

Abstract
Reactive astrogliosis is one of the pathological hallmarks of neurodegenerative diseases. Inflammatory cytokines, such as TNF-α and IL-1β, have been shown to mediate the reactive astrogliosis in neurodegenerative diseases; however, the molecular mechanism remains unclear. In this study, we investigated the role of transcription factor FOXO3a on astrocyte proliferation, one primary aspect of severe reactive astrogliosis. Our results confirmed that TNF-α and IL-1β increased astrocyte proliferation, as determined by Ki67 and BrdU immunostaining. Furthermore, we found that cytokine-mediated astrocyte proliferation was accompanied by an increase of the phosphorylation and reduced nuclear expression of FOXO3a. Intracranial injection of TNF-α and IL-1β induced astrocyte proliferation and hypertrophy, which was associated with reduced nuclear expression of Foxo3a in astrocytes. To determine the function of FOXO3a in astrocyte proliferation, wild type FOXO3a was overexpressed with adenovirus, which subsequently upregulated p27Kip1 and Gadd45α, and significantly inhibited cytokine-induced astrocyte proliferation. In contrast, overexpression of dominant negative FOXO3a decreased p27Kip1, upregulated cyclin D1 and promoted astrocyte proliferation. Along the same line, astrocytes isolated from Foxo3a-null mice have higher proliferative potential. In response to intracranial injection of cytokines, Foxo3a-null mice manifested severe astrogliosis in vivo. In conclusion, FOXO3a is important in restraining astrocyte proliferation during proinflammatory cytokine stimulation and loss of function of FOXO3a may be responsible for the proliferation of astrocytes in the severe form of reactive astrogliosis. Understanding the key regulatory role of FOXO3a in reactive astrogliosis may provide a novel therapeutic target during neuroinflammation.
Keywords: Astrogliosis, Proinflammatory cytokine, FOXO3a, Akt-1, Phosphorylation, Cyclin D1
Introduction
Reactive astrogliosis, which includes astrocytic hypertrophy and hyperplasia, process extension and interdigitation, and an increase in glia fibrillary acidic protein (GFAP), is a continuum of changes in astrocytes in response to all forms of CNS insult and disease(da Cunha et al. 1993; Eddleston and Mucke 1993; Sofroniew 2009). Although a growing body of information indicates that reactive astrocytes exert numerous essential functions, dysfunctions of reactive astrogliosis can contribute to CNS diseases(Barres 2008; De Keyser et al. 2008). In neurodegenerative diseases, reactive astrogliosis is one of the primary pathological features associated with neuronal damage and neurodegeneration(Fawcett 1997; Hirsch et al. 2005; Ridet et al. 1997; Rodriguez et al. 2008). At its extreme level of activation in response to overt tissue damage and inflammation, reactive astrogliosis is involved in scar formation that incorporates newly proliferated cells(Faulkner et al. 2004; Sofroniew 2009). Highly localized proliferative astroglial cells have been found in thalamic dementia or accumulate around the plaques in Alzheimer's disease, augmenting the neuroinflammation and neurodegeneration(Kiyota et al. 2009; Potts and Leech 2005; Rodriguez et al. 2008). Therefore, molecular dissection of reactive astrogliosis is imperative for the development of novel therapeutic strategies.
Proinflammatory cytokines, particularly TNF-α and IL-1β, have been shown to mediate astrocyte proliferation, as well as the permanent glial scar(Sriram et al. 2006; Woiciechowsky et al. 2004). Both cytokines are important in gliosis and neurodegeneration since they both initiate and sustain long-term features of astrogliosis(Buffo et al. 2009). Application of glucocorticoids or IL-10, which inhibit the proinflammatory response, has been shown to reduce reactive gliosis and the glia scar(Avola et al. 2004; Balasingam and Yong 1996; Muller et al. 2001). Although evidence suggests cell cycle regulation may be involved, the mechanisms underlying proinflammatory cytokine-mediated astrocyte proliferation are not fully understood(Di Giovanni et al. 2005; Hara et al. 2008; Zhu et al. 2007).
Mammalian forkhead members of the class O (FOXO) transcription factors, including FOXO1, FOXO3a, FOXO4, and FOXO6, are implicated in the regulation of multiple cellular processes in a cell-type- and environment-specific manner. Modulation of FOXO transcription factor activities can lead to a broad array of cellular outputs resulting in changes in proliferation, apoptosis, stress resistance, differentiation, and metabolic responses. Studies in mouse have shown that Foxos play significant roles in the homeostasis of the central nervous system. Specifically, Foxo3a plays a prominent role in maintaining the neural stem cell (NSC) pool by regulating genes involved in cellular proliferation, differentiation, and oxygen metabolism(Aranha et al. 2009; Paik et al. 2009; Renault et al. 2009). The regulation of the FOXO proteins is highly conserved. FOXO proteins are negatively regulated by the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB, also known as Akt) signaling pathway in response to growth factors and cytokines(Arden 2008; Tran et al. 2003). Akt-1 phosphorylates FOXO3a, sequestering FOXO3a in the cytoplasm and inhibiting FOXO3a activity(Brunet et al. 1999; Calnan and Brunet 2008). Previously we have demonstrated that dysfunction of FOXO3a induced apoptosis in HIV-1-infected macrophages(Cui et al. 2008). More recently, we have found that CXCL12 increases human neural progenitor cell proliferation through FOXO3a(Wu et al. 2009). Dysfunction of FOXO proteins is frequently observed in the proliferation disorders such as cancers(Cui et al. 2009; Maiese et al. 2009). Despite the multiple functional aspect of FOXO3a, its contribution to astrogliosis has not been elucidated to date.
To determine the regulation of FOXO3a and its functional roles in reactive astrogliosis, we used inflammatory cytokine stimulated astrocytes as models. TNF-α and IL-1β first activated Akt-1, subsequently phosphorylated FOXO3a, increased cyclin D1 expression, promoted cell cycle progression and augmented astrocyte proliferation. Intracranial injection of TNF-α and IL-1β induced an increased number and hypertrophy of astrocytes, which was associated with reduced nuclear presence of FOXO3a in astrocyte in vivo. Overexpression of FOXO3a upregulated p27Kip1 and Gadd45α expression as well as inhibited TNF-α and IL-1β induced astrocyte proliferation, while Dominant negative (DN) FOXO3a promoted astrocyte proliferation. Concordantly, astrocytes from Foxo3a null mice demonstrated higher proliferative potential both in in vitro and in vivo. These results suggest FOXO3a is a critical factor preventing inflammatory cytokine-mediated astrocyte proliferation. Modulation of FOXO3a may provide a therapeutic avenue for dysregulated reactive astrogliosis in neuroinflammatory diseases.
Material and methods
TNF-α and IL-1β intracranial injection
Animals were maintained in sterile microisolator cages under pathogen-free conditions in the Laboratory of Animal Medicine at UNMC in accordance with ethical guidelines for care of laboratory animals set forth by the National Institutes of Health. Four-week-old male FVB/NJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Foxo3a-null mice were purchased from Mutant Mouse Regional Resource Centers and bred in Laboratory of Animal Medicine at UNMC. Experiments involving Foxo3a null mice were performed with wild type mice from the same litter.
Intracranial injection was done as described previously(Persidsky et al. 1996). In brief, TNF-α (50 ng) and IL-1β (10 ng) in 5 μl PBS, or 5 μl PBS alone (as vehicle control) were injected into basal ganglia of left hemisphere in FVB/NJ mice. For experiments comparing Foxo3a null mice with wild type control, reduced concentrations of TNF-α (5 ng) and IL-1β (1 ng) were used. Four animals were included in each of the two groups. At 7 days after injection, mice were euthanized with isoflurane and the brains were removed. The brains were fixed with 4% paraformaldehyde for 48 h, and embedded in paraffin. For protein extraction, the mouse brain was sliced into 1.0 mm section, split into left and right hemisphere, and subjected to homogenization and protein extraction. For Western blotting of phosphorylated Foxo3a and total Foxo3a in mouse brain, the quantification was based on the densitometric determination of the blotting. Ratio of p-Foxo3a to total Foxo3a of left hemisphere (PBS injections or TNF-α/IL-1β injections) were used for statistical analysis. Right hemisphere was used as an anatomical reference.
Immunohistochemistry and image analysis
Blocks were cut to identify the injection site. For each mouse, 30∼100 serial (5-μm-thick) sections were cut from the injection. Sections were deparaffinized, and immunohistochemical staining followed a basic indirect immunostaining protocol using Ag retrieval by heating to 95°C in 0.01 M citrate buffer for 30 minutes. Incubation with 10% rabbit serum in phosphate-buffered saline was used to reduce non-specific binding and background staining. The anti-GFAP primary mouse monoclonal antibody (Sigma, G3893) and the anti-Foxo3a primary rabbit polyclonal antibody (Abcam, ab47409) were used at 1:500 dilution and 1:400 dilution respectively. Antibodies were applied onto the slides and incubated overnight at 4°C. Double-immunofluorescent staining was performed using Alexa Fluor 488 (green) and 647 (red) as secondary antibodies (Molecular Probes, Eugene, OR). Confocal pictures were taken at 10× to show big view and needle track. Pictures at 40× and 60× magnifications were taken in regions 200∼300 μm away in both directions from the needle track of brain sections. To allow semi-quantitive comparisons of fluorescence levels between two groups, all images were acquired using same microscopy setting. Four to five pictures per section were taken. All obtained images were imported into ImageJ-ProPlus, version 4.0 (Media Cybernetics, Sliver Spring, MD), for quantifying levels of GFAP- and Foxo3a-positive staining. The mean fluorescence intensity of GFAP in arbitrary unites was determined in 40× images. Twenty images from each group were analyzed. The mean fluorescence intensity of Foxo3a in arbitrary unites was determined at GFAP positive cells of 60× images. Three GFAP positive cells were randomly selected from each picture and twenty pictures were analyzed.
Statistical analyses
Data were expressed as means ± SD. The data were evaluated statistically by analysis of variance (ANOVA) followed by the Tukey's test for paired observations. Significance was considered as a p value of < 0.05. To account for any donor-specific differences, all experiments were performed with astrocytes from at least three donors. All assays were performed at least twice, with triplicate or quadruplicate samples in each experiment.
(For details of reagents, astrocyte culture, proliferation assays, Western blotting and adenoviral vectors, please see the supplemental data.)
Results
TNF-α and IL-1β increase human astrocyte proliferation
To study reactive astrogliosis, particularly the astrocyte proliferation in vitro, we used Ki67 immunocytochemical staining and bromodeoxyuridine (BrdU) incorporation in primary cultured human astrocytes following TNF-α and/or IL-1β stimulation. Astrocyte proliferation, as determined by Ki67 staining, increased after 3 days of TNF-α and/or IL-1β treatment (Fig. 1). By evaluating different doses of TNF-α and IL-1β on astrocyte proliferation, we found that a combination of TNF-α (50 ng/ml) and IL-1β (1 ng/ml) showed the most significant increase in cell proliferation (Fig. 1E). To confirm TNF-α- and IL-1β-induced astrocyte proliferation, BrdU incorporation assay was used and a similar result was found compared to Ki67 immunocytochemical staining (Fig. 1F). Treatment with serum, such as 10% FBS, has showed an increase of proliferation in astrocytes, and it served as positive control in our proliferation assays. These results establish an in vitro functional assay to detect TNF-α and IL-1β mediated astrocyte proliferation.
Fig. 1.
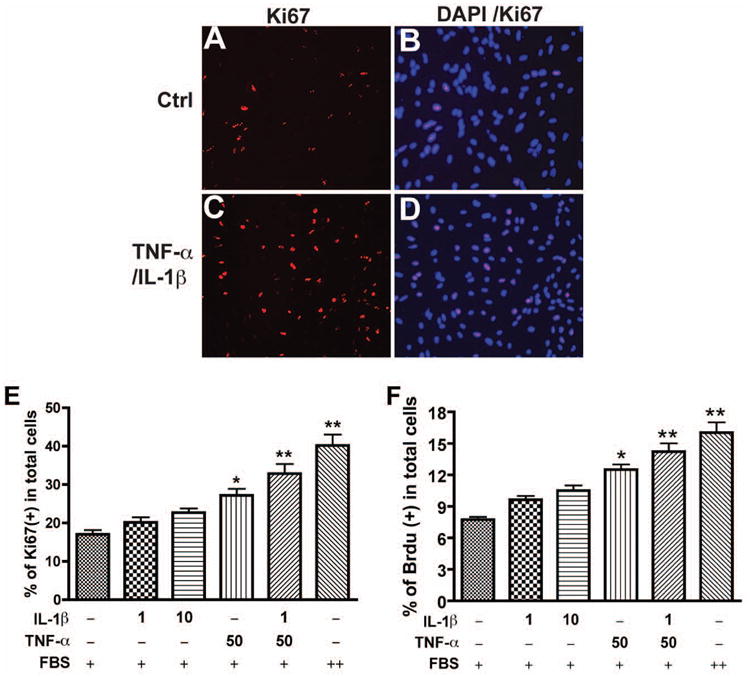
TNF-α and IL-1β promote human astrocyte proliferation. Human astrocytes were treated with different doses of IL-1β (1 ng/ml) and TNF-α (50ng/ml) for 3 days and then stained with antibody to Ki67 (red, A-D). 4′,6′-Diamidino-2-phenylindole (DAPI; blue) was used as a nuclear marker to count the total cell number (B, D). E-F, different doses of IL-1β (1 ng/ml, 10 ng/ml) and/or TNF-α 50ng/ml were used to treat astrocytes; proliferating cells were counted as the percent of Ki67+ cells in total DAPI-positive nuclei. F, at same treatment, cells were stained with antibody to BrdU conjugated with anti-mouse Alexa Fluor 488 nm secondary Ab. The DNA-binding fluorescent dye propidium iodide (PI) was used as a nuclear marker for the total cell number. BrdU+ cells were manually counted as the percent of PI-positive nuclei. + indicates 2% FBS-, and ++ indicates 10% FBS-containing in medium. *, p < 0.05; **, p < 0.01. Data are representative of three donors.
TNF-α and IL-1β induce cyclin D1 expression in human astrocytes
Cyclin D1 is essential for cell cycle transition from G1 to S phase and is upregulated when cell cycle progresses. To confirm the TNF-α and IL-1β mediated cell cycle progression, we determined the cyclin D1 protein levels with different doses of TNF-α and/or IL-1β treatment. Treatment with TNF-α or IL-1β alone induced a two-fold increase of cyclin D1 protein levels. Furthermore, treatment with TNF-α and IL-1β together induced a five-fold increase of cyclin D1 protein levels, which were similar to the cyclin D1 levels under serum treatment (p < 0.001, Fig. 2). The regulation of cyclin D1 by TNF-α and/or IL-1β treatment was in agreement with results from cell proliferation assays, which demonstrated that a combination of TNF-α (50 ng/ml) and IL-1β (1 ng/ml) induced significant increase in astrocyte proliferation (Fig. 1). Together, these results suggest that TNF-α and IL-1β promote astrocyte proliferation through cell cycle regulation.
Fig. 2.
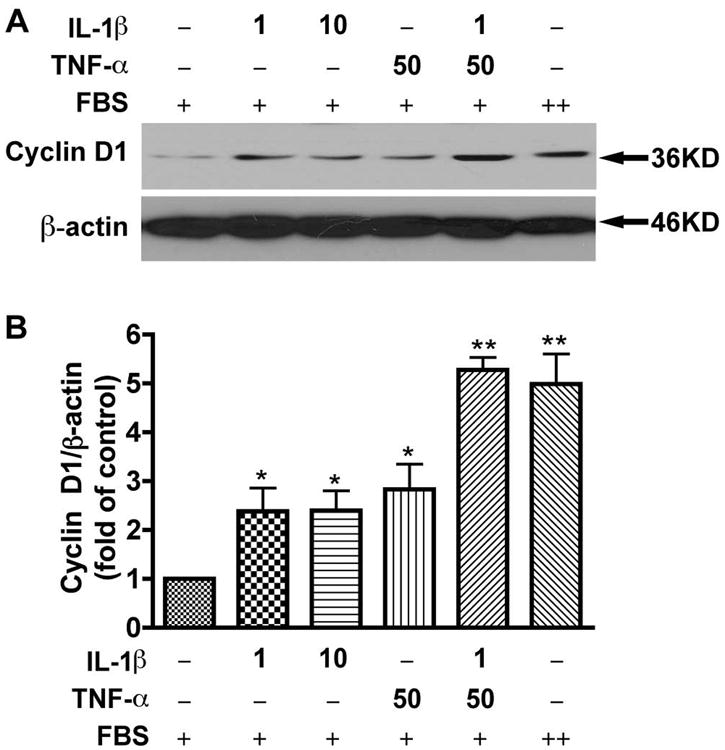
TNF-α and IL-1β induce cyclin D1 expression in human astrocytes. A, Human astrocytes were starved in DMEM/F12 medium for 6 h, then changed to condition medium (2% FBS in DMEM/F12) with the following treatments: IL-1β 1ng/ml, IL-1β 10ng/ml, TNF-α 50ng/ml, TNF-α 50ng/ml plus IL-1β 1ng/ml, and 10% FBS in DMEM/F12 for 24 h. Cell lysates were collected and subjected to Western blotting for cyclin D1. β-actin was used as a loading control. B, Results of A were normalized with β-actin and densimetrically quantified as fold of cyclin D1 in non-treated control. Average ± SD of three independent experiments from three donors were shown. + indicates 2% FBS and ++ indicates 10% FBS contained in medium. *, p < 0.05 compared to control; **, p < 0.01 compared to control. Data are representative of three donors.
TNF-α and IL-1β regulate FOXO3a through Akt-1 pathway in human astrocytes
In order to identify the signaling cascade that promotes cell cycle progression in astrocytes, we first examined the effect of TNF-α and IL-1β on the Akt-1 phosphorylation. TNF-α and IL-1β stimulation led to increased Akt-1 phosphorylation (Fig. 3A, B). Among all of the important substrates of Akt-1, FOXO transcription factors are essential to control cell cycle progression. Our microarray data have showed that FOXO3a is highly expressed in human astrocytes, as compared with other members of FOXO and FOXM that are important in cell cycle regulation (Supplemental Fig. 1). However, whether FOXO3a contributes to cytokine-mediated astrocyte proliferation has not been elucidated to date. We examined the phosphorylation of FOXO3a in the time course of TNF-α and IL-1β treatment in human astrocytes. Phosphorylation of FOXO3a reached a peak at 15 minutes following TNF-α and IL-1β treatment (Supplemental Fig. 2). After 24 h of treatment, the phosphorylation of FOXO3a remained high, while expression of total Akt-1 and FOXO3a remained unchanged (Fig. 3A, B and C). p27Kip1 and p21Waf1, members of a family of cyclin-dependent kinase inhibitors, are important downstream factors of FOXO3a that inhibit cyclin D1 expression(Rathbone et al. 2008; Zhang et al. 2009). p27Kip1 protein levels were significantly reduced after 24 h of TNF-α and IL-1β stimulation (Fig. 3A and D). In contrast, the levels of p21Waf1 remained unchanged after TNF-α and IL-1β stimulation (Data not shown). These results demonstrate that TNF-α and IL-1β phosphorylate Akt-1 and FOXO3a, suppressing p27Kip1, which may form a signaling cascade that promotes astrocyte proliferation.
Fig. 3.
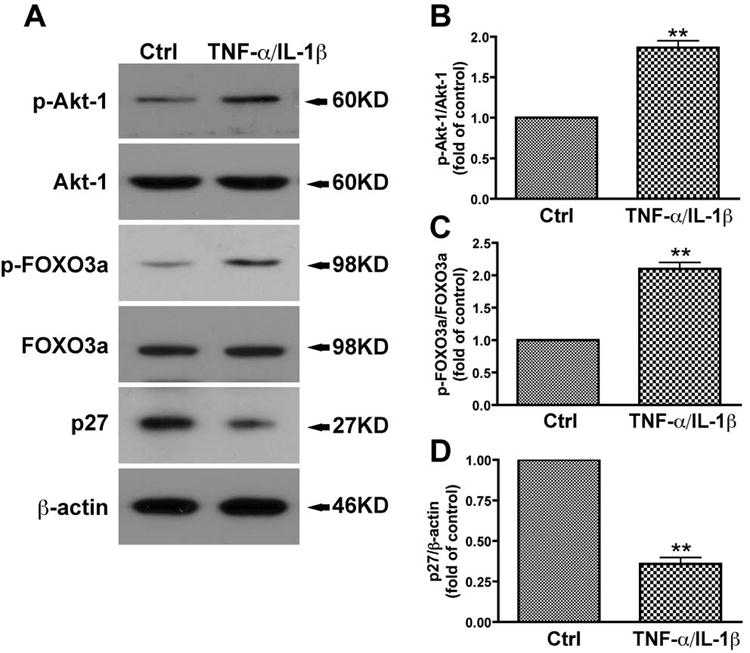
TNF-α and IL-1β activate the Akt-1 pathway in human astrocytes. A, Human astrocytes were starved overnight then treated with IL-1β (1ng/ml) and TNF-α (50ng/ml) in 2% FBS in DMEM/F12 for 24 h. Cell lysates were collected and subjected to Western blotting for p-Akt-1, Akt-1, p-FOXO3a, FOXO3a, and p27Kip1. β-actin was used as a loading control. B-D, results of A were normalized with β-actin and densimetrically quantified as fold of control. Average ± SD of three independent experiments from three donors were shown. **, p < 0.01 compared with control.
To verify Akt-1 as the main upstream kinase of FOXO3a in TNF-α- and IL-1β-induced astrocyte proliferation, an adenoviral vector was used to deliver constitutively active Akt-1 (myr-Akt) or dominant negative Akt-1 (DN-Akt). After 72 h of infection, overexpression of constitutively active Akt-1 was confirmed and the phosphorylation of FOXO3a was significantly increased (Fig. 4A). These data suggest that Akt-1 is sufficient to phosphorylate FOXO3a in astrocytes. To determine whether Akt-1 mediates FOXO3a phosphorylation in TNF-α- and IL-1β-stimulated astrocytes, we treated the astrocytes with TNF-α and IL-1β for 24 h following overexpression of DN-Akt-1. The overexpression of DN-Akt-1 abolished TNF-α/IL-1β-induced FOXO3a phosphorylation in astrocytes (Fig. 4B). Similarly, the PI3K inhibitor LY294002 pretreatment of human astrocytes, which is known to inhibit the Akt-1 phosphorylation, also inhibited TNF-α- and IL-1β-induced FOXO3a phosphorylation (Fig. 4C), suggesting that FOXO3a phosphorylation, in response to TNF-α and IL-1β stimulation, was dependent on the PI3K/Akt-1 pathway.
Fig. 4.
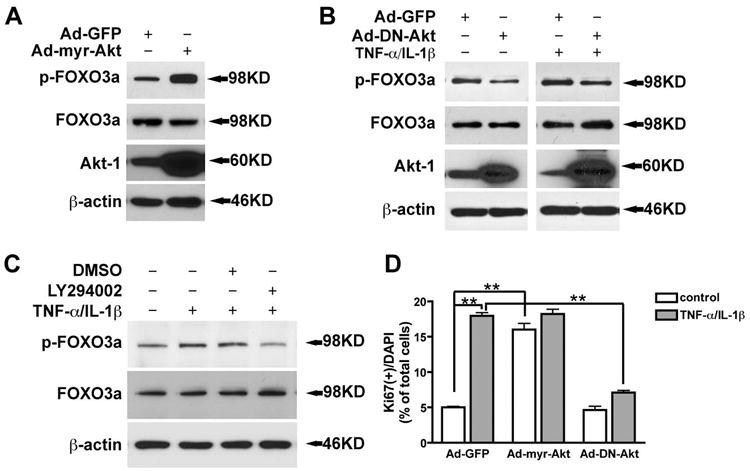
TNF-α and IL-1β induce astrocyte proliferation through Akt-1. A. Three days after Ad-myr-Akt infection, cells were lysated and subjected to Western blotting for p-FOXO3a, FOXO3a, and Akt-1. B, two days after Ad-DN-Akt-1 infection, human astrocytes were treated with IL-1β (1ng/ml) and TNF-α (50ng/ml) for 24 h. Cells were lysed and subjected to Western blotting for p-FOXO3a, FOXO3a, and Akt-1. C, cells were pre-treated with LY294002 for 4 h, and then changed to IL-1β (1ng/ml) and TNF-α (50ng/ml) for 24 h. Cell lysates were subjected to Western blotting for p-FOXO3a and FOXO3a. D, three days after adenovirus infection, cells were stained with Ki67 and DAPI. Proliferating cells were counted as the percent of Ki67+ in DAPI+ nuclei. **, p < 0.01.
To assess whether Akt-1 was necessary for TNF-α- and IL-1β-induced astrocyte proliferation, dominant negative Akt-1 and constitutively active Akt-1 were delivered to astrocytes by adenovirus. Ki67 immunocytochemical staining was used to measure cell proliferation. Compared with the GFP vector control, constitutively active Akt-1 (Ad-myr-Akt) dramatically increased cell proliferation in absence of TNF-α and IL-1β treatment. Furthermore, overexpression of dominant negative Akt-1 prevented TNF-α- and IL-1β-induced astrocyte proliferation (Fig. 4D). These results suggest that Akt-1 is critical upstream kinase of FOXO3a for TNF-α and IL-1β-induced astrocyte proliferation.
TNF-α and IL-1β reduce nuclear expression of FOXO3a in human astrocytes
FOXO3a is regulated by phosphorylation of conserved serine/threonine residues, which control FOXO subcellular localization. To assess the FOXO3a subcellular localization after cytokine treatment, we performed immunocytochemistry and confocal microscopy on astrocytes. Following serum starvation, most of the FOXO3a was localized in the nucleus (Fig. 5A-D); but with TNF-α and IL-1β stimulation, number of cells associated with positive nuclear staining of FOXO3a significantly decreased (Fig. 5E-H). 10% FBS treatment, which increased proliferation of astrocytes, showed a similar decrease of nuclear FOXO3a (Fig. 5I-L). Quantification showed that either TNF-α/IL-1β treatment or culture in complete medium contained 10% FBS reduced number of cells associated with positive nuclear staining of FOXO3a by more than 50% (Fig. 5M). These data further support that nuclear FOXO3a is regulated by the proinflammatory cytokines, which may play an important role in astrocyte proliferation.
Fig. 5.
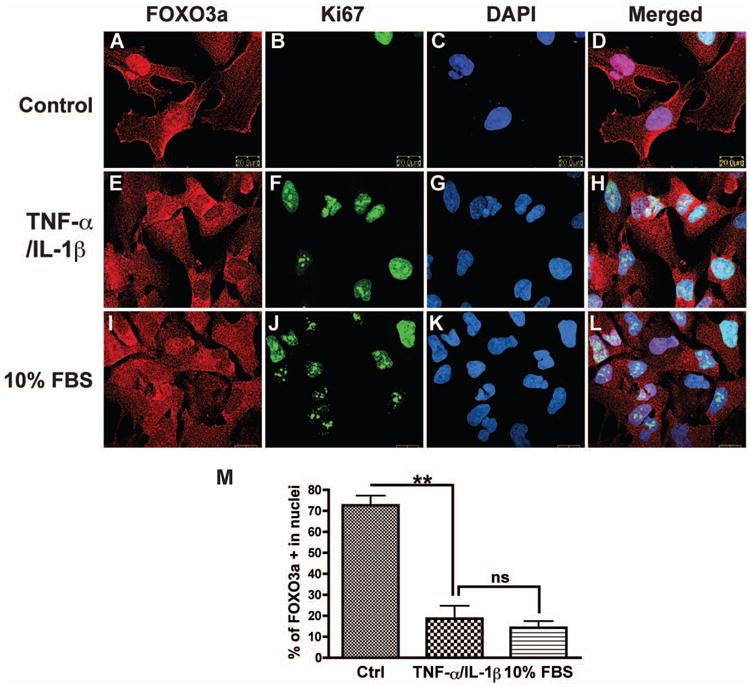
TNF-α and IL-1β decreases nuclear expression of FOXO3a in astrocytes. After 6 h of starvation, cells were treated with TNF-α and IL-1β in condition medium (2% FBS in DMEM/F12) or culture medium (10% FBS in DMEM/F12) for 24 h and then stained with FOXO3a (red) and Ki67 (green). DAPI (blue) was used as a nuclear marker. A-D, control. E-H, TNF-α and IL-1β treatment. I-L, 10% FBS in DMEM/F12. M, FOXO3a-positive nuclei were manually counted as the percent of DAPI-positive nuclei. Two hundred or more cells were counted for each group. **, p < 0.01. Data are representative of three donors.
Decreased nuclear FOXO3a expression in astrocytes during reactive astrogliosis in vivo
To test the subcelluar localization and regulation of FOXO3a during astrogliosis in vivo, we performed intracranial injection of TNF-α (50 ng) and IL-1β (10 ng). Mouse brain sections were examined at 2 or 7 days after injection. GFAP immunostaining was used as marker of astrocytes and indicator of reactive astrogliosis. Compared with the vehicle control (PBS injection, Fig. 6A and B), the TNF-α/IL-1β injection induced severe reactive astrogliosis (Fig. 6C and D), which showed a significant increase in the number and intensity of GFAP staining along the injection track (Fig. 6B, D, E and F). To determine the regulation of Foxo3a in reactive astrogliosis in vivo, we double-labeled the astrocytes with GFAP and Foxo3a, which enabled us to study Foxo3a expression in astrocytes because other cell types in the CNS also express Foxo3a (Fig. 7A, E). For both PBS and cytokines injection, the expression of the Foxo3a was more evident in the nucleus than in the cytosol along the injection track. However, after quantification, the fluorescent signal intensity of Foxo3a in astrocytes was significantly reduced in TNF-α/IL-1β injection compared with the PBS injection (Fig. 7C, G; Fig. 7I, p < 0.001). To confirm the protein changes of Foxo3a in vivo, brain tissues were obtained and protein analysis of Foxo3a was performed with Western blotting. Compared with its corresponding right hemisphere (no injection), left hemisphere (PBS injections or TNF-α/IL-1β injections) had higher levels of GFAP and phosphorylated Foxo3a (Fig. 7J). More importantly, the ratio of phospho-Foxo3a to total Foxo3a increased in TNF-α/IL-1β injection brain tissues when compared with PBS injection (Fig. 7K). These in vivo results indicate that Foxo3a is phosphorylated by inflammatory cytokines and its expression in astrocytes is reduced during brain inflammation
Fig. 6.
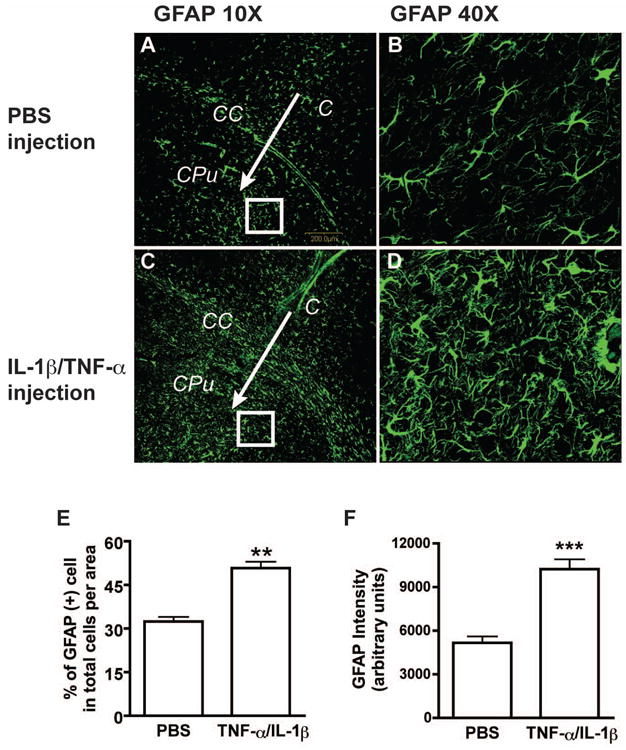
Intracranial injection of TNF-α and IL-1β induces astrogliosis in vivo. Seven days after TNF-α and IL-1β intracranial injection, reactive astrogliosis as detected by GFAP immunostaining was noted in the mouse brain (C and D), as compared to the phosphate buffered saline (PBS) injected brain (A and B). Arrows indicate the injection needle tracks. C= cortex, CC= corpus callosum, CPu= Caudate putamen. E, Data was shown as the percentage of GFAP-positive cell per area. F, GFAP expression were quantified by determining the GFAP-intensity around the needle track. Total 20 pictures from 4 mice were calculated. **, p < 0.005 in comparison to control. ***, p < 0.001 in comparison to control.
Fig. 7.
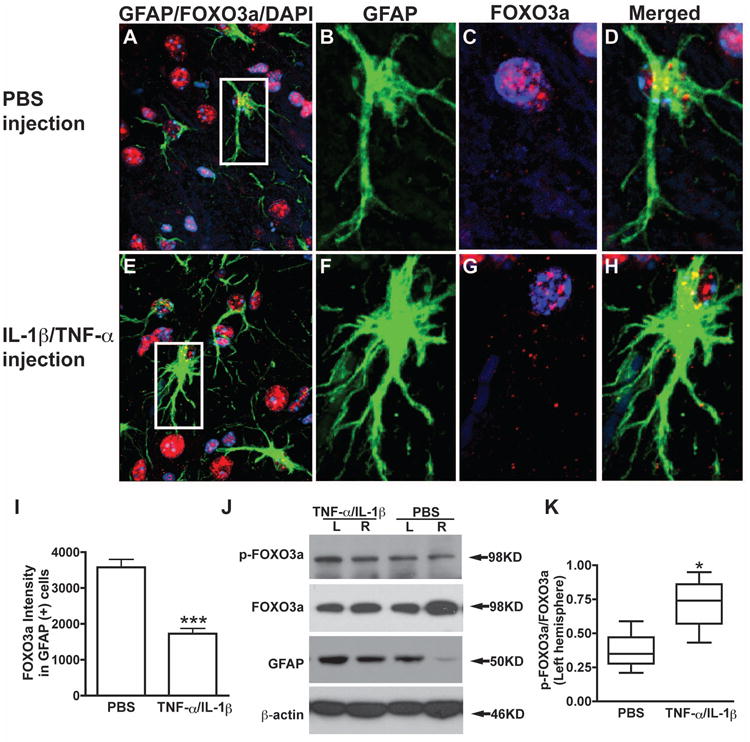
Reduced expression of Foxo3a in astrocytes during astrogliosis in vivo. After 7 days of injection, brain sections were taken for immunohistochemistry and brain tissue lysis were prepared for Western blotting. Representative fluorescence overlay micrographs from confocal pictures show the astrogliosis and Foxo3a expression (GFAP in green, Foxo3a in red) in PBS injection (A, B, C, and D) and TNF-α/IL-1β injection (E, F, G, and H). Nuclei were stained with DAPI (blue). I, Confocal pictures were imported and analyzed with ImageJ-ProPlus. Foxo3a expression was quantified by fluorescence intensity in 200 GFAP-positive cells random chose from 20 brain sections of four mice each group. J, Protein extracts from brain tissue contained injection site were subjected to Western blotting for p-Foxo3a, Foxo3a, and GFAP. β-actin was used as a loading control. K, The ratio of p-Foxo3a to total Foxo3a from left cerebral hemisphere (LH) was showed. Boxes encompass the interquartile ranges; error bars indicate the ranges of value. Horizontal lines, median value for each group (n=4). ***, p < 0.001 in comparison to PBS injection. *, p < 0.05 in comparison to PBS injection.
FOXO3a inhibits the inflammatory cytokine-mediated proliferation of human astrocytes
We next investigated how FOXO3a regulated TNF-α- and IL-1β-induced astrocyte proliferation. To manipulate FOXO3a function, replication-defective adenoviral gene-delivery systems were used to overexpress wild type FOXO3a (WT-FOXO3a) or dominant negative FOXO3a (DN-FOXO3a) in astrocytes. The DN-FOXO3a was constructed by deleting the transactivation domain from the C-terminus of FOXO3a, creating a protein with the molecular weight of about 40 KD, as shown in the immunoblotting (Supplemental Fig. 3B). The infection efficiency of the adenovirus was monitored by GFP fluorescent expression in astrocytes and was around 80% at 300 multiplicity of infection (MOI) (Supplemental Fig. 3A). The overexpression of WT-FOXO3a and DN-FOXO3a was confirmed by Western blotting (Supplemental Fig. 3B). Next, we determined cell proliferation with Ki67 immunocytochemical staining after adenovirus infection. Ki67 positive cells were counted against total GFP positive cells (Fig. 8). Overexpression of wild type FOXO3a inhibited astrocyte proliferation, as compared with the Ad-GFP control (Fig. 8A and I). TNF-α and IL-1β treatment significantly increased the astrocyte proliferation, as determined by Ki67 staining (p < 0.001, Fig. 8A, D, E, H, arrow indicates Ki67+/GFP+ cells). The increase of astrocyte proliferation by TNF-α and IL-1β treatment was completely blocked by overexpressed WT-FOXO3a, suggesting that ectopic expression of FOXO3a inhibited TNF-α- and IL-1β-induced astrocyte proliferation (Fig. 8Q). There were Ki67 positive cells in Ad-WT-FOXO3a group after TNF-α and IL-1β stimulation, however, those cells were likely untransduced with FOXO3a as indicated by GFP expression (Fig. 8L and P, arrow indicates Ki67+/GFP- cells). Together, these results demonstrate that TNF-α and IL-1β induce astrocyte proliferation through functionally repressing FOXO3a.
Fig. 8.
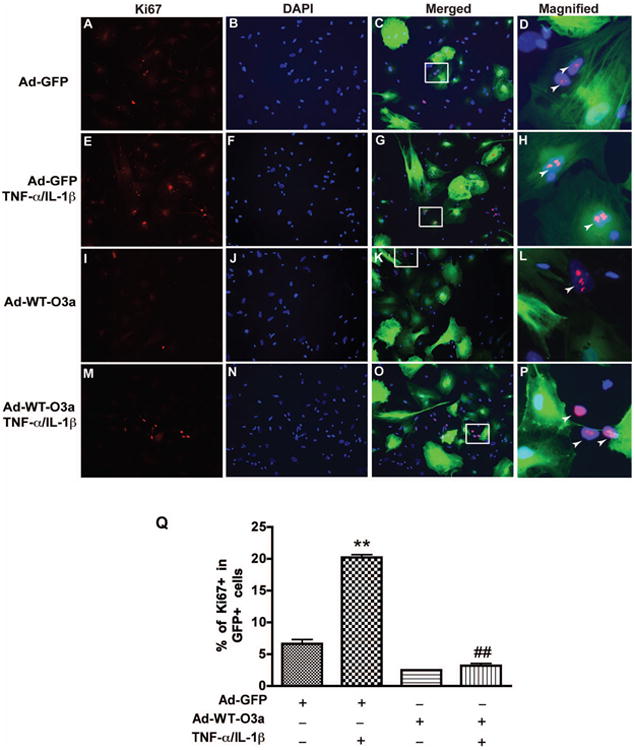
Overexpression of wild type of FOXO3a inhibits cytokine-mediated proliferation of human astrocyte. After 24 h of adenovirus infection, cells were treated with TNF-α and IL-1β for 3 days and then stained with Ki67. DAPI (blue) was used as a nuclear marker to count the total cell number. A-C, Ad-GFP infection without TNF-α and IL-1β treatment. E-G, Ad-GFP infection with TNF-α and IL-1β treatment. I-K, Ad-WT-FOXO3a infection without TNF-α and IL-1β treatment. M-O, Ad-WT-FOXO3a infection with TNF-α and IL-1β treatment. C, G, K, and O are KI67, DAPI and GFP merged images of each group. D, H, L, and O are magnified Images from outlined square from C, G, K, and O respectively. Arrow indicates the Ki67+/GFP+ cells in D and H, Ki67+/GFP- cells in L and O. Q, quantification of three donors. **, p < 0.01, compared with Ad-GFP without TNF-α and IL-1β treatment; ##, p < 0.01, compared with Ad-GFP in TNF-α and IL-1β treatment.
FOXO3a induces critical cell cycle regulatory factors in human astrocytes
Next, we explored several down stream genes of FOXO3a that are cell cycle regulatory factors in astrocytes. These genes include cyclin D1, p27Kip1, and Gadd45α. Cyclin D1 induction is a key event in G1 phase progression and is tightly regulated by p27Kip1. Gadd45α can induce cell cycle arrest in the G2-M phase. The protein levels of p27Kip1 were significantly increased in WT-FOXO3a overexpression and were reduced in DN-FOXO3a overexpression in astrocytes (Fig. 9A and B). p27Kip1 typically regulates cyclin D1 expression through the inhibition of CDKs in the G1 phase; thus, we tested the expression of cyclin D1. As expected, following Ad-WT-FOXO3a infection, p27Kip1 was increased whereas cyclin D1 was inhibited (Fig. 9C). Inversely, Ad-DN-FOXO3a infection decreased p27Kip1 and increased cyclin D1 (Fig. 9C). We also examined Gadd45α expression, which can interfere with the G2 phase of the cell cycle. Gadd45α was dramatically increased in Ad-WT-FOXO3a infection (Fig. 9D and E), suggesting that FOXO3a can also induce cell cycle arrest at the G2 phase. To further confirm the function of FOXO3a in cell cycle regulation, cell proliferation assay which was determined by Ki67 staining was performed. Ad-DN-FOXO3a group showed a significant increase of cell proliferation compared with vector control, and TNF-α and IL-1β treatment further potentiated this increase (Fig. 9F). These data indicate that FOXO3a functionally inhibits cell proliferation and prevents cell cycle progression in astrocytes through arresting cell cycle, either in the G1 or G2 phase, by induction of downstream factors p27Kip1 and Gadd45α.
Fig. 9.
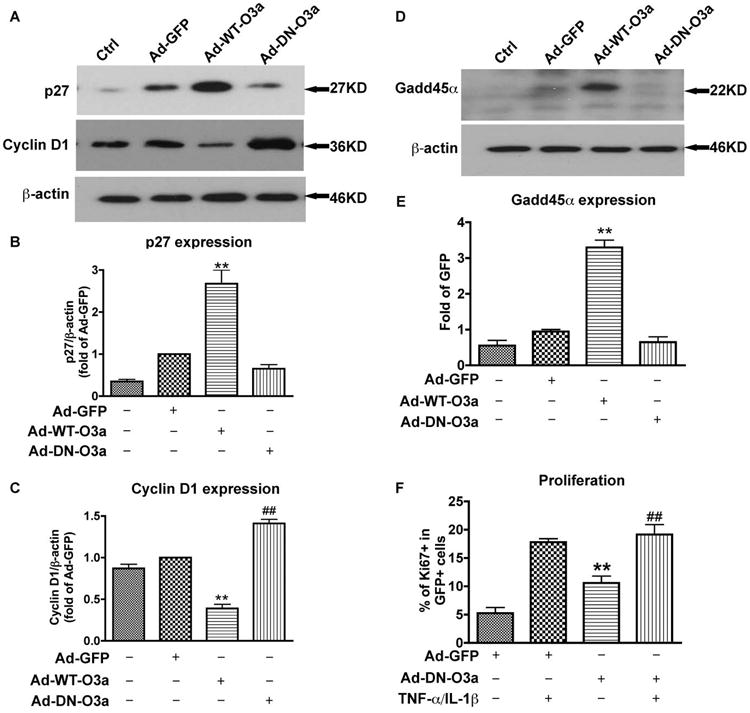
FOXO3a regulated cell cycle proteins and DN-FOXO3a promoted proliferation in human astrocytes. A and D, three days after adenovirus infection, human astrocytes were lysed and subjected to Western blotting for p27Kip1, cyclin D1 (A), and Gadd45α (D). β-actin was used as a loading control. B, C, and E, expression of p27Kip1, cyclin D1, and Gadd45α in A and D were normalized to β-actin and densimetrically quantified as fold of GFP control. F, after 24 h of adenovirus infection, cells were treated with TNF-α and IL-1β for 3 days and then stained with Ki67 and DAPI. Proliferating cells were counted as the percent of Ki67+ in DAPI+ nuclei. Average ± SD of three independent experiments from three donors were shown. **, p < 0.01, compared with Ad-GFP; ##, p < 0.01, compared with Ad-DN-FOXO3a without TNF-α and IL-1β treatment.
Astrocytes in Foxo3a-null mice have higher proliferative potential in vitro and in vivo
To further verify the function of FOXO3a in astrocytes, 4-week old Foxo3a null mice and wild type mice were subjected to brain dissection and astrocyte cultures were prepared. Total cell number was counted in each passage, which showed marked higher cell number in Foxo3a null astrocytes than those from wild type (Fig. 10A). After 3 passages, GFAP and DAPI staining were performed to verify the purity of astrocyte cultures. As shown in Fig. 10B, more than 98% cells were GFAP positive. Western blotting of Foxo3a from the astrocyte lysates confirmed the knockout status of mice. Furthermore, reduced levels of p27Kip1 and increased levels of cyclin D1 were observed in Foxo3a null astrocytes (Fig. 10C). Proliferation, as determined by BrdU staining, showed a significant increase in Foxo3a null astrocytes compared with wild type (Fig. 10D and E). To further investigate the role of FOXO3a in inflammatory cytokines-mediated reactive astrogliosis, low dose of TNF-α (5 ng) and IL-1β (1 ng) were intracranially injected into both Foxo3a null mice and their littermate wild type mice. Seven days after injection, mice were sacrificed and brain sections were stained with GFAP and DAPI to detect astrogliosis in vivo. Foxo3a null mice exhibited more severe astrogliosis compared with wild type (Fig. 11A). Both GFAP+ cell number and GFAP intensity showed significant increase along the injection track in Foxo3a null mice (Fig. 11 B and C). These data, in agreement with human astrocyte studies, suggesting that FOXO3a is a critical factor preventing inflammatory cytokine-mediated astrocyte proliferation.
Fig. 10.
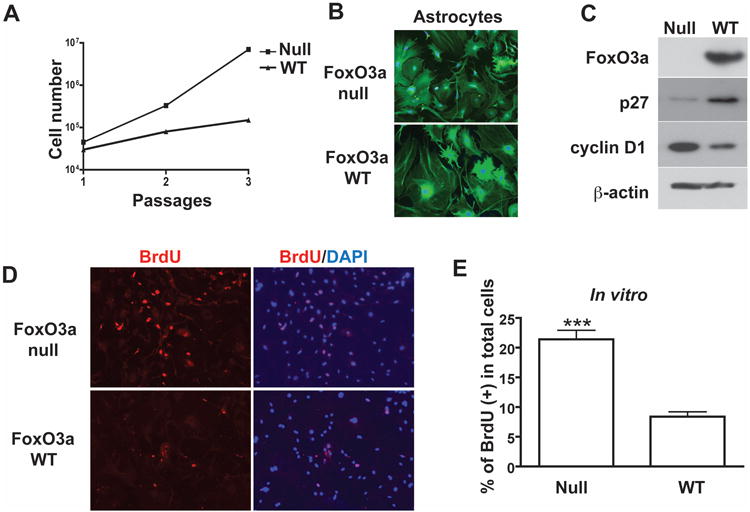
Astrocytes from Foxo3a-null mice exhibit higher proliferative potential in vitro. A, astrocytes were isolated from 4 week old of Foxo3a-null or Foxo3a-WT mouse brain and cultured in vitro. Total cell number was counted in each passage. B, after 3 passages, purity of astrocyte cultures was determined by GFAP and DAPI staining. C, astrocyte lysates were collected and subjected to Western blotting for Foxo3a, cyclin D1, and p27Kip1. β-actin was used as a loading control. D and E, mouse astrocytes were stained with BrdU and DAPI. Proliferating cells were counted as the percent of BrdU+ in DAPI+ nuclei. A total of 30 pictures from 4 mice were calculated. ***, p < 0.001 in comparison to control.
Fig. 11.
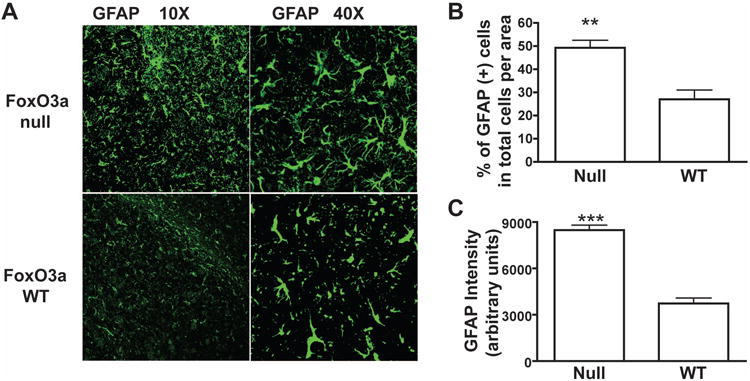
Inflammatory cytokines increase reactive astrogliosis in Foxo3a null mouse. A, at 7 days after TNF-α and IL-1β (5 ng and 1 ng) intracranial injection, reactive astrogliosis in Foxo3a null mouse was detected by GFAP immunostaining (10× and 40×), wild type mice from the same litter were used as control. GFAP positive cell was quantified in total cells per area (B), and GFAP expression were also quantified by determining the GFAP-intensity (C). Total 30 pictures from 4 mice were calculated. **, p < 0.005 in comparison to control. ***, p < 0.001 in comparison to control.
Discussion
Reactive astrogliosis is an underlying component associated with the CNS injury and neurodegenerative diseases, yet its molecular and cellular mechanisms are poorly understood. In this report, we demonstrated that inflammatory cytokine-mediated astrocyte proliferation is associated with reduced nuclear expression of FOXO3a both in vitro and in vivo. We determined that the function of FOXO3a in astrocytes is to inhibit cell proliferation. In addition, we have verified that TNF-α and IL-1β regulate FOXO3a through PI3K/Akt-1 pathway and FOXO3a induces critical cell cycle regulatory factors in human astrocytes. Foxo3a knockout further confirmed that Foxo3a negatively regulates cell proliferation. Lack of Foxo3a resulted in more severe forms of reactive astrogliosis in response to proinflammatory cytokine stimulation in vivo. Thus, we identify FOXO3a as a key transcription factor involved in cytokine-induced astrocyte proliferation. To our knowledge, this is the first report demonstrating that FOXO3a is a pivotal transcription factor involved in inflammatory cytokines-induced astrocyte proliferation. Identification of the signaling molecules that mediate proliferation aspect of astrogliosis will give valuable insight into the mechanisms of astrogliosis with the potential to find targets for therapeutic intervention of neurodegenerative diseases.
Proinflammatory cytokines are quickly induced and known to be critical mediators of astrogliosis(Hirsch et al. 2003; Woiciechowsky et al. 2004). For examples, IL-1β-null mice lack the increase of GFAP at 2 and 3 days after CNS injury that otherwise occurs in wild type controls(Herx and Yong 2001). Both TNF-α and IL-1β can induce IL-6 release into the CSF, which further increases GFAP expression in the cerebral cortex and hippocampus(Tanabe et al.; Woiciechowsky et al. 2004). In the present study, TNF-α and IL-1β stimulated astrocyte proliferation in vitro, and intracranial injection of TNF-α and IL-1β induced severe reactive astrogliosis, including increased astrocyte cell population, astrocyte hypertrophy and enhanced GFAP expression. While there were many aspects of reactive astrogliosis and our study only focus on the proliferation under proinflammatory conditions, the indication that FOXO3a function in the process potentially provides much-needed information for the molecular analyses of reactive astrogliosis.
FOXO transcription factors are critical regulators in diverse physiological processes, and their functions in the central nervous system have recently become of great interest. FoxOs regulate NSC homeostasis and are important in neuron polarity and neuron survival(de la Torre-Ubieta et al.; Paik et al. 2009; Renault et al. 2009; Yuan et al. 2009). FOXO3a, the main isoform of FOXO proteins in mammals, has been well studied in tumorigenesis, muscle atrophy, immune diseases, and stem cell homeostasis(Lin et al. 2004; Maiese et al. 2007; Senf et al. 2008; Yang et al. 2008). The regulatory and functional roles of FOXO3a are highly cell type-specific and dependent on context of the cellular environments. Gene knockout of Foxo3a in mice results in spontaneous tumorigenesis, lymphoproliferation, and depletion of the neural/hematopoietic stem cell pools, which suggests the multifaceted functions of FOXO3a in vivo(Renault et al. 2009; Tothova and Gilliland 2007). Particularly, a recent report by Paik et al. presented detailed data investigating the effect of gene knockout of FoxO(1, 3, 4) on neural stem cells, though no information was provided about the impact of FoxO knockout on astrocytes(Paik et al. 2009). In our mouse study, there is a significant reduction of Foxo3a staining in astrocytes (GFAP+ cells) in response to cytokines-stimulation. In agreement with this data, in vitro astrocyte cultures from Foxo3a null mice demonstrated increased proliferative potential, as well as concordant changes of key cell cycle regulatory protein p27Kip1 and cyclin D1. Similarly, Intracranial injection of low dose of TNF-α- and IL-1β showed more severe forms of reactive astrogliosis in FoxO3-null mice, as compared with the wild type mice. All of these results from wild type, Foxo3a-null mice, and cultured human astrocyte studies suggest that Foxo3a is a critical factor regulating astrocyte cell cycle and proliferation. We have noted that other cell types (neuron and microglia) express Foxo3a in the brain. In the injection area, neurons express high level of Foxo3a. However, we focus on astrocytes in this paper. Astrocytes outnumber these other cell types in the CNS. Therefore, the overall change of FOXO3a, found in our Western blotting of mid-brain lysates containing the injection site, should partially reflect levels of Foxo3a in astrocytes (Figure 6 J and K).
Phosphorylation of the FOXO3a is the most critical post-translational modification as it essentially regulates the translocation of FOXO3a proteins between the nucleus and cytoplasm(Skurk et al. 2004; Vogt et al. 2005). Akt-dependent phosphorylation of FOXO3a promotes FOXO3a export from the nucleus to the cytoplasm, thereby repressing the transcriptional activity of FOXO3a(Brunet et al. 1999; Kops et al. 1999). Our data demonstrated that TNF-α and IL-1β increased phosphorylation of Akt-1 and FOXO3a, whereas reduced nuclear expression of FOXO3a. The specific regulation of Akt-1/FOXO3a pathway by the cytokines is aiming to enhance the proliferation of human astrocytes. Although Akt-1 is dominating the regulation of FOXO3a, there are many kinases that also phosphorylate FOXO3a. These kinases include serum and glucocorticoid-regulated kinase (SGK), c-Jun N-terminal kinase (JNK), extracellular signal-related kinase (ERK), dual specificity tyrosine-phosphorylated and regulated kinase (DYRK1A), mammalian Ste20-like kinase-1 (MST1), and IκB kinase (IKK)(Huang and Tindall 2007; Vogt et al. 2005). It remains the subject for further investigation to determine whether ERK, IKKβ, or other kinases also regulate FOXO3a in astrocytes and interfere with cell cycle processes.
How downstream factors of FOXO3a are involved in TNF-α- and IL-1β-induced astrogliosis is also an interesting and important topic. FOXO3a has been reported to regulate p27Kip1 and p21Waf1 expression in cancer cells(Hauck et al. 2007; Hu et al. 2005). Both p27Kip1 and p21Waf1 are cyclin-dependent kinase (CDK) inhibitors, which block S-phase to G1-phase transition through repressing cyclin D expression(Rathbone et al. 2008; Zhang et al. 2009). Interestingly, in both human and mouse astrocytes, we found FOXO3a regulated p27Kip1 expression rather than p21Waf1. In Foxo3a null astrocytes, the levels of p27Kip1 decrease, while the levels of cyclin D1 and astrocyte proliferation increased. Thus, a signaling cascade involving FOXO3a/p27Kip1/cyclin D1 may exist in astrocyte to regulate cell cycle. Overexpression of WT-FOXO3a also upregulates Gadd45α (Fig. 9), which plays a key role in the G2/M checkpoint in response to DNA damage(Jin et al. 2002; Zhu et al. 2008). FOXO3a is known to induce G2/M cell cycle arrest and trigger the DNA repair process through upregulation of Gadd45α(Jin et al. 2002; Tran et al. 2002). It is unknown whether those down stream factors work in concert or in sequential order to regulate cell cycle and proliferation. The Identification of astrocyte-specific targets of FOXO3a and the detailed regulation of the factors are important and therefore warrant future investigation.
In summary, the evidence presented in this study demonstrated that marked astrocyte proliferation occurred in response to inflammatory cytokines stimulation. The cell cycle progression was associated with decreased nuclear exprssion of FOXO3a. FOXO3a negatively regulate astrocyte proliferation and attenuated cytokine-mediated astrocyte proliferation (Fig. 12). Understanding the critical role of FOXO3a in astrocyte proliferation may provide a potential target for therapeutic intervention in CNS diseases driven by dysfunctioned reactive astrogliosis.
Fig. 12.
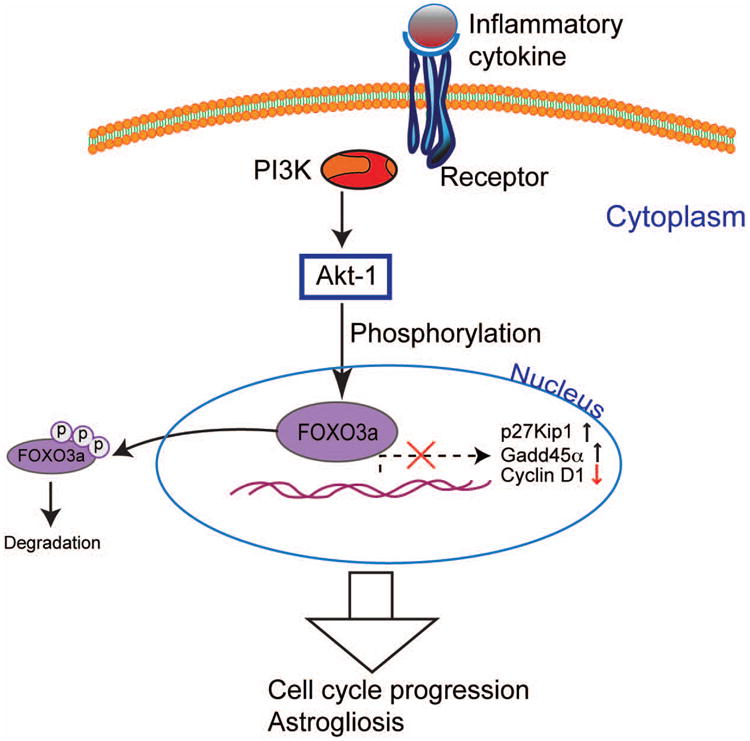
FOXO3a regulates cell cycle control of astrocyte during inflammation. Inflammatory cytokines TNF-α and IL-1β increase the phosphorylation and decrease the nuclear expression of FOXO3a via kinases such as protein kinase B (PKB, Akt). Phosphorylation of FOXO3a inhibits critical cell cycle regulatory genes p27Kip1 and Gadd45α, which promote cell-cycle arrest. Thus, functional repression of FOXO3a induces astrocyte cell cycle progression and astrogliosis.
Supplementary Material
Supplemental Fig. 1: Relative abundance of mRNA level of FOXO1, FOXO3a, FOXO4 and FOXM1 in microarray of human astrocytes. mRNA was extracted from pooled total RNA samples obtained from three donors.
Supplemental Fig. 2: Time course of FOXO3a phosphorylation after TNF-α/IL-1β treatment. Human astrocytes were starved in DMEM/F12 medium for 6 h, and then changed to condition medium (2% FBS in DMEM/F12) with TNF-α (50ng/ml) and IL-1β (1ng/ml) for different time points. Cell lysates were collected and subjected to Western blotting for phospho-FOXO3a and FOXO3a. β-actin was used as a loading control.
Supplemental Fig. 3: Adenoviral delivery of FOXO3a to human primary astrocyte. A, human astrocytes were infected with an adenovirus vector containing a GFP reporter system. Infection efficiencies were monitored by taking fluorescence images 24 h after adenovirus infection. Ad-GFP was used as a control adenovirus; Ad-WT-FOXO3a expressed wild type FOXO3a; Ad-DN-FOXO3a adenovirus expressed dominant-negative FOXO3a, without the transactivation domain C terminus. B, after 72 h of adenoviral infection at 300 MOI, FOXO3a overexpression was determined by immunoblotting. Wild type FOXO3a and endogenous FOXO3a both appeared at 98 kDa, while DN-FOXO3a appeared at 40 kDa.
Acknowledgments
We kindly acknowledge Dr. You Zhou, Beibei Jia, and Lijun Sun for providing technical support for this work. Tiffany R. Peña and Dr. Nicholas P. Whitney provided valuable comments and suggestions about the manuscript. Julie Ditter, Robin Taylor, Johna Belling, Na Ly, Myhanh Che, and Emilie Scoggins provided outstanding administrative support.
References
- Aranha MM, Sola S, Low WC, Steer CJ, Rodrigues CM. Caspases and p53 modulate FOXO3A/Id1 signaling during mouse neural stem cell differentiation. J Cell Biochem. 2009;107(4):748–58. doi: 10.1002/jcb.22172. [DOI] [PubMed] [Google Scholar]
- Arden KC. FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene. 2008;27(16):2345–50. doi: 10.1038/onc.2008.27. [DOI] [PubMed] [Google Scholar]
- Avola R, Di Tullio MA, Fisichella A, Tayebati SK, Tomassoni D. Glial fibrillary acidic protein and vimentin expression is regulated by glucocorticoids and neurotrophic factors in primary rat astroglial cultures. Clin Exp Hypertens. 2004;26(4):323–33. doi: 10.1081/ceh-120034137. [DOI] [PubMed] [Google Scholar]
- Balasingam V, Yong VW. Attenuation of astroglial reactivity by interleukin-10. J Neurosci. 1996;16(9):2945–55. doi: 10.1523/JNEUROSCI.16-09-02945.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60(3):430–40. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rolando C, Ceruti S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol. 2009 doi: 10.1016/j.bcp.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- Cui M, Huang Y, Zhao Y, Zheng J. Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macrophages. J Immunol. 2008;180(2):898–906. doi: 10.4049/jimmunol.180.2.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Huangu Y, Zhao Y, Zheng J. New Insights for FOXO and Cell-Fate Decision in HIV-Infection and HIV Associated Neurocognitive Disorder. In: Maiese K, editor. Forkhead Transcription factors: Vital Elements in Biology and Medicin. Landes Bioscience Springer; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha A, Jefferson JJ, Tyor WR, Glass JD, Jannotta FS, Vitkovic L. Control of astrocytosis by interleukin-1 and transforming growth factor- beta 1 in human brain. Brain Res. 1993;631(1):39–45. doi: 10.1016/0006-8993(93)91183-s. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Mostert JP, Koch MW. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J Neurol Sci. 2008;267(1-2):3–16. doi: 10.1016/j.jns.2007.08.044. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Gaudilliere B, Yang Y, Ikeuchi Y, Yamada T, DiBacco S, Stegmuller J, Schuller U, Salih DA, Rowitch D, et al. A FOXO-Pak1 transcriptional pathway controls neuronal polarity. Genes Dev. 24(8):799–813. doi: 10.1101/gad.1880510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci U S A. 2005;102(23):8333–8. doi: 10.1073/pnas.0500989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54(1):15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24(9):2143–55. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW. Astrocytic and neuronal factors affecting axon regeneration in the damaged central nervous system. Cell Tissue Res. 1997;290(2):371–7. doi: 10.1007/s004410050943. [DOI] [PubMed] [Google Scholar]
- Hara A, Saegusa M, Ichinoe M, Okayasu I. Diagnostic and prognostic significance of cyclin A expression in low-grade astrocytomas: comparison with astrogliosis and high-grade tumours. J Clin Pathol. 2008;61(3):287–92. doi: 10.1136/jcp.2007.051961. [DOI] [PubMed] [Google Scholar]
- Hauck L, Harms C, Grothe D, An J, Gertz K, Kronenberg G, Dietz R, Endres M, von Harsdorf R. Critical role for FoxO3a-dependent regulation of p21CIP1/WAF1 in response to statin signaling in cardiac myocytes. Circ Res. 2007;100(1):50–60. doi: 10.1161/01.RES.0000254704.92532.b9. [DOI] [PubMed] [Google Scholar]
- Herx LM, Yong VW. Interleukin-1 beta is required for the early evolution of reactive astrogliosis following CNS lesion. J Neuropathol Exp Neurol. 2001;60(10):961–71. doi: 10.1093/jnen/60.10.961. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP. The role of glial reaction and inflammation in Parkinson's disease. Ann N Y Acad Sci. 2003;991:214–28. doi: 10.1111/j.1749-6632.2003.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S, Hartmann A. Neuroinflammatory processes in Parkinson's disease. Parkinsonism Relat Disord. 2005;11(1):S9–S15. doi: 10.1016/j.parkreldis.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wang X, Zeng L, Cai DY, Sabapathy K, Goff SP, Firpo EJ, Li B. ERK phosphorylates p66shcA on Ser36 and subsequently regulates p27kip1 expression via the Akt-FOXO3a pathway: implication of p27kip1 in cell response to oxidative stress. Mol Biol Cell. 2005;16(8):3705–18. doi: 10.1091/mbc.E05-04-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120(Pt 15):2479–87. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- Jin S, Tong T, Fan W, Fan F, Antinore MJ, Zhu X, Mazzacurati L, Li X, Petrik KL, Rajasekaran B, et al. GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene. 2002;21(57):8696–704. doi: 10.1038/sj.onc.1206034. [DOI] [PubMed] [Google Scholar]
- Kiyota T, Yamamoto M, Schroder B, Jacobsen MT, Swan RJ, Lambert MP, Klein WL, Gendelman HE, Ransohoff RM, Ikezu T. AAV1/2-mediated CNS gene delivery of dominant-negative CCL2 mutant suppresses gliosis, beta-amyloidosis, and learning impairment of APP/PS1 mice. Mol Ther. 2009;17(5):803–9. doi: 10.1038/mt.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398(6728):630–4. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Lin L, Hron JD, Peng SL. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21(2):203–13. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Hou J, Shang YC. The “O” Class: Crafting Clinical Care with FoxO Transcription Factors. In: Maiese K, editor. Forkhead Transcription Factors: Vitl Elements in Biology and Medicine. Lands Bioscience and Spring; 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Shang YC. “Sly as a FOXO”: new paths with Forkhead signaling in the brain. Curr Neurovasc Res. 2007;4(4):295–302. doi: 10.2174/156720207782446306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJ, Holsboer F, Swaab DF. Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur J Neurosci. 2001;14(10):1603–12. doi: 10.1046/j.0953-816x.2001.01784.x. [DOI] [PubMed] [Google Scholar]
- Paik JH, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS, Chae SS, Zheng H, Ying H, Mahoney J, et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5(5):540–53. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet HS, Epstein L, Gelbard H, et al. Human immunodeficiency virus encephalitis in SCID mice [see comments] Am J Pathol. 1996;149(3):1027–53. [PMC free article] [PubMed] [Google Scholar]
- Potts R, Leech RW. Thalamic dementia: an example of primary astroglial dystrophy of Seitelberger. Clin Neuropathol. 2005;24(6):271–5. [PubMed] [Google Scholar]
- Rathbone CR, Booth FW, Lees SJ. FoxO3a preferentially induces p27Kip1 expression while impairing muscle precursor cell-cycle progression. Muscle Nerve. 2008;37(1):84–9. doi: 10.1002/mus.20897. [DOI] [PubMed] [Google Scholar]
- Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC, et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5(5):527–39. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20(12):570–7. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer's disease. Cell Death Differ. 2008 doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. Faseb J. 2008;22(11):3836–45. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279(2):1513–25. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32(12):638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. Faseb J. 2006;20(6):670–82. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Matsushima-Nishiwaki R, Yamaguchi S, Iida H, Dohi S, Kozawa O. Mechanisms of tumor necrosis factor-alpha-induced interleukin-6 synthesis in glioma cells. J Neuroinflammation. 7:16. doi: 10.1186/1742-2094-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007;1(2):140–52. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296(5567):530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO's road. Sci STKE. 2003;2003;(172):RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Jiang H, Aoki M. Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle. 2005;4(7):908–13. doi: 10.4161/cc.4.7.1796. [DOI] [PubMed] [Google Scholar]
- Woiciechowsky C, Schoning B, Stoltenburg-Didinger G, Stockhammer F, Volk HD. Brain-IL-1 beta triggers astrogliosis through induction of IL-6: inhibition by propranolol and IL-10. Med Sci Monit. 2004;10(9):BR325–30. [PubMed] [Google Scholar]
- Wu Y, Peng H, Cui M, Whitney NP, Huang Y, Zheng JC. CXCL12 increases human neural progenitor cell proliferation through Akt-1/FOXO3a signaling pathway. J Neurochem. 2009;109(4):1157–67. doi: 10.1111/j.1471-4159.2009.06043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10(2):138–48. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Lehtinen MK, Merlo P, Villen J, Gygi S, Bonni A. Regulation of neuronal cell death by MST1-FOXO1 signaling. J Biol Chem. 2009;284(17):11285–92. doi: 10.1074/jbc.M900461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Yang D, Fan Y, Xie P, Li H. Epigallocatechin-3-gallate enhances ischemia/reperfusion-induced apoptosis in human umbilical vein endothelial cells via AKT and MAPK pathways. Apoptosis. 2009;14(10):1245–54. doi: 10.1007/s10495-009-0391-1. [DOI] [PubMed] [Google Scholar]
- Zhu N, Shao Y, Xu L, Yu L, Sun L. Gadd45-alpha and Gadd45-gamma utilize p38 and JNK signaling pathways to induce cell cycle G2/M arrest in Hep-G2 hepatoma cells. Mol Biol Rep. 2008 doi: 10.1007/s11033-008-9419-9. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhang Q, Yu Z, Zhang L, Tian D, Zhu S, Bu B, Xie M, Wang W. Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia. 2007;55(5):546–58. doi: 10.1002/glia.20476. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1: Relative abundance of mRNA level of FOXO1, FOXO3a, FOXO4 and FOXM1 in microarray of human astrocytes. mRNA was extracted from pooled total RNA samples obtained from three donors.
Supplemental Fig. 2: Time course of FOXO3a phosphorylation after TNF-α/IL-1β treatment. Human astrocytes were starved in DMEM/F12 medium for 6 h, and then changed to condition medium (2% FBS in DMEM/F12) with TNF-α (50ng/ml) and IL-1β (1ng/ml) for different time points. Cell lysates were collected and subjected to Western blotting for phospho-FOXO3a and FOXO3a. β-actin was used as a loading control.
Supplemental Fig. 3: Adenoviral delivery of FOXO3a to human primary astrocyte. A, human astrocytes were infected with an adenovirus vector containing a GFP reporter system. Infection efficiencies were monitored by taking fluorescence images 24 h after adenovirus infection. Ad-GFP was used as a control adenovirus; Ad-WT-FOXO3a expressed wild type FOXO3a; Ad-DN-FOXO3a adenovirus expressed dominant-negative FOXO3a, without the transactivation domain C terminus. B, after 72 h of adenoviral infection at 300 MOI, FOXO3a overexpression was determined by immunoblotting. Wild type FOXO3a and endogenous FOXO3a both appeared at 98 kDa, while DN-FOXO3a appeared at 40 kDa.