

Abstract.
The epigenetic control of heterochromatin deposition is achieved through a network of protein interactions mediated by the heterochromatin protein 1 (HP1). In earlier studies, we showed that the CCAAT/enhancer-binding protein alpha (C/EBPα), a transcription factor that controls cell differentiation, localizes to heterochromatin, and interacts with HP1α. Here, deletion and mutagenesis are combined with live-cell imaging approaches to characterize these protein interactions. The results demonstrate that the basic region and leucine zipper (BZip) domain of C/EBPα is sufficient for the interaction with HP1α in regions of heterochromatin. Fluorescence correlation spectroscopy and cross-correlation (FCS and FCCS) revealed very different diffusion profiles for HP1α and the BZip protein, and co-expression studies indicated that the mobile fractions of these nuclear proteins diffuse independently of one another. The steady-state interactions of these proteins in regions of heterochromatin were monitored using Förster resonance energy transfer (FRET). A point mutation in HP1α, W174A, which disrupts the interactions with proteins containing the common PxVxL motif did not affect the interaction with the BZip protein. In contrast, the HP1α W41A mutation, which prevents binding to methylated histones, exhibited greatly reduced FRET efficiency when compared to the wild type HP1α or HP1αW174A. The functional significance of these interactions is discussed.
Keywords: fluorescence lifetime imaging microscopy, Förster resonance energy transfer microscopy, fluorescence correlation spectroscopy, fluorescent proteins, protein interactions
1. Introduction
There is a clear link between epigenetic dysregulation and the progression of many diseases including cancer, cardiovascular disease, diabetes, and obesity, but the mechanisms that control epigenetic signaling is poorly understood. The epigenetic modification of eukaryotic genomes is regulated by networks of nuclear protein interactions that control the formation of distinct domains of heterochromatin and euchromatin. For example, it is well established that the epigenetic control of heterochromatin deposition is achieved through a network of protein interactions mediated by heterochromatin protein 1 (HP1).1 While HP1 is principally known for directing the assembly and maintenance of heterochromatin through direct interactions with the histone methyltransferases (HMTs), it has become increasingly clear that HP1 has much broader roles in the regulation of gene expression.1–4 Recent studies suggest the HP1 proteins are positive regulators of gene transcription,3 and play roles in DNA replication5 and repair.6 A major issue in the field is to determine how the HP1 network interacts with sequence specific transcription factors, and how these interactions function to target chromatin remodeling activities to specific gene promoters.
In this regard, our earlier studies demonstrated that the CCAAT/enhancer-binding protein alpha (C/EBPα) interacts directly with HP1α.7–9 The C/EBP family of transcription factors direct programs of cellular differentiation, and C/EBPα plays key roles in the regulation of genes involved in energy metabolism.10,11 C/EBPα preferentially localizes to regions of centromeric heterochromatin as determined by immunocytochemical staining of differentiated mouse adipocyte cells,12 as well as by the expression of fluorescent protein (FP) labeled-C/EBPα in mouse pituitary cells.13 While the highly methylated DNA in heterochromatin is most typically associated with gene silencing, a surprising recent finding suggests that C/EBPα can localize to specific methylated promoters in heterochromatin, and that this results in the activation of a subset of differentiation specific genes.14 Together, these studies suggest that the interactions between C/EBPα and HP1α could function in the regulation of epigenetic signaling.
The HP1 family proteins are highly conserved nonhistone proteins containing a chromo-domain (CD), a disordered hinge region, and a chromoshadow domain (CSD; see Fig. 1). The CD binds directly to the methylated lysine 9 of histone H3,15,16 and a mutation changing the CD tryptophan at position 41 to alanine (W41A) blocks this binding, and consequently disrupts localization of the HP1 proteins to heterochromatin.15–18 The CSD, on the other hand, is necessary for homo-dimerization, and mediates the interactions with a variety of other proteins.19 Many proteins that directly interact with HP1α share the common peptide motif, PxVxL, which binds to the hydrophobic pocket that forms at the dimer interface between associated CSDs.18,20 This interaction is also thought to recruit the HP1-HMT complex to target gene promoters,21 and thus may represent a general mechanism by which transcription factors can modify the activities of the HP1 network. The interactions of the PxVxL motif proteins with HP1α can be specifically disrupted by a point mutation in the CSD, converting the tryptophan at position 174 to alanine (W174A; Fig. 1).18
Fig. 1.

Schematic representations of the HP1α and C/EBPα proteins indicating the relative locations of the different domains, and showing the positions of the point mutations of HP1α and the truncation of C/EBPα resulting in the BZip domain.
The biochemical methods and screening approaches that are typically used to characterize protein interaction networks cannot detect weak or transient interactions, nor can they identify the dynamics of protein interactions in discrete subcellular domains, information that can only be determined within the context of the intact cell. Therefore, the development of noninvasive quantitative imaging techniques to visualize the dynamic protein behavior inside living cells has become increasingly important for biomedical research. Coupled with the recent improvements in the photophysical qualities of the many different genetically encoded FPs, these noninvasive imaging approaches enable the detection of dynamic protein behaviors and interactions inside living cells.22 Here, frequency domain fluorescence lifetime imaging microscopy (FD FLIM) is utilized to detect protein interactions by Förster resonance energy transfer (FRET). This is used in combination with fluorescence correlation spectroscopy (FCS) to investigate the molecular mechanisms underpinning the interactions of C/EBPα and HP1α inside the living cell nucleus.
2. Materials and Methods
2.1. Expression Plasmids and Protein Purification
The FPs used in this study were chosen for optimal photophysical characteristics within their spectral class. The cDNA for Venus was obtained from Dr. Atsushi Miyawaki (RIKEN, Japan).23 The plasmids encoding the FPs mCerulean3, mTurquoise, mApple, and mRuby were obtained from Dr. Michael Davidson (FSU, Tallahassee, Florida). Standard recombinant DNA methods were used to generate the plasmids encoding mCerulean3-mRuby, as well as those for the indicated FPs linked in-reading frame to the sequence encoding either the basic region-leucine zipper (BZip) domain of the rat C/EBPα,24 or the human HP1α (American Type Cell Culture MGC-4985). The truncated BZip domain of C/EBPα begins with the methionine at position 237 of the full length C/EBPα protein (Fig. 1). The point mutations in HP1α (see Fig. 1) were introduced using specific primers and the QuikChange II site-directed mutagenesis kit (Stratagene). All plasmid inserts were confirmed by direct sequencing.
To generate the purified mCerulean3 protein, T7 cells (New England Biolabs) expressing the mCerulean3-6xHis proteins were grown in LB broth without antibiotics to an optical density of about 0.6. After addition of isopropyl βD-1 thiogalactopyranoside (IPTG) to 1 mM, the culture was placed in an orbital shaker at 16°C for 16 h, and the cells were recovered by centrifugation. The cells were lysed using CellLytic Express (Sigma) in 40 mL of 10 mM , (pH 7.5). After incubation at 37°C for 10 min, PMSF (phenylmethylsulfonyl fluoride, 0.1 mM) and Halt Protease Inhibitor (Thermo Scientific) were added and the cells were disrupted by sonication, and the supernatant was recovered by centrifugation at at 4°C for 10 min. The supernatant was mixed in an orbital shaker with Talon resin (Clontech) at 4°C for 1 h. The resin-bound mCerulean3 was resuspended in wash buffer and eluted according to the manufacturer’s protocol. The purified mCerulean3-6xHis was subsequently concentrated about 20-fold using an Amicon Ultra centrifugal filter (Millipore).
2.2. Tissue Culture and Transfection
The mouse pituitary GHFT1 cells25 are maintained in monolayer culture, and are harvested at 80% confluence. The cells are washed with phosphate buffered saline (PBS), briefly treated with trypsin (0.05% in 0.53 mM EDTA), and recovered by centrifugation in culture medium containing serum. The cells are washed two times by centrifugation in Dulbecco’s calcium-magnesium free PBS and resuspended in Dulbecco’s calcium-magnesium free PBS with 0.1% glucose and BioBrene Plus (Applied Biosystems Inc.) at a final concentration of approximately cells per ml. Exactly 400 μl of the cell suspension is transferred to each 0.2 cm gap electroporation cuvette containing the plasmid DNA(s). For FRET studies involving independently expressed donor- and acceptor-labeled proteins, the acceptor to the donor ratio influences the FRET efficiency. The average level of donor- and acceptor-labeled proteins expressed in the population of transfected cells is influenced by the ratio of plasmids mixed in the electroporation cuvette. Here, the amount of plasmid DNA encoding the donor- and the acceptor-labeled proteins was adjusted to favor acceptor expression, using a total of 10 μg of purified plasmid DNA per cuvette. For the fluorescence correlation spectroscopy (FCS) and fluorescence correlation and cross-correlation (FCCS) studies, 1 μg of each DNA per cuvette was used. The contents of the cuvettes are gently mixed, and then pulsed with 200 V at a capacitance of 1200 microfarads in a BTX ECM 830 electroporator (Harvard Apparatus), yielding pulse durations of about 10 ms. The cells are immediately recovered from the cuvette and diluted in phenol red-free tissue culture medium containing serum. The suspension is transferred to sterile 2 well-chambered coverglass (Lab-Tek II, Thermo Scientific), which are placed in an incubator (37°C and 5% ) prior to imaging the following day.
2.3. FD FLIM Measurements
The fluorescence lifetime measurements are made using the ISS Alba FastFLIM system (ISS Inc., Champagne, Illinois) coupled to an Olympus IX71 microscope equipped with a water-immersion objective lens. A Pathology Devices (Pathology Devices Inc.) stage-top environmental control system maintains temperature at 36°C and at 5%. For confocal imaging to analyze the co-localization of the different FP-labeled proteins, frame averaging is used to achieve at least 200 peak counts per pixel in the final images from both channels. For FLIM using the cyan and yellow FPs the 5 mW 448 nm diode laser is modulated by the Alba FastFLIM system at a fundamental frequency of 10 MHz with up to 13 sinusoidal harmonics. The modulated laser is coupled to the scanning system, which is controlled by the VistaVision software (ISS Inc., Champagne, Illinois). The fluorescence signals emitted from the specimen are routed by a 495 nm long pass beam splitter through the (acceptor emission) and (donor emission) band-pass emission filters, and the signals are detected using two identical avalanche photodiodes (APD). The phase delays, and modulation ratios of the emission relative to the excitation are measured at each pixel of an image for each frequency.
The system is calibrated with the 50 mM Coumarin 6 dissolved in ethanol (reference lifetime 2.5 ns) to provide the software with a standard to estimate the lifetime values from the experimental data.26 Additionally, a second reference standard, 10 mM HPTS (8-hydroxypyrene-1,3,6-trisulfonic acid from Santa Cruz Biotechnology Inc.) dissolved in phosphate buffer (PB) pH 7.8 (reference lifetime of 5.3 ns) is used to check that the system is accurately reporting the fluorescence lifetime of a known sample. A chambered coverglass with the dye samples is illuminated at sufficient laser power to achieve approximately 100,000 counts per second in the donor emission channel, and frame averaging is used to accumulate approximately 100 counts per pixel in the final image. The distribution of the lifetimes for all the pixels in the image is determined using the phasor (polar) plot method.26–28
For live-cell FLIM imaging, transfected cells are first identified using epifluorescence microscopy and then imaged by FD FLIM using the 448 nm laser line. The laser power at the specimen plane, determined with a digital laser power and energy meter (Thorlabs PM100D, Newton, New Jersey), was generally between 0.01 to 1 μW, and is adjusted to achieve the same count rate in the donor emission channel that was used for calibration. Using frame averaging, we generally acquire about 30 frames requiring approximately 45 s for a lifetime image with about 200 peak counts per pixel. No significant photobleaching of the samples was observed under these conditions. The FLIM images are analyzed with the VistaVision software (Build 143) by selecting regions of interest (ROI, typically 1 to ) with average intensity counts. Generally, 10 ROI are selected for analysis from each cell. The most accurate lifetime determinations are obtained by analyzing the first 12 modulation frequencies (10 to 120 MHz). The quality of the fit is judged by the reduced chi-square () values for the frequency response curves (phase and modulation versus modulation frequency) at the 12 frequencies. The ratio of the donor lifetimes determined in the absence () and in the presence of the acceptor () provides a direct estimate of FRET efficiency () by Eq. (1):
| (1) |
For intermolecular FRET measurements from independently expressed proteins, the ratio of the acceptor- to the donor-labeled proteins influences the .26 The two channel imaging system used here allows simultaneous measurement of the intensity of the donor emission () and the intensity of the acceptor emission (), which are detected by the two identical APDs. The mean intensity in both the donor and the acceptor channels are measured for each ROI, and this serves as a proxy for the ratio of the acceptor- to the donor-labeled proteins in each ROI. The FRET efficiency is calculated using Eq. (1) from the average quenched donor lifetime in each ROI, and the averaged results for each cell are plotted as a function of the measured acceptor to donor () ratio. The data are fitted by a nonlinear regression for saturation binding constraining the background to zero (there is no FRET without acceptor) using the GraphPad Prism software. The is then determined from the best-fit values.
2.4. Fluorescence Correlation Spectroscopy and Fluorescence Cross-Correlation Spectroscopy Measurements
The ISS Alba system is also configured for FCS and FCCS measurements. The system is equipped with a dual-color dichroic beamsplitter () to detect signals in two separate channels. The laser beam (448 or 561 nm) is focused to a confocal spot with small excitation geometry. The laser power was determined at the specimen plane as described above. The fluorescence signals emitted from the focal spot are routed via the beam splitter to the two band-pass emission filters ( and ). The emission signals are passed to the two identical APDs through adjustable pinholes that are set at 50 μm. The number of emission events per unit time is determined and used to generate autocorrelation and cross-correlation curves from the intensity traces and the fluctuations in the intensity . The curves are best fits for the general correlation function:
| (2) |
When , this returns the autocorrelation function, and when this measures the cross-correlation of signals between two different channels.
The autocorrelation data obtained from standard dyes with known diffusion coefficients () are used to determine the geometry of the confocal spot from the parameters and via Eq. (3):
| (3) |
The beam waist () is calculated using the relationship and beam length () from the relationship . The calibration standard used here is 2 nM Coumarin 6 in HPLC grade EtOH (448 nm laser line) and 10 nM Alexa 568 in PBS (561 laser line). Typically, 5 measurements of 30 s each are obtained to determine and . Following acquisition of the dye standard signal the data are fitted by a method that assumes a Gaussian point spread function, using the known diffusion coefficient ( for Coumarin6 in EtOH and for Alexa 568 at 25°C) to determine the parameters and . These parameters are not dependent on the laser power, so a calibration at one laser power suffices for other laser powers, and do not vary significantly over the course of a single session. From session to session, however, varied between 0.38 and 0.43 μm, and between 4.5 and 7.7, with slightly larger for the 561 laser line (red channel) than the 448 line (green channel).
For FCCS studies, the ISS Alba system allows independent adjustment of the radial placement of the pinholes, and axial positioning of the APD detectors for both the green and red channels. Once the pinhole for the green channel was optimized using 5 mM Coumarin 6, the pinhole for the red channel was optimized using 10 μM Alexa 568. The pinhole for the red channel was then rechecked using the Coumarin 6 to verify radial co-alignment of the red and green laser lines. A mixture of HPTS and Alexa 568 in PBS was then imaged and scans were performed using the red and green lasers singly and jointly to verify the axial alignment of the red and green pinholes. Two dye-labeled antibodies (donkey anti-goat Alexa488 and goat anti-rabbit Texas Red) were then mixed at a concentration of and diluted for FCCS measurements of cross-correlation. This allowed us to verify the radial overlap of the confocal volumes. In addition, whole cell lysates were prepared from cells transfected with the plasmid encoding a fusion protein consisting of mCerulean3 linked directly to mRuby (mCerulean3-mRuby). After 24 h in culture, the transfected cells were rinsed 3 times in PBS and lysed in radio-immunoprecipitation assay (RIPA) buffer with protease inhibitors (Aldrich) for 5 min on ice. The lysates are collected by gentle scraping, transferred to a tube and centrifuged 10 min at 13,000 g. The supernatant was kept on ice and analyzed by FCCS within 8 h of harvesting.
For FCS and FCCS in living cells, it is critical to establish conditions to achieve low concentrations of the FP-labeled proteins in the cells. The coverglass is scanned to identify healthy cells expressing low levels of the FP-labeled proteins. Once identified, a confocal image of the cell is acquired using the lowest laser power and the fastest acquisition time that allows the nucleus to be sufficiently resolved to identify regions of heterochromatin. The VistaVision software is used to set the location on the cell, and a profile is then obtained to accurately place the confocal spot in all three dimensions. For FCCS, the profile is also used to confirm the axial alignment of the red and green channels. For diffusion measurements the data were acquired using the 448 nm laser line at a power level at the specimen between 1.2 and 1.68 μW (a power density of up to ), with data collection times of 8 to 10 s. The mobile fraction was defined as the proteins whose diffusion is captured on an autocorrelation curve after an initial period of pre-bleaching (typically 2 s). An earlier study used a similar prebleaching approach for the FCS analysis of the mobile fraction of HP1α, and suggested that the very slow population corresponds to direction associations between HP1 proteins and chromatin.29 Here, a 2 s prebleach was used for the mCerulean3 fusion proteins, and the FCS analysis measured mobile proteins as slow as . For cross-correlation measurements in living cells, the data were acquired using the 448 laser line at 1.0 to 1.4 μW and the 561 laser line at 0.84 to 1.0 μW simultaneously with acquisition times from 10 to 20 s.
The diffusion coefficient for each labeled protein is determined from the autocorrelation curves using the known geometry of the confocal spot and Eq. (3). To test whether a sample contains two populations diffusing at different rates, a two-component autocorrelation fitting curve can be constructed that adds the contributions from two populations characterized by (, ) and (, ).30 The autocorrelation curve can also be fit with a model that includes a term for diffusion that is confined or restricted.30,31 This results in an autocorrelation curve with a coefficient as follows:
| (4) |
Further, it is also possible to model autocorrelation curves to account for additional species that may describe simple or anomalous diffusion, and to include an additional component to model binding events or correct for triplet conversion.32,33 For FCCS studies, the fraction of co-diffusing species relative to non-co-diffusing species determined in each channel ( or ) are measured directly from the cross-correlation and autocorrelation fitting curves at using the following relationship: and where is the concentration of species and is the value for in the ’th channel [, (green) or (cross)].34,35
3. Results
3.1. Measuring the Interactions Between HP1α and C/EBPα in Living Cells Using FRET-FLIM
In earlier studies, we demonstrated the interaction between HP1α and C/EBPα using co-immunoprecipitation studies, and by FRET analysis in living cells.7–9 Here, we used the FRET-FLIM approach to quantify the interactions between C/EBPα and HP1α in regions of heterochromatin inside the living cell nucleus. First, the lifetime for C/EBPα labeled with Cerulean3, a bright variant of Cerulean,36 was determined in regions of heterochromatin (typically 10 ROI per cell nucleus) for 10 different cells, yielding an average unquenched donor lifetime of . The Cerulean3-C/EBPα fusion protein was then co-expressed with the Venus-HP1α fusion protein (acceptor), and the lifetime of the Cerulean3-C/EBPα (donor) was measured in regions of heterochromatin and was determined using Eq. (1).
For these independently expressed proteins the acceptor to donor ratio varies for each transfected cell, and this ratio influences the . For each ROI, we determined the average intensity in the acceptor () and donor channels () as described in Sec. 2.3. As the increases, more of the Cerulean3-labeled proteins detected in the donor channel (480/40 nm) are associated with Venus-HP1α. The highest values () are achieved when the Venus- HP1α is in excess, saturating the available Cerulean3-C/EBPα proteins. This relationship is illustrated in Fig. 2, where , determined by Eq. (1), is plotted as a function of the for 23 different cells, with each point representing the average and average for the ROI analyzed in each cell. The of (Table 1 and Fig. 2) indicates a saturable association of C/EBPα and HP1α in the regions of heterochromatin.
Fig. 2.

FRET-FLIM analysis of the interactions between the C/EBPα and HP1α. The FD FLIM FRET approach was used to determine FRET efficiency (%) from the averaged donor lifetime acquired in multiple ROI for each cell using Eq. (1), and the results are plotted as a function of average measured in the same ROI. Each point represents the (see text in Sec. 2.3 for details). The images above the graph show intensity images of cell nuclei acquired in the acceptor and donor channels, and the fluorescence lifetime map acquired from representative cells with a low and intermediate ratios (1.8 and 7.1, respectively); the scale bar indicates 10 μm.
Table 1.
FRET-FLIM analysis of interactions between HP1α and C/EBPα.
| Expressed proteins | a | b | c (range) | d (range) | e |
|---|---|---|---|---|---|
| Cer3-C/EBPα + Venus-BZip | 23 | 184 | 1.8 to 21 | 1.3 to 22.3% | |
| Turquoise-BZip + Venus-HP1α | 13 | 128 | 2 to 35.2 | 2 to 22% | |
| Turquoise-BZip + Venus-HP1W174A | 12 | 117 | 3 to 34.7 | 3 to 20% | |
| Turquoise-BZip + Venus-HP1W41A | 12 | 120 | 3 to 19.8 | 0 to 15% | NDf |
| TurquoiseN1 + Venus-HP1α | 13 | 26 | 1.2 to 8.4 | to 1.5 | ND |
Number of individual cells analyzed for that FRET pair.
Total number of cellular ROI analyzed for that FRET pair.
The range of ratios measured in all ROI for that FRET pair.
The range of FRET efficiencies (%) measured in all ROI for that FRET pair. was determined using Eq. (1) (see text).
The was determined from the nonlinear regression for saturation binding (see Fig. 2);
ND: not determined.
The BZip family of proteins form obligate dimers through their leucine-zipper domains, and this allows the basic regions to adopt the necessary conformation to bind to specific DNA elements.11 Earlier, we showed that only the BZip domain of C/EBPα (see Fig. 1) was required for targeting to the centromeric heterochromatin.37 More recently, others showed that C/EBPβ, which shares a high degree of homology with C/EBPα in the basic and leucine zipper regions, but not in the transactivation domain, interacts with HP1α.38 That study demonstrated that the basic region of C/EBPβ interacted with HP1α. Therefore, we next determined whether the BZip domain of C/EBPα was sufficient for the interactions of HP1α. First, the lifetime for the BZip domain labeled with Turquoise, a bright variant of Cerulean,39 was determined in regions of heterochromatin (typically 10 ROI per cell nucleus) for 10 different cells, yielding an average unquenched donor lifetime of . The lifetime of the Turquoise-labeled BZip domain protein when co-expressed with Venus-HP1α was then measured in regions of heterochromatin, and the was determined for cells with different ratios (Fig. 3). The FRET efficiency increased with the increasing ratio, with an of determined by the nonlinear regression analysis (Fig. 3, Table 1). This result clearly shows that the BZip domain of C/EBPα is sufficient for the interaction with HP1α.
Fig. 3.

FRET-FLIM analysis of the interactions between the BZip domain and HP1α. The FD FLIM FRET approach was used to determine FRET efficiency (%) as described in the legend for Fig. 2. Each point represents the . The image insets show the fluorescence lifetime maps acquired of the nuclei of representative cells expressing the mTurquoise-BZip domain alone, or when co-expressed with Venus-HP1α; the scale bar indicates 10 μm.
3.2. Analysis of Mobile Populations of BZip and HP1α Proteins by FCS and FCCS
We then used FCS to measure the diffusion characteristics of the mobile fractions of BZip domain, or HP1α labeled with mCerulean3. First, we determined the suitability of mCerulean3 as a fluorophore for FCS studies by analyzing the diffusion characteristics of both the purified mCerulean3, as well as the protein expressed in live cells, comparing the results to earlier reports for GFP and ECFP.40 Representative autocorrelation curves for mCerulean3 in solution and for the protein expressed in living cells are shown in Fig. 4(a), and the statistical analyses derived using Eq. (3) are shown in Table 2. Brownian (simple) diffusion provided the best fit for mCerulean3 in solution, and the diffusion coefficient for the purified mCerulean3 is the same (within experimental error) as that reported by others for purified EGFP and ECFP, and is consistent with a monomeric fluorophore.40 Moreover, when the diffusion coefficient was measured at different laser powers, no buildup of a triplet state for mCerulean3 was detected, which, if present could introduce anomalies in measurements of the diffusion coefficients.33 The results in Table 2 show the diffusion coefficients for purified mCerulean3 determined at laser powers ranging from 0.37 to 3.1 μW, demonstrating that there was no change in the measured diffusion coefficient at the laser powers used in live cells (less than 1.7 μW power at the objective). Next, autocorrelation curves for mCerulean3 expressed in living GHFT1 cells were acquired, and a representative one component simple anomalous diffusion fit for mCerulean3 in living cells () is shown in Fig. 4(a). The average diffusion coefficient was (Table 2), which is consistent with the rate determined for ECFP and GFP in the cytoplasm of living cells.40 This confirmed that mCerulean3, like ECFP and GFP, displays the diffusion characteristic of a monomer and therefore is suitable for FCS studies in live cells.
Fig. 4.
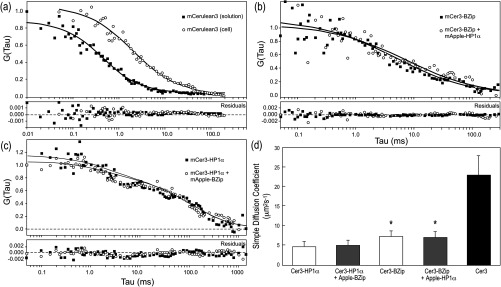
Autocorrelation curves and the normalized distance of each point from the best-fit (residuals) curves for mCerulean3 and the mCerulean3 fusion proteins. (a) Representatitive autocorrelation curves and residuals for mCerulean3 purified in solution, and mCerulean expressed in living GHFT1 cells. The autocorrelation curve for the purified mCerulean3 in solution is fitted to Eq. (3) and was acquired for 120 s. The autocorrelation curve for mCerulean3 in cells curve is fitted to Eq. (4) with and was acquired for 10 s. (b) The representative autocorrelation curves and residuals for mCerulean3-BZip expressed alone or when co-expressed with mApple-HP1α are fitted to Eq. (4) (one component anomalous) with . (c) The representative autocorrelation curves and residuals for mCerulean3-HP1α in the absence and presence of mApple-BZip are fitted to a two-component nonanomalous model with , and and 28, respectively. (d) The cumulative rates of diffusion as described by diffusion coefficients using Eq. (3). *P-values comparing the calculated diffusion coefficients for mCerulean3-labeled HP1α and BZip domain proteins show a significance level .
Table 2.
FCS analysis of the diffusion for mCerulean3 and mCerulean3 fusion proteins.
| Species | Power at the objective (μW)a | ()b | SD | Number of trialsc |
|---|---|---|---|---|
| mCer3 in solution | 0.37 | 79 | 6 | 6 |
| mCer3 in solution | 0.91 | 83 | 6 | 8 |
| mCer3 in solution | 1.68 | 82 | 7 | 7 |
| mCer3 in solution | 2.25 | 87 | 5 | 4 |
| mCer3 in solution | 3.11 | 89 | 4 | 8 |
| mCer3 in live cells | 1.68 | 23 | 5 | 28 |
| mCer3-BZip in the nucleus | 1.68 | 7.1 | 1.7 | 8 |
| mCer3-BZip + mApple-HP1α | 1.68 | 7 | 1.5 | 10 |
| mCer3-HP1α in the nucleus | 1.68 | 4.8 | 1.1 | 22 |
| mCer3-HP1α + mApple-BZip | 1.68 | 4.9 | 1.4 | 16 |
Power at the objective was determined as described in Sec. 2.4.
Coefficient of diffusion was determined using Eq. (3) by a Levenberg-Marquardt minimization of the autocorrelation curves after using a standard dye to determine beam waist and shape factor (as described in Sec. 2.4). For mCer3-BZip and mCer3-HP1α, Eq. (3) does not capture the complexity of the diffusion profiles, as discussed in the text.
The number of trials is the number of readings obtained for that experiment. For live cell experiments, these readings were obtained from a minimum of 3 cells. For trials involving two colors, the red channel was monitored to ensure that the concentration of co-expressed mApple proteins was equivalent to or greater than the concentration of mCerulean3 proteins being detected.
Previously, FCS was used to characterize mobile nuclear proteins, including HP1α, inside living cells.29,30 Here, FCS was used to measure the diffusion characteristics of the mCerulean3-labeled HP1α or BZip proteins inside the living cell nucleus, both alone or when co-expressed with either protein labeled with mApple, a bright red FP.41 The 448 and 561 nm laser lines were used to sequentially excite the expressed fusion proteins labeled with either mCerulean3 or mApple. The combination of these laser lines and appropriate filters allow mApple to be used as a marker for the co-expressed proteins, while measuring the FCS statistics for the proteins fused to mCerulean3. Here, measurements were obtained in regions of the nucleus away from heterochromatin (see Sec. 2.4).29 In contrast to the diffusion profile for mCerulean3, the autocorrelation curves representing mCerulean3-BZip and mCerulean3-HP1α are more complex, likely reflecting their known interactions with nuclear proteins and nucleic acids.
While many models are available to characterize these curves, the simplest model with the lowest value provides the most robust method of analysis when comparing two different species.29,30,32 Although neither mCerulean3-HP1α nor mCerulean3-BZip was adequately fit by the one-component simple diffusion model [Eq. (3)], this fitting method did provide the lowest values for both HP1α and BZip when compared to FCS data analyzed by 2 component simple diffusion, or 1 component anomalous diffusion. The results revealed that the best fit for the mobile fraction of mCerulean3-BZip diffused 30% faster than mCerulean3-HP1α (p-value ) [Table 2 and Fig. 4(d)]. The results in Fig. 4(b) show representative fitting curves for mCerulean3-BZip incorporating a model with a degree of anomalous diffusion [Eq. (4)]. The residuals show a random distribution above and below the baseline, indicating that this is an appropriate fit. In contrast, it was not possible to find an adequate fit for mCerulean3-HP1α using a one component fit with any degree of anomalous diffusion. Figure 4(c) shows mCerulean3-HP1α with a two component (simple) diffusion fit. This was found by setting one of the diffusion coefficients to , or essentially noninteracting, and fitting for the other diffusion coefficient. The second diffusion coefficient so fit was . When expressed independently, the autocorrelation curves for BZip and HP1α could not be fit with the same model, suggesting different nuclear interactions likely affect their diffusion.
The mCerulean3-labeled proteins were then co-expressed with the mApple-labeled protein partner, and the diffusion profiles for the mCerulean3 fusion proteins were reacquired. The cumulative results [Table 2, Fig. 4(d)] demonstrate that the diffusion of the mCerulean3-BZip domain protein was not significantly affected by the presence of the mApple-HP1α protein, nor was the diffusion of mCerulean3-HP1α significantly affected by the presence of mApple-BZip. Representative autocorrelation curves and the residuals to the different fits [one component anomalous for BZip in Fig. 4(b) and two component simple for Fig. 4(c)], as well as the cumulative results [Table 2 and Fig. 4(d)] demonstrate that the diffusion profiles of these proteins were not changed by the co-expression of the other protein. Together, these results suggest that mobile fractions of HP1α and the BZip domain protein are diffusing independently of one another.
Another method for analyzing protein co-diffusion is FCCS. We found that mApple had less than optimal behavior in FCS measurements, likely due to a fraction of the FP in a dark state, which is a problem associated with other mRFP derived proteins.42,43 Therefore, we switched to mRuby, another bright red FP,44 as the red channel label for FCCS studies. Here, the system was calibrated and aligned (see Sec. 2.4), and then verified by measuring the cross-correlation of a mixture of donkey anti-goat Alexa 488 and goat anti-rabbit Texas Red (Table 3). Next, whole cell lysates prepared from cells expressing the mCerulean3-mRuby fusion protein were analyzed by FCCS, and this was compared with FCCS measurements from the living cells expressing that fusion protein. Both in the lysate and in the living cells, the mCerulean3-mRuby construct showed a strong cross-correlation of and in lysates and living cells, respectively (Table 3). As a negative control, the unlinked mCerulean3 and mRuby proteins were co-expressed in live cells. The amplitude of the cross-correlation in cells expressing similar levels of mCerulean3 and mRuby was (Table 3). The cross-correlation amplitudes for both red and green species and the calculated diffusion coefficients for all the species are included in Table 3. Finally, mCerulean3-HP1α and mRuby-BZip were co-expressed and FCCS was used to determine the autocorrelation and cross-correlation in regions of the nucleus outside of heterochromatin as described above. A representative cross-correlation measurement is shown in Fig. 5(a) and the results for including the controls are summarized in Fig. 5(b). The amplitude of cross-correlation between mCerulean3-HP1α and mRuby-BZip was similar to that for the unfused FPs, with (Table 3), indicating no discernable cross-correlation in the mobile fraction.
Table 3.
FCCS analysis of co-expressed mCerulean3- and mRuby-linked proteins.
| Species | (green)a | (green) ()b | (red)a | (red) ()b | Number of trialsc |
|---|---|---|---|---|---|
| Donkey anti-Goat IgG Alexa 488 + Goat anti-Rabbit IgG Texas Red | 11 | ||||
| mCerulean3-mRuby (lysate) | 10 | ||||
| mCerulean3-mRuby (live cells) | 13 | ||||
| mCerulean3N1 + mRubyN1 | 19 | ||||
| mCerulean3HP1α + mRuby-BZip | 15 |
(green or red) as described in Sec. 2.4 is the amplitude of the cross-correlation or co-diffusion.
Coefficient of diffusion was determined using Eq. (3).
The number of trials is the number of readings obtained for that experiment. For live cell experiments, these readings were obtained from a minimum of three cells.
Fig. 5.

FCCS measurements of red and green fluorophores. (a) Representative cross-correlation measurement of mRuby-BZip co-expressed with mCerulean3-HP1α. (b) Mean and standard deviation of cross-correlation amplitudes found for different pairs of red and green fluorophores. The positive control for cross-correlation was the antibody-linked dyes (IgG Bound Dyes). All measurements from cells used the same red (mRuby) and green (mCerulean3) FP pair. The control for cross-correlation for the FPs was the linked fusion protein mCer3-mRuby, and this was measured in both cell lysates and in living cells. The co-expression of the individual FPs (mCer3 + mRuby) provided a negative control for cross-correlation, and this is compared to cells co-expressing the mCerulean3-HP1α and mRuby-BZip fusion proteins.
3.3. Effect of Point Mutations in HP1α on Its Co-Localization with the C/EBPα BZip Domain
We next determined the effect of the W174A and W41A point mutations in HP1α on its subnuclear localization and its co-localization with the C/EBPα BZip domain. Here, mCerulean3 was used to label HP1α or the indicated point mutants, and these were co-expressed with the BZip domain protein labeled with mApple to avoid signal crosstalk. The 448 nm laser line was used to excite the mCerulean3-labeled proteins, and images were acquired with the emission filter. Images of the mApple labeled BZip domain proteins were then acquired from the same field of view using 561 nm laser excitation in combination with the emission filter. Figure 6 (column 1) shows the subnuclear distribution of mCerulean3-HP1α (WT) [Fig. 6(a)], mCerulean3-HP1αW174A [Fig. 6(b)], and mCerulean3-HP1αW41A [Fig. 6(c)] when each was expressed alone. As anticipated, HP1α and HP1αW174A localized to heterochromatin, whereas HP1αW41A, which is defective in binding to the methylated lysine 9 of histone H3, did not. The mCerulean3-labeled proteins were then co-expressed with the mApple-BZip domain, and subnuclear distribution of the proteins is shown in Fig. 6(a)–6(c) (columns 2 to 4). As expected, there was strong co-localization of both HP1α and HP1αW174A with the co-expressed BZip domain. Surprisingly, when mCerulean3-HP1αW41A was co-expressed with the BZip domain, the mutant protein was also clearly localized to heterochromatin [Fig. 6(c), column 2 to 4]. This suggests that the W41A mutant, which does not bind to methylated histone H3 and fails to localize in heterochromatin on its own (Fig. 6, row c), can be recruited to heterochromatin by the BZip domain protein.
Fig. 6.

Co-localization of HP1α and its mutants, W174A and W41A, in the nuclei of GHFT1 cells. (a) to (c) Confocal images of Cerulean3-HP1α(WT), Cerulean3-HP1αW174A and Cerulean3-HP1αW41A expressed alone (column 1), or when co-expressed with mApple-BZip (columns 2 to 4). For two color imaging with no signal crosstalk, the Cerulean3 signal was obtained by excitation with the 448 nm laser line, while using the emission filter. The mApple signal was then acquired at the same focal plane by excitation with the 561 nm laser line, while using the emission filter; the scale bar is 5 μm.
3.4. FRET-FLIM Analysis of the Interactions Between the BZip Domain and the HP1α Mutants
FD-FLIM FRET was then used to determine how the point mutations in HP1α affected its interactions with the BZip domain of C/EBPα. The W174A mutation blocks access of the conserved PxVxL motif that is common to many proteins interacting with HP1α and there is a PxVxL-like sequence () within the BZip domain. The FD FLIM-FRET analysis of the Turquoise-BZip and Venus-HP1α W174A demonstrated FRET efficiencies very similar to that observed for the Venus-HP1α(WT) [Fig. 7(a) and Table 1] with a . This indicated that the disruption of the binding site for the PxVxL-motif proteins did not prevent the interaction between HP1α and the BZip domain. In contrast to HP1α W174A, the co-localization studies indicated that mCerulean3-HP1αW41A did not localize in regions of heterochromatin, but that it could be recruited to those regions by the co-expressed BZip domain [Fig. 6(c)]. The FD FLIM-FRET analysis using the co-expressed Turquoise-BZip and Venus-HP1αW41A over a similar range of demonstrated a greatly reduced FRET efficiency compared with HP1α(WT) or HP1αW174A. While there was a clear dependence of on the ratio, the linear regression model could not resolve an for this pair of proteins (Fig. 7 and Table 1). To demonstrate that the dependence of on the ratio was not the result of background signals (i.e., some form of crosstalk) detected in the donor channel, mTurquoise, which is localized to both the cytoplasm and nucleus, was co-expressed with mVenus-HP1α. Here, there was no trend toward increased with increasing ratio [Fig. 7(b) and Table 2]. However, we cannot rule out that the interactions between mCerulean3-HP1αW41A and the Venus-BZip domain in regions of heterochromatin are nonspecific. Taken together, the results of the FRET studies support the interaction of HP1α with the BZip domain of C/EBPα through a PxVxL-independent mechanism, and suggest that binding of HP1α to methylated chromatin likely plays a role in the interaction.
Fig. 7.

FRET-FLIM analysis of the interactions between the BZip domain and the W174A and W41A point mutants of HP1α. The FD FLIM approach was used to measure the average donor lifetime in multiple ROI for each cell, and this was used to determine the FRET efficiency for the indicated donor- and acceptor-labeled proteins. The FRET efficiency (%) is plotted as a function of average measured in the same ROI. Each point represents the . The inset panels show the lifetime maps of the nuclei for representative cells co-expressing mTurquoise-BZip and the indicated mVenus-HP1mutant; the scale bar indicates 10 μm.
4. Discussion
The distinct patterns of gene expression in differentiated cell lineages are due primarily to epigenetic mechanisms. A critical challenge for biomedical research is to identify the molecular mechanisms that function in the dynamic control of the epigenome. Therefore, it is important to understand how the key epigenetic modifier HP1 interacts with sequence specific transcription factors, and how these interactions might function to regulate the epigenome. In this regard our earlier studies demonstrated that the transcription factor C/EBPα localizes to regions of centromeric heterochromatin where it interacts directly with HP1α.7–9 Here, we used FD-FLIM FRET measurements and FCS analysis to characterize the steady-state and dynamic interactions between the BZip domain of C/EBPα and HP1α.
Recent studies using the bimolecular fluorescence complementation assay showed that HP1α interacted with C/EBPβ.38 While C/EBPα and share overlapping roles in the regulation of genes involved in energy metabolism, these isoforms have markedly different roles in the control of cellular differentiation and proliferation. The different roles of these BZip family proteins are reflected in the lack of sequence homology in their amino-terminal transactivation domains. However, the BZip family of proteins all share significant homology within their carboxyl-terminal basic and leucine zipper domains.11 The earlier bimolecular fluorescence complementation study showed that the CD and the hinge region of HP1α could interact with the basic region of C/EBPβ.38 This prompted us to focus our studies on the BZIP domain of C/EBPα.
Our earlier studies using co-immunoprecipitation showed that C/EBPα and HP1α could be isolated as part of a common protein complex.7 While it was clear that these nuclear proteins interacted while bound to chromatin, we did not know whether the mobile fractions of these proteins also interacted within the nuclear compartment. Here, FCS was used to determine the diffusion characteristics of BZip domain and HP1α in living cells, and then FCS and FCCS were used to probe for interactions between the mobile fractions of these proteins in living cells. It is important to point out that cross-correlation of signals from FPs in live cells are limited by their photophysical characteristics and biological factors that collectively decrease the maximum amplitude of cross-correlation and increase cross-talk. Nevertheless, fusion proteins containing a red and cyan FP expressed in live cells can achieve a significant degree of cross-correlation. The pitfalls of the cross-correlation method when using the FPs in living cells is discussed elsewhere.45
The FCS studies demonstrated that the proteins have distinctly different diffusion profiles, with mCerulean3-BZip diffusing faster than mCerulean3-HP1α. Second, our FCS and FCCS studies using the co-expressed proteins directly assessed whether the mobile fractions of HP1α and BZip were interacting. If a significant fraction of HP1α were to form a mobile complex with BZip in cells expressing both proteins, it would be expected that the diffusion profile including the rates of diffusion of one or both of the species, would change. However, within statistical significance the FCS studies detected no change in the diffusion profile or rates of diffusion of either protein with co-expression of the other protein, and the FCCS studies of the amplitude of cross-correlation did not detect co-diffusion. We cannot rule out the possibility that a small fraction of the co-expressed proteins are forming a mobile complex that might not be detected by FCS nor FCCS. Together, however, the results provide no evidence that these species interact away from chromatin.
In contrast to the FCS results, the FD-FLIM FRET analysis clearly demonstrated that steady-state interactions did occur between these proteins within regions of centromeric heterochromatin. This result suggests that HP1α and the C/EBPα BZip domain interact only when the proteins are bound to chromatin. Furthermore, we demonstrated that a specific mutation in HP1α that disrupted the PxVxL-motif binding site in the CSD (HP1α W174A) did not affect the steady-state interaction with the BZip domain. Together, these results indicate that the BZip domain of C/EBPα interacts with HP1α through a PxVxL-independent mechanism, but only when bound to chromatin.
The formation of heterochromatin involves signaling between the DNA methyl transferases that modify the DNA, and the HMTs that modify the chromatin.46,47 It is the reciprocal actions of HP1α and the HMTs, along with the direct communication with the DNA methyl transferases, that allows heterochromatin to spread linearly along the chromatin fiber and silence gene expression.48 It is possible that the interactions of specific transcription factors with these proteins could function to override this process, enabling specific genes previously inaccessible in heterochromatin to become transcriptionally active. To address the importance of the heterochromatin binding activity of HP1α to the interactions with C/EBPα, we used the HP1α W41A mutant that is defective in binding to the di- and tri-methylated lysine 9 of histone H3.17
Our results indicate that the HP1α W41A, which does not localize to regions of heterochromatin, could be recruited to these regions by the co-expressed BZip domain protein. This activity may reflect a direct interaction between BZip and HP1α W41A, or alternatively indicate the interactions of these proteins within a common complex. However, the FD FLIM FRET analysis demonstrated a reduced between BZip domain and HP1α W41A, suggesting that the interaction with the mutant protein was changed when compared with WT HP1α. This could be due to changes in the orientation of the FPs or changes in the binding interaction that move the FPs further apart.
Furthermore, the nonlinear regression analysis did not indicate saturation, so it is possible that the interactions between mCerulean3-HP1αW41A and the Venus-BZip domain in the regions heterochromatin are nonspecific. However, the results do suggest that HP1α binding to methylated histones likely plays an important role in mediating the association with the BZip domain protein. Interestingly, it was recently shown that C/EBPα binds to specific methylated promoters in heterochromatin, and that this may function in the activation of a subset of genes necessary for cellular differentiation.14 Our results argue that the functional interaction of HP1α with methylated chromatin is necessary for a strong interaction with BZip domain of C/EBPα, and this interaction could reflect a commonality in the activities of these proteins.
5. Conclusions
Here, we used the combination of FCS, FCCS, and FRET-FLIM to investigate the dynamic interactions between C/EBPα and HP1α in the nuclei of living pituitary cells. The results indicate that the BZip domain of C/EBPα alone is sufficient for the interaction with HP1α, as shown by FD FLIM FRET. The interactions between these proteins appear to occur only when the proteins were bound to chromatin, since FCS could not detect their interaction in the mobile fraction. Our analysis of the point mutants in HP1α revealed that the interaction with the BZip domain occurred through a PxVxL-independent mechanism, and that the binding of HP1α to methylated chromatin is likely necessary for a strong interaction. Interestingly, the co-localization studies demonstrated that the BZip domain recruited HP1α to heterochromatin through a mechanism that does not require the canonical binding of HP1α to methylated lysines on histone H3. Taken together, our results suggest a common interaction of the evolutionarily conserved BZip domain proteins with HP1α, and may point to a functional role for these transcription factors in epigenetic regulation. Further optical and biochemical studies are needed to more fully detail the BZip domain protein/HP1α interaction and determine the downstream effects on chromatin remodeling.
Acknowledgments
This research was supported by NIH 2RO1 DK43701, 3RO1 DK43701-15S1, and the Indiana University School of Medicine (RND). The cDNA for mCerulean3-6xHis was a gift from Dr. John Murray (Indiana University, Bloomington, Indiana). We dedicate this paper to the memory of Dr. Robert MacDonald Clegg: a gentleman, true scholar, enthusiastic teacher, and friend.
References
- 1.Grewal S. I., Jia S., “Heterochromatin revisited,” Nat. Rev. Genet. 8(1), 35–46 (2007). 10.1038/nrg2008 [DOI] [PubMed] [Google Scholar]
- 2.Meissner A., “Epigenetic modifications in pluripotent and differentiated cells,” Nat. Biotechnol. 28(10), 1079–1088 (2010). 10.1038/nbt.1684 [DOI] [PubMed] [Google Scholar]
- 3.Kwon S. H., et al. , “Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II,” Genes Dev. 24(19), 2133–2145 (2010). 10.1101/gad.1959110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwon S. H., Workman J. L., “The changing faces of HP1: from heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription,” BioEssays 33(4), 280–289 (2011). 10.1002/bies.201000138 [DOI] [PubMed] [Google Scholar]
- 5.Quivy J. P., et al. , “The HP1-p150/CAF-1 interaction is required for pericentric heterochromatin replication and S-phase progression in mouse cells,” Nat. Struct. Mol. Biol. 15(9), 972–979 (2008). 10.1038/nsmb.1470 [DOI] [PubMed] [Google Scholar]
- 6.Luijsterburg M. S., et al. , “Heterochromatin protein 1 is recruited to various types of DNA damage,” J. Cell Biol. 185(4), 577–586 (2009). 10.1083/jcb.200810035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demarco I. A., et al. , “Monitoring dynamic protein interactions with photoquenching FRET,” Nat. Methods 3(7), 519–524 (2006). 10.1038/nmeth889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y., et al. , “Three-color spectral FRET microscopy localizes three interacting proteins in living cells,” Biophys. J. 99(4), 1274–1283 (2010). 10.1016/j.bpj.2010.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel A. P., Hays N. M., Day R. N., “Dynamic nuclear protein interactions investigated using fluorescence lifetime and fluorescence fluctuation spectroscopy,” Proc. SPIE 8226, 82260B (2012). 10.1117/12.912883 [DOI] [Google Scholar]
- 10.Johnson P. F., “Molecular stop signs: regulation of cell-cycle arrest by C/EBP transcription factors,” J. Cell Sci. 118(12), 2545–2555 (2005). 10.1242/jcs.02459 [DOI] [PubMed] [Google Scholar]
- 11.Ramji D. P., Foka P., “CCAAT/enhancer-binding proteins: structure, function and regulation,” Biochem. J. 365(Pt. 3), 561–575 (2002). 10.1042/BJ20020508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Q. Q., Lane M. D., “Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation,” Genes Dev. 13(17), 2231–2241 (1999). 10.1101/gad.13.17.2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enwright J. F., III, et al. , “A PIT-1 homeodomain mutant blocks the intranuclear recruitment of the CCAAT/enhancer binding protein alpha required for prolactin gene transcription,” Mol. Endocrinol. 17(2), 209–222 (2003). 10.1210/me.2001-0222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rishi V., et al. , “CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes,” Proc. Natl. Acad. Sci. U. S. A. 107(47), 20311–20316 (2010). 10.1073/pnas.1008688107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bannister A. J., et al. , “Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain,” Nature 410(6824), 120–124 (2001) 10.1038/35065138 [DOI] [PubMed] [Google Scholar]
- 16.Lachner M., et al. , “Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins,” Nature 410(6824), 116–120 (2001). 10.1038/35065132 [DOI] [PubMed] [Google Scholar]
- 17.Jacobs S. A., Khorasanizadeh S., “Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail,” Science 295(5562), 2080–2083 (2002). 10.1126/science.1069473 [DOI] [PubMed] [Google Scholar]
- 18.Thiru A., et al. , “Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin,” EMBO J. 23(3), 489–499 (2004) 10.1038/sj.emboj.7600088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aasland R., Stewart A. F., “The chromo shadow domain, a second chromo domain in heterochromatin-binding protein 1, HP1,” Nucleic Acids Res. 23(16), 3168–3173 (1995). 10.1093/nar/23.16.3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brasher S. V., et al. , “The structure of mouse HP1 suggests a unique mode of single peptide recognition by the shadow chromo domain dimer,” EMBO J. 19(7), 1587–1597 (2000). 10.1093/emboj/19.7.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lomberk G., et al. , “Sequence-specific recruitment of heterochromatin protein 1 via interaction with Kruppel-like factor 11, a human transcription factor involved in tumor suppression and metabolic diseases,” J. Biol. Chem. 287(16), 13026–13039 (2012). 10.1074/jbc.M112.342634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day R. N., Davidson M. W., “The fluorescent protein palette: tools for cellular imaging,” Chem. Soc. Rev. 38(10), 2887–2921 (2009). 10.1039/b901966a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagai T., et al. , “A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications,” Nat. Biotechnol. 20(1), 87–90 (2002) 10.1038/nbt0102-87 [DOI] [PubMed] [Google Scholar]
- 24.Landschulz W. H., et al. , “Isolation of a recombinant copy of the gene encoding C/EBP,” Genes Dev. 2(7), 786–800 (1988). 10.1101/gad.2.7.786 [DOI] [PubMed] [Google Scholar]
- 25.Lew D., et al. , “GHF-1-promoter-targeted immortalization of a somatotropic progenitor cell results in dwarfism in transgenic mice,” Genes Dev. 7(4), 683–693 (1993). 10.1101/gad.7.4.683 [DOI] [PubMed] [Google Scholar]
- 26.Sun Y., et al. , “Monitoring protein interactions in living cells with fluorescence lifetime imaging microscopy,” Methods Enzymol. 504, 371–391 (2012). 10.1016/B978-0-12-391857-4.00019-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redford G. I., Clegg R. M., “Polar plot representation for frequency-domain analysis of fluorescence lifetimes,” J. Fluoresc. 15(5), 805–815 (2005). 10.1007/s10895-005-2990-8 [DOI] [PubMed] [Google Scholar]
- 28.Colyer R. A., Lee C., Gratton E., “A novel fluorescence lifetime imaging system that optimizes photon efficiency,” Microsc. Res. Tech. 71(3), 201–213 (2008). 10.1002/(ISSN)1097-0029 [DOI] [PubMed] [Google Scholar]
- 29.Schmiedeberg L., et al. , “High- and low-mobility populations of HP1 in heterochromatin of mammalian cells,” Mol. Biol. Cell 15(6), 2819–2833 (2004). 10.1091/mbc.E03-11-0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller K. P., et al. , “Multiscale analysis of dynamics and interactions of heterochromatin protein 1 by fluorescence fluctuation microscopy,” Biophys. J. 97(11), 2876–2885 (2009). 10.1016/j.bpj.2009.08.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malchus N., Weiss M., “Elucidating anomalous protein diffusion in living cells with fluorescence correlation spectroscopy—facts and pitfalls,” J. Fluoresc. 20(1), 19–26 (2010). 10.1007/s10895-009-0517-4 [DOI] [PubMed] [Google Scholar]
- 32.Stasevich T. J., et al. , “Cross-validating FRAP and FCS to quantify the impact of photobleaching on in vivo binding estimates,” Biophys. J. 99(9), 3093–3101 (2010). 10.1016/j.bpj.2010.08.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Widengren J., Rigler R., Mets Ü., “Triplet-state monitoring by fluorescence correlation spectroscopy,” J. Fluoresc. 4(3), 255–258 (1994). 10.1007/BF01878460 [DOI] [PubMed] [Google Scholar]
- 34.Schwille P., Meyer-Almes F. J., Rigler R., “Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution,” Biophys. J. 72(4), 1878–1886 (1997). 10.1016/S0006-3495(97)78833-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacia K., Schwille P., “Practical guidelines for dual-color fluorescence cross-correlation spectroscopy,” Nat. Protoc. 2(11), 2842–2856 (2007). 10.1038/nprot.2007.410 [DOI] [PubMed] [Google Scholar]
- 36.Markwardt M. L., et al. , “An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching,” PLoS One 6(3), e17896 (2011). 10.1371/journal.pone.0017896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day R. N., Periasamy A., Schaufele F., “Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus,” Methods 25(1), 4–18 (2001). 10.1006/meth.2001.1211 [DOI] [PubMed] [Google Scholar]
- 38.Susperreguy S., et al. , “Visualization by BiFC of different C/EBPbeta dimers and their interaction with HP1alpha reveals a differential subnuclear distribution of complexes in living cells,” Exp. Cell Res. 317(6), 706–723 (2011). 10.1016/j.yexcr.2010.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goedhart J., et al. , “Bright cyan fluorescent protein variants identified by fluorescence lifetime screening,” Nat. Methods 7(2), 137–139 (2010). 10.1038/nmeth.1415 [DOI] [PubMed] [Google Scholar]
- 40.Wang Z., et al. , “Fluorescence correlation spectroscopy investigation of a GFP mutant-enhanced cyan fluorescent protein and its tubulin fusion in living cells with two-photon excitation,” J. Biomed. Opt. 9(2), 395–403 (2004). 10.1117/1.1646416 [DOI] [PubMed] [Google Scholar]
- 41.Shaner N. C., et al. , “Improving the photostability of bright monomeric orange and red fluorescent proteins,” Nat. Methods 5(6), 545–551 (2008). 10.1038/nmeth.1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillesheim L. N., Chen Y., Muller J. D., “Dual-color photon counting histogram analysis of mRFP1 and EGFP in living cells,” Biophys. J. 91(11), 4273–4284 (2006). 10.1529/biophysj.106.085845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu B., Chen Y., Muller J. D., “Fluorescence fluctuation spectroscopy of mCherry in living cells,” Biophys. J. 96(6), 2391–2404 (2009). 10.1016/j.bpj.2008.12.3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kredel S., et al. , “mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures,” PLoS One 4(2), e4391 (2009). 10.1371/journal.pone.0004391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foo Y. H., et al. , “Factors affecting the quantification of biomolecular interactions by fluorescence cross-correlation spectroscopy,” Biophys. J. 102(5), 1174–1183 (2012). 10.1016/j.bpj.2012.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smallwood A., et al. , “Functional cooperation between HP1 and DNMT1 mediates gene silencing,” Genes Dev. 21(10), 1169–1178 (2007). 10.1101/gad.1536807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuks F., et al. , “The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase,” Nucleic Acids Res. 31(9), 2305–2312 (2003). 10.1093/nar/gkg332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stewart M. D., Li J., Wong J., “Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment,” Mol. Cell. Biol. 25(7), 2525–2538 (2005). 10.1128/MCB.25.7.2525-2538.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]