

Abstract
Although a number of the diabetic neuropathies may result in painful symptomatology, this review focuses on the most common: chronic sensorimotor distal symmetrical polyneuropathy (DSPN). It is estimated that 15–20% of diabetic patients may have painful DSPN, but not all of these will require therapy. In practice, the diagnosis of DSPN is a clinical one, whereas for longitudinal studies and clinical trials, quantitative sensory testing and electrophysiological assessment are usually necessary. A number of simple numeric rating scales are available to assess the frequency and severity of neuropathic pain. Although the exact pathophysiological processes that result in diabetic neuropathic pain remain enigmatic, both peripheral and central mechanisms have been implicated, and extend from altered channel function in peripheral nerve through enhanced spinal processing and changes in many higher centers. A number of pharmacological agents have proven efficacy in painful DSPN, but all are prone to side effects, and none impact the underlying pathophysiological abnormalities because they are only symptomatic therapy. The two first-line therapies approved by regulatory authorities for painful neuropathy are duloxetine and pregabalin. α-Lipoic acid, an antioxidant and pathogenic therapy, has evidence of efficacy but is not licensed in the U.S. and several European countries. All patients with DSPN are at increased risk of foot ulceration and require foot care, education, and if possible, regular podiatry assessment.
The neuropathies are the most common long-term microvascular complications of diabetes and affect those with both type 1 and type 2 diabetes, with up to 50% of older type 2 diabetic patients having evidence of a distal neuropathy (1). These neuropathies are characterized by a progressive loss of nerve fibers affecting both the autonomic and somatic divisions of the nervous system. The clinical features of the diabetic neuropathies vary immensely, and only a minority are associated with pain. The major portion of this review will be dedicated to the most common painful neuropathy, chronic sensorimotor distal symmetrical polyneuropathy (DSPN). This neuropathy has major detrimental effects on its sufferers, confirming an increased risk of foot ulceration and Charcot neuroarthropathy as well as being associated with increased mortality (1).
In addition to DSPN, other rarer neuropathies may also be associated with painful symptoms including acute painful neuropathy that often follows periods of unstable glycemic control, mononeuropathies (e.g., cranial nerve palsies), radiculopathies, and entrapment neuropathies (e.g., carpal tunnel syndrome). By far the most common presentation of diabetic polyneuropathy (over 90%) is typical DSPN or chronic DSPN.
The Toronto Diabetic Neuropathy Expert Group recently defined DSPN as “a symmetrical, length-dependent sensorimotor polyneuropathy attributable to metabolic and microvessel alterations as a result of chronic hyperglycemia exposure and cardiovascular risk covariates” (2). An abnormality of nerve conduction (NC) tests, which is frequently subclinical, appears to be the first objective quantitative indication of the condition. The occurrence of diabetic retinopathy and nephropathy in a given patient strengthen the case that the polyneuropathy is attributable to diabetes. DSPN results in insensitivity of the feet that predisposes to foot ulceration (1) and/or neuropathic pain (painful DSPN), which can be disabling.
Clinical features of painful DSPN
The onset of DSPN is usually gradual or insidious and is heralded by sensory symptoms that start in the toes and then progress proximally to involve the feet and legs in a stocking distribution. When the disease is well established in the lower limbs in more severe cases, there is upper limb involvement, with a similar progression proximally starting in the fingers. As the disease advances further, motor manifestations, such as wasting of the small muscles of the hands and limb weakness, become apparent. In some cases, there may be sensory loss that the patient may not be aware of, and the first presentation may be a foot ulcer. Approximately 50% of patients with DSPN experience neuropathic symptoms in the lower limbs including uncomfortable tingling (dysesthesia), pain (burning; shooting or “electric-shock like”; lancinating or “knife-like”; “crawling”, or aching etc., in character), evoked pain (allodynia, hyperesthesia), or unusual sensations (such as a feeling of swelling of the feet or severe coldness of the legs when clearly the lower limbs look and feel fine, odd sensations on walking likened to “walking on pebbles” or “walking on hot sand,” etc.). There may be marked pain on walking that may limit exercise and lead to weight gain. Painful DSPN is characteristically more severe at night and often interferes with normal sleep (3). It also has a major impact on the ability to function normally (both mental and physical functioning, e.g., ability to maintain work, mood, and quality of life [QoL]) (3,4).
In one study from the U.S., the burden of painful DSPN was found to be considerable, resulting in a persistent discomfort despite polypharmacy and high resource use and led to limitations in daily activities and poor satisfaction with treatments (4). The unremitting nature of the pain can be distressing, resulting in mood disorders including depression and anxiety (4). The natural history of painful DSPN has not been well studied, and there is a need for large, population-based, prospective studies looking at the natural history and potentially modifiable risk factors, if any. However, it is generally believed that painful symptoms may persist over the years (5), occasionally becoming less prominent as the sensory loss worsens (6).
Epidemiology
There have been relatively few epidemiological studies that have specifically examined the prevalence of painful DSPN, which range from 10–26% (7–9). In a recent study of a large cohort of diabetic patients receiving community-based health care in northwest England (n = 15,692), painful DSPN assessed using neuropathy symptom and disability scores was found in 21% (7). In one population-based study from Liverpool, U.K., the prevalence of painful DSPN assessed by a structured questionnaire and examination was estimated at 16% (8). Notably, it was found that 12.5% of these patients had never reported their symptoms to their doctor and 39% had never received treatment for their pain (8), indicating that there may be considerable underdiagnosis and undertreatment of painful neuropathic symptoms compared with other aspects of diabetes management such as statin therapy and management of hypertension.
Risk factors for DSPN per se have been extensively studied, and it is clear that apart from poor glycemic control, cardiovascular risk factors play a prominent role (10): risk factors for painful DSPN are less well known. Preliminary epidemiological studies suggest clinical correlates for painful DSPN compared with painless DSPN to include weight (11), obesity (12), waist circumference (13), peripheral arterial disease (11,13), and triglycerides (12). However, one limitation of these studies was that assessment of both DSPN and painful DSPN was carried out using screening methods (Michigan Neuropathy Screening Instrument) questionnaire (11,13) or the Douleur Neuropatique en 4 questions (12) for neuropathic pain, and the neurological examination of Michigan Neuropathy Screening Instrument (11,13) or Neuropen (12) for assessment of DSPN.
Diagnosing and assessing the severity of DSPN and painful DSPN
A broad spectrum of presentations may occur in patients with DSPN, ranging from one extreme of the patient with very severe painful symptoms but few signs, to the other when patients may present with a foot ulcer having lost all sensation without ever having any painful or uncomfortable symptoms; when pressed, such patients may admit to the feet feeling somewhat “numb” or “dead.” Thus, it is well recognized that the severity of symptoms may not relate to the severity of the deficit on clinical examination (1).
Diagnosing and assessing severity of DSPN
The American Diabetes Association Consensus Statement (14) recommended that the diagnosis of painful DSPN in clinical practice be a clinical one, relying on the patient’s description of pain and typical features of peripheral neuropathy manifesting in reduction of sensory modalities and absence/reduction of ankle/knee reflexes. Because DSPN is a diagnosis of exclusion, a careful clinical history and a peripheral neurological and vascular examination of the lower limbs are essential to exclude other causes of neuropathic pain and leg/foot pain such as peripheral vascular disease, arthritis, malignancy, alcohol abuse, spinal canal stenosis, etc. NC studies are rarely helpful in clinical practice but might help in excluding other causes of pain such as entrapment syndromes. Patients with asymmetrical symptoms and/or signs (such as loss of an ankle jerk in one leg only), rapid progression of symptoms, or predominance of motor symptoms and signs should be carefully assessed for other causes of the findings.
For longitudinal studies and clinical trials in which a more accurate quantification of neuropathy is required, the Toronto Diabetic Neuropathy Consensus Panel suggested a reliable objective and quantitative measure; that is, NC abnormality as the minimal criteria for the diagnosis of DSPN (2). When NC values have not been assessed, the Consensus Panel recommended that it is not possible to provide a “confirmed” diagnosis of DSPN—only a “possible” or “probable” diagnosis.
However, subjects with pure small-fiber neuropathy may be diagnosed by quantitative sensory testing (QST) to assess the psychophysical thresholds for cold and warm sensations, heat and cold pain, and pain to pin prick; or by cutaneous vasomotor function to measure skin blood flow as a marker of c-fiber neurovascular dysfunction (15). However, the gold standard for assessing small fiber function remains quantification of intraepidermal nerve fibers from punch skin biopsy immunostained by PGP9.5 (16). Indeed, QST has also been routinely used in some clinics to diagnose general neuropathy (e.g., the 10-g monofilament is widely used in diabetic clinics to screen for DSPN), and recently, the German Pain Network has developed QST to characterize the somatosensory phenotype of patients with neuropathic pain (17), though the Toronto Consensus Panel recognized its limitation of essentially being subjective and the potential for bias by perceptual/cognitive factors. Recent advances in small fiber neuropathy assessment include brain-evoked potentials with electrical and laser stimulation (laser-evoked potentials) (18), and contact heat–evoked potential—a measure of cerebral responses of A-delta fiber–mediated thermonociceptive stimuli (19).
There are several instruments that evaluate combinations of neuropathy symptoms, signs, and neurophysiological test abnormalities giving scores for severity of DSPN (20–24). However, clinical examination is not always reproducible, and recent studies emphasize the importance of the proficiency of the clinical neurological assessment (24). For controlled clinical trials of DSPN, the Toronto Consensus Panel advocated the use of an NC test as an early and reliable indicator of the occurrence of this neuropathy (2). The group also emphasized that to be reliable the test must be carried out rigorously, using appropriate reference values corrected for applicable variables (2).
Assessing neuropathic pain severity
The frequency and severity of painful symptoms can be assessed by a number of simple numeric rating scales (25). The use of simple scales such as the visual analog scale or the numerical rating scale, such as an 11-point Likert scale (0 = no pain to 10 = worst pain imaginable) is recommended. These scales can then be used to monitor response to treatment in clinical practice or research context. Other validated scales and questionnaires include 1) the modified Brief Pain Inventory Short Form (BPI-MSF) (26) recently used as the primary end point in pharmacological treatment trials of painful DSPN; 2) the neuropathic pain symptom inventory (27), which evaluates the different symptoms and dimensions of neuropathic pain; 3) the neuropathic pain questionnaire (28), which consists of 12 items, including 10 related to sensations and or sensory responses and 2 related to affect; 4) the LANNS pain scale (29), which contains five symptom and two clinical examination items and is easy to score within clinical settings; 5) painDETECT, a screening tool that incorporates an easy to use, patient based (self-report) questionnaire with nine items that do not require a clinical exam (30); and 6) the McGill Pain Questionnaire, which measures the sensory and affective components of pain, often used in its shortened format (31).
QoL might be assessed by generic instruments that may allow cross comparison with other chronic medical conditions. However, it is also important to use validated, neuropathy-specific measures of QoL, such as the NeuroQol (32), that reliably capture the key dimensions of patients’ experience of DSPN and is a valid tool for studying the impact of neuropathy and foot ulceration on QoL. Another neuropathy-specific measure of QoL is the Norfolk Quality of Life Scale (33), which is a validated patient-reported outcome measure, sensitive to the different features of diabetic neuropathy including small-fiber, large-fiber, and autonomic function. There are a number of scales that might assess pain behavior and sleep interference and the prediction of response to therapy (34). Finally, the impact of painful symptoms on mood can be assessed using the Hospital Anxiety and Depression Scale (35) and Beck’s Depression Inventory (36).
Mechanism of neuropathic pain in diabetes
We will now consider the changes at central and peripheral levels induced by DSPN that relate to the sensory changes experienced by patients and the mechanisms that treatments are thought to modulate.
The exact pathophysiological mechanisms of neuropathic pain in diabetes remain elusive although several mechanisms have been postulated (Table 1) (37). Other potential mechanisms include the association of increased blood glucose instability in the genesis of neuropathic pain (38), an increase in peripheral nerve epineurial blood flow (39), altered foot skin microcirculation (40), reduced intraepidermal nerve fiber density in the context of early neuropathy (41), increased thalamic vascularity (42), and autonomic dysfunction (43).
Table 1.
Mechanisms of neuropathic pain

The fact that diabetes induces neuropathy and that in a proportion of patients this is accompanied by pain despite the loss of input and numbness, suggests that marked changes occur in the processes of pain signaling in the peripheral and central nervous system. Neuropathic pain is characterized by ongoing pain together with exaggerated responses to painful and nonpainful stimuli, hyperalgesia, and allodynia. These combinations of loss and gain of function have been recently characterized by sensory testing by Maier et al. (44), and five subgroups of patients emerge with many of the signs and symptoms overlapping with those seen in patients with postherpetic neuralgia. This suggests common mechanisms but importantly, the subgroups may turn out to have different responses to pharmacological agents and so lead to a prediction of analgesic response. Nevertheless, the changes seen suggest altered peripheral signaling and central compensatory changes perhaps driven by the loss of input. Indeed, the descriptors used, such as electric shock, burning, and lancinating indicate that diabetes induces changes in the way peripheral signals are processed.
Capsaicin in chili peppers evokes burning pain due to activation of transient receptor potential channel (TRP) V1, a receptor situated on small peripheral fibers and part of the family of 27 TRP channels cloned in humans (45), some of which are thought to transduce thermal signals (46) and include TRPM8 and TRPA1, important in cold hypersensitivity (47). Local application of capsaicin can be analgesic in localized painful neuropathies, and polymorphisms in TRPV1 can explain some of the variation in pain seen after neuropathy in patients (48).
There are many other less understood or characterized peripheral sensors, including the P2× receptors for ATP, released from all damaged cells suggesting further potential targets for therapies (49). Very clear evidence points to the key role of changes in ion channels as a consequence of nerve damage and their roles in the disordered activity and transduction in damaged and intact fibers (50).
Sodium channels depolarize neurons and generate an action potential. Following damage to peripheral nerves, the normal distribution of these channels along a nerve is disrupted by the neuroma and “ectopic” activity results from the accumulation of sodium channels at or around the site of injury. Other changes in the distribution and levels of these channels are seen and impact upon the pattern of neuronal excitability in the nerve. Inherited pain disorders arise from mutated sodium channels, notably gain and loss of functions of the sodium channel Nav 1.7 (51,52), and polymorphisms in this channel impact on the level of pain in patients, indicating that inherited differences in channel function might explain some of the variability in pain between patients with DSPN (53). Following peripheral nerve damage, levels of another sodium channel, Nav 1.3, in dorsal root ganglia increase and are thought to support ectopic neuronal firing. Blockers of these pain-related channels and also the very important Nav1.8 have been produced but have yet to reach the clinic. Thus, treatments revolve around nonselective sodium channel blockers including local anesthetics such as lidocaine and the anticonvulsant carbamazepine in trigeminal neuralgia (54).
A recent study (55) proposes a mechanism for the observed neuropathic changes that follow diabetes. The levels of the glycolytic metabolite methylglyoxal distinguished between diabetic patients with pain and those without pain. Methylglyoxal activated peripheral nerves and altered the function of Nav 1.8, and Nav 1.7. In animals, the metabolite was found to slow NC, increase release of calcitonin gene-related peptide from nerves, and cause thermal and mechanical hyperalgesia. This hyperalgesia was reflected by functional changes in pain-related areas of the brain (55).
Where sodium channels act to generate action potentials, potassium channels serve as the molecular brakes of excitable cells, playing an important role in modulating neuronal hyperexcitability. The drug retigabine, a potassium channel opener acting on the channel (KV7, M-current) opener, blunts behavioral hypersensitivity in neuropathic rats (56) and also inhibits C and Aδ-mediated responses in dorsal horn neurons in both naïve and neuropathic rats (57), but has yet to reach the clinic as an analgesic although it has recently been approved by the FDA as an add-on treatment of partial seizures (58).
There are likely many other changes in potassium channels after neuropathy, and their importance is underscored by the fact that opioids act to open these channels and suppress activity in pain pathways by actions at both spinal and brain sites.
The third channel set in neuronal events is made up of voltage-gated Ca channels, which serve to release transmitter. In the case of afferent sensory fibers, these are glutamate and peptides such as substance P. Following nerve injury, α2δ subunits of these channels are slowly upregulated in the central terminals of peripheral nerves, so favoring increased release of these transmitters into the spinal cord (59). This will therefore compensate for the loss of afferent fibers produced by the neuropathy. Gabapentin and pregabalin target the α2δ subunit and by preventing its trafficking into the membrane reduce the abnormal transmitter release (59).
A further permissive factor in the actions of these drugs is activity in spinal 5-hydroxytryptamine 3 receptors, themselves driven by activity in descending pathways from the brain; this rapidly switching circuit may not only explain the actions of α2δ ligands after nerve injury but also their acute effects such as those seen after surgery (60).
Central excitatory mechanisms
Aδ and C fibers terminate primarily in the superficial laminae of the dorsal horn where the large majority of neurons are nociceptive specific (Fig. 1). Some of these neurons gain low threshold inputs after neuropathy and these cells project predominantly to limbic brain areas such as the periaqueductal gray, lateral parabrachial nucleus, thalamus, nucleus tractus solitarius, and the medullary reticular formation. Within deeper lamina V, neurons are wide-dynamic range, responding to both innocuous and noxious stimuli, and so candidates for abnormal coding of low-threshold inputs and hence allodynia, exhibiting wind-up and projecting to sensory areas of the brain such as the thalamus and thence the cortex, where the sensory component of pain is represented (Fig. 2). Thus, spinal cord neurons provide parallel outputs to the affective and sensory areas of the brain. Changes induced in these neurons by repeated noxious inputs underpin central sensitization where the resultant hyperexcitability of neurons leads to greater responses to all subsequent inputs—innocuous and noxious—expanded receptive fields and enhanced outputs to higher levels of the brain (Fig. 2). These changes may then alter descending controls (Fig. 2). The spinal mechanisms involve the actions of glutamate and its coreleased transmitter substance P permitting N-methyl-d-aspartate (NMDA) receptor activation, resulting in the wind-up of dorsal horn neurons. The analgesic effects of ketamine result from its ability to modulate this activity through blocks of the NMDA receptor (Fig. 1). However, similar mechanistic events underlie memory, cognitive, and related functions and so the side effects of ketamine result from a lack of selectivity for spinal-pain related NMDA functions (61).
Figure 1.
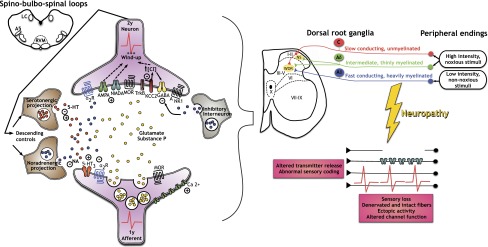
A representation of the central spinal and peripheral changes that accompany neuropathy. The existence of a lesion or disease of a peripheral sensory nerve alters the conduction and transmission of sensory messages. The normal transfer of modalities (top right) onto spinal nociceptive specific (NS) and wide-dynamic range (WDR) neurons is changed by ectopic activity, sensory loss, and changes in ion channels. Spinal cord neurons are subject to many receptor-mediated events, but increases in Ca channel function lead to increased transmitter release so that glutamate causes an enhanced activation of AMPA and NMDA receptors. Substance P acts on neurokinin 1 (NK1) receptors to add to the excitation. Reduced spinal inhibition through γ-aminobutyric acid (GABA) and the transporter potassium-chloride transporter member 5 (KCC2) aids enhanced pain messages. Reduced noradrenaline (NA) descending inhibition via α-2 adrenoceptors and increased 5-hydroxytryptamine (5HT) descending excitation via 5HT3 receptors add to the dominance of excitatory transmission. μ-Opioid receptors (MOR) are found on this circuitry.
Figure 2.

An overview of the ascending (left) and descending (right) pain pathways. From the spinal cord there are spinoreticular projections and dorsal column pathways to the cuneate nucleus (CN) and nucleus gracilis (NG). Other limbic projections relay in the parabrachial nucleus (PB) and then project to the hypothalamus (Hyp) and amygdala (Am), where central autonomic function and fear/anxiety are processed. Spinothalamic pathways run to the ventrobasal medial (VPM) and lateral (VPL) areas and then run to the somatosensory part of the cerebral cortex (CC) where the location and the sensory components of pain are generated. Limbic brain areas (Am and Hyp) project down to the periqueductal gray (PAG) and to the locus coereleus (LC), A5 and A7 nuclei, and the rostroventral medial medulla (RVM). Thence, descending noradrenaline (NA) inhibition via α-2 adrenoceptors and increased 5-hydroxytryptamine (5HT) descending excitation via 5HT3 receptors modulates spinal cord activity. The changes induced by peripheral neuropathy on these brain functions are depicted together with comorbidities, and genotypic and phenotypic factors are shown.
As a consequence of these changes in the sending of nociceptive information within the peripheral nerve and then the spinal cord, the information sent to the brain becomes amplified so that pain ratings become higher. Alongside this, the persistent input into the limbic brain areas such as the amygdala are likely to be causal in the comorbidities that patients often report due to ongoing painful inputs disrupting normal function and generating fear, depression, and sleep problems (Fig. 2). Of course, many patients report that their pains are worse at night, which may be due to nocturnal changes in these central pain processing areas. Finally, the neuropathic pain alters function and activity in the descending controls, which relay information from higher brain centers via the midbrain and brainstem to the spinal cord. In healthy volunteers and patients, a loss of inhibition or gain of facilitation promotes pain whereas evoking inhibition reduces pain (60). Preclinical studies suggest that the bases for this are descending noradrenergic, mostly inhibitory, and certain serotonergic controls that facilitate pain (Figs. 1 and 2). These monoamine descending controls regulate spinal neuronal activity bidirectionally and underlie the efficacy of antidepressants for the treatment of pain (60).
The increased analgesic effect of tapentadol, recently shown to be effective in animals and patients with diabetic neuropathy, likely resides in synergistic interactions between its weak opioid actions at spinal and supraspinal sites coupling to enhancing noradrenergic controls and so reducing opioid load (62). Interestingly, changes in the balance of control from the brain—the gain in descending facilitation coupled with a loss of inhibition—occur in the later stages after peripheral injury suggestive of a role in maintaining but not initiating the pain state. Furthermore, animal studies suggest that the recruitment of descending inhibitions may be protective and so can override the pain initiated by the neuropathic damage to the peripheral nerve (63).
So, overall, the mechanisms of pain in diabetic neuropathy extend from altered channel function in peripheral nerves through enhanced spinal processing and finally to changes in many higher centers (Fig. 2).
Management of painful DSPN
The assessment and treatment of painful DSPN should ideally involve a multidisciplinary team that may include a diabetologist, a neurologist, the pain clinic team, specialist nurses, podiatrists, psychologists, physiotherapists, occupational therapists, and others. However, in most clinical settings this is not possible, and the management falls mainly to the diabetes physician, the primary care physician, or the neurologist. When treatment is started, a realistic objective would be to achieve around 50% reduction in pain intensity. However, being “realistic” shouldn’t be interpreted as less aggressive pursuit of maximum pain relief. Secondary objectives should include restoration or improvement in functional measures, QoL, sleep, and mood. Although it is hoped that improvement in pain will be followed by improvement in functionality, this may not be the case as many of these patients may have other comorbidities. Moreover, the multidisciplinary team should discuss potential interventions in addition to pharmacotherapy to help patients optimize function in the presence of residual pain.
Although strong evidence implicates poor glycemic control as a pathogenetic mechanism in the etiology of DSPN, there is no proof from randomized, controlled trials that this is the case for neuropathic pain in diabetes. However, as increased blood glucose flux has been reported to contribute to pain in DSPN (38), there is a general consensus that good blood glucose control should be the first step in the management of any form of diabetic neuropathy. Additionally, as cardiovascular disease is common in patients with DSPN (10) and vascular risk factors (hypertriglyceridemia, hypertension, visceral obesity etc.) appear to be implicated in the pathogenesis of DSPN (10,64), there is a good rationale for management of vascular risk factors beyond glycemic control.
Pharmacological treatment
Several pharmacological treatments have proven efficacy in the management of painful DSPN, although only duloxetine and pregabalin are approved for the treatment of neuropathic pain in diabetes by both the Food and Drugs Administration of the U.S. and the European Medicines Agency.
Pharmacological treatment of painful DSPN is not entirely satisfactory because currently available drugs are often ineffective and complicated by adverse events. Tricyclic compounds (TCAs) have been used as first-line agents for many years, but their use is limited by frequent side effects that may be central or anticholinergic, including dry mouth, constipation, sweating, blurred vision, sedation, and orthostatic hypotension (with the risk of falls particularly in elderly patients). For this reason, low-dose amitriptyline or imipramine 10 mg taken at night may be started. Depending upon efficacy and side effects, the dose can gradually be increased to 75 mg/day and on occasions even up to 150 mg/day (1). Higher doses have been associated with an increased risk of sudden cardiac death, and caution should be taken in any patient with a history of cardiovascular disease (65).
The selective serotonin noradrenalin reuptake inhibitors (SNRI) duloxetine and venlafaxine have been used for the management of painful DSPN (65). SNRIs relieve pain by increasing synaptic availability of 5-hydroxytryptamine and noradrenalin in the descending pathways that inhibit pain impulses. The efficacy of duloxetine in painful DSPN has been investigated in three identical trials, and pooled data from these shows that the 60 mg/day and 120 mg/day doses are effective in relieving painful symptoms, starting within 1 week and lasting the full treatment period of 12 weeks (66). The main side effects include nausea, somnolence, dizziness, constipation, dry mouth, and reduced appetite, although these tend to be mild to moderate and are transient. It is advisable to start at 30 mg/day taken with food for the first week and then increase to the standard dose of 60 mg/day. Venlafaxine (150–225 mg/day) is also effective in relieving painful DSPN, although cardiovascular adverse events limit its use in diabetes (67).
The anticonvulsant gabapentin, which binds to the α2δ subunit of the calcium channel thereby reducing neurotransmitter release in the hyperexcited neuron, gradually titrated from 100 mg t.i.d. to 3,600 mg/day is also effective (68). More recently, there have been several clinical trials involving pregabalin in painful DSPN, and these showed clear efficacy in management of painful DSPN (69). Unlike gabapentin, pregabalin has linear pharmacokinetics, doesn’t require a long titration period, and is started at 75 mg b.i.d. for about a week and increased to 150 mg b.i.d. maintenance dose with a maximum dose of 600 mg/day (55). The side effects include dizziness, somnolence, peripheral edema, headache, and weight gain.
Other effective but generally considered second line drugs (65) for painful DSPN include other anticonvulsants,in particular carbamazepine (65), although it has troublesome side effects including dizziness, somnolence and gait disturbance; tramadol, a weak opioid and weak inhibitor of noradrenaline and serotonin reuptake (65); the strong opioid oxycodone controlled release (65); and topical treatments including the substance-P depleter, topical capsaicin and the lidocaine patch (65). Refractory cases of patients with painful DSPN may be treated with intravenous lignocaine (5 mg/kg over 30 min) (65). Of the pathogenetically oriented treatments for painful DSPN only the antioxidant, α-lipoic acid administered intravenously over 3 weeks (600 mg i.v. per day) has been proven to be efficacious (2,70).
Recent guidelines for pharmacological treatment
The European Federation of Neurological Societies proposed that first-line treatments might comprise of TCAs, SNRIs, gabapentin, or pregabalin (71). The U.K. National Institute for Health and Care Excellence guidelines on the management of neuropathic pain in nonspecialist settings proposed that duloxetine should be the first-line treatment with amitriptyline as an alternative, and pregabalin as a second-line treatment for painful DSPN (72). However, this recommendation of duloxetine as the first-line therapy was not based on efficacy but rather cost-effectiveness. More recently, the American Academy of Neurology recommended that pregabalin is “established as effective and should be offered for relief of [painful DSPN] (Level A evidence)” (73), whereas venlafaxine, duloxetine, amitriptyline, gabapentin, valproate, opioids, and capsaicin were considered to be “probably effective and should be considered for treatment of painful DSPN (Level B evidence)” (63). However, this recommendation was primarily based on achievement of greater than 80% completion rate of clinical trials, which in turn may be influenced by the length of the trials. Finally, the International Consensus Panel on Diabetic Neuropathy recommended TCAs, duloxetine, pregabalin, and gabapentin as first-line agents having carefully reviewed all the available literature regarding the pharmacological treatment of painful DSPN (65), the final drug choice tailored to the particular patient based on demographic profile and comorbidities.
Tailoring treatment to individual requirements
The initial selection of a particular first-line treatment will be influenced by the assessment of contraindications, evaluation of comorbidities (including sleep disturbance, mood disorders, and other chronic medical/diabetes complications), and cost (65). For example, in diabetic patients with a history of heart disease, elderly patients on other concomitant medications such diuretics and antihypertensives, and patients with comorbid orthostatic hypotension TCAs have relative contraindications. In patients with liver disease, duloxetine should not be prescribed, and in those with peripheral edema, pregabalin or gabapentin should be avoided. Moreover, although pharmaceutical companies may recommend a particular starting dose for their drugs based on their clinical trials, one has to appreciate that the clinical practice scenario is different from clinical trial scenario because many elderly patients with multiple comorbidities would have been excluded from trials. Therefore, treatment has to be individualized to take patient comorbidities including occupation, renal impairment, etc. into account, and caution is advised to start at lower than recommended doses and titrate gradually.
Comparator and combination trials
A major deficiency in the area of the treatment of neuropathic pain in diabetes is the relative lack of comparative or combination studies. Virtually all previous trials have been of active agents against placebo, whereas there is a need for more studies that compare a given drug with an active comparator and indeed lower-dose combination treatments (64). These issues have been highlighted by recent consensus guidelines from international institutions that have emphasized the need for large comparative and combination treatment trials in painful DSPN as a matter of priority (74,75).
Comparator trials.
Bansal et al. (76) compared amitriptyline with pregabalin in painful DSPN in a small, randomized, double-blind, crossover trial. This study confirmed that whereas there was little difference in efficacy, pregabalin was the preferred drug because of a superior adverse event profile. However, a major drawback of this study was its small size, involving 51 patients only with many patients failing to complete the study.
Another recent small crossover study from the same group as the above study has compared duloxetine with amitriptyline (77). The study found that both drugs were equally efficacious although of the reported adverse events, dry mouth was more common with amitriptyline than duloxetine (55 vs. 24%; P < 0.01). Numerically, more patients preferred duloxetine although this was not statistically significant (48 vs. 36%; P = 0.18).
The lack of direct comparator studies led to an indirect comparison of the efficacy and tolerability of duloxetine with that of pregabalin and gabapentin in participants with painful DSPN, using placebo as a common comparator (78). Efficacy criteria were reduction in 24-h pain severity for all three treatments, and treatment response rate (≥50% pain reduction), and overall health improvement (as measured on the Patient Global Impression of Improvement/Change questionnaire) for duloxetine and pregabalin only. Indirect comparison between duloxetine and gabapentin found no statistically significant differences. Comparing duloxetine with pregabalin, the authors found significant differences in overall health improvement, favoring pregabalin, and in dizziness, favoring duloxetine. There was no significant difference in 24-h pain severity between duloxetine and pregabalin (78).
Combination trials.
Gilron et al. (79) studied nortriptyline and gabapentin either in combination or alone in a randomized trial and confirmed that when given together, they were more efficacious than either drug given alone. In another crossover study by the same group, low-dose combination therapy with gabapentin and morphine was significantly more effective than higher doses of either (80).
COMBO-DN study.
The COMBO-DN study that has just been completed (81) is the largest combination trial in painful DSPN and assessed whether combining standard doses of duloxetine and pregabalin is superior to increasing each drug to its maximum recommended dose in patients with incomplete pain relief. Patients with painful DSPN with a daily pain score of at least 4 (scale 0–10) were randomly assigned in a 1:1:1:1 ratio to one of four groups. For the 8-week initial treatment period, patients in groups 1 and 2 were treated with 60 mg duloxetine/day; patients in groups 3 and 4 received 300 mg pregabalin/day. Thereafter, only nonresponders (<30% improvement in pain relief) received double-blind treatment for further 8 weeks of the combination versus high-dose monotherapy treatment period with duloxetine 120 mg/day for group 1, duloxetine 60 mg/day + pregabalin 300 mg/day for groups 2 and 3, and pregabalin 600 mg/day for group 4. The primary outcome was change in the 24-h average pain (an item from BPI-MSF) after combination versus high-dose monotherapy period (groups 1, 4 pooled versus groups 2, 3 pooled).
Eight-hundred and four patients were evaluated in the initial period and 339 in the combination versus high-dose monotherapy treatment period, respectively. The difference between combination and monotherapy in the mean change of BPI-MSF average pain during combination versus high-dose monotherapy treatment period was not statistically significant (combination, −2.35; monotherapy, −2.16; P = 0.37) (81). Proportions of patients with treatment emergent adverse events were however similar: 36.7% (Combination) and 33.5% (Monotherapy). As a secondary end point the COMBO-DN study also compared the efficacy of standard doses of duloxetine and pregabalin as initial treatment for painful DSPN, and duloxetine was found to have superior efficacy compared with pregabalin, without any safety findings of concern. At the end of the combination versus high-dose monotherapy treatment period, although the groups are no longer randomized, 50% pain relief was found in 46.9% of subjects on 600 mg/day pregabalin compared with 28.4% on 120 mg/day of duloxetine.
Taken together, even though the primary end point was not met, the COMBO-DN study demonstrated that at standard doses duloxetine has better efficacy than pregabalin as an initial treatment for painful DSPN, without any safety findings of concern. However, pregabalin catches up with duloxetine in terms of efficacy as the doses are increased to maximum.
Conclusion
A simple algorithm was suggested by the Toronto International Neuropathy Consensus meeting (Fig. 3) to help practitioners in the management of patients with painful DSPN (2,65). After exclusion of other causes and optimization of glycemic control, first-line therapies would include either an antidepressant (a tricyclic or duloxetine) or an anticonvulsant (gabapentin or pregabalin). These therapies all have level A evidence for efficacy and a clear pathway of progression if initial therapies fail is provided. Those patients with the severest neuropathic pain unresponsiveness to antidepressant or anticonvulsant therapy might require short-term treatment with opioid or opioid-like drugs such as tramadol or controlled release oxycodone. Finally, it must always be emphasized that all patients with any form of diabetic neuropathy are at increased risk of foot ulceration and require education in self foot care and, if possible, regular podiatry assessment and treatment.
Figure 3.

Treatment algorithm for painful DSPN. Adapted with permission from Tesfaye et al. (65).
Acknowledgments
S.T. has received honoraria for invited lectures from Eli Lilly and Pfizer. A.J.M.B. has received honoraria from Eli Lilly and Pfizer and has served on an advisory board for Pamlab. A.H.D. is a speaker and is on the advisory board panels for Astellas, Grunenthal, and Pfizer and Lilly. A.H.D. is supported by the Wellcome Trust London Pain Consortium. No other potential conflicts of interest relevant to this article were reported.
The authors gratefully acknowledge the production of figures by Dr. Shafaq Sikandar.
References
- 1.Boulton AJM, Malik RA, Arezzo JC, Sosenko JM. Diabetic somatic neuropathies. Diabetes Care 2004;27:1458–1486 [DOI] [PubMed] [Google Scholar]
- 2.Tesfaye S, Boulton AJM, Dyck PJ, et al. Toronto Diabetic Neuropathy Expert Group Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010;33:2285–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zelman DC, Brandenburg NA, Gore M. Sleep impairment in patients with painful diabetic peripheral neuropathy. Clin J Pain 2006;22:681–685 [DOI] [PubMed] [Google Scholar]
- 4.Gore M, Brandenburg NA, Dukes E, Hoffman DL, Tai KS, Stacey B. Pain severity in diabetic peripheral neuropathy is associated with patient functioning, symptom levels of anxiety and depression, and sleep. J Pain Symptom Manage 2005;30:374–385 [DOI] [PubMed] [Google Scholar]
- 5.Boulton AJM, Armstrong WD, Scarpello JHB, Ward JD. The natural history of painful diabetic neuropathy—a 4-year study. Postgrad Med J 1983;59:556–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benbow SJ, Chan AW, Bowsher D, MacFarlane IA, Williams G. A prospective study of painful symptoms, small-fibre function and peripheral vascular disease in chronic painful diabetic neuropathy. Diabet Med 1994;11:17–21 [DOI] [PubMed] [Google Scholar]
- 7.Abbott CA, Malik RA, van Ross ER, Kulkarni J, Boulton AJ. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 2011;34:2220–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daousi C, McFarlane IA, Woodward A, Nurmikko TJ, Bendred PE, Benbow SJ. Chronic painful peripheral neuropathy in an urban community: a control comparison of people with and without diabetes. Diabet Med 2004;21:976–982 [DOI] [PubMed] [Google Scholar]
- 9.Davies M, Brophy S, Williams R, Taylor A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care 2006;29:1518–1522 [DOI] [PubMed] [Google Scholar]
- 10.Tesfaye S, Chaturvedi N, Eaton SEM, et al. EURODIAB Prospective Complications Study Group Vascular risk factors and diabetic neuropathy. N Engl J Med 2005;352:341–350 [DOI] [PubMed] [Google Scholar]
- 11.Ziegler D, Rathmann W, Dickhaus T, Meisinger C, Mielck A, KORA Study Group Neuropathic pain in diabetes, prediabetes and normal glucose tolerance: the MONICA/KORA Augsburg Surveys S2 and S3. Pain Med 2009;10:393–400 [DOI] [PubMed] [Google Scholar]
- 12.Van Acker K, Bouhassira D, De Bacquer D, et al. Prevalence and impact on quality of life of peripheral neuropathy with or without neuropathic pain in type 1 and type 2 diabetic patients attending hospital outpatients clinics. Diabetes Metab 2009;35:206–213 [DOI] [PubMed] [Google Scholar]
- 13.Ziegler D, Rathmann W, Meisinger C, Dickhaus T, Mielck A, KORA Study Group Prevalence and risk factors of neuropathic pain in survivors of myocardial infarction with pre-diabetes and diabetes. The KORA Myocardial Infarction Registry. Eur J Pain 2009;13:582–587 [DOI] [PubMed] [Google Scholar]
- 14.Boulton AJ, Vinik AI, Arezzo JC, et al. American Diabetes Association Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005;28:956–962 [DOI] [PubMed] [Google Scholar]
- 15.Vinik AI, Erbas T, Park TS, Pierce KK, Stansberry KB. Methods for evaluation of peripheral neurovascular dysfunction. Diabetes Technol Ther 2001;3:29–50 [DOI] [PubMed] [Google Scholar]
- 16.Malik R, Veves A, Tesfaye S, et al. on behalf of the Toronto Consensus Panel on Diabetic Neuropathy Small fiber neuropathy: role in the diagnosis of diabetic sensorimotor polyneuropathy. Diabetes Metab Res Rev 2011;27:678–684 [DOI] [PubMed] [Google Scholar]
- 17.Rolke R, Baron R, Maier C, et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain 2006;123:231–243 [DOI] [PubMed] [Google Scholar]
- 18.Ragé M, Van Acker N, Knaapen MW, et al. Asymptomatic small fiber neuropathy in diabetes mellitus: investigations with intraepidermal nerve fiber density, quantitative sensory testing and laser-evoked potentials. J Neurol 2011;258:1852–1864 [DOI] [PubMed] [Google Scholar]
- 19.Chao C-C, Tseng MT, Lin YJ, et al. Pathophysiology of neuropathic pain in type 2 diabetes: skin denervation and contact heat-evoked potentials. Diabetes Care 2010;33:2654–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young MJ, Boulton AJM, MacLeod AF, Williams DR, Sonksen PH. A multicentre study of the prevalence of diabetic peripheral neuropathy in the United Kingdom hospital clinic population. Diabetologia 1993;36:150–154 [DOI] [PubMed] [Google Scholar]
- 21.Bril V, Perkins BA. Validation of the Toronto Clinical Scoring System for diabetic polyneuropathy. Diabetes Care 2002;25:2048–2052 [DOI] [PubMed] [Google Scholar]
- 22.Feldman EL, Stevens MJ, Thomas PK, Brown MB, Canal N, Greene DA. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes Care 1994;17:1281–1289 [DOI] [PubMed] [Google Scholar]
- 23.Dyck PJ. Detection, characterization, and staging of polyneuropathy: assessed in diabetics. Muscle Nerve 1988;11:21–32 [DOI] [PubMed] [Google Scholar]
- 24.Dyck PJ, Overland CJ, Low PA, et al. Signs and symptoms versus nerve conduction studies to diagnose diabetic sensorimotor polyneuropathy: Cl vs. NPhys trial. Muscle Nerve 2010;42:157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruccu G, Sommer C, Anand P, et al. EFNS guidelines on neuropathic pain assessment: revised 2009. Eur J Neurol 2010;17:1010–1018 [DOI] [PubMed] [Google Scholar]
- 26.Zelman DC, Gore M, Dukes E, Tai KS, Brandenburg N. Validation of a modified version of the brief pain inventory for painful diabetic peripheral neuropathy. J Pain Symptom Manage 2005;29:401–410 [DOI] [PubMed] [Google Scholar]
- 27.Bouhassira D, Attal N, Fermanian J, et al. Development and validation of the Neuropathic Pain Symptom Inventory. Pain 2004;108:248–257 [DOI] [PubMed] [Google Scholar]
- 28.Backonja MM, Krause SJ. Neuropathic pain questionnaire—short form. Clin J Pain 2003;19:315–316 [DOI] [PubMed] [Google Scholar]
- 29.Bennett M. The LANSS Pain Scale: the Leeds assessment of neuropathic symptoms and signs. Pain 2001;92:147–157 [DOI] [PubMed] [Google Scholar]
- 30.Freynhagen R, Baron R, Gockel U, Tölle TR. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin 2006;22:1911–1920 [DOI] [PubMed] [Google Scholar]
- 31.Melzack R. The short-form McGill pain questionnaire. Pain 1987;30:191–197 [DOI] [PubMed] [Google Scholar]
- 32.Vileikyte L, Peyrot M, Bundy C, et al. The development and validation of a neuropathy- and foot ulcer-specific quality of life instrument. Diabetes Care 2003;26:2549–2555 [DOI] [PubMed] [Google Scholar]
- 33.Vinik EJ, Hayes RP, Oglesby A, et al. The development and validation of the Norfolk QOL-DN, a new measure of patients’ perception of the effects of diabetes and diabetic neuropathy. Diabetes Technol Ther 2005;7:497–508 [DOI] [PubMed] [Google Scholar]
- 34.Vinik A. The approach to the management of the patient with neuropathic pain. J Clin Endocrinol Metab 2010;95:4802–4811 [DOI] [PubMed] [Google Scholar]
- 35.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361–370 [DOI] [PubMed] [Google Scholar]
- 36.Beck AT, Steer RA, Brown GK. Manual for the Beck Depression Inventory-II San Antonio, TX, Psychological Corporation, 1996, p. 38 [Google Scholar]
- 37.Tesfaye S, Kempler P. Painful diabetic neuropathy. Diabetologia 2005;48:805–807 [DOI] [PubMed] [Google Scholar]
- 38.Oyibo SO, Prasad YDM, Jackson NJ, Jude EB, Boulton AJ. The relationship between blood glucose excursions and painful diabetic peripheral neuropathy: a pilot study. Diabet Med 2002;19:870–873 [DOI] [PubMed] [Google Scholar]
- 39.Eaton SE, Harris ND, Ibrahim S, et al. Increased sural nerve epineurial blood flow in human subjects with painful diabetic neuropathy. Diabetologia 2003;46:934–939 [DOI] [PubMed] [Google Scholar]
- 40.Quattrini C, Harris ND, Malik RA, Tesfaye S. Impaired skin microvascular reactivity in painful diabetic neuropathy. Diabetes Care 2007;30:655–659 [DOI] [PubMed] [Google Scholar]
- 41.Sorensen L, Molyneaux L, Yue DK. The relationship among pain, sensory loss, and small nerve fibers in diabetes. Diabetes Care 2006;29:883–887 [DOI] [PubMed] [Google Scholar]
- 42.Selvarajah D, Wilkinson ID, Gandhi R, Griffiths PD, Tesfaye S. Microvascular perfusion abnormalities of the Thalamus in painful but not painless diabetic polyneuropathy: a clue to the pathogenesis of pain in type 1 diabetes. Diabetes Care 2011;34:718–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gandhi RA, Marques JLB, Selvarajah D, Emery CJ, Tesfaye S. Painful diabetic neuropathy is associated with greater autonomic dysfunction than painless diabetic neuropathy. Diabetes Care 2010;33:1585–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maier C, Baron R, Tölle TR, et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain 2010;150:439–450 [DOI] [PubMed] [Google Scholar]
- 45.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 2007;76:387–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997;389:816–824 [DOI] [PubMed] [Google Scholar]
- 47.Colburn RW, Lubin ML, Stone DJ, Jr, et al. Attenuated cold sensitivity in TRPM8 null mice. Neuron 2007;54:379–386 [DOI] [PubMed] [Google Scholar]
- 48.Binder A, May D, Baron R, et al. Transient receptor potential channel polymorphisms are associated with the somatosensory function in neuropathic pain patients. PLoS One 2011; 6:e17387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honore P, Kage K, Mikusa J, et al. Analgesic profile of intrathecal P2X(3) antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain 2002;99:11–19 [DOI] [PubMed] [Google Scholar]
- 50.Dickenson AH, Matthews EA, Suzuki R. Neurobiology of neuropathic pain: mode of action of anticonvulsants. Eur J Pain 2002;6(Suppl. A):51–60 [DOI] [PubMed] [Google Scholar]
- 51.Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet 2004;41:171–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006;444:894–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reimann F, Cox JJ, Belfer I, et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proc Natl Acad Sci USA 2010;107:5148–5153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harvey VL, Dickenson AH. Mechanisms of pain in nonmalignant disease. Curr Opin Support Palliat Care 2008;2:133–139 [DOI] [PubMed] [Google Scholar]
- 55.Bierhaus A, Fleming T, Stoyanov S, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med 2012;18:926–933 [DOI] [PubMed] [Google Scholar]
- 56.Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol 2003;460:109–116 [DOI] [PubMed] [Google Scholar]
- 57.Passmore GM, Selyanko AA, Mistry M, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 2003;23:7227–7236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harris JA, Murphy JA. Retigabine (ezogabine) as add-on therapy for partial-onset seizures: an update for clinicians. Ther Adv Chronic Dis 2011;2:371–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bauer CS, Nieto-Rostro M, Rahman W, et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci 2009;29:4076–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bannister K, Bee LA, Dickenson AH. Preclinical and early clinical investigations related to monoaminergic pain modulation. Neurotherapeutics 2009;6:703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D’Mello R, Dickenson AH. Spinal cord mechanisms of pain. Br J Anaesth 2008;101:8–16 [DOI] [PubMed] [Google Scholar]
- 62.Bee LA, Bannister K, Rahman W, Dickenson AH. Mu-opioid and noradrenergic α(2)-adrenoceptor contributions to the effects of tapentadol on spinal electrophysiological measures of nociception in nerve-injured rats. Pain 2011;152:131–139 [DOI] [PubMed] [Google Scholar]
- 63.De Felice M, Sanoja R, Wang R, et al. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain 2011;152:2701–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ziegler D. Current concepts in the management of diabetic polyneuropathy. Curr Diabetes Rev 2011;7:208–220 [DOI] [PubMed] [Google Scholar]
- 65.Tesfaye S, Vileikyte L, Rayman G, et al. Toronto Expert Panel on Diabetic Neuropathy Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metab Res Rev 2011;27:629–638 [DOI] [PubMed] [Google Scholar]
- 66.Kajdasz DK, Iyengar S, Desaiah D, et al. Duloxetine for the management of diabetic peripheral neuropathic pain: evidence-based findings from post hoc analysis of three multicenter, randomized, double-blind, placebo-controlled, parallel-group studies. Clin Ther 2007;29(Suppl.):2536–2546 [DOI] [PubMed] [Google Scholar]
- 67.Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain 2004;110:697–706 [DOI] [PubMed] [Google Scholar]
- 68.Backonja MM, Beydoun A, Edwards KR, et al. Gabapentin for the symptomatic treatment of painful neuropathy in patients with diabetes mellitus: a randomized controlled trial. JAMA 1998;280:1831–1836 [DOI] [PubMed] [Google Scholar]
- 69.Freeman R, Durso-Decruz E, Emir B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes Care 2008;31:1448–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ziegler D, Nowak H, Kempler P, Vargha P, Low PA. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a meta-analysis. Diabet Med 2004;21:114–121 [DOI] [PubMed] [Google Scholar]
- 71.Attal N, Cruccu G, Baron R, et al. European Federation of Neurological Societies EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 2010;17:1113–e88 [DOI] [PubMed] [Google Scholar]
- 72.NICE Clinical Guideline 96: Neuropathic Pain. The pharmacological management of neuropathic pain in adults in non-specialist settings. March 2010. Available from www.nice.org.uk [PubMed]
- 73.Bril V, England J, Franklin GM, et al. American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation Evidence-based guideline: Treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology 2011;76:1758–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vorobeychik Y, Gordin V, Mao J, Chen L. Combination therapy for neuropathic pain: a review of current evidence. CNS Drugs 2011;25:1023–1034 [DOI] [PubMed] [Google Scholar]
- 75.O’Connor AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. Am J Med 2009;122(Suppl.):S22–S32 [DOI] [PubMed] [Google Scholar]
- 76.Bansal D, Bhansali A, Hota D, Chakrabarti A, Dutta P. Amitriptyline vs. pregabalin in painful diabetic neuropathy: a randomized double blind clinical trial. Diabet Med 2009;26:1019–1026 [DOI] [PubMed] [Google Scholar]
- 77.Kaur H, Hota D, Bhansali A, Dutta P, Bansal D, Chakrabarti A. A comparative evaluation of amitriptyline and duloxetine in painful diabetic neuropathy: a randomized, double-blind, cross-over clinical trial. Diabetes Care 2011;34:818–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quilici S, Chancellor J, Löthgren M, et al. Meta-analysis of duloxetine vs. pregabalin and gabapentin in the treatment of diabetic peripheral neuropathic pain. BMC Neurol 2009;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gilron I, Bailey JM, Tu D, Holden RR, Jackson AC, Houlden RL. Nortriptyline and gabapentin, alone and in combination for neuropathic pain: a double-blind, randomised controlled crossover trial. Lancet 2009;374:1252–1261 [DOI] [PubMed] [Google Scholar]
- 80.Gilron I, Bailey JM, Tu D, Holden RR, Weaver DF, Houlden RL. Morphine, gabapentin, or their combination for neuropathic pain. N Engl J Med 2005;352:1324–1334 [DOI] [PubMed] [Google Scholar]
- 81.Tesfaye S, Wilhelm S, Lledo A, et al. Duloxetine and pregabalin: high-dose monotherapy or their combination? The “COMBO-DN study” - a multinational, randomized, double-blind, parallel-group study in patients with diabetic peripheral neuropathic pain. Pain. 31 May 2013 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]