

Abstract
Prion diseases, also called transmissible spongiform encephalopathies (TSEs), lead to neurological dysfunction in animals and are fatal. Infectious prion proteins are causative agents of many mammalian TSEs, including scrapie (in sheep), chronic wasting disease (in deer and elk), bovine spongiform encephalopathy (BSE; in cattle), and Creutzfeldt–Jakob disease (CJD; in humans). BSE, better known as mad cow disease, is among the many recently discovered zoonotic diseases. BSE cases were first reported in the United Kingdom in 1986. Variant CJD (vCJD) is a disease that was first detected in 1996, which affects humans and is linked to the BSE epidemic in cattle. vCJD is presumed to be caused by consumption of contaminated meat and other food products derived from affected cattle. The BSE epidemic peaked in 1992 and decreased thereafter; this decline is continuing sharply owing to intensive surveillance and screening programs in the Western world. However, there are still new outbreaks and/or progression of prion diseases, including atypical BSE, and iatrogenic CJD and vCJD via organ transplantation and blood transfusion. This paper summarizes studies on prions, particularly on prion molecular mechanisms, BSE, vCJD, and diagnostic procedures. Risk perception and communication policies of the European Union for the prevention of prion diseases are also addressed to provide recommendations for appropriate government policies in Korea.
Keywords: prion, risk perception, surveillance, transmissible spongiform encephalopathy, variant Creutzfeldt–Jakob disease
1. Introduction
Transmissible spongiform encephalopathy (TSE) is a general term for misfolded proteins, also called prions-related diseases, which had been discussed only among a few scientists until bovine spongiform encephalopathy (BSE), better known to the public as mad cow disease, became a public concern. It became widely known in the 1980s due to a sudden increase in the incidence of BSE in Europe; in the 1990s, it drew people’s attention as variant Creutzfeldt–Jakob disease (vCJD), which is also known as mad cow disease for humans [1].
BSE caused by pathogenic prions is an emerging zoonotic disease that can induce vCJD in humans. Considering that the prevalence of BSE in the United Kingdom (UK) hasmuchtodowithsheep farming, this new cross-species epidemic appears to be related to the current conditions involving frequent human contact with animals, enlarged livestock markets, increasing interchange/trade, and global warming [2,3]. The first case of BSE was reported in the mid-1980s, starting with 16 cattle; it has since increased dramatically to about 190,000 cases worldwide [4–6], concentrated in Europe. Thus, European countries are taking precautionary measures against the disease, and the prevalence of BSE has been decreasing since the mid-1990s [7].
However, many problems remain from both social and preventive medical perspectives since TSEs can spread through food and blood transfusions with very low concentrations of pathogenic prions, which current technologies cannot detect. In addition, since they have long incubation periods similar to other degenerative chronic diseases, more scientific investigations must be performed to identify the overall pathogeneses of BSE and vCJD, and to develop treatment strategies for them. Although the uncertainty surrounding prion diseases must still be clarified, they are controllable through intensive precautionary actions, as there are many existing studies on prions and their basic pathogenesis.
Reviews of prion diseases are already available elsewhere [8–11]; thus, this paper will give an overview of prion studies including not only prion diseases and diagnostic methods, but also the perception and communication of risk in the European Union (EU), which has successfully established their policy against BSE and vCJD as zoonotic diseases. This is for providing recommendations to governments for developing appropriate policies, based on scientific policy to free BSE.
1.1. Causes and prevalence of prion diseases
Prions, which were first proposed by Dr Prusiner at the University of California, San Francisco, became a hot topic since they do not have genes, unlike bacteria or viruses, and are able to replicate, unlike toxic elements [4]. Eventually, he scientifically answered a number of questions and suggested that these gene-less proteins can replicate in the body, induce the disease, and be subsequently transmitted to other animals; he received a Nobel Prize for his work in 1997. This type of prion-only hypothesis is recognized as a new pathogenic mechanism of neurodegenerative disorders [12].
The word “prion”, distinguished from virus or virion, was coined by Prusiner to refer to the scrapie pathogen in sheep; prion means a proteinaceous infectious particle [13]. PrPSc, a scrapie form of a prion known to be pathogenic and misfolded, does not always induce clinical symptoms; therefore, the PrPSc that induces clinical symptoms is marked as a disease prion: PrPd. However, prions already exist in animals and humans in the cellular form of the protein (PrPC), which does not have any pathogenic properties. The primary amino acid sequences and the state of modification in both isoforms of PrPSc and PrPC are identical; however, they have different three-dimensional structures, which give them distinct biochemical and biophysical properties. Also, alterations in amino acid sequences change the conformation of these proteins, resulting in a thermodynamically stable protein variant (PrPSc) that can cause diseases in both animals and humans [14].
When PrPC comes in contact with PrPSc, PrPC changes into a thermodynamically stable PrPSc via protein folding; then, PrPSc converts PrPC into another PrPSc. After this process is repeated over time, PrPSc accumulates in the body and results in induction of TSE [15]. Although it is not certain whether TSEs are caused by PrPSc alone or by a complex reaction with PrPSc and other factors, such as other proteins, nucleic acids, or pathogens [16], it is certain that the main causative agent is PrPSc. However, to explain the replication of PrPSc within the body, two thermodynamically stable PrPSc molecules must be separated and combined with other PrPCs. The animal’s genetic type and other additional factors should be considered in the process [17]. Therefore, scientists assume that the hypothetical macromolecule—designated protein X—may play a role in the conversion from PrPC to PrPSc and continue to search for candidates. At present, dozens of proteins in the cytosol, plasma membrane, extracellular matrix, and lipid rafts are known to interact with PrPC and/or PrPSc; however, strong evidence for the identity of protein X has not been revealed yet [18]. Identifying the existence and role of protein X, prion diseases can be prevented and/or treated. Normal PrPC, which is encoded on the gene locus PRNP in the genome of hosts, is a glyco-protein found in neuron cell membranes in animals and humans. PrPC has a ∼40% α-helical and ∼3% β-sheet conformation, whereas PrPSc has a ∼30% α-helical and ∼40% β-sheet conformation. The conformational transition from the helices and hydrophobic regions of PrPC is a major cause of the increase of β-sheet composition in PrPSc (Figure 1) [19,20]. Since the conformational alteration from PrPC to PrPSc is not immunogenic, the immune system of the organism can hardly distinguish the normal protein structure from the infectious prion structure, except with respect to its stability [21]. Unlike bacteria or viruses, pathogenic PrPSc cannot be removed by regular alcohol or formalin sterilization processes, and cannot be decomposed by proteolytic enzymes. Moreover, it is resistant to heat, ultraviolet rays, and chemicals. Under three times the atmospheric pressure the materials for prion should be sterilized for more than 18 minutes at 134–138° C; surfaces should be sterilized for over an hour with 2% sodium hypochlorite and 2N sodium hydroxide at 20°C, and equipment should be sterilized for more than 12 hours with the same solution. In laboratories, materials should be disinfected for more than 4.5 hours at 132°C or for 1 hour at 134–138°Cby steam sterilization under pressure. Because it responds well to alkaline conditions, highly concentrated sodium hydroxide or phenol is used to decontaminate PrPSc[22].
Figure 1.
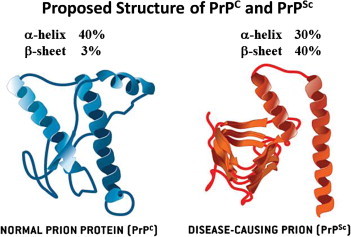
Normal and disease-causing prions structures [20].
1.2. Specific risk material and the species barrier
PrPSc in infected animals is concentrated in specific areas. These areas are called specific risk material (SRM) and include the brain, eyes, spinal cord, skull, vertebral column, tonsils, and distal ileum; these are the most crucial areas for disease management and control. The disease is transmittable via surgical tools that came into contact with SRM and via blood transfusion. Since blood rarely contains the prion, it was considered safe until a death caused by vCJD from a blood transfusion was reported in the UK, which was alarming to the public. From this case, the UK spent more than £200 million on a preventive process to protect surgical tools against prion transmission. This shows that the occurrence of BSE or vCJD can cause an enormous amount of indirect expenses, although they do not occur frequently.
The species barrier makes it difficult for infectious diseases to be transmitted from one species to another. The value of the species barrier for prion disease transmission between humans and cattle has been estimated to be 4000, based on the study of BSE zoonosis. However, a precautionary principle assumes that there is only one value of species barrier between humans and cattle, and suggests that the same dose that causes the disease in cattle may affect humans in the same way [23]. The experimental disease-inducing amount of SRM in a single administration is 0.001 g injected orally; 10 g can lead to BSE in all administered cattle [24]. The amount required to induce the disease is very small; a scientific report submitted to the British Council in 2001 states that an amount as small as one speck of pepper may cause the disease [25]. Five grams of an oral inoculum with brain homogenate from a BSE-infected cattle in a primate (Cynomolgus macaques) resulted in the development of a vCJD-like neurological disease 60 months after exposure [26].
Since such a small dose can cause the disease affecting both humans and animals and there is currently no cure, experiments on the PrPSc agent transmissible to humans are supposed to be performed in Biosafety Level 3 in the same manner as biologically strong infectious agents (e.g., anthrax bacterium, Severe acute respiratory syndrome, and the West Nile virus). It is notable that in the study of pathogenic prions, the protein particles have been observed to have many different strains, which are under investigation [27]. In fact, the discovery of new strains is related to the species barrier and has many implications in disease control in relation to the adaptation and progression of TSE [28].
In that respect, the EU, which has conducted much research on BSE and vCJD, defines any beef that has had contact with any SRM as SRM itself and advises people not to use any cosmetic or food items with SRM, although transmission via cosmetics or food has not been reported yet. In BSE-infected cattle, PrPSc is also found on peripheral nerves. Consequently, the whole body of the infected cattle is disposed of according to the EU regulation [29].
It is noteworthy that Payer’s patch tissue, which is the most essential factor for PrPSc absorption, is mostly on the ileum in humans; however, similar tissues are predominantly found in the whole intestine including mesentery in cattle [30]. Therefore, the EU defines the whole intestine as SRM, the evidence of which is verified annually [31]. Recently, Switzerland submitted a request (EFSA-Q-2009-00226) to the European Food Safety Authority (EFSA) to reassess the use of bovine intestines for stuffing sausage. The request was declined, which demonstrates a careful approach to the consumption of bovine intestine by global organizations. Similarly, Koreans also need to take precautionary action in consuming bovine intestines.
2. Types of Prion Diseases
2.1. Scrapie
In 1732, scrapie—a disease among sheep—was first reported in the UK, affecting the wool industry. The official name for the disease (scrapie) was used from 1853 onward. The name scrapie is derived from one of the clinical signs of the condition, wherein the affected flock will compulsively scrape off their fleece against rocks, trees, or fences. The disease apparently causes an itching sensation in the animals. Other clinical signs include excessive lip smacking, altered gaits, and convulsive collapse [32]. Fortunately, other livestock did not have such symptoms; therefore, it was only a concern among sheep-farming communities, and not among other people or other animal farmers.
In the 1900s, farmers in the UK started to feed cows with internal organs or bones of sheep to benefit economically from the increase in milk and meat production. By the 1930s, other European countries and the United States (USA) had adopted this practice. Based on the findings of epidemiological studies on the origin of BSE, this later became the main cause of prion disease transmission from sheep to cows across the species barrier [33].
As the PrPSc-inducing disease in sheep and cows is also found in other animals, TSE can be considered an inclusive term for the disease. TSE is subdivided into BSE for bovines, vCJD for humans, scrapie for sheep, chronic wasting disease (CWD) for deer, and transmissible mink encephalopathy for mink (Table 1); TSE is found in 26 species, including goats, cats, and wild ruminants [1,34,35]. It is noteworthy that prion diseases among sheep and deer can be transmitted horizontally by saliva, unlike BSE or vCJD [36]; CWD can even be transmitted via aerosols, according to a recent animal experiment report [37].
Table 1.
| Disease name | Animal species | Reported year |
|---|---|---|
| Scrapie | Sheep | 1732 |
| Creutzfeldt–Jakob disease | Humans | 1920 |
| Transmissible mink encephalopathy | Mink | 1947 |
| Chronic wasting disease | Deer | 1967 |
| Bovine spongiform encephalopathy | Cows | 1986 |
| Feline spongiform encephalopathy | Cats | 1990 |
| Variant Creutzfldte–Jakob disease | Humans | 1996 |
2.2. Bovine spongiform encephalopathy
There are several theories regarding the cause of the first reported case of BSE in the mid-1980s; some insist that the BSE pathogen (PrPSc) formed naturally and others claim that the disease was caused by the cow feed made from sheep infected with scrapie. By an extensive epidemiologic investigation, the main cause for BSE turned out to be a meat and bone meal (MBM) made from the discarded bones and intestines of slaughtered cows and sheep. In the UK, in particular, cow intestines have been used in MBM as a protein supplement since 1972, which accelerated the increase of the occurrence of BSE [1,38].
BSE has occurred in European countries that import MBM from the UK; according to statistics from the World Organization for Animal Health (Office International des Epizooties; OIE), there have been 190,628 BSE cases in 25 countries worldwide as of August 30, 2012 (http://www.oie.int). Most reported cases are from the UK, peaking in 1992, and in other countries the epidemic peaked in 2002 or 2003; from then the number started to decrease sharply.
BSE is a chronic degenerative neurological disease in cows; part of the brain becomes sponge like, and exhibits many different kinds of neurotic symptoms and paralysis, eventually leading to death [39]. In BSE, nerve cells and central nerve tissues take on a sponge-like form. After approximately 2–5 years of incubation, the animal dies within approximately 2 weeks to 6 months of development of the disease. Clinical symptoms include extreme sensitivity to external stimuli such as light and sound, neurotic changes (depression and nervousness), positional imbalance, inability to stand straight or move, paralysis in the hind legs, and paralysis of the whole body before death [40].
At present, BSE is under surveillance by the OIE; in Korea, it is classified as a second category of animal epidemics along with scrapie and CWD. BSE has no effect on the cattle younger than 7 months old; by the time cows reach 24 months of age, there are many variant prions in the body. Most occurrences of BSE are in cows older than 36 months. Therefore, the OIE examines the occurrence of BSE in 24-month-old cows.
In the UK, more than 184,000 cases of BSE have been reported and more than 3 million cows were destroyed to stop the spread of the disease; hence, the UK strictly banned MBM. Owing to their efforts, the occurrence of BSE was dramatically reduced. However, since the 2000s, the disease has been spreading worldwide, including in the USA, Japan, Israel, and various African countries. Determining the precise number of occurrences is challenging, as some infected animals do not exhibit any particular symptoms. Without total inspection and surveillance, it is difficult to research the actual status of the disease [41].
Therefore, the EU places much emphasis on active monitoring and surveillance systems, such as total inspection, thorough removal of SRM (where 99% of the pathogenic prions exist), banning MBM, and monitoring animal feed. Through such actions, BSE has become manageable, but it is still not eradicated. The USA also started to emphasize the development of an effective animal-monitoring system over concerns for human health [42].
However, some BSE cases have been reported even after stricter surveillance was put in place, which means that the disease is not controllable by monitoring animal feed alone. Some scientific evidence is given regarding this: pathogenic prions from the feces of TSE-infected animal can be absorbed into the soil [43,44], can combine with minerals in the soil, and can become stable [45]. Although BSE does not seem to be transmitted horizontally within species, such findings suggest that more precautionary actions and approaches should be performed in epidemiologic investigations, including studying the possibility of transmission via a contaminated environment [46].
BSE-infected cows show the possibility of self-mutation of the BSE prion, since the prion gene that causes vCJD in humans, which had some mutations, was found in the brains of affected cows. This implies that a wide range of monitoring systems in DNA and/or protein levels is necessary in addition to a strict animal feed policy. Considering the transmission of BSE to humans, control SRM is the most important step to take. Based on recent findings on the relationship between SRM and the occurrence of the disease, the EU developed some guidelines in April 2008 for its member countries to follow regarding SRM. According to these guidelines, the tonsils, whole intestine, and mesenterium are all vulnerable to prions across all ages; the brains, eyes, spinal cords, and skulls of cows that are older than 12 months are considered to be SRM.
Some older cows have an atypical form of BSE (BASE), which differs from typical BSE with respect to its molecular and biochemical properties; it appears to be a sporadic BSE, although more precise etiological studies need to be performed for confirmation. Recently, attention has been given to BASE [47–49] because of its infectivity and relation to vCJD.
2.3. Variant CJD
Clinical symptoms similar to those of CJD that were reported in the 1920s were displayed in a patient who consumed contaminated beef (affected with BSE); the disease was called vCJD, which was first described in 1996. There are four types of CJDs: two nontransmittable CJDs, including sporadic CJD (sCJD) and familial or genetic CJD (implying a genetic cause), and two transmittable CJDs, including iatrogenic CJD and vCJD (Table 2).
Table 2.
Various types of Creutzfeldt–Jakob disease [50]
| Type of disease | Cause |
|---|---|
| Sporadic CJD | Sporadic. The cause is not known; it is not common but is found worldwide and is generally found in old people; one in 1 million spontaneous conversion |
| Familial CJD | Genetic. It is caused by mutation of a prion gene; 5–10% of all cases of TSE. It is also called genetic CJD |
| Iatrogenic CJD | Acquired. It is transmitted through medical procedures including surgery, transplantation, and blood transfusion |
| Variant CJD | Acquired. It was discovered in 1996; it is assumed that it is acquired by the consumption of pathogenic prions from BSE-infected beef (it was initially called new variant CJD) |
BSE = bovine spongiform encephalopathy; CJD = Creutzfeldt–Jakob disease.
An endemic disease similar to CJD was found in cannibal tribes (such as the Fore, Gimi, and Yate in Papua New Guinea) who used to eat the dead bodies of their relatives as part of their rituals. This endemic disease, called kuru (meaning “shiver” in Uruna or Guzigli, among other tribes), was first reported in 1957. At that time, the cause could not be identified, so people assumed that the disease was caused by an unknown virus. However, ever since cannibalism was banned, the incidence of kuru decreased sharply.
Meanwhile, many studies have sought to identify the association between specific genotypes and the occurrence of the disease; a correlation between vCJD and methionine homozygote (MM type) at codon 129 in the human prion suggests that the MM type is highly related and susceptible to prion infection [51], and is clearly documented as a significant genetic risk factor [52]. A study on kuru among cannibal tribes shows that the incubation period of prion diseases differs among individuals based on genotype [53]. The MM type at codon 129 of the prion is most common in Korean people and has the shortest incubation period, with death following shortly after disease development; the methionine–valine heterozygote (MV type) appears to be most resistant to the disease, and a case with 40 years of incubation has also been reported. At present, all vCJD patients in Europe (including patients affected through transfusion [54]) have the MM type except for one case [55]. Therefore, Aguzzi at University Hospital of Zurich in Switzerland warns that patients should be observed for at least 40 years since the number of patients with the MV type may increase in the future. The Spongiform Encephalopathy Advisory Committee appointed by ministers from the UK assumes that there may be 4000–10000 infected persons with no symptoms in the UK; however, valine homozygote- (VV type) or MV-type patients may die of different causes due to the long incubation period after they have been exposed to the pathogenic prion.
Since vCJD was first reported in 1996, a total of 224 patients with this disease from 12 countries have been identified worldwide, as shown in Table 3; the main symptoms include unstable emotions; abnormal senses; paralysis in linguistic, visual, and other senses; as well as the inability to move and cognitive disability before death [56,57]. While sCJD is common in older people, vCJD can occur in young people after a short incubation period [58]. There is much epidemiological and laboratory evidence of a strong correlation between variants of CJD and BSE; vCJD differs from other CJDs clinically and histopathologically, and the initial extended exposure of the population to potentially BSE-contaminated food (1984–1986) was geographically and chronologically consistent with the initial onset of vCJD cases (1994–1996), considering the incubation period [1,59]. The facts that sCJD occurs in old people at the ratio of one to two out of 1 million people and vCJD is frequent in people in their 20s and 30s have led the public to believe that the dietary habits of the younger generation (who tend to eat more fast food such as hamburgers) may be the cause of the disease. However, recent findings suggest that age-related body conditions are the causative factors of the disease [60]. The EU has been establishing countermeasures against the transmission of vCJD via blood transfusion. Current studies are focusing on the possibility of transmission via dental treatment [61,62].
Table 3.
Incidence of vCJD until August 2012
| Country | Number of primary cases | Number of secondary cases: blood transfusion | Cumulative residence in the UK for >6 months during 1980–1996 |
|---|---|---|---|
| Canada | 2 | 0 | 1 |
| France | 27 | 0 | 1 |
| Italy | 2 | 0 | 0 |
| Japan | 1 | 0 | 0 |
| Netherland | 3 | 0 | 0 |
| Portugal | 2 | 0 | 0 |
| Republic of Ireland | 4 | 0 | 2 |
| Saudi Arabia | 1 | 0 | 0 |
| Spain | 5 | 0 | 0 |
| Taiwan | 1 | 0 | 1 |
| UK | 173 | 3 | 176 |
| USA | 3 | 0 | 2 |
| Total | 224 | 3 | 183 |
VCD = variant Creutzfeldt–Jakob disease.
3. Diagnostic Methods
At present, a reliable diagnosis of prion disease is possible only through autopsy since there is no approved method for detecting prion levels, which are too low to be detected by any test, in the peripheral nervous systems of live animals or humans. Thus, tissues of the central nervous system, including the brain and spinal cord, obtained at autopsy are used for prion diagnostic tests using immunology-based techniques such as enzyme-linked immunosorbent assay (ELISA), immunohistochemistry, and immunoblotting; a histopathological test is then performed for confirmation. However, the final confirmation is obtained by performing a bioassay to assess the infectivity of the pathogen; this is the most sensitive test and uses a transgenic mouse with the human prion gene to detect pathogenic PrPd by observing the infection. Moreover, DNA sequencing for determination of genetic variations is also considered.
The standard diagnosis procedures for BSE suggested by the OIE include ELISA, Western blotting, and immunohistochemical methods to test brain tissues (Table 4). However, simple immunological tests to detect BSE cannot distinguish PrPSc from PrPC. Therefore, the target specimen should be processed by proteinase K (PK) first, and then the remaining PK-resistant prions such as PrPSc are detected. For rapid testing, 15 rapid diagnosis kits are available; the tests most frequently used around the world are shown in Table 5.
Table 4.
| Method | Specimen | Detection of |
|---|---|---|
| Rapid test using ELISA | Fresh brain tissue | PrPSc antigen |
| Histopathological test | Formalin-fixed brain tissue | Spongiform in brain tissue |
| Immunohistochemistry | Formalin-fixed brain tissue | PrPSc antigen |
| Western blot | Fresh brain tissue | PrPSc antigen |
| Electron microscopy | Fresh brain tissue | Scrapie-associated fibril |
| Bioassay | Fresh tissue | PrPSc and infectivity |
BSE = bovine spongiform encephalopathy; ELISA = enzyme-linked immunosorbent assay.
Table 5.
| Product name (company) | Diagnostic method | Authorized country | Note |
|---|---|---|---|
| Prionics-Check | Western blot using the proteinase–processed specimen using an antigen to prion | EU | Specimen: 0.5 g obex tissue |
| Western test (Prionics AG) | USA | Limit: ∼5.0–20 pmol | |
| Test period: ∼6–8 h | |||
| Prionics-Check LIA (Prionics AG) | Sandwich ELISA | EU | Specimen: 0.5 g obex tissue |
| USA | Limit: ∼1.0–5.0 pmol | ||
| Test period: 4 h | |||
| Bio-Rad test (Bio-Rad) | Sandwich ELISA | EU | Specimen: 0.35 g obex tissue |
| USA | Limit: ∼0.5–2.0 pmol | ||
| Japan | Test period: 6 h | ||
| Enfer test (Abbott Labs) | Simple ELISA | EU | Limit: ∼1–10 pmol |
| USA | Test period: 4 h | ||
| Japan | |||
| CDI test (Impro Biotechnology) | Immunity analysis based on the structural differences | EU | Limit: ∼0.5–5.0 pmol |
| USA | Test period: 8 h | ||
| Japan | No proteinase K | ||
| Herd-check (IDEXX Laboratories) | Immunity analysis after separating the variant prion (PrPSc) from brain tissue specimens | USA | Test period: ∼4–5hours |
| No proteinase K |
ELISA = enzyme-linked immunosorbent assay.
However, authorized tests that use antigen–antibody reactions are good for highly concentrated specimens, such as the brain and spinal cord, since the amount detectable by ELISA-based procedures is so low that they cannot be applied to actual blood specimens, through which the disease can be transmitted. Therefore, many scientific efforts have been made to overcome this shortcoming, such as protein misfolding cyclic amplification, which uses the replication of the conformation of a protein [65], and real-time immuno-polymerase chain reaction (PCR), which utilizes a combination of antibody features and the sensitivity of PCR. Recently, real-time quaking-induced conversion has been developed for an assay in which disease-associated prion protein initiates a rapid conformational transition in recombinant prion protein, resulting in the formation of amyloid that can be monitored in real time [66]. Nevertheless, although these new diagnostic methods have more than 100 times the sensitivity of the conventional ELISA method, they still have unstable backgrounds or give false positives, making it difficult to use them as authorized tests.
However, diagnostic methods to determine pathogenic PrPd based on PK resistance are limited, since PK-sensitive prions with pathogenic characteristics have been found [67]. Conventional tests for BSE or vCJD cannot detect PK-sensitive prions. Furthermore, some cases diagnosed as dementia may actually be cases of vCJD [68]. Therefore, there is an urgent need to develop novel diagnostic tests that are faster and more accurate in detecting new types of prions [69].
3.1. Risk perception, communication, and management of prion diseases
The occurrence of BSE or vCJD in any country is beyond a simple outbreak of an incurable disease; it affects the economy, society, and the meat market due to changes in policies for exportation and importation. In these situations, scientists should provide accurate information to the public [70]. However, despite the guidelines suggested by the OIE, the fact that Canada, the USA, and the EU, which have had BSE outbreaks, have different countermeasures and policies indicates that science is not the only thing that needs to be considered in risk management of BSE.
Consumers’ trust in the government and industry can affect the degree of how science should be reflected in policy. Thus, when policies are re-established, scientific findings should be clearly communicated among interest groups through open discussion and public opinion in advance. In this process, the risk perception of the government and food industry should be treated and considered equally as the overall risk perception of the public. Therefore, policy makers must consider risk communication to come up with strategies and plans.
Two of the conclusions and recommendations with respect to the TSE roadmap from the workshop for the EU countries on risk perception and communication among interest groups are described below [29]. First, SRM control and feed bans should be considered, by all countries, as the most important policies for BSE risk regulation; any relaxation of these policies should be made with extreme caution, based on solid scientific knowledge and accompanied by an effective communication strategy toward stakeholders as well as the general public. Second, surveillance systems are also important, although most countries consider testing regimens merely as tools for epidemiological monitoring of the disease. In that respect, active surveillance systems should be retained for some more time, although the current regulatory design can be modified to be more flexible when all stakeholders are in a consensus.
The EU has separate organizations for evaluation and management that aid in controlling BSE and vCJD and public understanding, thus putting a fair and accurate control system in place. Risk assessment is performed by the EFSA, while risk management is carried out by the Directorate General for Health and Consumer Affairs. Two different organizations can achieve a balance in a politically sensitive issue and prevent each other from distorting the scientific truth, which is a very desirable situation in controlling zoonotic diseases such as BSE.
The communication channel for prevention of the epidemic is important. Most of the misunderstandings regarding BSE are due to the absence of or misconception about risk communication and risk management [71] in societies. It is very risky to mention this disease without considering the difference between laboratory findings and field applications in quarantine, particularly when establishing policies that are relevant to daily life [72]. The EU maintains estimations of cow age through a history tracking system called traceability, in which the electronic ear tag of the cow must match its history document before it can be slaughtered. Because of such strict management, BSE outbreak is decreasing in the EU. However, there are still some nonscientific opinions based on the distorted understanding; some people believe that it does not matter to assume a cow’s age by examining its teeth while importing SRM and that this disease itself is going to disappear soon.
The USA banned the import of human sperm for in vitro fertilization from Northern Europe due to vCJD risk. Although vCJD infection via sperm is not reported yet, the USA took this strong action to protect its people from Europe where prion diseases are more prevalent. Among scientists, the possibility of the occurrence of vCJD in the USA has been discussed continuously [73]. Such strict risk management implies how government policy should reflect both public opinions and scientific truth through careful communication, considering that there was a meaningless probability controversy over the transmission of disease via the importation of beef.
Furthermore, farming and slaughtering systems of importing countries are lagging behind those of the exporting countries such as the EU and the USA, so it is reasonable to demand strict management and quarantine to exporting countries. From the perspective of preventive medicines, such opinions are very risky and irresponsible. Poor farming and slaughtering conditions in importing countries mean more favorable conditions for the disease to spread even when there is only a small threat to the nation. According to the EU’s geographical BSE-risk analysis for the risk control of BSE, more strict policies and actions should be taken to prevent BSE from spreading to countries with poorer conditions [74].
4. Concluding Remarks
All emerging zoonotic diseases are not simple diseases and involve sociocultural issues because of their threatening and fearful characteristics. In that respect, BSE and vCJD are typical emerging epidemics along with AIDS; these diseases are still progressive and being studied. However, new information on the pathogenesis of these diseases is found every year; it is hoped, on the basis of the experience in the EU, that BSE and vCJD can be controlled, and their incidence be lowered gradually. Nevertheless, for risk reduction, strict precautionary principles should be applied since prevention is the most effective way of controlling this emerging disease that has not been experienced before.
Although OIE regulations are expected to be relaxed in time to keep up with the free-trade era, managing SRM is the most important aspect in controlling this disease. In one case, it took 5 years to change the age regulation of the EU on SRM (up to 6 months). The different regulations of the OIE and the EU regarding SRM standards cause confusion among people. SRM standards set by the OIE are “conditions or guidelines for trade” that can prevent the disease from spreading from one country to another. Based on these criteria, countries are supposed to establish their own trade regulations in which their industry structures, conditions for controlling the disease, and dietary habits should be considered. In other words, OIE regulations are the necessary conditions for all countries to follow to prevent the spread of the disease. However, SRM standards set by the EU are “scientifically sufficient conditions” that participating countries with different cultural and industrial backgrounds can utilize [75].
In Korea, it is critical to implement a traceability system or active total inspection to prevent prion diseases as soon as possible. In fact, the possibility of BSE and vCJD occurring naturally in Korea is extremely low. The major reason for this is that there are not enough preconditions for such diseases to occur. Korea has historically had few sheep farms. A British national institution reports that one of the main causes of the BSE outbreak in the UK was feeding cows with sheep intestines [25]. However, Korea has not developed sheep farming, so the precondition of the BSE and vCJD pandemic does not exist there. The second reason is that Koreans used to consume cow intestines. For the epidemic to spread, there should be at least some infected entities; after the tipping point (threshold), it becomes an epidemic [76,77]. Prion diseases have species barriers; within one species, the diseases spread more easily and quickly. However, a step for the multiplication of prions within a species is blocked since Koreans consume cow intestines themselves rather than feeding them to cows. In that respect, it is almost impossible for BSE or vCJD to occur naturally in Korea. If a BSE outbreak is reported in Korea, the cause would likely be from outside the country.
Acknowledgments
We are thankful to Bo-Ran Choi and Sangho Choi for their assistance in preparing the manuscript. We have cited more recent and easy-to-understand references in this review due to the large number of available literature. We have also cited personal opinions communicated with other scientists and previous review articles. This work was supported by Korea Centers for Disease Control and Prevention (Project number 2011E5300600), Ministry of Health and Welfare, Republic of Korea.
References
- 1.Prusiner SB. Prion biology and diseases. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- 2.Jones KE, Patel NG, Levy MA, et al. Global trends in emerging infectious diseases. Nature. 2008 Feb;451(7181):990–3. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stark KD, Regula G, Hernandez J, et al. Concepts for risk-based surveillance in the field of veterinary medicine and veterinary public health: review of current approaches. BMC Health Serv Res. 2006;6:20. doi: 10.1186/1472-6963-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prusiner SB. Molecular biology of prion diseases. Science. 1991 Jun;252(5012):1515–22. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 5.Legname G, Baskakov IV, Nguyen HO, et al. Synthetic mammalian prions. Science. 2004 Jul;305(5684):673–6. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 6.de Pedro-Cuesta J, Glatzel M, Almazan J, et al. Human transmissible spongiform encephalopathies in eleven countries: diagnostic pattern across time, 1993–2002. BMC Public Health. 2006;6:278. doi: 10.1186/1471-2458-6-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prusiner SB, DeArmond SJ. Prion diseases and neurodegeneration. Annu Rev Neurosci. 1994;17:311–39. doi: 10.1146/annurev.ne.17.030194.001523. [DOI] [PubMed] [Google Scholar]
- 8.Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: a phenotype under challenge. Prion. 2007 Apr;1(2):83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pezza JA, Serio TR. Prion propagation: the role of protein dynamics. Prion. 2007 Jan;1(1):36–43. doi: 10.4161/pri.1.1.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacs GG, Budka H. Molecular pathology of human prion diseases. Int J Mol Sci. 2009 Mar;10(3):976–99. doi: 10.3390/ijms10030976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abid K, Soto C. The intriguing prion disorders. Cell Mol Life Sci. 2006 Oct;63(19–20):2342–51. doi: 10.1007/s00018-006-6140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prusiner SB. Prions. Sci Am. 1984 Oct;251(4):50–9. doi: 10.1038/scientificamerican1084-50. [DOI] [PubMed] [Google Scholar]
- 13.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982 Apr;216(4542):136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 14.Moore RA, Taubner LM, Priola SA. Prion protein misfolding and disease. Curr Opin Struct Biol. 2009 Feb;19(1):14–22. doi: 10.1016/j.sbi.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liemann S, Glockshuber R. Transmissible spongiform encephalopathies. Biochem Biophys Res Commun. 1998 Sep;250(2):187–93. doi: 10.1006/bbrc.1998.9169. [DOI] [PubMed] [Google Scholar]
- 16.Bremer J, Heikenwalder M, Haybaeck J, et al. Repetitive immunization enhances the susceptibility of mice to peripherally administered prions. PLoS One. 2009;4(9):e7160. doi: 10.1371/journal.pone.0007160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd SE, Maytham EG, Pota H, et al. HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet. 2009 Feb;5(2):e1000383. doi: 10.1371/journal.pgen.1000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ryou C. Prions and prion diseases: fundamentals and mechanistic details. J Microbiol Biotechnol. 2007 Jul;17(7):1059–70. [PubMed] [Google Scholar]
- 19.Novakofski J, Brewer MS, Mateus-Pinilla N, Killefer J, McCusker RH. Prion biology relevant to bovine spongiform encephalopathy. J Anim Sci. 2005 Jun;83(6):1455–76. doi: 10.2527/2005.8361455x. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner SB. Detecting mad cow disease. Sci Am. 2004 Jul;291(1):86–93. doi: 10.1038/scientificamerican0704-86. [DOI] [PubMed] [Google Scholar]
- 21.Bruederle CE, Hnasko RM, Kraemer T, et al. Prion infected meat-and-bone meal is still infectious after biodiesel production. PLoS One. 2008;3(8):e2969. doi: 10.1371/journal.pone.0002969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutton JM, Dickinson J, Walker JT, Raven ND. Methods to minimize the risks of Creutzfeldt–Jakob disease transmission by surgical procedures: where to set the standard? Clin Infect Dis. 2006 Sep;43(6):757–64. doi: 10.1086/507030. [DOI] [PubMed] [Google Scholar]
- 23.Gale P. BSE risk assessments in the UK: a risk tradeoff? J Appl Microbiol. 2006 Mar;100(3):417–27. doi: 10.1111/j.1365-2672.2006.02878.x. [DOI] [PubMed] [Google Scholar]
- 24.Wells GA, Konold T, Arnold ME, et al. Bovine spongiform encephalopathy: the effect of oral exposure dose on attack rate and incubation period in cattle. J Gen Virol. 2007 Apr;88(Pt 4):1363–73. doi: 10.1099/vir.0.82421-0. [DOI] [PubMed] [Google Scholar]
- 25.Phillips NL, Bridgeman J, Ferguson-Smith MA. 2000. The BSE inquiry: return to an order of the Honourable House of Commons dated October 2000 for the report, evidence and supporting papers of the inquiry into the emergence and identification of bovine spongi-form encephalopathy (BSE) and variant Creutzfeldt–Jakob disease (vCJD) and the action taken in response to it up to 20 March 1996.
- 26.Lasmezas CI, Comoy E, Hawkins S, et al. Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet. 2005 Feb-Mar;365(9461):781–3. doi: 10.1016/S0140-6736(05)17985-9. [DOI] [PubMed] [Google Scholar]
- 27.Aguzzi A. Unraveling prion strains with cell biology and organic chemistry. Proc Natl Acad Sci U S A. 2008 Jan;105(1):11–2. doi: 10.1073/pnas.0710824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beringue V, Vilotte JL, Laude H. Prion agent diversity and species barrier. Vet Res. 2008 Jul-Aug;39(4):47. doi: 10.1051/vetres:2008024. [DOI] [PubMed] [Google Scholar]
- 29.Kerstin Dressel AP, Ru Giuseppe, Van Wassenhove Wim. TSE roadmap—a comparative study of the risk perceptions and risk communications of stakeholders within European countries. Brussels, EC: Nov 23, 2009. The NeuroPrion Project. [Google Scholar]
- 30.van Keulen LJ, Bossers A, van Zijderveld F. TSE pathogenesis in cattle and sheep. Vet Res. 2008 Jul-Aug;39(4):24. doi: 10.1051/vetres:2007061. [DOI] [PubMed] [Google Scholar]
- 31.TAFS . TAFS Position Paper on Specified Risk Materials. 2009. Swiss. [Google Scholar]
- 32.Foster JD, Parnham D, Chong A, Goldmann W, Hunter N. Clinical signs, histopathology and genetics of experimental transmission of BSE and natural scrapie to sheep and goats. Vet Rec. 2001 Feb;148(6):165–71. doi: 10.1136/vr.148.6.165. [DOI] [PubMed] [Google Scholar]
- 33.Wilesmith JW, Ryan JB, Atkinson MJ. Bovine spongiform encephalopathy: epidemiological studies on the origin. Vet Rec. 1991 Mar;128(9):199–203. doi: 10.1136/vr.128.9.199. [DOI] [PubMed] [Google Scholar]
- 34.Vaccari G, Panagiotidis CH, Acin C, et al. State-of-the-art review of goat TSE in the European Union, with special emphasis on PRNP genetics and epidemiology. Vet Res. 2009 Sep-Oct;40(5):48. doi: 10.1051/vetres/2009031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998 Nov;95(23):13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller MW, Williams ES. Prion disease: horizontal prion transmission in mule deer. Nature. 2003 Sep;425(6953):35–6. doi: 10.1038/425035a. [DOI] [PubMed] [Google Scholar]
- 37.Denkers ND, Seelig DM, Telling GC, Hoover EA. Aerosol and nasal transmission of chronic wasting disease in cervidized mice. J Gen Virol. 2010 Jun;91(Pt 6):1651–8. doi: 10.1099/vir.0.017335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Public health issues related to animal and human spongiform encephalopathies: memorandum from a WHO meeting. Bull World Health Organ. 1992;70(2):183–90. [PMC free article] [PubMed] [Google Scholar]
- 39.Hope J, Reekie LJ, Hunter N, et al. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature. 1988 Nov;336(6197):390–2. doi: 10.1038/336390a0. [DOI] [PubMed] [Google Scholar]
- 40.Horiuchi M, Nakamitsu S. [Prion diseases in animals–bovine spongiform encephalopathy] Nippon Rinsho. 2007 Aug;65(8):1513–20. [PubMed] [Google Scholar]
- 41.Wilesmith JW. Preliminary epidemiological analyses of the first 16 cases of BSE born after July 31, 1996, in Great Britain. Vet Rec. 2002 Oct;151(15):451–2. doi: 10.1136/vr.151.15.451. [DOI] [PubMed] [Google Scholar]
- 42.Sapkota AR, Lefferts LY, McKenzie S, Walker P. What do we feed to food-production animals? A review of animal feed ingredients and their potential impacts on human health. Environ Health Perspect. 2007 May;115(5):663–70. doi: 10.1289/ehp.9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safar JG, Lessard P, Tamguney G, et al. Transmission and detection of prions in feces. J Infect Dis. 2008 Jul;198(1):81–9. doi: 10.1086/588193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kruger D, Thomzig A, Lenz G, Kampf K, McBride P, Beekes M. Faecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Vet Res. 2009 Jan-Feb;40(1):4. doi: 10.1051/vetres:2008042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies P, Brown DR. Manganese enhances prion protein survival in model soils and increases prion infectivity to cells. PLoS One. 2009;4(10):e7518. doi: 10.1371/journal.pone.0007518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saunders SE, Bartelt-Hunt SL, Bartz JC. Prions in the environment: occurrence, fate and mitigation. Prion. 2008 Oct;2(4):162–9. doi: 10.4161/pri.2.4.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lombardi G, Casalone C, D’Angelo A, et al. Intraspecies transmission of BASE induces clinical dullness and amyotrophic changes. PLoS Pathog. 2008 May;4(5):e1000075. doi: 10.1371/journal.ppat.1000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capobianco R, Casalone C, Suardi S, et al. Conversion of the BASE prion strain into the BSE strain: the origin of BSE? PLoS Pathog. 2007 Mar;3(3):e31. doi: 10.1371/journal.ppat.0030031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Comoy EE, Casalone C, Lescoutra-Etchegaray N, et al. Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS One. 2008;3(8):e3017. doi: 10.1371/journal.pone.0003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hörnlimann B, Riesner D, Kretzschmar HA. Prions in humans and animals. Berlin, New York: Walter de Gruyter; 2007. [Google Scholar]
- 51.Zeidler M, Stewart G, Cousens SN, Estibeiro K, Will RG. Codon 129 genotype and new variant CJD. Lancet. 1997 Aug 30;350(9078):668. doi: 10.1016/s0140-6736(05)63366-1. [DOI] [PubMed] [Google Scholar]
- 52.Bishop MT, Pennington C, Heath CA, Will RG, Knight RS. PRNP variation in UK sporadic and variant Creutzfeldt–Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med Genet. 2009;10:146. doi: 10.1186/1471-2350-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collinge J, Whitfield J, McKintosh E, et al. A clinical study of kuru patients with long incubation periods at the end of the epidemic in Papua New Guinea. Philos Trans R Soc Lond B Biol Sci. 2008 Nov;363(1510):3725–39. doi: 10.1098/rstb.2008.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ponte ML. Insights into the management of emerging infections: regulating variant Creutzfeldt–Jakob disease transfusion risk in the UK and the US. PLoS Med. 2006 Oct;3(10):e342. doi: 10.1371/journal.pmed.0030342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bishop MT, Hart P, Aitchison L, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006 May;5(5):393–8. doi: 10.1016/S1474-4422(06)70413-6. [DOI] [PubMed] [Google Scholar]
- 56.Zeidler M, Johnstone EC, Bamber RW, et al. New variant Creutzfeldt–Jakob disease: psychiatric features. Lancet. 1997 Sep;350(9082):908–10. doi: 10.1016/s0140-6736(97)03148-6. [DOI] [PubMed] [Google Scholar]
- 57.Zeidler M, Stewart GE, Barraclough CR, et al. New variant Creutzfeldt–Jakob disease: neurological features and diagnostic tests. Lancet. 1997 Sep;350(9082):903–7. doi: 10.1016/s0140-6736(97)07472-2. [DOI] [PubMed] [Google Scholar]
- 58.Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet. 1996 Apr;347(9006):921–5. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 59.Smith PG. The epidemics of bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease: current status and future prospects. Bull World Health Organ. 2003;81(2):123–30. [PMC free article] [PubMed] [Google Scholar]
- 60.Lefrere JJ, Hewitt P. From mad cows to sensible blood transfusion: the risk of prion transmission by labile blood components in the United Kingdom and in France. Transfusion. 2009 Apr;49(4):797–812. doi: 10.1111/j.1537-2995.2008.02044.x. [DOI] [PubMed] [Google Scholar]
- 61.Bourvis N, Boelle PY, Cesbron JY, Valleron AJ. Risk assessment of transmission of sporadic Creutzfeldt–Jakob disease in endodontic practice in absence of adequate prion inactivation. PLoS One. 2007;2(12):e1330. doi: 10.1371/journal.pone.0001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonetti D, Young L, Black I, Cassie H, Ramsay CR, Clarkson J. Can’t do it, won’t do it! Developing a theoretically framed intervention to encourage better decontamination practice in Scottish dental practices. Implement Sci. 2009;4:31. doi: 10.1186/1748-5908-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gavier-Widen D, Stack MJ, Baron T, Balachandran A, Simmons M. Diagnosis of transmissible spongiform encephalopathies in animals: a review. J Vet Diagn Invest. 2005 Nov;17(6):509–27. doi: 10.1177/104063870501700601. [DOI] [PubMed] [Google Scholar]
- 64.Soto C. Diagnosing prion diseases: needs, challenges and hopes. Nat Rev Microbiol. 2004 Oct;2(10):809–19. doi: 10.1038/nrmicro1003. [DOI] [PubMed] [Google Scholar]
- 65.Soto C, Saborio GP, Anderes L. Cyclic amplification of protein misfolding: application to prion-related disorders and beyond. Trends Neurosci. 2002 Aug;25(8):390–4. doi: 10.1016/s0166-2236(02)02195-1. [DOI] [PubMed] [Google Scholar]
- 66.Wilham JM, Orrú CD, Bessen RA, et al. Rapid end-point quantitation of prion seeding activity with sensitive comparable to bioassay. PLoS Pathog. 2010;6(12):e1001217. doi: 10.1371/journal.ppat.1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008 Jun;63(6):697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zou WQ, Gambetti P. Prion: the chameleon protein. Cell Mol Life Sci. 2007 Dec;64(24):3266–70. doi: 10.1007/s00018-007-7380-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A. A versatile prion replication assay in organotypic brain slices. Nat Neurosci. 2008 Jan;11(1):109–17. doi: 10.1038/nn2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wilson K, Code C, Dornan C, Ahmad N, Hebert P, Graham I. The reporting of theoretical health risks by the media: Canadian newspaper reporting of potential blood transmission of Creutzfeldt–Jakob disease. BMC Public Health. 2004 Jan;4:1. doi: 10.1186/1471-2458-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chou WY, Hunt YM, Beckjord EB, Moser RP, Hesse BW. Social media use in the United States: implications for health communication. J Med Internet Res. 2009;11(4):e48. doi: 10.2196/jmir.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hanney SR, Gonzalez-Block MA, Buxton MJ, Kogan M. The utilisation of health research in policy-making: concepts, examples and methods of assessment. Health Res Policy Syst. 2003 Jan;1(1):2. doi: 10.1186/1478-4505-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holman RC, Belay ED, Christensen KY, et al. Human prion diseases in the United States. PLoS One. 2010;5(1):e8521. doi: 10.1371/journal.pone.0008521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giovannini A, Savini L, Conte A, Fiore GL. Comparison of BSE prevalence estimates from EU countries for the period July to December 2001 to the OIE and EU GBR classifications. J Vet Med B Infect Dis Vet Public Health. 2005 Aug;52(6):262–71. doi: 10.1111/j.1439-0450.2005.00862.x. [DOI] [PubMed] [Google Scholar]
- 75.Goossens B. Personal communication with Dr. Goossens, a Senior Scientific Officer of European Food Safety Authority. Prion 2009 Conference; Sep 23, 2009. [Google Scholar]
- 76.Marsh DR, Gilroy KE, Van de Weerdt R, Wansi E, Qazi S. Community case management of pneumonia: at a tipping point? Bull World Health Organ. 2008 May;86(5):381–9. doi: 10.2471/BLT.07.048462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khumalo-Sakutukwa G, Morin SF, Fritz K, et al. Project Accept (HPTN 043): a community-based intervention to reduce HIV incidence in populations at risk for HIV in sub-Saharan Africa and Thailand. J Acquir Immune Defic Syndr. 2008 Dec;49(4):422–31. doi: 10.1097/QAI.0b013e31818a6cb5. [DOI] [PMC free article] [PubMed] [Google Scholar]