies? Optics Lett. 2002c;27:333–335. doi: 10.1364/ol.27.000333. [DOI] [PubMed] [Google Scholar]
- 267.Ntziachristos V, Tung CH, Bremer C, Weissleder R. Fluorescence molecular tomography resolves protease activity in vivo. Nat Med. 2002b;8:757–760. doi: 10.1038/nm729. [DOI] [PubMed] [Google Scholar]
- 268.Citrin D, Lee AK, Scott T, Sproull M, Menard C, Tofilon PJ, Camphausen K. In vivo tumor imaging in mice with near-infrared labeled endostatin. Mol Cancer Ther. 2004;3:481–488. [PubMed] [Google Scholar]
- 269.Cheng Z, Wu Y, Xiong Z, Gambhir SS, Chen X. Near-infrared fluorescent RGD peptides for optical imaging of integrin alphavbeta3 expression in living mice. Bioconjug Chem. 2005;16:1433–1441. doi: 10.1021/bc0501698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Ye Y, Bloch S, Xu B, Achilefu S. Design, synthesis, and evaluation of near infrared fluorescent multimeric RGD peptides for targeting tumors. J Med Chem. 2006;49:2268–2275. doi: 10.1021/jm050947h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kelly KA, Nahrendorf M, Yu AM, Reynolds F, Weissleder R. In vivo phage display selection yields atherosclerotic plaque targeted peptides for imaging. Mol Imaging Biol. 2006;8:201–207. doi: 10.1007/s11307-006-0043-6. [DOI] [PubMed] [Google Scholar]
- 272.Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. Multivalent effects of RGD peptides obtained by nanoparticle display. J Med Chem. 2006;49:6087–6093. doi: 10.1021/jm060515m. [DOI] [PubMed] [Google Scholar]
- 273.Kobayashi H, Kawamoto S, Saga T, Sato N, Hiraga A, Konishi J, Togashi K, Brechbiel MW. Micro-MR angiography of normal and intratu-moral vessels in mice using dedicated intravascular MR contrast agents with high generation of polyamidoamine dendrimer core: Reference to pharmacokinetic properties of dendrimer-based MR contrast agents. J Magn Reson Imaging. 2001;14:705–713. doi: 10.1002/jmri.10025. [DOI] [PubMed] [Google Scholar]
- 274.Kobayashi H, Brechbiel MW. Dendrimer-based nanosized MRI contrast agents. Curr Pharm Biotechnol. 2004;5:539–549. doi: 10.2174/1389201043376571. [DOI] [PubMed] [Google Scholar]
- 275.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Viral nanoparticles as tools for intravital vascular imaging. Nat Med. 2006;12:354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Voisin P, Ribot EJ, Miraux S, Bouzier-Sore AK, Lahitte JF, Bouchaud V, Mornet S, Thiaudiere E, Franconi JM, Raison L, Labrugere C, Delville MH. Use of lanthanide-grafted inorganic nanoparticles as effective contrast agents for cellular uptake imaging. Bioconjug Chem. 2007;18:1053–1063. doi: 10.1021/bc060269t. [DOI] [PubMed] [Google Scholar]
- 277.Bertorelle F, Wilhelm C, Roger J, Gazeau F, Menager C, Cabuil V. Fluorescence-modified superparamagnetic nanoparticles: Intracellular uptake and use in cellular imaging. Langmuir. 2006;22:5385–5391. doi: 10.1021/la052710u. [DOI] [PubMed] [Google Scholar]
- 278.Sosnovik D, Weissleder R. Magnetic resonance and fluorescence based molecular imaging technologies. Prog Drug Res. 2005;62:83–115. doi: 10.1007/3-7643-7426-8_3. [DOI] [PubMed] [Google Scholar]
- 279.Bradbury M, Hricak H. Molecular MR imaging in oncology. Magn Reson Imaging Clin N Am. 2005;13:225–240. doi: 10.1016/j.mric.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 280.Neves AA, Brindle KM. Assessing responses to cancer therapy using molecular imaging. Biochim Biophys Acta. 2006;766:242–261. doi: 10.1016/j.bbcan.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 281.Desai N, Trieu V, Yao Z, Louie L, Ci S, Yang A, Tao C, De T, Beals B, Dykes D, Noker P, Yao R, Labao E, Hawkins M, Soon-Shiong P. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res. 2006;12:1317–1324. doi: 10.1158/1078-0432.CCR-05-1634. [DOI] [PubMed] [Google Scholar]
- 282.John TA, Vogel SM, Minshall RD, Ridge K, Tiruppathi C, Malik AB. Evidence for the role of alveolar epithelial gp60 in active transalveolar albumin transport in the rat lung. J Physiol. 2001;533:547–559. doi: 10.1111/j.1469-7793.2001.0547a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kharb V, Bhatia M, Dureja H, Kaushik D. Nanoparticle technology for the delivery of poorly water-soluble drugs. Pharm Technol. 2006;30:82–92. [Google Scholar]
- 284.Muller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47:3–19. doi: 10.1016/s0169-409x(00)00118-6. [DOI] [PubMed] [Google Scholar]
- 285.Frazza EJ, Schmitt EE. A new absorbable suture. J Biomed Mater Res. 1971;1:43–55. doi: 10.1002/jbm.820050207. [DOI] [PubMed] [Google Scholar]
- 286.Gilding DK. Biodegradable polymers. In: Williams DF, editor. Biocompatibility of clinical implant materials. CRC Press; Boca Raton, FL: 1981. pp. 209–232. [Google Scholar]
- 287.Vert M, Mauduit J, Li S. Biodegradation of PLA/GA polymers: Increasing complexity. Biomaterials. 1994;15:1209–1213. doi: 10.1016/0142-9612(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 288.Bazile DV, Ropert C, Huve P, Verrecchia T, Marlard M, Frydman A, Veillard M, Spenlehauer G. Body distribution of fully biodegradable [14C]-poly(lactic acid) nanoparticles coated with albumin after parenteral administration to rats. Biomaterials. 1992;13:1093–1102. doi: 10.1016/0142-9612(92)90142-b. [DOI] [PubMed] [Google Scholar]
- 289.Venier-Julienne MC, Benoit JP. Preparation, purification and morphology of polymeric nanoparticles as drug carriers. Pharm Acta Helv. 1996;71:121–128. doi: 10.1016/0031-6865(95)00059-3. [DOI] [PubMed] [Google Scholar]
- 290.Bala I, Hariharan S, Kumar MN. PLGA nanoparticles in drug delivery: The state of the art. Crit Rev Ther Drug Carrier Syst. 2004;21:387–422. doi: 10.1615/critrevtherdrugcarriersyst.v21.i5.20. [DOI] [PubMed] [Google Scholar]
- 291.Couvreur P, Kante B, Roland M, Guiot P, Bauduin P, Speiser P. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: Preparation, morphological and sorptive properties. J Pharm Pharmacol. 1979;31:331–332. doi: 10.1111/j.2042-7158.1979.tb13510.x. [DOI] [PubMed] [Google Scholar]
- 292.Lenaerts V, Couvreur P, Christiaens-Leyh D, Joiris E, Roland M, Rollman B, Speiser P. Degradation of poly (isobutyl cyanoacrylate) nanoparticles. Biomaterials. 1984;5:65–68. doi: 10.1016/0142-9612(84)90002-4. [DOI] [PubMed] [Google Scholar]
- 293.Vauthier C, Dubernet C, Fattal E, Pinto-Alphandary H, Couvreur P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv Drug Deliv Rev. 2003;55:519–548. doi: 10.1016/s0169-409x(03)00041-3. [DOI] [PubMed] [Google Scholar]
- 294.Merle P, Si-Ahmed S, Habersetzer F, Abergel A, Taieb J, Bonyhay L, Constantini D, Dufour-Lamartinie J, Trépo C. Phase 1 study of intra-arterial hepatic (IAH) delivery of doxorubicin-transdrug® (DT) for patients with advanced hepatocellular carcinoma (HCC) J Clin Virol. 2006;36:S179–S179. [Google Scholar]
- 295.(a) Carlesso G, Kozlov E, Prokop A, Unutmaz D, Davidson JM. Nanoparticulate system for efficient gene transfer into refractory cell targets. Biomacromol. 2003;6:1185–1192. doi: 10.1021/bm0492531. [DOI] [PubMed] [Google Scholar]; (b) Anonymous: http://www.bioalliancepharma.-com/clinicaltrials.asp.; (c) Kattan J, Droz JP, Couvreur P, Marino JP, Boutan-Laroze A, Rougier P, Brault P, Vranckx H, Grognet JM, Morge X, Sancho-Garnier H. Phase I clinical trial and pharmacokinetic evaluation of doxorubicin carried by polyisohexylcyanoacrylate nanoparticles. Invest New Drugs. 1992;10:191–199. doi: 10.1007/BF00877245. [DOI] [PubMed] [Google Scholar]
- 296.Prokop A, Kozlov E, Newman GW, Newman MJ. Water-based nanoparticulate polymeric system for protein delivery: Permeability control and vaccine application. Biotechnol Bioeng. 2000c;78:459–466. doi: 10.1002/bit.1200. [DOI] [PubMed] [Google Scholar]
- 297.Fernandez-Urrusuno R, Calvo P, Remunan-Lopez C, Vila-Jato JL, Alonso MJ. Enhancement of nasal absorption of insulin using chitosan nanoparticles. Pharm Res. 1999;16:1576–1581. doi: 10.1023/a:1018908705446. [DOI] [PubMed] [Google Scholar]
- 298.Calvo P, Remunan-Lopez C, Vila-Jato JL, Alonso MJ. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Pol Sci. 1997;63:125–132. [Google Scholar]
- 299.Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed Nanotechnol Biol Med. 2006;2:8–21. doi: 10.1016/j.nano.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 300.Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 301.Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev. 2003;55:403–419. doi: 10.1016/s0169-409x(02)00226-0. [DOI] [PubMed] [Google Scholar]
- 302.Oishi M, Nagatsugi F, Sasaki S, Nagasaki Y, Kataoka K. Smart polyion complex micelles for targeted intracellular delivery of PEGylated antisense oligonucleotides containing acid-labile linkages. Chembiochem. 2005;6:718–725. doi: 10.1002/cbic.200400334. [DOI] [PubMed] [Google Scholar]
- 303.Nakanishi T, Fukushima S, Okamoto K, Suzuki M, Matsumura Y, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Development of the polymer micelle carrier system for doxorubicin. J Control Release. 2001;74:295–302. doi: 10.1016/s0168-3659(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 304.Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: Tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem. 2005;16:122–130. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
- 305.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93:678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Hamaguchi T, Matsumura Y, Suzuki M, Shimizu K, Goda R, Nakamura I, Nakatomi I, Yokoyama M, Kataoka K, Kakizoe T. NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer. 2005;92:1240–1246. doi: 10.1038/sj.bjc.6602479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Lavasanifar A, Samuel J, Kwon GS. Poly(-ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 2002;54:169–190. doi: 10.1016/s0169-409x(02)00015-7. [DOI] [PubMed] [Google Scholar]
- 308.Liu X, Gao C, Shen J, Mohwald H. Multilayer microcapsules as anti-cancer drug delivery vehicle: Deposition, sustained release, and in vitro bioactivity. Macromol Biosci. 2005;5:1209–1219. doi: 10.1002/mabi.200500176. [DOI] [PubMed] [Google Scholar]
- 309.Ai H, Pink JJ, Shuai X, Boothman DA, Gao J. Interactions between self-assembled polyelectrolyte shells and tumor cells. J Biomed Mater Res A. 2005;73:303–312. doi: 10.1002/jbm.a.30289. [DOI] [PubMed] [Google Scholar]
- 310.Berg MC, Zhai L, Cohen RE, Rubner MF. Controlled drug release from porous polyelectrolyte multilayers. Biomacromolecules. 2006;7:357–364. doi: 10.1021/bm050174e. [DOI] [PubMed] [Google Scholar]
- 311.Garza JM, Jessel N, Ladam G, Dupray V, Muller S, Stoltz JF, Schaaf P, Voegel JC, Lavalle P. Polyelectrolyte multilayers and degradable polymer layers as multicompartment films. Langmuir. 2005;21:12372–12377. doi: 10.1021/la051465b. [DOI] [PubMed] [Google Scholar]
- 312.Zhang J, Fredin NJ, Janz JF, Sun B, Lynn DM. Structure/property relationships in erodible multilayered films: Influence of polycation structure on erosion profiles and the release of anionic polyelectrolytes. Langmuir. 2006a;22:239–245. doi: 10.1021/la052360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Haynie DT, Zhang L, Rudra JS, Zhao W, Zhong Y, Palath N. Polypeptide multilayer films. Biomacromol. 2005;6:2895–2913. doi: 10.1021/bm050525p. [DOI] [PubMed] [Google Scholar]
- 314.Morgan JR, Cloninger MJ. Heterogeneously functionalized dendrimers. Curr Opin Drug Discov Devel. 2002;5:966–973. [PubMed] [Google Scholar]
- 315.Majoros IJ, Thomas TP, Mehta CB, Baker JR., Jr. Poly(amidoamine) dendrimer-based multi-functional engineered nanodevice for cancer therapy. J Med Chem. 2005;48:5892–5899. doi: 10.1021/jm0401863. [DOI] [PubMed] [Google Scholar]
- 316.Bhadra D, Bhadra S, Jain S, Jain NK. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int J Pharm. 2003;257:111–124. doi: 10.1016/s0378-5173(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 317.Andre JP, Geraldes CF, Martins JA, Merbach AE, Prata MI, Santos AC, de Lima JJ, Toth E. Lanthanide(III) complexes of DOTA-glycoconjugates: A potential new class of lectin-mediated medical imaging agents. Chemistry. 2004;10:5804–5816. doi: 10.1002/chem.200400187. [DOI] [PubMed] [Google Scholar]
- 318.Khopade AJ, Shenoy DB, Khopade SA, Jain NK. Phase structures of a hydrated anionic phos pholipid composition containing cationic dendrimers and pegylated lipids. Langmuir. 2004;20:7368–7373. doi: 10.1021/la049682k. [DOI] [PubMed] [Google Scholar]
- 319.Duncan R, Izzo L. Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev. 2005;57:2215–2237. doi: 10.1016/j.addr.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 320.Majoros IJ, Myc A, Thomas T, Mehta CB, Baker JR., Jr. PAMAM dendrimer-based multifunctional conjugate for cancer therapy: Synthesis, characterization, and functionality. Biomacromol. 2006;7:572–579. doi: 10.1021/bm0506142. [DOI] [PubMed] [Google Scholar]
- 321.Jiang YH, Emau P, Cairns JS, Flanary L, Morton WR, McCarthy TD, Tsai CC. PL7013 gel as a topical microbicide for prevention of vaginal transmission of SH IV89.6P in macaques. AIDS Res Hum Retroviruses. 2005;21:207–213. doi: 10.1089/aid.2005.21.207. [DOI] [PubMed] [Google Scholar]
- 322.Blume G, Cevc G. Liposomes for the sustained drug release in vivo. Biochim Biophys Acta. 1990;1029:91–97. doi: 10.1016/0005-2736(90)90440-y. [DOI] [PubMed] [Google Scholar]
- 323.Crosasso P, Ceruti M, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J Control Release. 2000;63:19–30. doi: 10.1016/s0168-3659(99)00166-2. [DOI] [PubMed] [Google Scholar]
- 324.Cattel L, Ceruti M, Dosio F. From conventional to stealth liposomes: A new frontier in cancer chemotherapy. J Chemother. 2004;16:94–97. doi: 10.1179/joc.2004.16.Supplement-1.94. [DOI] [PubMed] [Google Scholar]
- 325.Ishida O, Maruyama K, Sasaki K, Iwatsuru M. Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int J Pharm. 1999;190:49–56. doi: 10.1016/s0378-5173(99)00256-2. [DOI] [PubMed] [Google Scholar]
- 326.Bakouche O, Lachman LB. Antigen presentation by liposomes bearing class II MHC and membrane IL-1. Yale J Biol Med. 1990;63:95–107. [PMC free article] [PubMed] [Google Scholar]
- 327.Fries LF, Gordon DM, Richards RL, Egan JE, Hollingdale MR, Gross M, Silverman C, Alving CR. Liposomal malaria vaccine in humans: A safe and potent adjuvant strategy. Proc Natl Acad Sci USA. 1992;89:358–362. doi: 10.1073/pnas.89.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Campos S. Liposomal anthracyclines: Adjuvant and neoadjuvant therapy for breast cancer. Oncologist. 2003;8:10–16. doi: 10.1634/theoncologist.8-suppl_2-10. [DOI] [PubMed] [Google Scholar]
- 329.Song LY, Ahkong QF, Rong Q, Wang Z, Ansell S, Hope MJ, Mui B. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim Biophys Acta. 2002;1558:1–13. doi: 10.1016/s0005-2736(01)00399-6. [DOI] [PubMed] [Google Scholar]
- 330.Monck MA, Mori A, Lee D, Tam P, Wheeler JJ, Cullis PR, Scherrer P. Stabilized plasmidlipid particles: Pharmacokinetics and plasmid delivery to distal tumors following intravenous injection. J Drug Target. 2000;7:439–452. doi: 10.3109/10611860009102218. [DOI] [PubMed] [Google Scholar]
- 331.Kamps JA, Morselt HW, Scherphof GL. Uptake of liposomes containing phosphatidylserine by liver cells in vivo and by sinusoidal liver cells in primary culture: In vivo-in vitro differences. Biochem Biophys Res Commun. 1999;256:57–62. doi: 10.1006/bbrc.1999.0290. [DOI] [PubMed] [Google Scholar]
- 332.Goyal P, Goyal K, Vijaya Kumar SG, Singh A, Katare OP, Mishra DN. Liposomal drug delivery systems-clinical applications. Acta Pharm. 2005;55:1–25. [PubMed] [Google Scholar]
- 333.Drummond DC, Zignani M, Leroux J. Current status of pH-sensitive liposomes in drug delivery. Prog Lipid Res. 2000;39:409–460. doi: 10.1016/s0163-7827(00)00011-4. [DOI] [PubMed] [Google Scholar]
- 334.Ruponen M, Yla-Herttuala S, Urtti A. Interactions of polymeric and liposomal gene delivery systems with extracellular glycosaminoglycans: physicochemical and transfection studies. Biochim Biophys Acta. 1999;1415:331–341. doi: 10.1016/s0005-2736(98)00199-0. [DOI] [PubMed] [Google Scholar]
- 335.Lee KD, Hong K, Papahadjopoulos D. Recognition of liposomes by cells: In vitro binding and en docytosis mediated by specific lipid headgroups and surface charge density. Biochim Biophys Acta. 1992;1103:185–197. doi: 10.1016/0005-2736(92)90086-2. [DOI] [PubMed] [Google Scholar]
- 336.Bummer PM. Physical chemical considerations of lipid-based oral drug delivery–solid lipid nanoparticles. Crit Rev Ther Drug Carrier Syst. 2004;21:1–20. [PubMed] [Google Scholar]
- 337.Manjunath K, Reddy JS, Venkateswarlu V. Solid lipid nanoparticles as drug delivery systems. Methods Find Exp Clin Pharmacol. 2005;27:127–144. doi: 10.1358/mf.2005.27.2.876286. [DOI] [PubMed] [Google Scholar]
- 338.Saupe A, Wissing SA, Lenk A, Schmidt C, Muller RH. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC). Structural investigations on two different carrier systems. Biomed Mater Eng. 2005;15:393–402. [PubMed] [Google Scholar]
- 339.Rensen PC, de Vrueh RL, Kuiper J, Bijsterbosch MK, Biessen EA, van Berkel TJ. Recombinant lipoproteins: Lipoprotein-like lipid particles for drug targeting. Adv Drug Deliv Rev. 2001;47:251–276. doi: 10.1016/s0169-409x(01)00109-0. [DOI] [PubMed] [Google Scholar]
- 340.West KR, Otto S. Reversible covalent chemistry in drug delivery. Curr Drug Discov Technol. 2005;2:123–160. doi: 10.2174/1570163054866882. [DOI] [PubMed] [Google Scholar]
- 341.Miyamoto Y, Oda T, Maeda H. Comparison of the cytotoxic effects of the high- and low-molecular-weight anticancer agents on multidrug-resistant Chinese hamster ovary cells in vitro. Cancer Res. 1990;50:1571–1575. [PubMed] [Google Scholar]
- 342.St'astny M, Strohalm J, Plocova D, Ulbrich K, Rihova B. A possibility to overcome P-glycoprotein (PGP)-mediated multidrug resistance by antibody-targeted drugs conjugated to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer carrier. Eur J Cancer. 1999;35:459–466. doi: 10.1016/s0959-8049(98)00373-6. [DOI] [PubMed] [Google Scholar]
- 343.Goren D, Horowitz AT, Tzemach D, Tarshish M, Zalipsky S, Gabizon A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin Cancer Res. 2000;6:1949–1957. [PubMed] [Google Scholar]
- 344.Katragadda S, Budda B, Anand BS, Mitra AK. Role of efflux pumps and metabolising enzymes in drug delivery. Expert Opin Drug Deliv. 2005;2:683–705. doi: 10.1517/17425247.2.4.683. [DOI] [PubMed] [Google Scholar]
- 345.Mamot C, Drummond DC, Hong K, Kirpotin DB, Park JW. Liposome-based approaches to overcome anticancer drug resistance. Drug Resist Updat. 2003;6:271–279. doi: 10.1016/s1368-7646(03)00082-7. [DOI] [PubMed] [Google Scholar]
- 346.de Verdiere AC, Dubernet C, Nemati F, Soma E, Appel M, Ferte J, Bernard S, Puisieux F, Couvreur P. Reversion of multidrug resistance with polyalkylcyanoacrylate nanoparticles: Towards a mechanism of action. Br J Cancer. 1997;76:198–205. doi: 10.1038/bjc.1997.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Garcion E, Lamprecht A, Heurtault B, Paillard A, Aubert-Pouessel A, Denizot B, Menei P, Benoit JP. A new generation of anticancer, drug-loaded, colloidal vectors reverses multidrug resistance in glioma and reduces tumor progression in rats. Mol Cancer Ther. 2006;5:1710–1722. doi: 10.1158/1535-7163.MCT-06-0289. [DOI] [PubMed] [Google Scholar]
- 348.Wong HL, Rauth AM, Bendayan R, Manias JL, Ramaswamy M, Liu Z, Erhan SZ, Wu XY. A new polymer-lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm Res. 2006;23:1574–1585. doi: 10.1007/s11095-006-0282-x. [DOI] [PubMed] [Google Scholar]
- 349.Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ. Cancer Research Campaign Phase I/II Clinical Trials committee. Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- 350.Satchi-Fainaro R, Puder M, Davies JW, Tran HT, Sampson DA, Greene AK, Corfas G, Folkman J. Targeting angiogenesis with a conjugate of HPMA copolymer and TNP-470. Nat Med. 2004;10:255–261. doi: 10.1038/nm1002. [DOI] [PubMed] [Google Scholar]
- 351.Qiu LY, Bae YH. Polymer architecture and drug delivery. Pharm Res. 2006;23:1–30. doi: 10.1007/s11095-005-9046-2. [DOI] [PubMed] [Google Scholar]
- 352.Zorko M, Langel U. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 353.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 354.Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637–651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 355.Marty C, Meylan C, Schott H, Ballmer-Hofer K, Schwendener RA. Enhanced heparan sulfate proteoglycan-mediated uptake of cell-penetrating peptide-modified liposomes. Cell Mol Life Sci. 2004;61:1785–1794. doi: 10.1007/s00018-004-4166-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Oehlke J, Lorenz D, Wiesner B, Bienert M. Studies on the cellular uptake of substance P and lysine-rich, KLA-derived model peptides. J Mol Recognit. 2005;18:50–59. doi: 10.1002/jmr.691. [DOI] [PubMed] [Google Scholar]
- 357.Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, Takehashi M, Tanaka S, Ueda K, Simpson JC, Jones AT, Sugiura Y, Futaki S. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 358.Dokka S, Toledo-Velasquez D, Shi X, Wang L, Rojanasakul Y. Cellular delivery of oligonucleotides by synthetic import peptide carrier. Pharm Res. 1997;14:1759–1764. doi: 10.1023/a:1012188014919. [DOI] [PubMed] [Google Scholar]
- 359.Fernandez-Carneado J, Van Gool M, Martos V, Castel S, Prados P, de Mendoza J, Giralt E. Highly efficient, nonpeptidic oligoguanidinium vectors that selectively internalize into mitochondria. J Am Chem Soc. 2005;127:869–874. doi: 10.1021/ja044006q. [DOI] [PubMed] [Google Scholar]
- 360.Yanagishita M, Hascall VC. Cell surface heparan sulfate proteoglycans. J Biol Chem. 1992;267:9451–9454. [PubMed] [Google Scholar]
- 361.Sonvico F, Dubernet C, Colombo P, Couvreur P. Metallic colloid nanotechnology, applications in diagnosis and therapeutics. Curr Pharm Des. 2005b;11:2095–2105. doi: 10.2174/1381612054065738. [DOI] [PubMed] [Google Scholar]
- 362.Loo C, Lowery A, Halas N, West J, Drezek R. Immunotargeted nanoshells for integrated cancer imaging and therapy. Nano Lett. 2005;5:709–711. doi: 10.1021/nl050127s. [DOI] [PubMed] [Google Scholar]
- 363.Martin CR, Kohli P. The emerging field of nanotube biotechnology. Nat Rev Drug Discov. 2003;2:29–37. doi: 10.1038/nrd988. [DOI] [PubMed] [Google Scholar]
- 364.Kam NW, O'Connell M, Wisdom JA, Dai H. Carbon nanotubes as multifunctional biological transporters and near-infrared agents for selective cancer cell destruction. Proc Natl Acad Sci USA. 2005;102:11600–11605. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Jain TK, Morales MA, Sahoo SK, Leslie-Pelecky DL, Labhasetwar V. Iron oxide nanoparticles for sustained delivery of anticancer agents. Mol Pharm. 2005;2:194–205. doi: 10.1021/mp0500014. [DOI] [PubMed] [Google Scholar]
- 366.Maitra A. Calcium phosphate nanoparticles: Second-generation nonviral vectors in gene therapy. Expert Rev Mol Diagn. 2005;5:893–905. doi: 10.1586/14737159.5.6.893. [DOI] [PubMed] [Google Scholar]
- 367.Gemeinhart RA, Luo D, Saltzman WM. Cellular fate of a modular DNA delivery system mediated by silica nanoparticles. Biotechnol Prog. 2005;21:532–537. doi: 10.1021/bp049648w. [DOI] [PubMed] [Google Scholar]
- 368.Klumpp C, Kostarelos K, Prato M, Bianco A. Functionalized carbon nanotubes as emerging nanovectors for the delivery of therapeutics. Biochim Biophys Acta. 2005;1758:404–412. doi: 10.1016/j.bbamem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 369.Wang H, Wang J, Deng X, Sun H, Shi Z, Gu Z, Liu Y, Zhao Y. Biodistribution of carbon single-wall carbon nanotubes in mice. J Nanosci Nanotechnol. 2004;4:1019–1024. doi: 10.1166/jnn.2004.146. [DOI] [PubMed] [Google Scholar]
- 370.Pantarotto D, Singh R, McCarthy D, Erhardt M, Briand JP, Prato M, Kostarelos K, Bianco A. Functionalized carbon nanotubes for plasmid DNA gene delivery. Angew Chem Int Ed Engl. 2004;43:5242–5246. doi: 10.1002/anie.200460437. [DOI] [PubMed] [Google Scholar]
- 371.Bianco A. Carbon nanotubes for the delivery of therapeutic molecules. Expert Opin Drug Deliv. 2004;1:57–65. doi: 10.1517/17425247.1.1.57. [DOI] [PubMed] [Google Scholar]
- 372.Constancis A, Meyrueix R, Bryson N, Huille S, Grosselin JM, Gulik-Krzywicki T, Soula G. Macromolecular colloids of diblock poly(amino acids) that bind insulin. J Colloid Interface Sci. 1999;217:357–368. doi: 10.1006/jcis.1999.6383. [DOI] [PubMed] [Google Scholar]
- 373.Nowak AP, Breedveld V, Pine DJ, Deming TJ. Unusual salt stability in highly charged diblock co-polypeptide hydrogels. J Am Chem Soc. 2003;125:15666–15670. doi: 10.1021/ja0381050. [DOI] [PubMed] [Google Scholar]
- 374.Kreiss P, Cameron B, Rangara R, Mailhe P, Aguerre-Charriol O, Airiau M, Scherman D, Crouzet J, Pitard B. Plasmid DNA size does not affect the physicochemical properties of lipoplexes but modulates gene transfer efficiency. Nucleic Acids Res. 1999;27:3792–3798. doi: 10.1093/nar/27.19.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Escriou V, Carriere M, Scherman D, Wils P. NLS bioconjugates for targeting therapeutic genes to the nucleus. Adv Drug Deliv Rev. 2003;55:295–306. doi: 10.1016/s0169-409x(02)00184-9. [DOI] [PubMed] [Google Scholar]
- 376.Vacik J, Dean BS, Zimmer WE, Dean DA. Cell-specific nuclear import of plasmid DNA. Gene Ther. 1999;6:1006–1014. doi: 10.1038/sj.gt.3300924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Lechardeur D, Lukacs GL. Intracellular barriers to non-viral gene transfer. Curr Gene Ther. 2002;2:183–194. doi: 10.2174/1566523024605609. [DOI] [PubMed] [Google Scholar]
- 378.Rosenberg SA, Blaese RM, Brenner MK, Deisseroth AB, Ledley FD, Lotze MT, Wilson JM, Nabel GJ, Cornetta K, Economou JS, Freeman SM, Riddell SR, Brenner M, Oldfield E, Gansbacher B, Dunbar C, Walker RE, Schuening FG, Roth JA, Crystal RG, Welsh MJ, Culver K, Heslop HE, Simons J, Wilmolt RW, Boucher RC, Siegler HF, Barranger JA, Karlsson S, Kohn D, Galpin JE, Raffel C, Hesdorffer C, Ilan J, Cassileth P, O'Shaughnessy J, Kun LE, Das TK, Wong-Staal F, Sobol RE, Haubrich R, Sznol M, Rubin J, Sorcher EJ, Rosenblatt J, Walker R, Brigham K, Vogelzang N, Hersh E, Eck SL. Human gene marker/therapy clinical protocols. Hum Gene Ther. 2000;11:919–979. doi: 10.1089/10430340050015536. [DOI] [PubMed] [Google Scholar]
- 379.Dyer MR, Herrling PL. Progress and potential for gene-based medicines. Mol Ther. 2000;1:213–224. doi: 10.1006/mthe.2000.0044. [DOI] [PubMed] [Google Scholar]
- 380.Trubetskoy VS, Wong SC, Subbotin V, Budker VG, Loomis A, Hagstrom JE, Wolff JA. Recharging cationic DNA complexes with highly charged polyanions for in vitro and in vivo gene delivery. Gene Ther. 2003;10:261–271. doi: 10.1038/sj.gt.3301888. [DOI] [PubMed] [Google Scholar]
- 381.Sagara K, Kim SW. A new synthesis of galactose-poly(ethylene glycol)-polyethylenimine for gene delivery to hepatocytes. J Control Release. 2002;79:271–281. doi: 10.1016/s0168-3659(01)00555-7. [DOI] [PubMed] [Google Scholar]
- 382.Kichler A. Gene transfer with modified polyethylenimines. J Gene Med. 2004;6:S3–10. doi: 10.1002/jgm.507. [DOI] [PubMed] [Google Scholar]
- 383.Simonson OE, Svahn MG, Tornquist E, Lundin KE, Smith CI. Bioplex technology: Novel synthetic gene delivery pharmaceutical based on peptides anchored to nucleic acids. Curr Pharm Des. 2005;11:3671–3680. doi: 10.2174/138161205774580813. [DOI] [PubMed] [Google Scholar]
- 384.Piskin E, Dincer S, Turk M. Gene delivery: Intelligent but just at the beginning. J Biomater Sci Polym Ed. 2004;15:1181–1202. doi: 10.1163/1568562041753016. [DOI] [PubMed] [Google Scholar]
- 385.Brown MD, Schatzlein A, Brownlie A, Jack V, Wang W, Tetley L, Gray AI, Uchegbu IF. Preliminary characterization of novel amino acid based polymeric vesicles as gene and drug delivery agents. Bioconj Chem. 2000;11:880–891. doi: 10.1021/bc000052d. [DOI] [PubMed] [Google Scholar]
- 386.Cui Z, Mumper RJ. Plasmid DNA-entrapped nanoparticles engineered from microemulsion precursors: In vitro and in vivo evaluation. Bioconjug Chem. 2002;13:1319–1327. doi: 10.1021/bc0255586. [DOI] [PubMed] [Google Scholar]
- 387.Bozkir A, Saka OM. Chitosan-DNA nanoparticles: Effect on DNA integrity, bacterial transformation and transfection efficiency. J Drug Target. 2004;12:281–288. doi: 10.1080/10611860410001714162. [DOI] [PubMed] [Google Scholar]
- 388.Prabha S, Labhasetwar V. Nanoparticle-mediated wild-type p53 gene delivery results in sustained antiproliferative activity in breast cancer cells. Mol Pharm. 2004;1:211–219. doi: 10.1021/mp049970+. [DOI] [PubMed] [Google Scholar]
- 389.Haas J, Ravi Kumar MN, Borchard G, Bakowsky U, Lehr CM. Preparation and characterization of chitosan and trimethyl-chitosan-modified poly-(epsilon-caprolactone) nanoparticles as DNA carriers. AAPS Pharm Sci Tech. 2005;6:E22–E30. doi: 10.1208/pt060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Banerjee P, Reichardt W, Weissleder R, Bogdanov A., Jr. Novel hyperbranched dendron for gene transfer in vitro and in vivo. Bioconjug Chem. 2004;15:960–968. doi: 10.1021/bc0342128. [DOI] [PubMed] [Google Scholar]
- 391.O'Brien J, Lummis SC. An improved method of preparing microcarriers for biolistic transfection. Brain Res Brain Res Protoc. 2002;10:12–15. doi: 10.1016/s1385-299x(02)00175-7. [DOI] [PubMed] [Google Scholar]
- 392.Eming SA, Krieg T, Davidson JM. Gene transfer in tissue repair: Status, challenges and future directions. Expert Opin Biol Ther. 2004;4:1373–1386. doi: 10.1517/14712598.4.9.1373. [DOI] [PubMed] [Google Scholar]
- 393.Roelvink PW, Mi Lee G, Einfeld DA, Kovesdi I, Wickham TJ. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science. 1999;286:1568–1571. doi: 10.1126/science.286.5444.1568. [DOI] [PubMed] [Google Scholar]
- 394.Lin E, Nemunaitis J. Oncolytic viral therapies. Cancer Gene Ther. 2004a;11:643–664. doi: 10.1038/sj.cgt.7700733. [DOI] [PubMed] [Google Scholar]
- 395.Mizuguchi H, Nakagawa T, Morioka Y, Imazu S, Nakanishi M, Kondo T, Hayakawa T, Mayumi T. Cytoplasmic gene expression system enhances the efficiency of cationic liposome-mediated in vivo gene transfer into mouse brain. Biochem Biophys Res Commun. 1997;234:15–18. doi: 10.1006/bbrc.1997.6568. [DOI] [PubMed] [Google Scholar]
- 396.Patil SD, Rhodes DG, Burgess DJ. DNA-based therapeutics and DNA delivery systems: A comprehensive review. AAPS J. 2005;7:E61–E77. doi: 10.1208/aapsj070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Irache JM, Merodio M, Arnedo A, Camapanero MA, Mirshahi M, Espuelas S. Albumin nanoparticles for the intravitreal delivery of anticytomegaloviral drugs. Mini Rev Med Chem. 2005;5:293–305. doi: 10.2174/1389557053175335. [DOI] [PubMed] [Google Scholar]
- 398.Tousignant JD, Gates AL, Ingram LA, Johnson CL, Nietupski JB, Cheng SH, Eastman SJ, Scheule RK. Comprehensive analysis of the acute toxicities induced by systemic administration of cationic lipid:plasmid DNA complexes in mice. Hum Gene Ther. 2000;11:2493–2513. doi: 10.1089/10430340050207984. [DOI] [PubMed] [Google Scholar]
- 399.Ruiz FE, Clancy JP, Perricone MA, Bebok Z, Hong JS, Cheng SH, Meeker DP, Young KR, Schoumacher RA, Weatherly MR, Wing L, Morris JE, Sindel L, Rosenberg M, van Ginkel FW, McGhee JR, Kelly D, Lyrene RK, Sorscher EJ. A clinical inflammatory syndrome attributable to aerosolized lipid-DNA administration in cystic fibrosis. Hum Gene Ther. 2001;12:751–761. doi: 10.1089/104303401750148667. [DOI] [PubMed] [Google Scholar]
- 400.Verma IM, Somia N. Gene therapy—Promises, problems and prospects. Nature. 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 401.Wiethoff CM, Middaugh CR. Barriers to nonviral gene delivery. J Pharm Sci. 2003;92:203–217. doi: 10.1002/jps.10286. [DOI] [PubMed] [Google Scholar]
- 402.Wikswo JP, Prokop A, Baudenbacher F, Cliffel D, Csukas B, Velkovsky M. The engineering challenges of BioMEMS: The integration of microfluidics, micro- and nano-devices, models, and external control for systems biology. IEE Proc Nanobiotech. 2006;153:81–101. doi: 10.1049/ip-nbt:20050045. [DOI] [PubMed] [Google Scholar]
- 403.Hummel JN. Naturlicher Ablauf der Entropievermehrung und die Lebenserschinungen. Akad Verlagsgesell Becker u. Erler; Leipzig: 1942. [Google Scholar]
- 404.Prokop A. Systems-analysis and synthesis in biology and biotechnology. Int J Gen Syst. 1982;8:7–31. [Google Scholar]
- 405.Rashevsky N. Mathematical foundations of general biology. Ann N Y Acad Sci. 1962;96:1105–1116. [PubMed] [Google Scholar]
- 406.Miller JG. Living systems. McGraw-Hill; New York: 1978. [Google Scholar]
- 407.(a) Papin JA, Palsson BO. Topological analysis of mass-balanced signaling networks: A framework to obtain network properties including crosstalk. J Theor Biol. 2004;227:283–297. doi: 10.1016/j.jtbi.2003.11.016. [DOI] [PubMed] [Google Scholar]; (b) Papin JA, Stelling J, Price ND, Klamt S, Schuster S, Palsson BO. Comparison of network-based pathway analysis methods. Trends Biotechnol. 2004;22:400–405. doi: 10.1016/j.tibtech.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 408.Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- 409.Kholodenko BN, Kiyatkin A, Bruggeman FJ, Sontag E, Westerhoff HV, Hoek JB. Untangling the wires: A strategy to trace functional interactions in signaling and gene networks. Proc Natl Acad Sci USA. 2002;99:15245. doi: 10.1073/pnas.192442699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Kholodenko BN, Hoek JB, Westerhoff HV, Brown GC. Quantification of information transfer via cellular signal transduction pathways. FEBS Lett. 1997;414:430–434. doi: 10.1016/s0014-5793(97)01018-1. Erratum in: FEBS Lett 1997, 419: 150. [DOI] [PubMed] [Google Scholar]
- 411.Hornberg JJ, Bruggeman FJ, Westerhoff HV, Lankelma J. Cancer: A systems biology disease. Biosystems. 2006;83:81–90. doi: 10.1016/j.biosystems.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 412.Radivoyevitch T, Loparo KA, Jackson RC, Sedwick WD. On systems and control approaches to therapeutic gain. BMC Cancer. 2006;6:104. doi: 10.1186/1471-2407-6-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Ahmed A, Gohlke H. Multiscale modeling of macromolecular conformational changes combining concepts from rigidity and elastic network theory. Proteins. 2006;63:1038–1151. doi: 10.1002/prot.20907. [DOI] [PubMed] [Google Scholar]
- 414.Botta M, Corelli F, Manetti F, Tafi A. Molecular modeling as a powerful technique for understanding small-large molecules interactions. Farmaco. 2002;57:153–165. doi: 10.1016/s0014-827x(01)01184-3. [DOI] [PubMed] [Google Scholar]
- 415.Balakin KV, Savchuk NP, Tetko IV. In silico approaches to prediction of aqueous and DMSO solubility of drug-like compounds: Trends, problems and solutions. Curr Med Chem. 2006;13:223–241. doi: 10.2174/092986706775197917. [DOI] [PubMed] [Google Scholar]
- 416.Stenberg P, Bergstrom CA, Luthman K, Artursson P. Theoretical predictions of drug absorption in drug discovery and development. Clin Pharmacokinet. 2002;41:877–899. doi: 10.2165/00003088-200241110-00005. [DOI] [PubMed] [Google Scholar]
- 417.Braccini I, Grasso RP, Perez S. Conformational and configurational features of acidic polysaccharides and their interactions with calcium ions: A molecular modeling investigation. Carbohydr Res. 1999;317:119–130. doi: 10.1016/s0008-6215(99)00062-2. [DOI] [PubMed] [Google Scholar]
- 418.Makabe K, McElheny D, Tereshko V, Hilyard A, Gawlak G, Yan S, Koide A, Koide S. Atomic structures of peptide self-assembly mimics. Proc Natl Acad Sci USA. 2006;103:17753–17758. doi: 10.1073/pnas.0606690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Tieleman DP. Computer simulations of transport through membranes: Passive diffusion, pores, channels and transporters. Clin Exp Pharmacol Physiol. 2006;33:893–903. doi: 10.1111/j.1440-1681.2006.04461.x. [DOI] [PubMed] [Google Scholar]
- 420.Kanjickal DG, Lopina ST. Modeling of drug release from polymeric delivery systems–A review. Crit Rev Ther Drug Carrier Syst. 2004;21:345–386. doi: 10.1615/critrevtherdrugcarriersyst.v21.i5.10. [DOI] [PubMed] [Google Scholar]
- 421.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: Current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 422.Boscolo R, Sabatti C, Liao JC, Roychowdhury VP. A generalized framework for network component analysis. IEEE/ACM Trans Comput Biol Bioinform. 2005;2:289–301. doi: 10.1109/TCBB.2005.47. [DOI] [PubMed] [Google Scholar]
- 423.Maeda YT, Sano M. Regulatory dynamics of synthetic gene networks with positive feedback. J Mol Biol. 2006;359:1107–1124. doi: 10.1016/j.jmb.2006.03.064. [DOI] [PubMed] [Google Scholar]
- 424.Teusink B, Passarge J, Reijenga CA, Esgalhado E, van der Weijden CC, Schepper M, Walsh MC, Bakker BM, van Dam K, Westerhoff HV, Snoep JL. Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur J Biochem. 2000;267:5313–5329. doi: 10.1046/j.1432-1327.2000.01527.x. [DOI] [PubMed] [Google Scholar]
- 425.Oliveira JS, Bailey CG, Jones-Oliveira JB, Dixon DA, Gull DW, Chandler ML. A computational model for the identification of biochemical pathways in the krebs cycle. J Comput Biol. 2003;10:57–82. doi: 10.1089/106652703763255679. [DOI] [PubMed] [Google Scholar]
- 426.Namjoshi AA, Hu WS, Ramkrishna D. Unveiling steady-state multiplicity in hybridoma cultures: The cybernetic approach. Biotechnol Bioeng. 2003;81:80–91. doi: 10.1002/bit.10447. [DOI] [PubMed] [Google Scholar]
- 427.Uygun K, Matthew HW, Huang Y. Investigation of metabolic objectives in cultured hepatocytes. Biotechnol Bioeng. 2006;97:622–637. doi: 10.1002/bit.21237. [DOI] [PubMed] [Google Scholar]
- 428.Balaban RS. Modeling mitochondrial function. Am J Physiol Cell Physiol. 2006;291:C1107–C1113. doi: 10.1152/ajpcell.00223.2006. [DOI] [PubMed] [Google Scholar]
- 429.Schwartz JM, Kanehisa M. Quantitative elementary mode analysis of metabolic pathways: The example of yeast glycolysis. BMC Bioinformatics. 2006;7:186. doi: 10.1186/1471-2105-7-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Clarke MF, Fuller M. Stem cells and cancer: Two faces of eve. Cell. 2006;124:1111–11115. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 431.(a) Ganguly R, Puri IK. Mathematical model for the cancer stem cell hypothesis. Cell Prolif. 2006;39:3–14. doi: 10.1111/j.1365-2184.2006.00369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dasika MS, Burgard A, Maranas CD. A computational framework for the topological analysis and targeted disruption of signal transduction networks. Biophys J. 2006;91:382–398. doi: 10.1529/biophysj.105.069724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Geho DH, Petricoin EF, Liotta LA, Araujo RP. Modeling of protein signaling networks in clinical proteomics. Cold Spring Harb Symp Quant Biol. 2005;70:517–524. doi: 10.1101/sqb.2005.70.022. [DOI] [PubMed] [Google Scholar]
- 433.Orton RJ, Sturm OE, Vyshemirsky V, Calder M, Gilbert DR, Kolch W. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem J. 2005;392:249–261. doi: 10.1042/BJ20050908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Schoeberl B, Eichler-Jonsson C, Gilles ED, Muller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol. 2002;20:370–375. doi: 10.1038/nbt0402-370. [DOI] [PubMed] [Google Scholar]
- 435.Sasagawa S, Ozaki Y, Fujita K, Kuroda S. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat Cell Biol. 2005;7:365–373. doi: 10.1038/ncb1233. [DOI] [PubMed] [Google Scholar]
- 436.Hua F, Hautaniemi S, Yokoo R, Lauffenburger DA. Integrated mechanistic and data-driven modelling for multivariate analysis of signalling pathways. J R Soc Interface. 2005;3:515–526. doi: 10.1098/rsif.2005.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Klamt S, Saez-Rodriguez J, Lindquist JA, Simeoni L, Gilles ED. A methodology for the structural and functional analysis of signaling and regulatory networks. BMC Bioinformatics. 2006;7:56. doi: 10.1186/1471-2105-7-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Zhu D, Lennon SP, Peters MH, Finney WC, Singh M. Brownian diffusion and surface kinetics of liposome and viral particle uptake by human lung cancer cells in-vitro. Ann Biomed Eng. 2006;34:1573–1586. doi: 10.1007/s10439-006-9158-9. [DOI] [PubMed] [Google Scholar]
- 439.Lund KA, Opresko LK, Starbuck C, Walsh BJ, Wiley HS. Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J Biol Chem. 1990;265:15713–15723. [PubMed] [Google Scholar]
- 440.(a) Wustner D. Quantification of polarized trafficking of transferrin and comparison with bulk membrane transport in hepatic cells. Biochem J. 2006;400:267–280. doi: 10.1042/BJ20060626. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wustner D. Mathematical analysis of hepatic high density lipoprotein transport based on quantitative imaging data. J Biol Chem. 2005;280:6766–6779. doi: 10.1074/jbc.M413238200. [DOI] [PubMed] [Google Scholar]
- 441.Heckman CA, Runyeon CS, Wade JG, Seubert S. Mathematical modeling of marker influx and efflux in cells. Bull Math Biol. 2001;63:431–449. doi: 10.1006/bulm.2001.0216. [DOI] [PubMed] [Google Scholar]
- 442.Enderling H, Anderson AR, Chaplain MA, Munro AJ, Vaidya JS. Mathematical modelling of radiotherapy strategies for early breast cancer. J Theor Biol. 2006;241:158–171. doi: 10.1016/j.jtbi.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 443.Ribba B, Colin T, Schnell S. A multiscale mathematical model of cancer, and its use in analyzing irradiation therapies. Theor Biol Med Model. 2006;243:532–541. doi: 10.1186/1742-4682-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Sinek JP, Frieboes HB, Silvaraman B, Sanga S, Cristi V. Mathematical and computational modeling: Towards the development and application of nanodevices for drug delivery. In: Kumar C, editor. Series: Nanotechnology for Life Scences 2006, Biofunctionalization of nanomaterials. Vol. 4. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 445.Wein LM, Wu JT, Ianculescu AG, Puri RK. A mathematical model of the impact of infused targeted cytotoxic agents on brain tumours: Implications for detection, design and delivery. Cell Prolif. 2002;35:343–361. doi: 10.1046/j.1365-2184.2002.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Levasseur LM, Slocum HK, Rustum YM, Greco WR. Modeling of the time-dependency of in vitro drug cytotoxicity and resistance. Cancer Res. 1998;58:5749–5761. [PubMed] [Google Scholar]
- 447.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–466. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 448.Araujo RP, Petricoin EF, Liotta LA. A mathematical model of combination therapy using the EGFR signaling network. Biosystems. 2005;80:57–69. doi: 10.1016/j.biosystems.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 449.Ramsay EC, Dos Santos N, Dragowska WH, Laskin JJ, Bally MB. The formulation of lipid-based nanotechnologies for the delivery of fixed dose anticancer drug combinations. Curr Drug Deliv. 2005;2:341–351. doi: 10.2174/156720105774370294. [DOI] [PubMed] [Google Scholar]
- 450.Novak B, Tyson JJ. A model for restriction point control of the mammalian cell cycle. J Theor Biol. 2004;230:563–579. doi: 10.1016/j.jtbi.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 451.Frieboes HB, Zheng X, Sun CH, Tromberg B, Gatenby R, Cristini V. An integrated computational/experimental model of tumor invasion. Cancer Res. 2006;66:1597–1604. doi: 10.1158/0008-5472.CAN-05-3166. [DOI] [PubMed] [Google Scholar]
- 452.Michor F, Nowak MA, Iwasa Y. Stochastic dynamics of metastasis formation. J Theor Biol. 2006;240:521–530. doi: 10.1016/j.jtbi.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 453.Colijn C, Mackey MC. A mathematical model of hematopoiesis–I. Periodic chronic myelogenous leukemia. J Theor Biol. 2005;237:117–132. doi: 10.1016/j.jtbi.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 454.McDougall SR, Anderson AR, Chaplain MA. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: Clinical implications and therapeutic targeting strategies. J Theor Biol. 2006;241:564–589. doi: 10.1016/j.jtbi.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 455.Chaplain M, Anderson A. Mathematical modelling of tumour-induced angiogenesis: Network growth and structure. Cancer Treat Res. 2004;117:51–75. doi: 10.1007/978-1-4419-8871-3_3. [DOI] [PubMed] [Google Scholar]
- 456.Spinelli L, Torricelli A, Ubezio P, Basse B. Modelling the balance between quiescence and cell death in normal and tumour cell populations. Math Biosci. 2006;202:349–370. doi: 10.1016/j.mbs.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 457.Bentele M, Lavrik I, Ulrich M, Stosser S, Heermann DW, Kalthoff H, Krammer PH, Eils R. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J Cell Biol. 2004;166:839–851. doi: 10.1083/jcb.200404158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458.(a) Anderson AR, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–915. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]; (b) Sinek J, Frieboes H, Zheng X, Cristini V. Two-dimensional chemotherapy simulations demonstrate fundamental transport and tumor response limitations involving nanoparticles. Biomed Microdevices. 2004;6:297–309. doi: 10.1023/B:BMMD.0000048562.29657.64. [DOI] [PubMed] [Google Scholar]
- 459.Furlani EP, Ng KC. Analytical model of magnetic nanoparticle transport and capture in the microvasculature. Phys Rev E Stat Nonlin Soft Matter Phys. 2006;73:061919. doi: 10.1103/PhysRevE.73.061919. [DOI] [PubMed] [Google Scholar]
- 460.Ghaghada KB, Saul J, Natarajan JV, Bellamkonda RV, Annapragada AV. Folate targeting of drug carriers: A mathematical model. J Control Release. 2005;104:113–128. doi: 10.1016/j.jconrel.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 461.Chappell MJ, Thomas GD, Godfrey KR, Bradwell AR. Optimal tumor targeting by antibodies: Development of a mathematical model. J Pharmacokinet Biopharm. 1991;19:227–260. doi: 10.1007/BF01073870. [DOI] [PubMed] [Google Scholar]
- 462.He Y, Shirazaki M, Liu H, Himeno R, Sun Z. A numerical coupling model to analyze the blood flow, temperature, and oxygen transport in human breast tumor under laser irradiation. Comput Biol Med. 2006;36:1336–1350. doi: 10.1016/j.compbiomed.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 463.Decuzzi P, Causa F, Ferrari M, Netti PA. The effective dispersion of nanovectors within the tumor microvasculature. Ann Biomed Eng. 2006;34:633–641. doi: 10.1007/s10439-005-9072-6. [DOI] [PubMed] [Google Scholar]
- 464.Graff CP, Wittrup KD. Theoretical analysis of antibody targeting of tumor spheroids: Importance of dosage for penetration, and affinity for retention. Cancer Res. 2003;63:1288–1296. [PubMed] [Google Scholar]
- 465.Maseide K, Rofstad EK. Mathematical modeling of chronical hypoxia in tumors considering potential doubling time and hypoxic cell lifetime. Radiother Oncol. 2000;54:171–177. doi: 10.1016/s0167-8140(99)00154-1. [DOI] [PubMed] [Google Scholar]
- 466.Reddy ST, Berk DA, Jain RK, Swartz MA. A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles. J Appl Physiol. 2006;101:1162–1169. doi: 10.1152/japplphysiol.00389.2006. [DOI] [PubMed] [Google Scholar]
- 467.Bondareva IB, Parfenov AS. A nonlinear mathematical model for the in vivo evaluation of the RES phagocytic function. Medinfo. 1995;8:1091. [PubMed] [Google Scholar]
- 468.Wang G, Krueger GR, Buja LM. A simplified and comprehensive computational model to study the behavior of T cell populations in the thymus during normal maturation and in infection with mouse moloney leukemiavirus. In Vivo. 2003;17:225–228. [PubMed] [Google Scholar]
- 469.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. IV. A microscopic model of the perivascular distribution. Microvasc Res. 1991;41:252–272. doi: 10.1016/0026-2862(91)90026-8. [DOI] [PubMed] [Google Scholar]
- 470.Stachowska-Pietka J, Waniewski J, Flessner MF, Lindholm B. Distributed model of peritoneal fluid absorption. Am J Physiol Heart Circ Physiol. 2006;291:H1862–H1874. doi: 10.1152/ajpheart.01320.2005. [DOI] [PubMed] [Google Scholar]
- 471.Green AJ, Johnson CJ, Adamson KL, Begent RH. Mathematical model of antibody targeting: Important parameters defined using clinical data. Phys Med Biol. 2001;46:1679–1693. doi: 10.1088/0031-9155/46/6/307. [DOI] [PubMed] [Google Scholar]
- 472.Yates JW. Structural identifiability of physiologically based pharmacokinetic models. J Pharmacokinet Pharmacodyn. 2006;33:421–439. doi: 10.1007/s10928-006-9011-7. [DOI] [PubMed] [Google Scholar]
- 473.Pond SM, Tozer TN. First-pass elimination. Basic concepts and clinical consequences. Clin Pharmacokinet. 1984;9:1–25. doi: 10.2165/00003088-198409010-00001. [DOI] [PubMed] [Google Scholar]
- 474.Mano Y, Suzuki H, Terasaki T, Iwahashi T, Ono K, Naito M, Tsuruo T, Sugiyama Y. Kinetic analysis of the disposition of MRK16, an anti-P-glycoprotein monoclonal antibody, in tumors: comparison between in vitro and in vivo disposition. J Pharmacol Exp Ther. 1997;283:391–401. [PubMed] [Google Scholar]
- 475.Varner JD. Systems biology and the mathematical modelling of antibody-directed enzyme prodrug therapy (ADEPT) Syst Biol (Stevenage) 2005;152:291–302. doi: 10.1049/ip-syb:20050047. [DOI] [PubMed] [Google Scholar]
- 476.Christopher R, Dhiman A, Fox J, Gendelman R, Haberitcher T, Kagle D, Spizz G, Khalil IG, Hill C. Data-driven computer simulation of human cancer cell. Ann N Y Acad Sci. 2004;1020:132–153. doi: 10.1196/annals.1310.014. [DOI] [PubMed] [Google Scholar]
- 477.Quaranta V, Weaver AM, Cummings PT, Anderson AR. Mathematical modeling of cancer: The future of prognosis and treatment. Clin Chim Acta. 2005;357:173–179. doi: 10.1016/j.cccn.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 478.el-Kareh AW, Secomb TW. Theoretical models for drug delivery to solid tumors. Crit Rev Biomed Eng. 1997;25:503–571. doi: 10.1615/critrevbiomedeng.v25.i6.20. [DOI] [PubMed] [Google Scholar]
- 479.Gadkar KG, Gunawan R, Doyle FJ., III Iterative approach to model identification of biological networks. BMC Bioinformatics. 2005;6:155. doi: 10.1186/1471-2105-6-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Fischer HP. Towards quantitative biology: Integration of biological information to elucidate disease pathways and to guide drug discovery. Biotechnol Annu Rev. 2005;11:1–168. doi: 10.1016/S1387-2656(05)11001-1. [DOI] [PubMed] [Google Scholar]
- 481.Abbott RG, Forrest S, Pienta KJ. Simulating the hallmarks of cancer. Artif Life. 2006;12:617–634. doi: 10.1162/artl.2006.12.4.617. [DOI] [PubMed] [Google Scholar]
- 482.Novak B. Intelligent systems in medical diagnosis. Stud Health Technol Inform. 1999;68:700–702. [PubMed] [Google Scholar]
- 483.Ulrich CM, Nijhout HF, Reed MC. Mathematical modeling: Epidemiology meets systems biology. Cancer Epidemiol Biomarkers Prev. 2006;15:827–829. doi: 10.1158/1055-9965.EPI-06-0252. [DOI] [PubMed] [Google Scholar]
- 484.Cho CR, Labow M, Reinhardt M, van Oostrum J, Peitsch MC. The application of systems biology to drug discovery. Curr Opin Chem Biol. 2006;10:294–302. doi: 10.1016/j.cbpa.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 485.(a) Csukas B, Bankuti G. Hybrid, quantitative and qualitative process simulation by the generic bi-layered net model,” in 2003 IEEE International Symposium on Intelligent Signal Processing. IEEE; Piscataway, NJ: 2003. pp. 283–288. [Google Scholar]; (b) Atri M. New technologies and directed agents for applications of cancer imaging. J Clin Oncol. 2006;24:3299–3308. doi: 10.1200/JCO.2006.06.6159. [DOI] [PubMed] [Google Scholar]
- 486.Agrawal A. New institute to study systems biology. Nat Biotechnol. 2005;17:743–744. doi: 10.1038/11667. [DOI] [PubMed] [Google Scholar]
- 487.Khalil IG, Hill C. Systems biology for cancer. Curr Opin Oncol. 2005;17:44–48. doi: 10.1097/01.cco.0000150951.38222.16. [DOI] [PubMed] [Google Scholar]
- 488.Mahmud A, Lavasanifar A. The effect of block copolymer structure on the internalization of polymeric micelles by human breast cancer cells. Colloids Surf B Biointerfaces. 2005;45:82–89. doi: 10.1016/j.colsurfb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 489.Hong S, Leroueil PR, Janus EK, Peters JL, Kober MM, Islam MT, Orr BG, Baker JR, Jr, Banaszak Holl MM. Interaction of polycationic polymers with supported lipid bilayers and cells: nanoscale hole formation and enhanced membrane permeability. Bioconjug Chem. 2006;17:728–734. doi: 10.1021/bc060077y. [DOI] [PubMed] [Google Scholar]
- 490.Manunta M, Nichols BJ, Tan PH, Sagoo P, Harper J, George AJ. Gene delivery by dendrimers operates via different pathways in different cells, but is enhanced by the presence of caveolin. J Immunol Methods. 2006;314:134–146. doi: 10.1016/j.jim.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 491.Shukla R, Thomas TP, Peters JL, Desai AM, Kukowska-Latallo J, Patri AK, Kotlyar A, Baker JR., Jr HER2 specific tumor targeting with dendrimer conjugated anti-HER2 mAb. Bioconjug Chem. 2006;17:1109–1115. doi: 10.1021/bc050348p. [DOI] [PubMed] [Google Scholar]
- 492.Manunta M, Tan PH, Sagoo P, Kashefi K, George AJ. Gene delivery by dendrimers operates via a cholesterol dependent pathway. Nucleic Acids Res. 2004;32:2730–2739. doi: 10.1093/nar/gkh595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Nan A, Ghandehari H, Hebert C, Siavash H, Nikitakis N, Reynolds M, Sauk JJ. Water-soluble polymers for targeted drug delivery to human squamous carcinoma of head and neck. J Drug Target. 2005;13:189–197. doi: 10.1080/10611860500065187. [DOI] [PubMed] [Google Scholar]
- 494.Pujals S, Fernandez-Carneado J, Lopez-Iglesias C, Kogan MJ, Giralt E. Mechanistic aspects of CPP-mediated intracellular drug delivery: Relevance of CPP self-assembly. Biochim Biophys Acta. 2006;1758:264–279. doi: 10.1016/j.bbamem.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 495.Remy-Kristensen A, Clamme JP, Vuilleumier C, Kuhry JG, Mely Y. Role of endocytosis in the transfection of L929 fibroblasts by polyethyleni-mine/DNA complexes. Biochim Biophys Acta. 2001;1514:21–32. doi: 10.1016/s0005-2736(01)00359-5. [DOI] [PubMed] [Google Scholar]
- 496.Miglietta A, Cavalli R, Bocca C, Gabriel L, Gasco MR. Cellular uptake and cytotoxicity of solid lipid nanospheres (SLN) incorporating doxorubicin or paclitaxel. Int J Pharm. 2000;210:61–67. [Google Scholar]
- 497.Bremer E, van Dam G, Kroesen BJ, de Leij L, Helfrich W. Targeted induction of apoptosis for cancer therapy: Current progress and prospects. Trends Mol Med. 2006;12:382–393. doi: 10.1016/j.molmed.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 498.Huth US, Schubert R, Peschka-Suss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110:490–504. doi: 10.1016/j.jconrel.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 499.Phelps MA, Foraker AB, Swaan PW. Cytoskeletal motors and cargo in membrane trafficking: Opportunities for high specificity in drug intervention. Drug Discov Today. 2003;8:494–502. doi: 10.1016/s1359-6446(03)02707-7. [DOI] [PubMed] [Google Scholar]
- 500.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 501.Bhattacharya R, Kang-Decker N, Hughes DA, Mukherjee P, Shah V, McNiven MA, Mukhopadhyay D. Regulatory role of dynamin-2 in VEGFR-2/KDR-mediated endothelial signaling. FASEB J. 2005;19:1692–1694. doi: 10.1096/fj.05-3889fje. [DOI] [PubMed] [Google Scholar]
- 502.Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–537. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 503.Haucke V. Cargo takes control of endocytosis. Cell. 2006;127:35–37. doi: 10.1016/j.cell.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 504.Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur J Pharm Sci. 2003;18:113–120. doi: 10.1016/s0928-0987(02)00251-8. [DOI] [PubMed] [Google Scholar]
- 505.Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3:785–796. doi: 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]
- 506.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Krauze MT, Mcknight TR, Yamashita Y, Bringas J, Noble CO, Saito R, Geletneky K, Forsayeth J, Berger MS, Jackson P, Park JW, Bankiewicz KS. Real-time visualization and characterization of liposomal delivery into the monkey brain by magnetic resonance imaging. Brain Res Brain Res Protoc. 2005;16:20–26. doi: 10.1016/j.brainresprot.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 508.Mortensen MW, Bjorkdahl O, Sorensen PG, Hansen T, Jensen MR, Gundersen HJ, Bjornholm T. Functionalization and cellular uptake of boron carbide nanoparticles. The first step toward T cell-guided boron neutron capture therapy. Bioconjug Chem. 2006;17:284–290. doi: 10.1021/bc050206v. [DOI] [PubMed] [Google Scholar]
- 509.Langer R, Tirrell D. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 510.Alexander C. Synthetic polymer systems in drug delivery. Expert Opin Emerg Drugs. 2001;6:345–363. doi: 10.1517/14728214.6.2.345. [DOI] [PubMed] [Google Scholar]
- 511.Venkatesh S, Byrne ME, Peppas NA, Hilt JZ. Applications of biomimetic systems in drug delivery. Expert Opin Drug Deliv. 2005;2:1085–1096. doi: 10.1517/17425247.2.6.1085. [DOI] [PubMed] [Google Scholar]
- 512.Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Sahoo SK, Labhasetwar V. Nanotech approaches to drug delivery and imaging. Drug Discov Today. 2003;8:1112–1120. doi: 10.1016/s1359-6446(03)02903-9. [DOI] [PubMed] [Google Scholar]
- 514.Allen TM, Cullis PR. Drug delivery systems: Entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 515.Nijhara R, Balakrishnan K. Bringing nanomedicines to market: Regulatory challenges, opportunities, and uncertainties. Nanomed Nanotechnol Biol Med. 2006;2:127–136. doi: 10.1016/j.nano.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 516.Chavanpatil MD, Khdair A, Panyam J. Nanoparticles for cellular drug delivery: Mechanisms and factors influencing delivery. J Nanosci Nanotechnol. 2006;6:2651–2663. doi: 10.1166/jnn.2006.443. [DOI] [PubMed] [Google Scholar]
- 517.Szekerke M, Wade R, Whisson ME. The use of macromolecules as carriers of cytotoxic groups (part I) conjugates of nitrogen mustards with proteins, polypeptidyl proteins and polypeptides. Neoplasma. 1972;19:199–209. [PubMed] [Google Scholar]
- 518.Petrak K. Nanotechnology and site-taregted drug delivery. J Biomater Sci Polymer Edn. 2006;17:1209–1219. doi: 10.1163/156856206778667497. [DOI] [PubMed] [Google Scholar]