

Abstract
OBJECTIVE
Insulin receptor substrate 1 (IRS1) is central to insulin signaling pathways. This study aimed to examine the association of IRS1 variants with insulin resistance (IR) and related phenotypes, as well as potential modification by diet.
RESEARCH DESIGN AND METHODS
Two IRS1 variants (rs7578326 and rs2943641) identified by genome-wide association studies as related to type 2 diabetes were tested for their associations with IR and related traits and interaction with diet in the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study (n = 820) and the Boston Puerto Rican Health Study (BPRHS) (n = 844).
RESULTS
Meta-analysis indicated that rs7578326 G-allele carriers and rs2943641 T-allele carriers had a lower risk of IR, type 2 diabetes, and metabolic syndrome (MetS). Significant interactions on IR and MetS were found for these two variants and their haplotypes with diet. In GOLDN, rs7578326 G-allele carriers and rs2943641 T-allele carriers and their haplotype G-T carriers had a significantly lower risk of IR and MetS than noncarriers only when the dietary saturated fatty acid-to-carbohydrate ratio was low (≤0.24). In both GOLDN (P = 0.0008) and BPRHS (P = 0.011), rs7578326 G-allele carriers had a lower risk of MetS than noncarriers only when dietary monounsaturated fatty acids were lower than the median intake of each population.
CONCLUSIONS
IRS1 variants are associated with IR and related traits and are modulated by diet in two populations of different ancestries. These findings suggest that IRS1 variants have important functions in various metabolic disorders and that dietary factors could modify these associations.
The prevalence of type 2 diabetes continues to increase, accounting for >10% of U.S. adults and >6% of adults worldwide in 2010 (1,2). Insulin resistance is not only a hallmark of type 2 diabetes but also one of the risk factors of metabolic syndrome (MetS), which is defined by a combination of conditions that includes hypertension, dyslipidemia, impaired glucose tolerance, and obesity (3). Identifying genetic and environmental risk factors for type 2 diabetes and insulin resistance is a key step for the prevention of these diseases. With the broad adoption of genome-wide association studies (GWAS), a growing number of genetic loci related to type 2 diabetes and insulin resistance have been identified (4). However these loci explain only ∼10% of the diabetes heritability (5), and thus the influence of environmental factors and their interaction with genotypes have garnered more attention (6).
Among the loci recently identified by GWAS of type 2 diabetes is IRS1 (5,7), which encodes insulin receptor substrate 1 (IRS1), a protein central to insulin signaling pathways. Insulin signaling is initiated by insulin binding to its receptor to activate tyrosine kinase. This enzyme phosphorylates select tyrosine residues of the IRS1 protein to activate the downstream phosphatidylinositol 3-kinase (PI3K) pathway, leading to glucose uptake and glycogen synthesis (8,9). Rodent models and cell culture experiments have provided solid evidence that dysregulation of IRS1 expression is related to insulin resistance (10–12), and IRS1 knockout mice show reduced insulin-induced glucose transport in insulin-responsive tissues, such as skeletal muscle and adipose tissue (11,12). In humans, two genetic variants (rs7578326 and rs2943641) near IRS1 were identified by GWAS to be associated with type 2 diabetes (5,7). However, these results are limited exclusively to European populations and still lack confirmation in populations of different ancestries. In addition, one potentially functional IRS1 variant (rs2943641) (7) showed interaction with dietary carbohydrate and fat for insulin resistance in an intervention study (13) and for type 2 diabetes in an observational study (14). These gene-diet interactions lack replications. Furthermore, MetS is closely related to insulin resistance and type 2 diabetes (15,16) and is also influenced by gene-diet interactions (17,18).
The aim of the current study was to examine the associations of two GWAS-identified IRS1 variants with insulin resistance, type 2 diabetes, and MetS and the interactions of these variants with diet in two populations of different ancestries: the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study and the Boston Puerto Rican Health Study (BPRHS).
RESEARCH DESIGN AND METHODS
Study populations
The GOLDN participants were predominantly of European ancestry and recruited from two genetically homogeneous centers (Minneapolis, MN, and Salt Lake City, UT). In this study, only 820 participants (406 men and 414 women) of European ancestry were included in our analyses. The primary aim of GOLDN was to examine the influence of genetic and dietary factors on the response of individuals to fenofibrate. Baseline data obtained from subjects before they entered the intervention were selected for this analysis. The study details and related methodology of GOLDN have been described (19). Dietary intake was collected using a diet history questionnaire, which was developed by the National Cancer Institute and was validated in two studies (20,21). Calculation of dietary glycemic load (GL) and glycemic index (GI) was according to the method described previously (22). The protocol was approved by the institutional review boards at the University of Alabama, the University of Minnesota, the University of Utah, and Tufts University.
The BPRHS is a longitudinal cohort study of stress, nutrition, health, and aging, for which study participants were self-identified as Puerto Rican and living in the Boston and metropolitan area (23). The ancestry composition of the BPRHS is 57.2% European, 27.4% African, and 15.4% Native American (24). For this study, we included 844 participants (239 men and 605 women) with complete genotype and dietary data. Dietary intake was assessed by a validated food frequency questionnaire (FFQ) that was designed for and validated in this population (25). Dietary GL and GI were calculated per the method previously used in this population (26). The study protocol was approved by the institutional review boards at Tufts University and Northeastern University.
Biochemistry and anthropometric measurements
Blood samples were drawn after an overnight fast. In GOLDN, fasting insulin was obtained using a radioimmunoassay by a commercial kit (Linco Research), and fasting glucose was measured using a hexokinase-mediated reaction on the Hitachi commercial kit (Roche Diagnostics). Measurements of blood lipids, including triglycerides and HDL cholesterol (HDL-C), have been described (27). In the BPRHS, fasting insulin was measured using an Immulite 1000 Insulin Kit (LKIN1) on the Immulite 1000 (Seimens Medical Solution Diagnostics), and the Olympus Au400e with Olympus glucose reagents (Olympus America Inc.) were used to measure fasting glucose. Fasting triglycerides and HDL-C were measured with Olympus HDL-C reagents (OSR6195) and Olympus triglyceride reagents (OSR6033).
For both GOLDN and the BPRHS, homeostasis model assessment of insulin resistance (HOMA-IR, calculated as fasting glucose × fasting insulin/22.5) was used to represent insulin resistance. Type 2 diabetes was defined as fasting glucose ≥126 mg/dL or use of diabetes medication. Normal fasting glucose was defined as individuals without diabetes and with fasting glucose <100 mg/dL, and impaired fasting glucose (IFG) was defined as individuals without diabetes but with 100≤ fasting glucose <126 mg/dL. As the prevalence of type 2 diabetes in GOLDN was low, thereby limiting the power to detect the main association and gene-diet interaction, IFG/T2D was defined as the combined IFG and type 2 diabetes in GOLDN; IFG/T2D was also treated as an outcome in the BPRHS to be comparable with GOLDN. MetS was defined as having at least three of the following five criteria: waist circumference ≥102 cm for men or ≥88 cm for women, elevated triglycerides ≥150 mg/dL or drug treatment for elevated triglycerides, low HDL-C (<40 mg/dL for men or <50 mg/dL for women) or drug treatment for reduced HDL-C, high blood pressure (systolic ≥130 mmHg or diastolic ≥85 mmHg) or antihypertensive medication, and elevated fasting glucose ≥100 mg/dL or drug treatment for elevated glucose (3).
DNA isolation, genotyping, and haplotype analysis
DNA was obtained from blood samples with Gentra Puregene Blood Kits (Gentra Systems) in GOLDN and with QIAamp DNA Blood Mini Kits (Qiagen) in the BPRHS. For GOLDN, Affymetrix Genome-Wide Human SNP Array 6.0 was used for genome-wide genotyping; for the BPRHS, Illumina HumanOmni1-Quad GWAS arrays were used to conduct the genome-wide genotyping. Genotypes of two IRS1 single nucleotide polymorphisms (SNPs) (rs7578326 and rs2943641) were selected for these analyses in both populations. Haplotype frequencies were estimated by the expectation-maximization algorithm, using the haplo.stats package in R (version 2.15.0).
Population admixture of the BPRHS population was calculated by selecting 100 SNPs as ancestry-informative markers (24). We adjusted for population admixture for all analyses in the BPRHS.
Statistical analyses
All continuous dependent variables were Box-Cox transformed to obtain normal distribution before statistical analysis (28). χ2 tests were conducted to examine the Hardy-Weinberg equilibrium for IRS1 variants. Dietary factors, including carbohydrate, monounsaturated fatty acid (MUFA), saturated fatty acid (SFA), total fat, and SFA-to-carbohydrate ratio were all expressed as percentages of total energy intake and dichotomized based on the median intake of each population for the interaction analysis. In GOLDN, the GENMOD procedure in SAS was used to adjust for family relationships, and a generalized estimating equation approach with exchangeable correlation structure was used in GENMOD. A multivariate interaction model was used to assess the interactions of IRS1 variants with dietary factors, while adjusting for potential confounders, including age, sex, waist circumference, study center, smoking status, alcohol drinking, type 2 diabetes, physical activity, and family relationships. In the BPRHS, multivariate logistic regression models were used to assess the association of IRS1 variants with binary outcomes, and the interaction of these variants with diet. For continuous outcomes, multivariate linear regression models were used, with control for age, sex, waist circumference, smoking status, alcohol drinking, type 2 diabetes, physical activity, and population structure. All statistical analyses were conducted using SAS 9.2 (SAS Institute Inc., Cary, NC).
Meta-analysis was conducted with the Meta-Analysis Helper (METAL) (http://www.sph.umich.edu/csg/abecasis/metal/) under fixed-effects models. For binary outcomes, we used meta-analysis to combine the effect size estimates (β coefficients) from GOLDN and the BPRHS, weighted by the inverse of the corresponding standard errors. For continuous outcomes, meta-analysis was conducted, combining the z statistics across the two populations, weighted by sample size.
RESULTS
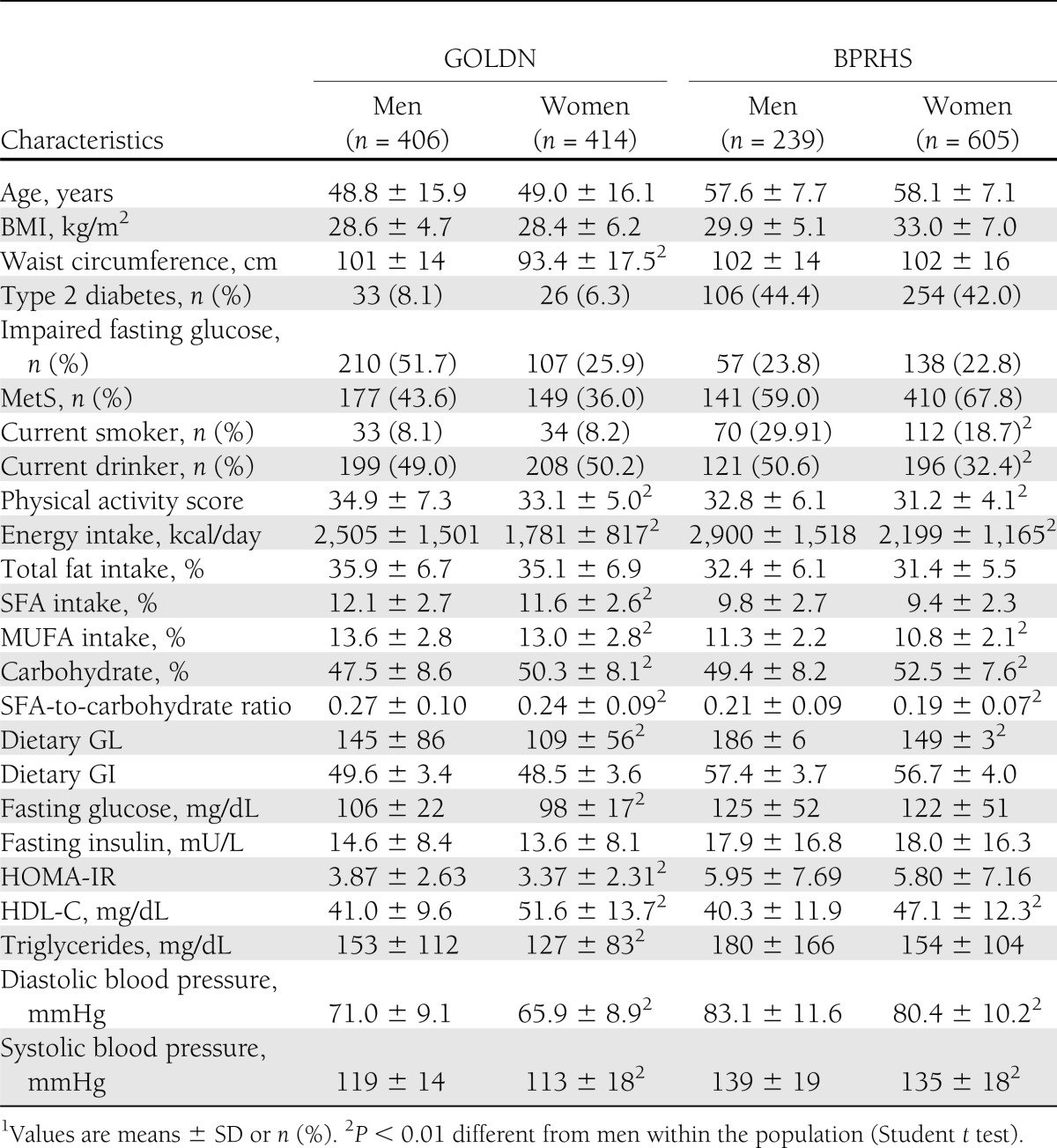
Characteristics of the study populations and IRS1 variants
In both GOLDN and BPRHS populations, men had a significantly higher physical activity score, total energy and MUFA intake, SFA-to-carbohydrate ratio, dietary GL, and diastolic and systolic blood pressure than women, whereas HDL-C and dietary carbohydrate intake were lower in men than women (Table 1). Minor allele frequencies of the two IRS1 variants, rs7578326 (G allele) and rs2943641 (T allele), were 0.328 and 0.355 in GOLDN and 0.364 and 0.328 in the BPRHS. Neither IRS1 variant deviated from the Hardy-Weinberg equilibrium expectation in either population (P > 0.05). This pair of IRS1 variants was in strong linkage disequilibrium (LD) in both GOLDN (r2 = 0.714) and the BPRHS (r2 = 0.458).
Table 1.
Characteristics of participants in the GOLDN and BPRHS populations1
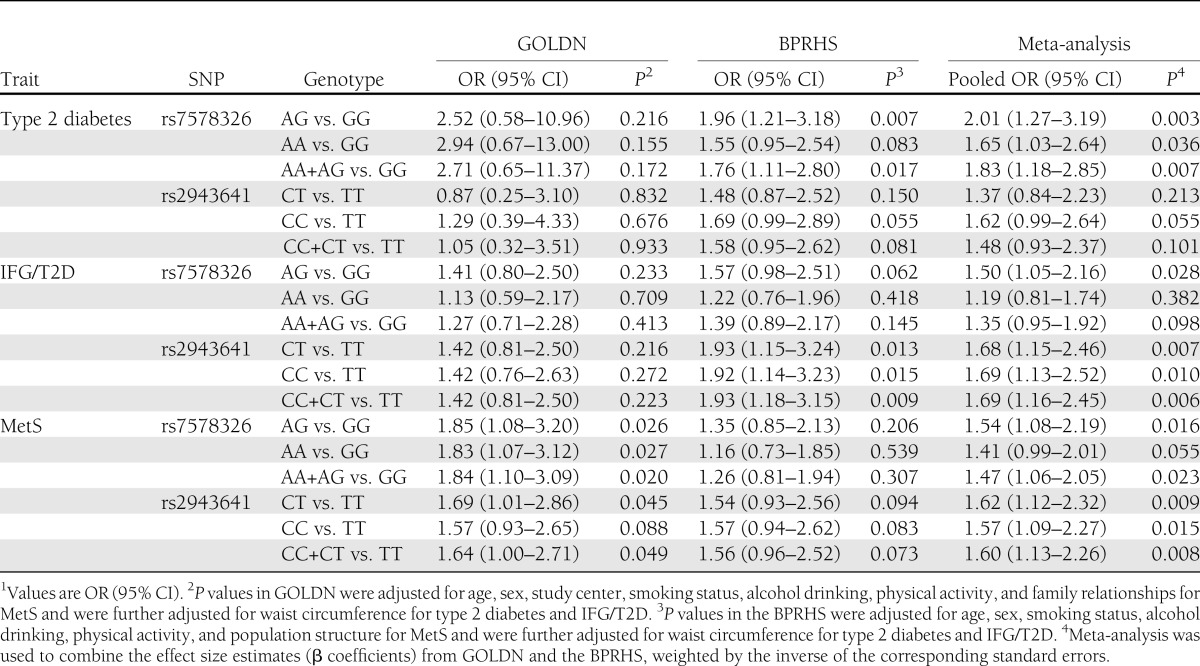
Meta-analysis of IRS1 variants with HOMA-IR, fasting insulin, type 2 diabetes, IFG/T2D, and MetS
For SNP rs7578326, G-allele carriers had significantly lower HOMA-IR (z = −3.102, P = 0.002) and fasting insulin (z = −3.648, P = 0.0003) than A-allele homozygotes (Supplementary Table 1). For SNP rs2943641, T-allele carriers had a significantly lower HOMA-IR (z = −3.08, P = 0.002) and fasting insulin (z = −2.932, P = 0.003) than C-allele homozygotes. No significant heterogeneity was observed (P heterogeneity >0.1).
The pooled odds ratios (ORs) of type 2 diabetes (pooled OR 1.83 [95% CI 1.18–2.85], P = 0.007) and MetS (1.47 [1.06–2.05], P = 0.023) were both statistically significant for the rs7578326 A-allele carriers compared with G-allele homozygotes (Table 2). For SNP rs2943641, C-allele carriers had a higher risk of IFG/T2D (1.69 [1.16–2.45], P = 0.006) and MetS (1.60 [1.13–2.26], P = 0.008), compared with T-allele homozygotes. No significant heterogeneity was observed (P heterogeneity >0.1).
Table 2.
Associations of IRS1 variants with risk of type 2 diabetes, IFG/T2D, and MetS1
Interaction of IRS1 variants with diet for HOMA-IR and fasting insulin
All dietary factors were dichotomized based on the median intake of each population for the interaction analysis. Meta-analysis was not performed for the gene-diet interaction between the two populations, as the food frequency questionnaires used and dietary intake ranges were different between the two populations. In GOLDN, both rs7578326 and rs2943641 significantly interacted with dietary MUFA (rs7578326: P = 0.024; rs2943641: P = 0.008) (Fig. 1), SFA (rs7578326: P = 0.019; rs2943641: P = 0.01), total fat (rs7578326: P = 0.038; rs2943641: P = 0.01), carbohydrate (rs7578326: P = 0.009; rs2943641: P = 0.002), and SFA-to-carbohydrate ratio (rs7578326: P = 0.003; rs2943641: P = 0.003) for HOMA-IR (Table 3). SNP rs7578326 G-allele carriers and rs2943641 T-allele carriers, compared with noncarriers, had significantly lower HOMA-IR when consuming low MUFA, low total fat, or low SFA-to-carbohydrate ratio. SNP rs7578326 also interacted with dietary carbohydrate (P = 0.027) and SFA-to-carbohydrate ratio (P = 0.017) for fasting insulin, whereas rs2943641 interacted with MUFA (P = 0.033), carbohydrate (P = 0.004), and SFA-to-carbohydrate ratio (P = 0.014) for fasting insulin (data not shown). To further explore the potential influence of carbohydrate quality, we examined the interactions of IRS1 variants with dietary GL and GI for HOMA-IR and fasting insulin, but with no significant results (data not shown).
Figure 1.

Interaction of IRS1 variant with dietary MUFA on insulin resistance in the GOLDN and BPRHS populations. Dietary MUFA interacted significantly (P = 0.024) with IRS1 variant rs7578326 on insulin resistance in GOLDN and marginally significantly (P = 0.07) in BPRHS. In both populations, G-allele carriers of rs7578326 had significantly lower HOMA-IR than noncarriers only when dietary MUFA intake was low (≤median intake of each population), but not when MUFA intake was high. P values in GOLDN were adjusted for age, sex, waist circumference, study center, smoking status, alcohol drinking, type 2 diabetes, physical activity, and family relationships. P values in the BPRHS were adjusted for age, sex, waist circumference, smoking status, alcohol drinking, type 2 diabetes, physical activity, and population structure. Number inside the bar indicates the number of subjects in that group. Values are means ± SEM.
Table 3.
Interaction between IRS1 variants and diet on HOMA-IR and risk of MetS in the GOLDN population1
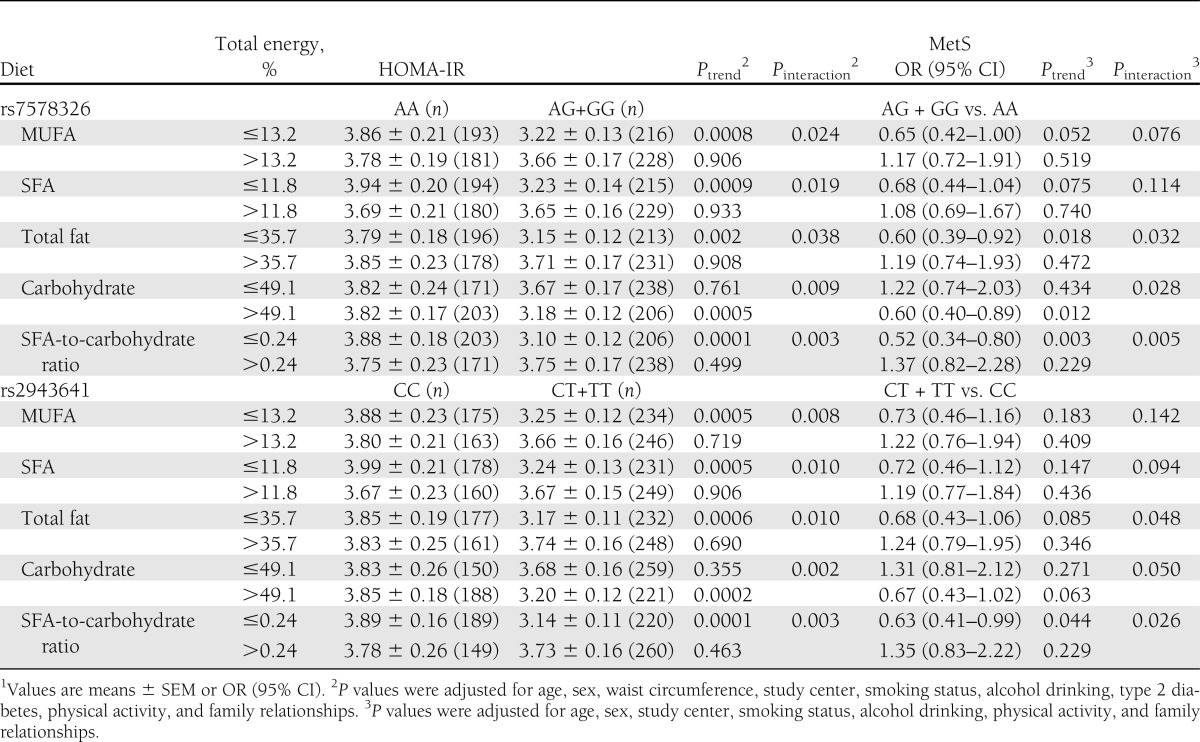
In the BPRHS, rs7578326 tended to interact with dietary MUFA for HOMA-IR (P = 0.07) (Fig. 1), and rs7578326 G-allele carriers showed lower HOMA-IR compared with A-allele homozygotes only when MUFA intake was low (≤11.0% energy, P = 0.011), but not when it was high (>11.0% energy). In addition, this SNP significantly interacted with dietary GL on HOMA-IR (P = 0.038) and fasting insulin (P = 0.014). HOMA-IR for G-allele carriers of rs7578326 was significantly lower than noncarriers when dietary GL was low (≤141.2, P = 0.007), but not for high GL (>141.2). No significant interactions with GI or other dietary factors were observed for rs7578326. For rs2943641, no significant gene-diet interaction was observed (data not shown).
Interaction of IRS1 variants with diet for type 2 diabetes, IFG/T2D, and MetS
In the GOLDN population, no significant interaction between IRS1 variants and dietary factors for the risk of type 2 diabetes or IFG/T2D was observed, whereas both rs7578326 and rs2943641 significantly interacted with dietary total fat, carbohydrate, and SFA-to-carbohydrate ratio to modulate risk of MetS (Table 3). Only when dietary SFA-to-carbohydrate ratio was low (≤0.24) did subjects with rs7578326 G allele have a lower risk of MetS compared with AA carriers (OR 0.52 [95% CI 0.34–0.80]), and only when the ratio was low (≤0.24) did rs2943641 T-allele carriers, compared with the CC carriers, have a lower risk of MetS (0.63 [0.41–0.99]). There was no significant interaction for dietary GL or GI for these outcomes in GOLDN. For the BPRHS population, no significant interaction was observed for IRS1 variants and dietary factors for type 2 diabetes, IFG/T2D, or MetS.
Haplotype analyses for IRS1 variants
Main genetic associations.
For rs7578326 and rs2943641, four haplotypes were observed in both GOLDN and the BPRHS, with the frequencies ranging from 0.016 to 0.629 in GOLDN, and from 0.056 to 0.581 in the BPRHS (Supplementary Table 2). Meta-analysis indicated that haplotype G-T carriers had lower HOMA-IR (z = 2.817, P = 0.005) and fasting insulin (z = 3.072, P = 0.002) than noncarriers (Supplementary Table 3). Haplotype A-C carriers had a higher risk of type 2 diabetes (pooled OR 1.62 [95% CI 1.10–2.38], P = 0.014), IFG/T2D (1.46 [1.06–2.01], P = 0.02), and MetS (1.46 [1.09–1.96], P = 0.012), compared with noncarriers (Supplementary Table 2). No significant heterogeneity was observed for the meta-analysis (Pheterogeneity > 0.1).
Haplotype-diet interaction.
For GOLDN, haplotype G-T significantly interacted with SFA (P = 0.031), carbohydrate (P = 0.007), and the SFA-to-carbohydrate ratio (P = 0.005) on HOMA-IR (Supplementary Table 4). Haplotype G-T also significantly interacted with dietary total fat (P = 0.032), carbohydrate (P = 0.008), and the SFA-to-carbohydrate ratio (P = 0.002), influencing the risk of MetS. Haplotype A-C interacted with total fat intake (P = 0.048) and GL (P = 0.006) for risk for MetS (P = 0.048). Subjects not carrying haplotype A-C had a lower risk of MetS compared with A-C carriers when dietary GL was low (≤111.5, OR 0.38 [95% CI 0.18–0.63]), but not with high GL (>111.5, 1.36 [0.10–2.84]) (Supplementary Table 5). No interaction for type 2 diabetes or IFG/T2D was observed.
For the BPRHS, haplotype A-C marginally interacted with dietary GL for HOMA-IR (P = 0.065) and fasting insulin (P = 0.065) (data not shown). Subjects not carrying haplotype A-C had lower HOMA-IR than carriers only when dietary GL was low (≤141.2, P = 0.007), but not for high dietary GL. Haplotype A-C also interacted with dietary GI for MetS risk (P = 0.034) (Supplementary Table 4). Subjects not carrying haplotype A-C had a lower risk of MetS than A-C carriers only with low dietary GI (≤57.1, OR 0.50 [95% CI 0.28–0.87]). In addition, haplotype G-T had a marginally significant interaction with dietary carbohydrate (P = 0.051) for type 2 diabetes risk. Haplotype G-T carriers had a lower risk of type 2 diabetes compared with noncarriers when consuming high carbohydrate (>51.5, 0.65 [0.43–1.00]), but not when consuming low carbohydrate (≤ 51.5, 1.22 [0.80–1.87]). No significant interaction between IRS1 haplotypes and other dietary factors for either outcome was observed in this population.
CONCLUSIONS
In the current study, we found that genetic variants at IRS1 were associated with insulin resistance, fasting insulin, type 2 diabetes, IFG/T2D, and MetS. Haplotype analyses further confirmed these associations. Our findings are consistent with previous GWAS (5,7) in European populations. Rung et al. (7) reported that the C allele of rs2943641 was associated with insulin resistance, hyperinsulinemia, and a higher risk of diabetes in French, Danish, and Finnish populations. For rs7578326, the A allele was associated with a higher risk of diabetes in populations of European ancestry (5). The results from previous GWAS were successfully replicated not only in GOLDN (a white population of European descent), but also in the BPRHS, whose genetic background is quite different from the European populations (24). In addition, to our knowledge, this was the first study to reveal that these two IRS1 variants were also associated with the risk of MetS in two independent populations.
Impaired regulation of insulin signaling is considered to be a major contributor to insulin resistance and type 2 diabetes, and phosphorylation of IRS plays a key role in the insulin signaling pathways (9). IRS1 and IRS2 are major IRS proteins associated with glucose homeostasis, and IRS1 is the major protein initiating the stimulation of glucose transport in both muscle and adipose tissues (10). In addition to the well-established role of the IRS1 protein in insulin signaling, previous evidence supports a link between the IRS1 genotype and dysregulation of glucose metabolism. For example, the diabetogenic C allele of rs2943641 was associated with decreased IRS1 protein expression in Danish twins (7). The same study reported that, after in vivo insulin infusion, the rs2943641 C allele was associated with reduced IRS1-associated PI3K-activity and with reduced insulin sensitivity (7). Therefore, the associations between rs2943641 and insulin resistance and type 2 diabetes observed in the current study may be attributed to the dysregulation of IRS1 protein expression and impaired insulin signaling. Similarly, we observed that the C allele of rs2943641 was associated with a higher risk of MetS, which is plausible because insulin resistance is a component of MetS, and type 2 diabetes is also closely related to MetS (16). These findings are consistent with a previous study (29). A missense mutation at IRS1, rs1801278 (G972R), was associated with MetS (29), and this SNP was also associated with insulin resistance and type 2 diabetes (30,31). However, these two SNPs, rs2943641 and rs1801278, are 567 kbp apart and not in LD (7). The mechanisms for their associations with insulin resistance, type 2 diabetes, and MetS may be quite different and need further clarification. Another study (32) indicated that the T allele of the IRS1 variant rs2943650 (in complete LD [r2=1] with rs2943641 in white populations) was associated with a decreased risk for several MetS components, including body fat, insulin resistance, and dyslipidemia. Therefore, our results confirmed the prior findings and suggested a decreased risk of MetS associated with the rs2943641 T allele. The second SNP (rs7578326) tested in the current study is in strong LD with rs2943641 in both GOLDN and the BPRHS. Therefore, rs7578326 may regulate insulin signaling through rs2943641, or both SNPs combined may represent new causal genetic variant at IRS1 affecting insulin resistance and related phenotypes.
In addition to our analyses of genetic associations, we also explored interactions between dietary intake and IRS1 SNPs. SNPs rs7578326 G allele and rs2943641 T allele showed more beneficial effects on HOMA-IR, fasting insulin, and MetS than the CC genotype only when the SFA-to-carbohydrate ratio was low. These results were further confirmed by the haplotype analyses. Our findings, consistent with another observational study (14), suggested a protective effect of a diet high in carbohydrate or low in fat on diabetes for men with the rs2943641 T allele. In contrast, rs2943641 showed a different interaction pattern with dietary carbohydrate and fat for HOMA-IR and fasting insulin in an intervention study (13). Specifically, the CC genotype carriers had a greater improvement of insulin and HOMA-IR than the other genotypes when consuming high-carbohydrate and low-fat diets. The inconsistencies between the current study and the previous intervention study may be attributed to the different ranges of dietary intake and the study designs. For example, average carbohydrate intake in the high-carbohydrate, low-fat dietary group was 65% energy in the previous intervention study (13), whereas the median carbohydrate intake was only 49.1% energy in GOLDN and 51.5% energy in the BPRHS. Of concern, high-quality carbohydrate-rich foods with a low GI were used in that study (13), whereas combined sources of carbohydrate intake were evaluated in our study, and we, for the first time, reported significant interactions between dietary GL and GI and IRS1 variants for insulin resistance and related phenotypes. Therefore, carbohydrate quantity and quality may be the most relevant sources of inconsistencies between the current study and the previous one. However, the precise mechanism for the observed inconsistencies still needs further investigation. In addition, the previous intervention study (13) did not explicitly evaluate macronutrients separately because fat and carbohydrate were both altered simultaneously. Our study clearly shows that the dietary SFA-to-carbohydrate ratio and carbohydrate quantity and quality were the important dietary factors contributing to the interactions with IRS1 SNPs. In addition, dietary MUFA was found to interact with IRS1 variants for insulin resistance in both populations. Our results provide consistent evidence that the T allele of rs2943641 and the G allele of rs7578326 were associated with lower levels of insulin resistance or its related phenotypes under certain dietary conditions, including a low SFA-to-carbohydrate ratio, low MUFA intake, or low GL or GI .
The potential mechanisms for these interactions may be related to lipid-induced insulin resistance (33). A high-fat diet was associated with a reduction in tyrosine phosphorylation and an increase in serine phosphorylation of IRS1, thus leading to the suppression of downstream PI3K activity and decreased insulin sensitivity (33). When dietary fat intake was low, reduced levels of IRS1 protein associated with carrying the risk allele C of rs2943641 could still suppress the downstream PI3K activity (7), thereby increasing insulin resistance. In contrast, the T allele tended to be protective and associated with the enhancement of PI3K activity. These hypotheses may provide a plausible explanation for our results. Insulin resistance for subjects carrying the rs2943641T allele and with low fat (MUFA, total fat, or SFA) intake was lower compared with subjects carrying the CC genotype or compared with subjects carrying either genotype with high fat intake. In addition, the interaction of IRS1 variants with GL or GI was also plausible, as high-GI foods could induce higher blood glucose and be associated with insulin resistance and diabetes (34). It might be that the protective effect of the IRS1 nonrisk allele was enhanced only when dietary GL or GI was low. However, the precise mechanism for the interaction remains to be clarified. Another concern is whether the interaction of IRS1 with dietary fat on MetS was confounded by the correlation between triglycerides, HDL-C, and dietary fat. However, our analyses ensured that the significant interaction was independent of the main effect of dietary fat, and no significant interaction for triglycerides, HDL-C, or other MetS components was found.
The current study has several limitations. First, the GOLDN and BPRHS populations have quite different ancestries and lifestyles. For example, the dietary intakes differ significantly, and this could explain the different forms of gene-diet interactions observed. However, we found consistent trends across the two populations in terms of the main genetic associations and gene-diet interactions, and these relationships are all biologically plausible. Second, moderate sample sizes for GOLDN and BPRHS limited the statistical power. Nevertheless, to our knowledge, this is the first study revealing the interactions between IRS1 variants (rs7578326 and rs2943641) and dietary factors to modulate insulin resistance, risk of type 2 diabetes, and MetS in observational studies. More replications in other populations are clearly warranted.
In conclusion, IRS1 variants rs7578326 G allele and rs2943641 T allele were associated with a lower risk of insulin resistance, type 2 diabetes, and MetS in two independent populations of different ancestries. Notably, these associations appeared to be modulated by dietary factors, especially the dietary SFA-to-carbohydrate ratio, MUFA, and carbohydrate quantity and quality. If replicated, these results may eventually provide useful information for the prevention of insulin resistance, type 2 diabetes, and MetS and could help develop effective dietary recommendations in different populations.
Acknowledgments
This study was funded by the China Scholarship Council, the National Basic Research Program of China (973 Program: 2011CB504002), the National Heart, Lung, and Blood Institute (grants HL54776 and HL078885), the National Institutes of Health (P01AG023394 and P50HL105185), and the U.S. Department of Agriculture (USDA), Agriculture Research Service (contracts 53-K06-5-10 and 58-1950-9-001).
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. The USDA is an equal opportunity provider and employer.
No potential conflicts of interest relevant to this article were reported.
J.-S.Z. analyzed data, wrote the manuscript, and was primarily responsible for the final content. D.K.A., K.L.T., and J.M.O. designed and conducted the research. L.D.P., C.E.S., and D.L. wrote the manuscript. I.B.B. designed the research C.-Q.L. designed and conducted the research, analyzed data, wrote the manuscript, and was primarily responsible for the final content. All authors read and approved the final manuscript. C.-Q.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc12-2607/-/DC1.
References
- 1.Centers for Disease Control and Prevention. National Diabetes Fact Sheet: United States 2011 Atlanta, Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, 2011 [Google Scholar]
- 2.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010;87:4–14 [DOI] [PubMed] [Google Scholar]
- 3.Grundy SM, Cleeman JI, Daniels SR, et al. American Heart Association. National Heart, Lung, and Blood Institute Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005;112:2735–2752 [DOI] [PubMed] [Google Scholar]
- 4.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009;106:9362–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voight BF, Scott LJ, Steinthorsdottir V, et al. MAGIC investigators. GIANT Consortium Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 2010;42:579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornelis MC, Hu FB. Gene-environment interactions in the development of type 2 diabetes: recent progress and continuing challenges. Annu Rev Nutr 2012;32:245–259 [DOI] [PubMed] [Google Scholar]
- 7.Rung J, Cauchi S, Albrechtsen A, et al. Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet 2009;41:1110–1115 [DOI] [PubMed] [Google Scholar]
- 8.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002;296:1655–1657 [DOI] [PubMed] [Google Scholar]
- 9.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab 2009;296:E581–E591 [DOI] [PubMed] [Google Scholar]
- 10.Thirone AC, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab 2006;17:72–78 [DOI] [PubMed] [Google Scholar]
- 11.Yamauchi T, Tobe K, Tamemoto H, et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol Cell Biol 1996;16:3074–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaburagi Y, Satoh S, Tamemoto H, et al. Role of insulin receptor substrate-1 and pp60 in the regulation of insulin-induced glucose transport and GLUT4 translocation in primary adipocytes. J Biol Chem 1997;272:25839–25844 [DOI] [PubMed] [Google Scholar]
- 13.Qi Q, Bray GA, Smith SR, Hu FB, Sacks FM, Qi L. Insulin receptor substrate 1 gene variation modifies insulin resistance response to weight-loss diets in a 2-year randomized trial: the Preventing Overweight Using Novel Dietary Strategies (POUNDS LOST) trial. Circulation 2011;124:563–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ericson U, Rukh G, Stojkovic I, et al. Sex-specific interactions between the IRS1 polymorphism and intakes of carbohydrates and fat on incident type 2 diabetes. Am J Clin Nutr 2013;97:208–216 [DOI] [PubMed] [Google Scholar]
- 15.Lorenzo C, Okoloise M, Williams K, Stern MP, Haffner SM, San Antonio Heart Study The metabolic syndrome as predictor of type 2 diabetes: the San Antonio heart study. Diabetes Care 2003;26:3153–3159 [DOI] [PubMed] [Google Scholar]
- 16.Wilson PWF, D’Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation 2005;112:3066–3072 [DOI] [PubMed] [Google Scholar]
- 17.Shen J, Arnett DK, Peacock JM, et al. Interleukin1beta genetic polymorphisms interact with polyunsaturated fatty acids to modulate risk of the metabolic syndrome. J Nutr 2007;137:1846–1851 [DOI] [PubMed] [Google Scholar]
- 18.Ordovas JM, Shen J. Gene-environment interactions and susceptibility to metabolic syndrome and other chronic diseases. J Periodontol 2008;79(Suppl):1508–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corella D, Arnett DK, Tsai MY, et al. The -256T>C polymorphism in the apolipoprotein A-II gene promoter is associated with body mass index and food intake in the genetics of lipid lowering drugs and diet network study. Clin Chem 2007;53:1144–1152 [DOI] [PubMed] [Google Scholar]
- 20.Subar AF, Thompson FE, Kipnis V, et al. Comparative validation of the Block, Willett, and National Cancer Institute food frequency questionnaires : the Eating at America’s Table Study. Am J Epidemiol 2001;154:1089–1099 [DOI] [PubMed] [Google Scholar]
- 21.Thompson FE, Subar AF, Brown CC, et al. Cognitive research enhances accuracy of food frequency questionnaire reports: results of an experimental validation study. J Am Diet Assoc 2002;102:212–225 [DOI] [PubMed] [Google Scholar]
- 22.Salmerón J, Manson JE, Stampfer MJ, Colditz GA, Wing AL, Willett WC. Dietary fiber, glycemic load, and risk of non-insulin-dependent diabetes mellitus in women. JAMA 1997;277:472–477 [DOI] [PubMed] [Google Scholar]
- 23.Tucker KL, Mattei J, Noel SE, et al. The Boston Puerto Rican Health Study, a longitudinal cohort study on health disparities in Puerto Rican adults: challenges and opportunities. BMC Public Health 2010;10:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai CQ, Tucker KL, Choudhry S, et al. Population admixture associated with disease prevalence in the Boston Puerto Rican health study. Hum Genet 2009;125:199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker KL, Bianchi LA, Maras J, Bermudez OI. Adaptation of a food frequency questionnaire to assess diets of Puerto Rican and non-Hispanic adults. Am J Epidemiol 1998;148:507–518 [DOI] [PubMed] [Google Scholar]
- 26.van Rompay MI, McKeown NM, Castaneda-Sceppa C, Falcón LM, Ordovás JM, Tucker KL. Acculturation and sociocultural influences on dietary intake and health status among Puerto Rican adults in Massachusetts. J Acad Nutr Diet 2012;112:64–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai MY, Hanson NQ, Straka RJ, et al. Effect of influenza vaccine on markers of inflammation and lipid profile. J Lab Clin Med 2005;145:323–327 [DOI] [PubMed] [Google Scholar]
- 28.Box GEP, Cox DR. An analysis of transformations. J R Stat Soc Series B Stat Methodol 1964;26:211–252 [Google Scholar]
- 29.Pérez MS, Tellechea ML, Aranguren F, et al. The rs1801278 G>A polymorphism of IRS-1 is associated with metabolic syndrome in healthy nondiabetic men. Modulation by cigarette smoking status. Diabetes Res Clin Pract 2011;93:e95–e97 [DOI] [PubMed] [Google Scholar]
- 30.Jellema A, Zeegers MPA, Feskens EJM, Dagnelie PC, Mensink RP. Gly972Arg variant in the insulin receptor substrate-1 gene and association with type 2 diabetes: a meta-analysis of 27 studies. Diabetologia 2003;46:990–995 [DOI] [PubMed] [Google Scholar]
- 31.Baroni MG, Leonetti F, Sentinelli F, et al. The G972R variant of the insulin receptor substrate-1 (IRS-1) gene is associated with insulin resistance in “uncomplicated” obese subjects evaluated by hyperinsulinemic-euglycemic clamp. J Endocrinol Invest 2004;27:754–759 [DOI] [PubMed] [Google Scholar]
- 32.Kilpeläinen TO, Zillikens MC, Stančákova A, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet 2011;43:753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 2010;375:2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willett W, Manson J, Liu S. Glycemic index, glycemic load, and risk of type 2 diabetes. Am J Clin Nutr 2002;76:274S–280S [DOI] [PubMed] [Google Scholar]