

Two fast energy calibration procedures have been established: (i) an in situ calibration tracks the energies scan-by-scan for observing tiny spectral shifts; (ii) a quick-switching calibration reduces the calibration time dramatically and is suitable for normal NRVS measurements.
Keywords: nuclear resonant vibrational spectroscopy, high-resolution monochromator, in situ energy calibration, quick-switching energy calibration, small energy shift, energy scale, energy position
Abstract
The conventional energy calibration for nuclear resonant vibrational spectroscopy (NRVS) is usually long. Meanwhile, taking NRVS samples out of the cryostat increases the chance of sample damage, which makes it impossible to carry out an energy calibration during one NRVS measurement. In this study, by manipulating the 14.4 keV beam through the main measurement chamber without moving out the NRVS sample, two alternative calibration procedures have been proposed and established: (i) an in situ calibration procedure, which measures the main NRVS sample at stage A and the calibration sample at stage B simultaneously, and calibrates the energies for observing extremely small spectral shifts; for example, the 0.3 meV energy shift between the 100%-57Fe-enriched [Fe4S4Cl4]= and 10%-57Fe and 90%-54Fe labeled [Fe4S4Cl4]= has been well resolved; (ii) a quick-switching energy calibration procedure, which reduces each calibration time from 3–4 h to about 30 min. Although the quick-switching calibration is not in situ, it is suitable for normal NRVS measurements.
1. Introduction
Nuclear resonant vibrational spectroscopy (NRVS) scans an extremely monochromatic (∼1 meV) X-ray beam through the nuclear resonance (e.g. 57Fe at 14.4 keV) and measures the corresponding creation or annihilation of phonons (Seto et al., 1995 ▶, 2009 ▶; Sturhahn et al., 1995 ▶; Yoda et al., 2001 ▶, 2012 ▶). It is a relatively new X-ray spectroscopy that became available because of the development of third-generation synchrotron sources, insertion devices and advanced X-ray optics. It has several distinguished advantages in comparison with traditional infrared (IR) and resonant Raman (RR) spectroscopies (Smith et al., 2005 ▶) and has become an excellent pin-point tool in recent years for studying iron-specific inorganic and bioinorganic chemistry. For example, it revealed and resolved Fe-S/P/Cl and Fe-CO/CN/NO vibrational modes inside various chemical or enzymatic molecules (Smith et al., 2005 ▶; Xiao et al., 2005 ▶, 2006 ▶; Tinberg et al., 2010 ▶; Do et al., 2011 ▶; Kamali et al., 2013 ▶).
The energy of the high-resolution monochromator (HRM) used for NRVS can be scanned (or altered involuntarily) by changing: (i) the relative scattering angles among the HRM crystals; and/or (ii) the temperature of one back-reflection crystal (Zhao & Sturhahn, 2012 ▶). The accuracy for the energy scale therefore relies on the high-precision measurements (and controls) on the angle, the temperature, as well as the thermal gradients for the HRM crystals. The error is usually about a few percent of the HRM energy. For higher accuracy, additional calibration is required (Zhao & Sturhahn, 2012 ▶). NRVS, especially on biological samples, has a very weak signal level and needs about 24–48 h (Kamali et al., 2013 ▶) to obtain a quality spectrum for a dilute sample. During such a long period, the energy position/scale becomes a particular issue for obtaining good statistics for the targeted weak spectral peak(s), and should be frequently calibrated. In addition, a careful and scan-by-scan calibration is absolutely necessary for observing small shifts between different NRVS spectra.
A conventional NRVS calibration is performed by measuring a standard calibration sample (CS) in the main NRVS chamber. The procedure includes: (i) warming up the cryostat base to 140–200 K (Wang et al., 2012 ▶); (ii) taking the existing NRVS sample out of the chamber (cryostat); (iii) loading in the CS; (iv) cooling down the CS (e.g. to 10 K; sensor reading); and (v) measuring the CS. In practice, the first two NRVS spectra have slightly irreproducible peak positions, which could be due to the gradual transition of the real sample temperature (Wang et al., 2012 ▶). The overall procedure usually takes about 3–4 h. This time-consuming nature makes a frequent energy calibration impractical.
Meanwhile, samples under X-ray irradiation could have stored energy inside the molecule. The ‘radiation damage’ may be ‘frozen’ under cryogenic temperatures (LT) (Garman & Nave, 2009 ▶). Taking the samples off the cryostat base means warming up the sample and increasing the chance of radiation damage, which should be avoided if possible. This makes it impossible to perform an energy calibration during one NRVS measurement, which could be as long as 48 h for a dilute sample [e.g. on NiFe hydrogenase (Kamali et al., 2013 ▶)]. Thus, alternative calibration procedures without moving the NRVS sample out of the cryostat must be explored for biological NRVS measurements.
In a recent publication, Zhao & Sturhahn (2012 ▶) reported a successful procedure to calibrate HRM energy using the detailed-balance between the phonon annihilation and the phonon creation in an iron foil (or in any samples), with minor changes in the NRVS measurement set-up. This is a sample-independent method and does not require a specified standard sample. It also provides an accurate energy ruler (including the higher-order corrections) for an interested HRM. However, their procedure takes about 4 h (Zhao & Sturhahn, 2012 ▶), which still limits its application as a frequent energy checkup during regular NRVS measurements.
In this study, by manipulating the 14.4 keV beam through the main measurement chamber [Figs. 1(a) and 1(b) ▶] without moving the main sample out of the cryostat, two alternative calibration procedures have been tested and developed: (i) an in situ calibration procedure, which measures the main NRVS sample and the calibration sample simultaneously; (ii) a quick-switching energy calibration procedure, which reduces each calibration time dramatically and makes a frequent energy calibration possible.
Figure 1.

(a) Schematic illustration of SPring-8 BL09XU beamline with its main measurement chamber at cryogenic temperature at stage A and the calibration set-up at room temperature at stage B. (b) Photograph of the NRVS measurement hutch at BL09XU, showing the stages A and B. The thick yellow arrow indicates the E = 14.4 keV and ΔE ≃ 1 meV X-ray beam. (c) Schematic and cross-section view (against the beam direction) of the main measurement chamber at stage A. The red bar represents the beam position relative to the NRVS sample for a Z scan (c 1, and c 2 with an expanded view) and an X scan (c 3).
2. Experiment details
2.1. NRVS measurements at stage A
The NRVS beamline at SPring-8, BL09XU, has been described in detail elsewhere (Seto et al., 2009 ▶; Yoda et al., 2012 ▶) and is re-illustrated in Fig. 1(a) ▶. The synchrotron-radiation-produced X-rays were first converted into a beam with E = 14.4 keV and ΔE = 1 eV by a high-heat-load monochromator; it was then further converted into a beam with E = 14.4 keV and ΔE ≃ 1 meV by a HRM. The photon flux at 14.4 keV was then ∼1.4 × 109 photons s−1 with a 0.8 meV energy bandwidth, or ∼2.5 × 109 photons s−1 with a 1.1 meV energy resolution. The beam size was about 0.6 mm (height) × 1 mm (width). The stage A is for NRVS measurements and the stage B is for energy calibration measurements. The measurement hutch for stage A and stage B is shown in Fig. 1(b) ▶.
At stage A, NRVS spectra were recorded using a standard experimental procedure on a standard set-up with a liquid-helium (LHe) flow cryostat (Smith et al., 2005 ▶; Xiao et al., 2005 ▶). The spectra were generally measured from −30 meV to 70–100 meV, depending on the sample. Delayed nuclear fluorescence and Fe Kα fluorescence were recorded with a 2 × 2 avalanche photodiode (APD) detector array and processed with data-acquisition electronics. The maximum resonant peaks were different from sample to sample (e.g. 100–2000 counts s−1). NRVS spectral analysis was performed using the PHOENIX software package (Sturhahn et al., 1995 ▶), where the observed raw NRVS spectra were calibrated (to the nuclear resonant peak), normalized (to the I 0 variation), summed and converted to the 57Fe partial vibrational density of states (PVDOS). Either NRVS or PVDOS in the following text actually means a PVDOS spectrum unless it is specified with raw NRVS.
2.2. Passing beam through stage A
At stage A, there is a ∼1 mm space between the sample’s top surface and the main chamber’s beryllium window (Wang et al., 2012 ▶), as illustrated in Figs. 1(c 1) and 1(c 2) ▶. Part of the 14.4 keV beam can pass through the sample at stage A and part of it can pass through the ∼1 mm space to reach the sample at stage B. The simultaneous NRVS measurements on samples at A and B constitute an in situ calibration.
Figs. 2(a) and 2(b) ▶ exhibit the signal levels (counts s−1) for Fe NRVS measured at B under several circumstances: (i) there is about a 2.7 mm space for the beam passage when an empty cryostat base is in the main chamber at stage A (black curve, between positions Z a and Z d); (ii) there is about a 1.8 mm space with an empty Lucite sample holder (blue, between Z b and Z d); (iii) the space is reduced to ∼0.4 mm and the signal level drops to ∼50% with a real [Fe4S4Cl4]= sample at A (red, between Z c and Z d). These sizes are in general consistent with the real structural sizes. The positions Z a, Z b, Z c and Z d are the same as in Fig. 1(c 2) ▶. The green curve shows the NRVS signal of [Fe4S4Cl4]= at A.
Figure 2.
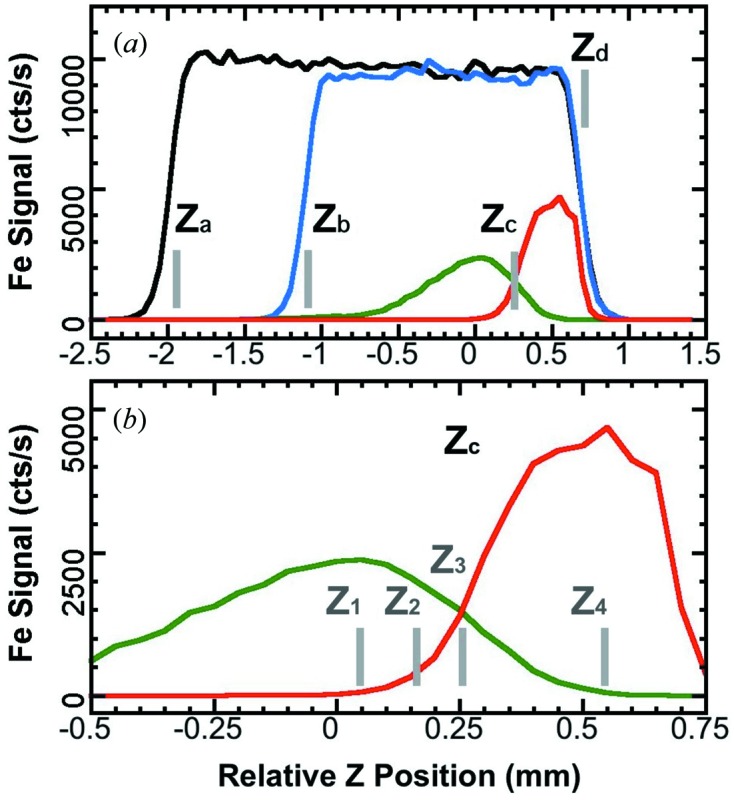
(a) Fe NRVS signal at B (black, blue and red) versus the relative beam positions (Z): the black curve is for an empty cryostat base; the blue is for an empty sample holder; the red is for a real sample ([Fe4S4Cl4]=) at A. Letters Z a, Z b, Z c and Z d indicate the cut-off at the cryostat base, sample holder’s inner bottom, sample’s top surface and vacuum chamber’s top window, respectively. (b) Blow-up of (a). The bars Z 1, Z 2, Z 3 and Z 4 are several possible choices for an in situ calibration. The green line represents the [Fe4S4Cl4]= NRVS signal at A.
There are also spaces on the left-hand and right-hand sides of the main sample at A, as illustrated in Fig. 1(c
3) ▶ (positions 1 or 3). These spaces can also be utilized to allow the X-ray beam pass. In our practice, the best result was obtained by moving the stage A to the right for X = 4 mm [Fig. 1(c
3) ▶, position 1]. Stage A’s height (Z) and tilting angle (θ) also need minor optimization for a complete beam passage. The positions for the NRVS measurement (at A) and for the energy calibration (at B) can be switched back and forth [(X
1, Z
1, θ1)  (X
2, Z
2, θ2)] in 1 or 2 mins rather than in 1 h for a sample change process. Therefore the second procedure is called the quick-switching calibration procedure.
(X
2, Z
2, θ2)] in 1 or 2 mins rather than in 1 h for a sample change process. Therefore the second procedure is called the quick-switching calibration procedure.
2.3. NRVS measurements at stage B
At approximately 1 m behind stage A, stage B was set up to use the beam passing through A [Figs. 1(a) and 1(b) ▶] for energy calibration measurements. The NRVS measurement procedure at B is the same as at A, but with a single-element APD. Powder 57Fe or 57Fe-enriched Fe2O3 was used as the calibration samples for our new calibration measurements. The calibration measurements at B were performed at room temperature (RT).
2.4. Samples
Powder sample [Et4N][57FeCl4] ([FeCl4]− for short) (Smith et al., 2005 ▶) was synthesized at the University of Michigan and was used as the standard sample in a conventional energy calibration procedure at stage A (Smith et al., 2005 ▶; Xiao et al., 2005 ▶, 2006 ▶). Powders of 57Fe or 57Fe labeled Fe2O3 were purchased from Isoflex (San Francisco, CA, USA), and were used for our new calibration procedures at stage B. For associated experiments, 57Fe(edt)(CO)2(PMe3)2 (Guo et al., 2008 ▶) was synthesized at the University of Illinois at Urbana Champaign; (NH4)2Mg57FeII(CN)6 {or [MgFe(CN)6]=} (Chumakov & Rüffer, 1998 ▶) was measured at ESRF BL18 and cited here. All the above samples are 57Fe labeled. Natural abundance KGdFeII(CN)6 and KEuFeII(CN)6 {or [EuFe(CN)6]− and [GdFe(CN)6]−} (Shokouhimehr et al., 2010 ▶; Huang et al., 2010 ▶) were synthesized at Kent State University. The 100%-57Fe and 10%-57Fe/90%-54Fe labeled [Fe4S4Cl4](Ph4P)2 samples ([Fe4S4Cl4]= and [XFe4S4Cl4]= for short in the following text) were prepared at the University of California at Davis.
Details for non-calibration samples measured at stage A are omitted.
3. Results and discussions
3.1. A conventional calibration
Omitting the higher-order corrections (Zhao & Sturhahn, 2012 ▶), the real energy position can be calculated with E real = (K E obs+ΔE), where E real stands for the real energy, E obs stands for the observed energy, ΔE is the energy position drift and K is the energy scale. The energy calibration thus includes: (i) aligning the nuclear resonant peak to E = 0 to correct for ΔE; (ii) rescaling the energy axis to obtain the correct K value. Part (i) is in general obtained automatically during the PHOENIX analyses. At BL09XU, the scanning software also includes a procedure to automatically re-align the E = 0 position prior to each scan. For (ii), K is calculated by comparing the observed and the published [FeCl4]− NRVS spectrum (Smith et al., 2005 ▶). The published standard spectrum has the same peaks in its IR spectrum (Smith et al., 2005 ▶). The above procedure is outlined figuratively in Fig. S1(a) and S1(b) in the supplementary information, while a series of [FeCl4]− calibrated K values are also illustrated in Fig. S2.1 The obtained K values appear to be between 0.938 and 0.976, a span of about 4%, which is not unexpected for a HRM (Zhao & Sturhahn, 2012 ▶).
As a conventional calibration takes time, the practical calibration interval was about 72 h. For more frequent energy calibrations, alternative procedures have to be developed.
3.2. An in situ calibration
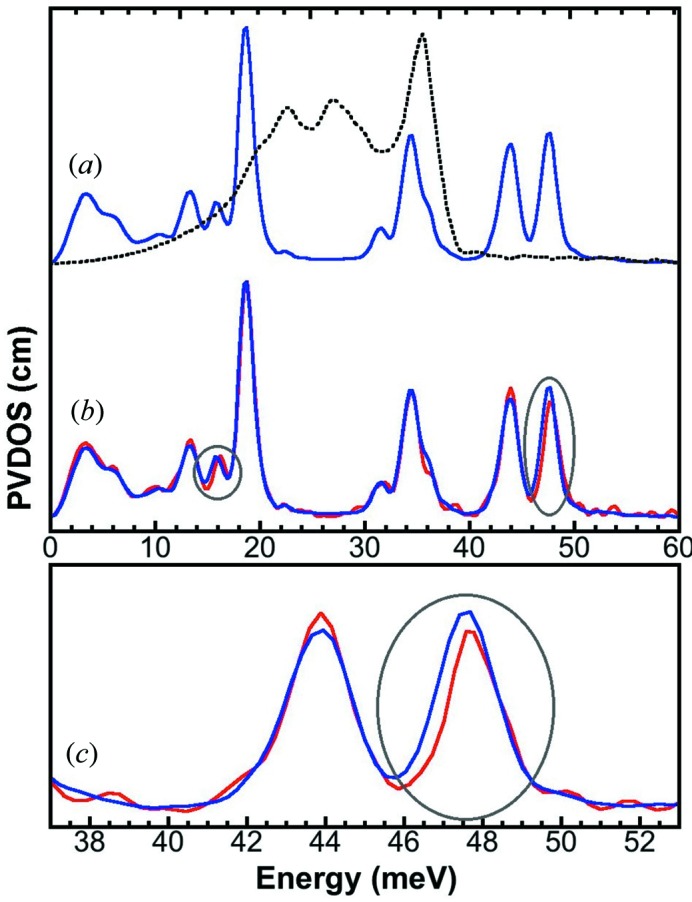
There are two types of energy calibration procedures in general: (i) scan-by-scan calibrations and (ii) several calibrations during one beam time. An in situ calibration is a scan-by-scan calibration to track well the energy position/scale. As shown in Fig. 3(a) ▶, it seems impossible to distinguish the spectra with a 0.1% difference in their energy scale. On the other hand, it is clear to distinguish the spectra with a 0.3% difference (Fig. 3b ▶). Although it is not rigid, 0.2–0.3% could be used as a practical guide for a clear identification of a spectral shift. The expanded spectra in Figs. 3(c) and 3(d) ▶ provide a better illustration of this practical limit.
Figure 3.
Calibrated NRVS of Fe (red) and [FeCl4]− (orange) versus their copies with 99.9% (a) and 99.7% (b) scaled energy axis (blue and green). There is no observable difference between the original and scaled NRVS in (a), while there is a clear difference between the two spectra in (b). (c) and (d) are the expanded versions of (a) and (b), respectively.
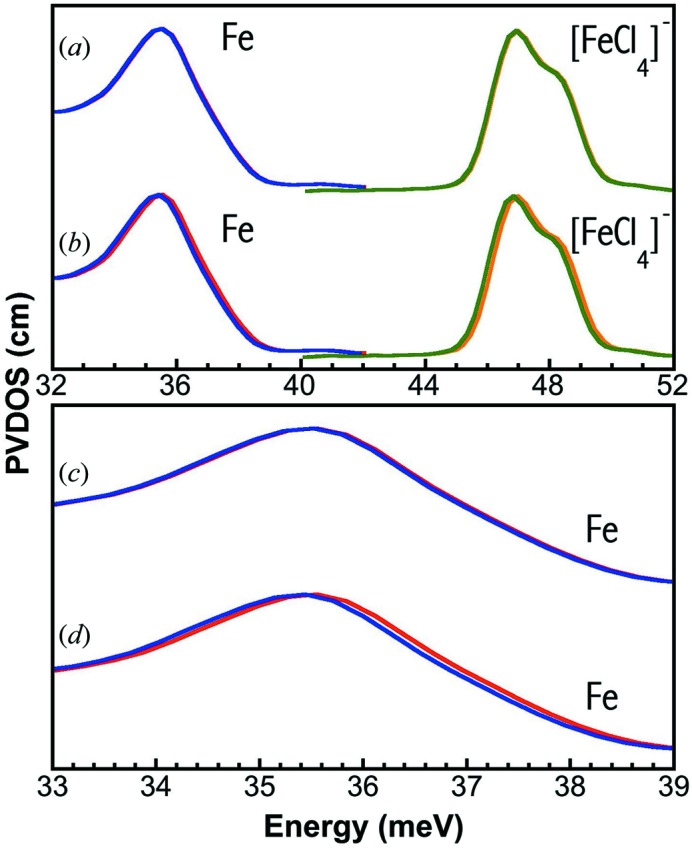
The first task for our in situ calibration is to establish the correlation between the new (Fe or Fe2O3) and the old ([FeCl4]−) standard spectra. The results are shown in Figs. 4(a) and 4(b) ▶. A sample selected for the calibrations at stage B needs to have several properties: (i) it must be chemically stable in air at RT; (ii) its NRVS or PVDOS spectrum at RT needs to have sharp feature(s); and (iii) it must have a high signal level (counts s−1). Powder Fe has a prominent peak at 35.6 meV in both raw NRVS and converted PVDOS. Powder Fe2O3 has multiple peaks, including ones at 35.7, 41.0, 46.9, 50.1 meV, in its PVDOS. Under current experimental conditions, Fe has about 10000 counts s−1 while Fe2O3 has about 15000 counts s−1. The higher counts s−1 for Fe2O3 is probably due to the wider absorption width (the larger magnetic splitting in the case of thick samples). Therefore both samples are good for the new calibration procedures. At RT, [FeCl4]− does not have a sharp peak, and it has only 800 counts s−1.
Figure 4.
PVDOS measured at stages A and B simultaneously: (a) observed [FeCl4]− at A (×0.5, dashed black) versus observed Fe at B (solid red); (b) observed [FeCl4]− at A (×0.5, dashed black) versus observed Fe2O3 at B (×2, solid blue). The solid purple curve is the standard PVDOS for [FeCl4]− (Smith et al., 2005 ▶).
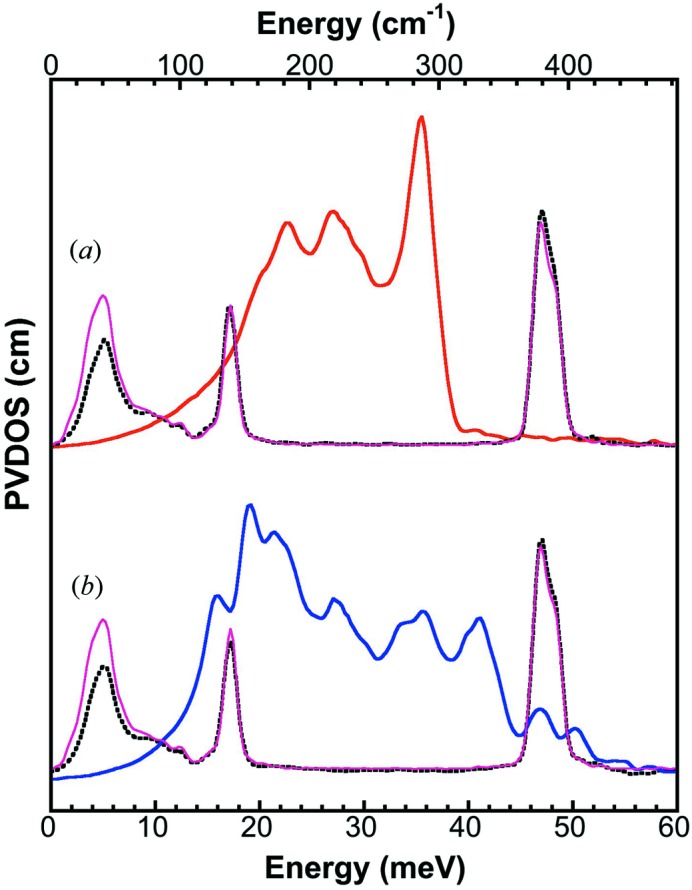
The second task is to observe extremely small energy shifts. Owing to a careful in situ calibration, such as the one shown in Fig. 5(a) ▶, a small isotopic shift of 47.5 → 47.8 meV (0.3 meV, or 0.63%) was observed and resolved for [Fe4S4Cl4]= → [XFe4S4Cl4]= (Fig. 5b ▶). The expanded NRVS spectra are presented in Fig. 5(c) ▶ to better illustrate this 0.3 meV shift. As discussed in detail in the supplementary information, a [XFe4S4Cl4]= complex has a combination of 57Fe and 54Fe sites, and a mixed NRVS signal from [57Fe2 54Fe2S4Cl4]= (25%) and [57Fe54Fe3S4Cl4]= (75%) species. A simple Fe—S stretching estimation indicates a 0.33 meV spectral shift between [Fe4S4Cl4]= and [XFe4S4Cl4]=, which is consistent with the observed 0.3 meV.
Figure 5.
(a) PVDOS of [Fe4S4Cl4]= at A (solid blue) versus Fe powder at B (dashed black). (b) Fe calibrated PVDOS of [Fe4S4Cl4]= (blue) versus [XFe4S4Cl4]= (red). (c) Expanded spectra of (b).
Small isotopic shifts are often useful for identifying metal-involved vibration(s). For example, (i) in a RR experiment (Mascarenhas et al., 1989 ▶), the 1.8 cm−1 down-shift at 148.6 cm−1 by substituting 65Cu for 64Cu concluded that the peak is the Cu-involved vibration(s) in YBa2Cu2O7–X, while the invariable 112.5 cm−1 peak does not involve Cu at all; (ii) with IR (Zhou et al., 2013 ▶), the 2 to 3 cm−1 energy difference between 100Mo/92Mo labeled Mo homocitrate complexes identified the Mo=O and Mo—O bands inside the complex; and (iii) in NRVS, the small 57Fe/54Fe isotopic shift between [Fe4S4Cl4]= and [XFe4S4Cl4]= can help pin-point the Fe—S vibrations as well (Mitra et al., 2011 ▶).
As mentioned in §2.2, the measurements at A and at B cannot be optimized simultaneously. More signal for a NRVS measurement at A means less signal for the energy calibration at B and vice versa. Thus a balanced compromise has to be reached between the two samples. In measuring [Fe4S4Cl4]=, which has a maximum signal level of 2350 counts s−1, the signal for A ([Fe4S4Cl4]=, green) and for B (Fe, red) are well balanced at position Z 3 if the energy calibration is the focus, as illustrated in Fig. 2(b) ▶. At this position, both samples have ∼1400 counts s−1, corresponding to 62% of the [Fe4S4Cl4]= peak value and 31% of the Fe’s maximum (the maximum under the circumstance of an in situ calibration). In measuring [XFe4S4Cl4]= (maximum NRVS signal = 255 counts s−1), the sample at A has 155 counts s−1 while Fe powder at B still has 1400 counts s−1.
3.3. A semi in situ calibration
When the NRVS measurement rather than energy calibration is the focus, one no longer has the luxury to sacrifice 38% of the NRVS signal. In these cases, Z 2 (Fig. 2b ▶) becomes an alternative position, which sacrifices ∼15% of the NRVS signal and has a much weaker (350 counts s−1 or ∼7.5% of its maximum level) and noisier calibration signal. Although single scans may not be good for tracking the energy scale scan-by-scan, few-scan-averaged spectra are still good for energy calibration. For example, as shown in Fig. 6(a) ▶, (i) a five-scan-averaged semi in situ calibration spectrum (dashed red) overlaps well with the one observed with a quick-switching calibration (solid green); and (ii) the sharp peak at 35.6 meV is still clear. To further improve statistics, the centroid, instead of the peak position, were used in the calibration procedure. We call this procedure semi in situ calibration.
Figure 6.

(a) Raw 57Fe powder NRVS measured with a quick-switching procedure (solid green) and a semi in situ procedure (using five-scan-averaged spectra, dashed red). X1 and X2 signify the integration range for the Fe 35.6 meV peak (32–39 meV). (b) 57Fe powder monitored semi in situ energy scales K versus time. The open symbols are analyzed from single Fe scans and the red solid line is from the five-scan-averaged spectra.
Semi in situ calibrated K values versus the time for one beam time are illustrated in Fig. 6(b) ▶. The single-scan-calculated K values (black circles) look scattered, and probably do not reflect the real K values from scan to scan. On the other hand, the K values obtained from the five-scan-averaged spectra (red line) seem reasonably stable. The single-scan-derived data vary between 0.955 and 0.975 (2.0%) for a period of about 110 h while the five-scan-probed values vary from 0.957 to 0.973 (1.6%). The maximum difference between the neighboring K values is 0.4%.
The K variation trend seems to be more or less following a 24 h circle. This is consistent with the period of the environmental temperature circle.
3.4. A quick-switching calibration
As mentioned above, an in situ calibration either sacrifices the signal level in the NRVS measurement at A (§3.2) or results in a poor signal level for the energy calibration at B (§3.3). Therefore, a quick-switching calibration procedure was developed to swap between the two events without changing NRVS samples. As the calibration sample at B is always at one temperature (RT), the systematic irreproducible peak positions for the first two scans (with a conventional calibration) were never observed. Consequently, a single scan is good enough for one energy calibration. With this procedure, NRVS measurements and energy calibrations can use the full beam flux alternatively.
Although it is not an in situ calibration method, the less required calibration scans and the fast switching time reduce each energy calibration time from 3–4 h to about 30 min and thus the calibration interval from 72 h to as short as 6 h. A series of Fe2O3 calibrated K values during one particular beam time are illustrated in Fig. 7(a) ▶ (blue circles). The overall variation is between 0.951 and 0.965, about 1.4% difference. Another series of Fe calibrated K values for a separate beam time are shown in Fig. 7(b) ▶ (0.948–0.958 or 0.8%). The maximum calibration-to-calibration difference is 0.8% (only one case) and the average difference is 0.4% using all our quick-switching calibrated K values.
Figure 7.
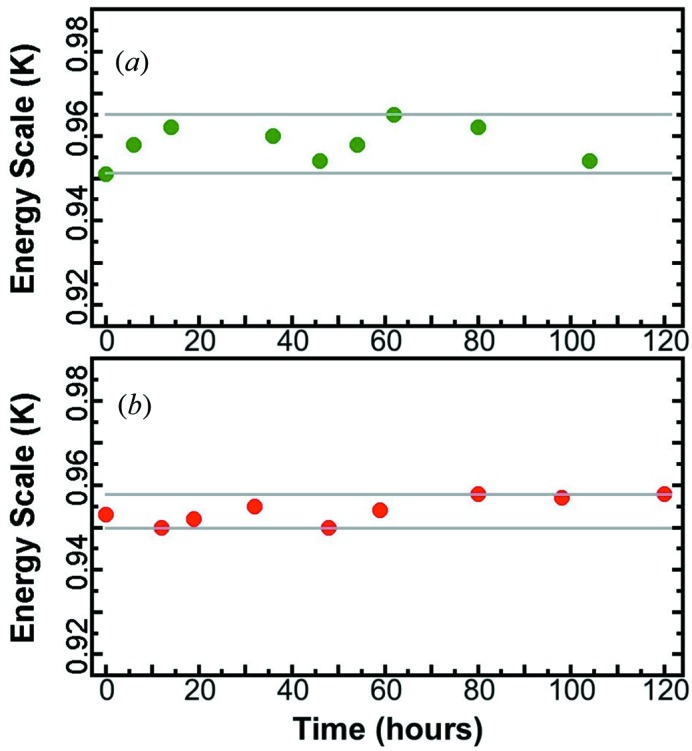
Energy scales (K values) obtained with Fe2O3 PVDOS (a) and with Fe PVDOS (b) using a quick-switching calibration procedure for two different beamtimes.
A 6 h interval is recommended because: (i) we noticed that all the calibrations with a 6 h interval have a maximum calibration-to-calibration difference of 0.6% (instead of 0.8%); (ii) it will cover the possible minima and maxima in the K values, assuming a 24 h thermal circle. When a 6 h calibration is not possible, an alternating 12–18–12–18 h interval is recommended instead. Incidents, such as entering the monochromator hutch or having a long time beam disruption, could change a HMR crystal’s temperature by over 0.1° and produce a few percent change in the HRM’s energy scale (K). This effect may need hours to recover. During the recovery period, more frequent calibrations are recommended to monitor the real HRM energy.
Is a 0.4–0.8% calibration precision good for an ordinary NRVS measurement? Let us see a few examples. Assuming a standalone iron–ligand stretch, Table 1 ▶ summarizes some common isotopic shifts, such as 57Fe—12C/13C (Kamali et al., 2013 ▶), 14N/15N (Tonzetich et al., 2010 ▶), 16O/18O (Do et al., 2011 ▶) and 32S/36S (Mitra et al., 2011 ▶). Figs. 8(a)–8(c) ▶ also illustrate several observed examples of isotope shifts between 14N/15N, 16O/18O and 32S/36S. These theoretical and observed isotope shifts are of the order of 3–4% of the vibrational peak positions. Fig. 8(d) ▶ illustrates an example of a redox shift in Fe—S, which is ∼4.6%. Therefore a quick-switching calibration is quite suitable for observing the above spectral shifts.
Table 1. Sizes of some common isotopic shifts in iron-related Fe—X stretching modes.
| 57Fe—N X stretches | 57Fe—C | 57Fe—N | 57Fe—O | 57Fe—S |
|---|---|---|---|---|
| Isotopes (N X) | 12C/13C | 14N/15N | 16O/18O | 32S/36S |
| ΔE (estimated) | 3.2% | 2.7% | 3.9% | 3.6% |
| ΔE (observed) | 3.8% | 2.2% | 5.7% | 3.8% |
Figure 8.

Various spectral shifts in typical NRVS: (a) the 14N/15N shifts in (Et4N)[Fe4S3(NO)7] (Tonzetich et al., 2010 ▶); (b) the 16O/18O shifts in [Fe(μ-O)(N-EtHPTB)(PhCO)]+ (Do et al., 2011 ▶); (c) the 32S/36S shifts in P. furiosis ferredoxin (containing a Fe4S4 cluster) (Mitra et al., 2011 ▶); (d) the redox shift for P. furiosis ferredoxin (Mitra et al., 2011 ▶).
3.5. Future perspectives
A higher-energy calibration peak will lead to a lower calibration error bar. In addition, sectional scans are often useful to resolve critical but weak features, e.g. the Fe—CO/CN in a NiFe hydrogenase enzyme (Kamali et al., 2013 ▶). To calibrate sectional scans in situ, a calibration sample with a prominent peak in the same energy section (e.g. 60–80 meV) becomes necessary. Complex [MgFe(CN)6]= has a sharp peak at ∼75 meV (or 600 cm−1) (Fig. 9a ▶) and can be used to calibrate high-energy sectional scans.
Figure 9.
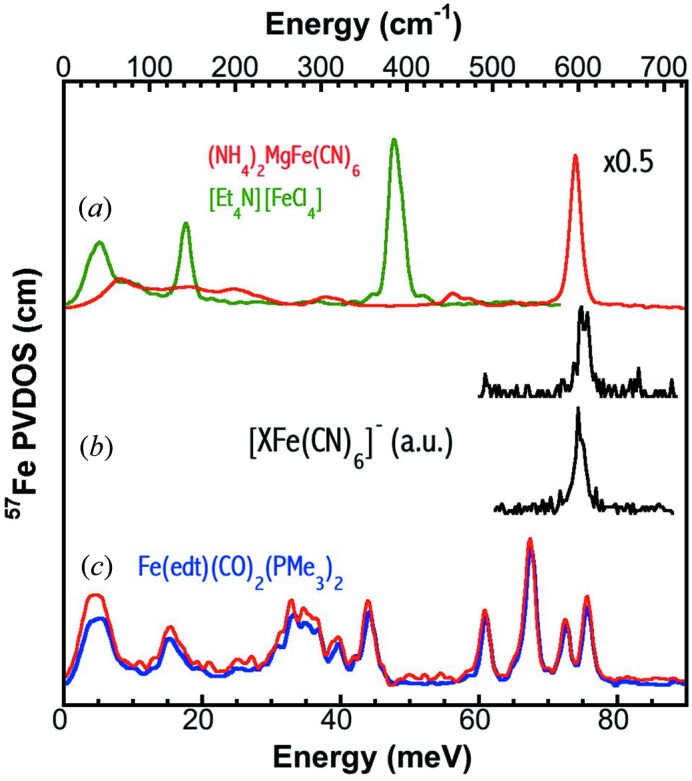
(a) PVDOS of [MgFe(CN)6]= (red) at RT versus [FeCl4]− at LT (green); (b) raw NRVS (a.u.) for non-enriched [XFe(CN)6]− (X = Gd or Eu) at RT; (c) PVDOS of Fe(edt)(CO)2(PMe3)2 six years saved under LN2 (red, at 140 K) versus the fresh sample (blue, at 100 K).
Many similar complexes also exist. For example, potential medical MRI or cellular imaging agents [GdFe(CN)6]− and [EuFe(CN)6]− (Shokouhimehr et al., 2010 ▶; Huang et al., 2010 ▶) also have the Fe—CN peak roughly around 75 meV (Fig. 9b ▶). The small peak drifts in these spectra without the elastic peak, in turn, illustrate the need for a standard sample with feature(s) in the same region to calibrate the energy axis (Fig. 9b ▶).
A LT environment will also extend our choices for the calibration samples because many complexes are not stable at RT, but are very stable at LT. Fig. 9(c) ▶ illustrates the PVDOS for a model complex Fe(edt)(CO)2(PMe3)2. The blue curve shows the initial spectrum measured in 2006 (Guo et al., 2008 ▶) [sensor reading T = 10 K, real T = 100 K (Wang et al., 2012 ▶)] while the red curve shows the spectrum measured in 2012 (sensor reading T = 120 K, real T = 140 K). The comparison shows that Fe(edt)(CO)2(PMe3)2 is stable after six years stored in LN2. This sample should be a good sample to calibrate the Fe—CO region if a LT chamber is available at B.
As calibration precision also depends on the signal level, it will be gratifying to see a further increase in the calibration signal at A without sacrificing more NRVS signal at A. Under the current experimental conditions, use of a multiple-element APD array is an immediate option; a LT environment, using a LHe cryostat (Xiao et al., 2005 ▶), a LN2 cryostat (77 K) (Dong et al., 2013 ▶), or a thermoelectric cooling device (120 K) (Monroe, 2013 ▶), should also be useful to make spectral peaks sharper and background cleaner. In addition, more beam flux will be available in the future due to the continuous development in synchrotron radiation sources, insertion devices and beamline optics.
4. Summary
Two alternative energy calibration procedures have been successfully established. Neither of these requires removing NRVS samples out of the cryostat. An in situ calibration procedure tracks the energies scan-by-scan and is best for observing an extremely small spectral shift, such as the 0.3 meV difference between [Fe4S4Cl4]= and [XFe4S4Cl4]=. A quick-switching calibration reduces the calibration time and calibration interval dramatically, and is suitable for observing general isotopic or redox shift, which is of the order of a few percent of the vibrational peak energy.
While Fe and Fe2O3 are good for calibrating regular vibrational modes (e.g. Fe—S at 14–54 meV), several [XFe(CN)6]– (X = metal) complexes are found useful for calibrating high-energy features (e.g. Fe—CO at ∼70 meV).
Supplementary Material
Supplementary material file. DOI: 10.1107/S0909049513021201/mo5055sup1.pdf
Acknowledgments
This work was funded by NIH grants GM-65440, EB-001962, and the DOE Office of Biological and Environmental Research (all to Professor Stephen P. Cramer at UC Davis). The work at Kent State University was supported by NIH-NCI (1R21CA143408-01A1, to SDH). NRVS spectra were measured at SPring-8 BL09XU with the approval of JASRI (Proposal No. 2011A/B0032 and 2012A/B0032). We also thank Professor Cramer (at UC Davis) for the overall support, Dr Ilya Sergeev/Dr Aleksandr Chumakov (at ESRF/ID18) for assistance in obtaining the [MgFe(CN)6]= NRVS, and Dr Jiyong Zhao (at APS 03ID) for discussion on HRM calibrations.
Footnotes
Supplementary data for this paper are available from the IUCr electronic archives (Reference: MO5055). Services for accessing these data are described at the back of the journal.
References
- Chumakov, A. & Rüffer, R. (1998). Hyperfine Interact. 113, 59–79.
- Do, L., Wang, H., Tinberg, C. E., Dowty, E., Yoda, Y., Cramer, S. P. & Lippard, S. J. (2011). Chem. Commun. 47, 10945. [DOI] [PMC free article] [PubMed]
- Dong, W., He, P., Wang, J., Zhou, Z. & Wang, H. (2013). Infrared Phys. Technol. 56, 51–56.
- Garman, E. F. & Nave, C. (2009). J. Synchrotron Rad. 16, 129–132.
- Guo, Y., Wang, H., Xiao, Y., Vogt, S., Thauer, R. K., Shima, S., Volkers, P. I., Rauchfuss, T. B., Pelmenschikov, V., Case, D. A., Alp, E. E., Sturhahn, W., Yoda, Y. & Cramer, S. P. (2008). Inorg. Chem. 47, 3969–3977. [DOI] [PMC free article] [PubMed]
- Huang, S. D., Li, Y. & Shokouhimehr, M. (2010). US 2010/0215587A1.
- Kamali, S., Wang, H., Mitra, D., Ogata, H., Lubitz, W., Manor, B. C., Rauchfuss, T. B., Byrne, D., Bonnefoy, V., Jenney, F. E., Adams, M. W. W., Yoda, Y., Alp, E., Zhao, J. & Cramer, S. P. (2013). Angew. Chem. Int. Ed. 52, 724–728. [DOI] [PMC free article] [PubMed]
- Mascarenhas, A., Katayama-Yoshida, H., Pankove, J. & Deb, S. (1989). Phys. Rev. B, 39, 4699–4700. [DOI] [PubMed]
- Mitra, D., Pelmenschikov, V., Guo, Y., Case, D. A., Wang, H., Dong, W., Tan, M. L., Ichiye, T., Jenney, F. E., Adams, M. W., Yoda, Y., Zhao, J. & Cramer, S. P. (2011). Biochemistry, 50, 5220–5235. [DOI] [PMC free article] [PubMed]
- Monroe, D. (2013). Physics, 6, 63.
- Seto, M., Masuda, R., Higashitaniguchi, S., Kitao, S., Kobayashi, Y., Inaba, C., Mitsui, T. & Yoda, Y. (2009). Phys. Rev. Lett. 102, 217602. [DOI] [PubMed]
- Seto, M., Yoda, Y., Kikuta, S., Zhang, X. W. & Ando, M. (1995). Phys. Rev. Lett. 74, 3828–3831. [DOI] [PubMed]
- Shokouhimehr, M., Soehnlen, E. S., Hao, J., Griswold, M., Flask, C., Fan, X., Basilion, J. P., Basu, S. & Huang, S. D. (2010). J. Mater. Chem. 20, 5251.
- Smith, M. C., Xiao, Y., Wang, H., George, S. J., Coucouvanis, D., Koutmos, M., Sturhahn, W., Alp, E. E., Zhao, J. & Cramer, S. P. (2005). Inorg. Chem. 44, 5562–5570. [DOI] [PubMed]
- Sturhahn, W., Toellner, T. S., Alp, E. E., Zhang, X., Ando, M., Yoda, Y., Kikuta, S., Seto, M., Kimball, C. W. & Dabrowski, B. (1995). Phys. Rev. Lett. 74, 3832–3835. [DOI] [PubMed]
- Tinberg, C. E., Tonzetich, Z. J., Wang, H., Do, L. H., Yoda, Y., Cramer, S. P. & Lippard, S. J. (2010). J. Am. Chem. Soc. 132, 18168–18176. [DOI] [PMC free article] [PubMed]
- Tonzetich, Z. J., Wang, H., Mitra, D., Tinberg, C. E., Do, L. H., Jenney, F. E., Adams, M. W., Cramer, S. P. & Lippard, S. J. (2010). J. Am. Chem. Soc. 132, 6914–6916. [DOI] [PMC free article] [PubMed]
- Wang, H., Yoda, Y., Kamali, S., Zhou, Z.-H. & Cramer, S. P. (2012). J. Synchrotron Rad. 19, 257–263. [DOI] [PMC free article] [PubMed]
- Xiao, Y., Fisher, K., Smith, M. C., Newton, W. E., Case, D. A., George, S. J., Wang, H., Sturhahn, W., Alp, E. E., Zhao, J., Yoda, Y. & Cramer, S. P. (2006). J. Am. Chem. Soc. 128, 7608–7612. [DOI] [PMC free article] [PubMed]
- Xiao, Y., Wang, H., George, S. J., Smith, M. C., Adams, M. W., Jenney, F. E., Sturhahn, W., Alp, E. E., Zhao, J., Yoda, Y., Dey, A., Solomon, E. I. & Cramer, S. P. (2005). J. Am. Chem. Soc. 127, 14596–14606. [DOI] [PubMed]
- Yoda, Y., Imai, Y., Kobayashi, H., Goto, S., Takeshita, K. & Seto, M. (2012). Hyperfine. Interact. 206, 83–86.
- Yoda, Y., Yabashi, M., Izumi, K., Zhang, X. W., Kishimoto, S., Kitao, S., Seto, M., Mitsui, T., Harami, T., Imai, Y. & Kikuta, S. (2001). Nucl. Instrum. Methods Phys. Res. A, 467, 715–718.
- Zhao, J. Y. & Sturhahn, W. (2012). J. Synchrotron Rad. 19, 602–608. [DOI] [PMC free article] [PubMed]
- Zhou, Z. H., Wang, H., Yu, P., Olmstead, M. M. & Cramer, S. P. (2013). J. Inorg. Biochem. 118, 100–106. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material file. DOI: 10.1107/S0909049513021201/mo5055sup1.pdf