

Abstract
Since the invention of hybridoma technology, methods for generating affinity reagents that bind specific target molecules have revolutionized biology and medicine.[1] In the postgenomic era, there is a pressing need to accelerate the pace of ligand discovery to elucidate the functions of a rapidly growing number of newly characterized molecules and their modified states.[2] Nonimmunoglobulin-based proteins such as DARPins, affibodies, and monobodies represent attractive alternatives to traditional antibodies as these are small, soluble, disulfide-free, single-domain scaffolds that can be selected from combinatorial libraries and expressed in bacteria.[3] For example, monobodies—highly stable scaffolds based on the immunoglobulin VH-like 10th fibronectin type III (10Fn3) domain of human fibronectin[4]—have yielded antibody mimetics that bind to numerous targets for applications including intracellular inhibition,[5,6] therapeutics,[7] and biosensors.[6,8] These 10Fn3-based ligands can be derived from highly diverse libraries using techniques such as phage, ribosome, mRNA, bacterial, and yeast displays.[9]
Keywords: antibodies, directed evolution, mRNA, ligand design, selection methods
Among these techniques, mRNA display has the advantage of being an entirely in vitro method that uniquely pairs a covalent, monovalent linkage between genotype and phenotype with a relatively high fusion yield.[10,11] Furthermore, it becomes possible to use very large combinatorial libraries because of the lack of an obligate in vivo step during iterative rounds of enrichment, and mRNA-display libraries can encompass 103–105 -fold more unique sequences than typical phage- or cell-based display experiments.[11] Larger libraries often produce higher-affinity binders,[12] and can also yield diverse pools of target-specific ligands with unique properties.[5] Large libraries are especially beneficial for evolving rare functionalities including enzymatic activity,[13] and the capacity to target nonstructured biological targets.[6] However, effective isolation of desired molecules from high-complexity mRNA-display libraries typically requires many rounds of selection, which is often resource intensive. Many iterative selection cycles may also result in the enrichment of suboptimal ligands as a result of compounding biases from selective constraints other than binding efficiency (e.g. PCR or translation efficiencies). Thus, novel technologies that can accelerate and automate the selection process are urgently needed.
We report herein a rapid, low-cost, highly efficient method for generating high-affinity antibody mimetics using small-scale, continuous-flow magnetic separation (CFMS). Unlike previous microfluidic approaches, which have required fabrication of specialized devices,[14] CFMS can be performed within a small section of perfluoroalkoxy (PFA) tubing to achieve highly stringent selection with minimal background. This low background directly contributes to the efficiency of selection, and continuous flow improves the washing efficiency while promoting selection for low off-rates; together, these factors contribute to the rapid convergence of high-affinity ligands.
We also describe the implementation of an improved 10Fn3 library with enhanced expression (e10Fn3). Previously, we utilized an in vivo expression screen based on a green fluorescence protein (GFP) folding reporter[15] to improve the expression of a phospho-specific IκBα-binding Fn3 variant.[6] We demonstrate here that these framework mutations plus an additional rational mutation enhance expression of four unrelated 10Fn3 variants both in vivo and in vitro (see Figure S1 in the Supporting Information). The enhancement in expression may be due to the replacement of three solvent exposed hydrophobic residues, which are localized on the three-dimensional structure, with polar residues, as well as replacing a buried Leu for Ile, which may enhance stability because of a significantly higher β-sheet-forming propensity.[16,17] The substantial 2–4-fold expression increase for individual e10Fn3 clones in rabbit reticulocyte lysate (Figure S1c) is also seen for the naïve e10Fn3 library as a whole as expected (data not shown). One additional change includes limiting variation of the final BC loop random position to the hydrophobic residues Leu, Ile, and Val, as this position is a buried core residue in the wild-type 10Fn3 structure and may interact in the transition-state folding nucleus.[18]
We chose to target interleukin 6 (IL-6) as a model to generate high-affinity e10Fn3-based ligands using CFMS mRNA display. This cytokine contributes to the regulation of the immune response and hematopoiesis,[19] and aberrant IL-6 serum levels are implicated in various inflammatory diseases and cancers.[20] We show that CFMS selection offers significantly improved (ca. 30-fold) partition efficiencies compared to conventional methods, and report the generation of a high-affinity IL-6 ligand (KD = 21 nM) with an excellent off-rate (8.8 × 10−4 s−1). This high-affinity IL-6 ligand is capable of inhibiting signaling through gp130, thus indicating the molecules potential value and demonstrating the effectiveness of CFMS for rapidly identifying clinically relevant molecules.
A key advantage of our method is the ability to perform highly stringent molecular selections on very small scales using readily available materials while maintaining high recovery of target binders. The selection process is shown in Figure 1A, including standard mRNA-protein fusion preparation.[5,6] Briefly, after incubating the target-coated beads with the library, the beads are efficiently captured by the NdFeB rare-earth magnets in the PFA tubing, and stringently washed under continuous flow conditions. Almost all of the beads are recovered after selection by cutting the section of the tubing containing the beads and eluting directly into the PCR buffer. We performed numerical simulations to determine optimal flow conditions for washing and dissociation of weakly bound ligands, and found that a flow rate of 30 mLh−1 (ca. 16 cm min−1) through tubing having a diameter of approximately 1 mm would deplete a 500 μL suspension containing 1 × 106 beads by more than four orders of magnitude after magnetic capture (Figure 1B). We modeled capture using a convection-diffusion equation, with convective particle velocity accounting for the magnetic force on the beads (calculation details provided in the Supporting Information). We also calculated the magnetic field gradient horizontally across the magnet surface, and confirmed that captured beads would remain trapped at high flow rates (Figure 1C). Even at 30 μL above the tube wall, regions exist where the magnetic field gradient is suitable for bead capture.
Figure 1.

CFMS mRNA display selection. A) In vitro selection of mRNA-display libraries using CFMS. 10Fn3 scaffold: blue; random regions: red, gold, and purple. The 10Fn3 scaffold is represented as greek key motif with random BC and FG loops. Selections begin with PCR amplification (a) to produce copies of approximately one trillion unique sequences, in vitro run-off transcription (b), splint-mediated ligation of a 3′ puromycin-containing oligonucleotide (c), in vitro translation and fusion formation (d), and subsequent reverse transcription (e), pool binding (f), and affinity enrichment. Target-coated beads are captured within the PFA tubing by three small NdFeB magnets (g) and washed under continuous flow. Bead recovery is achieved by cutting a minimal length of tubing containing the beads and eluting directly into the PCR mix (h). B) Simulation of the depletion in bead concentration resulting from vertical magnetic capture. C) Simulation showing the regions in the tube where the magnetic field is sufficient to retain beads attracted to the tube surface against the fluid flow. In the regions shown in red, the magnetic force in the negative horizontal direction exceeds the maximum Stokes drag force experienced by a stationary bead in the flow field within the tube. We identified four regions that extend to a distance of 10–30 microns into the tubing. D) Bead recovery analysis using FACS. All but 0.01% of beads applied to the device were successfully trapped.
Using CFMS, trapping, washing, and bead recovery can be performed in less than 10 minutes, and highly efficient recovery is possible within a minimal volume (less than 20 μL). We demonstrated high recovery using FACS by passing 1 × 106 biotin-phycoerythrin-labeled beads through the system; only 0.01% of the beads were detected in the flow-through (Figure 1D). Some residual beads remained in the microcentrifuge tube, which is in part due to the concentration of the beads at the descending buffer surface, but we could minimize loss with a single rinse step.
To demonstrate the utility of CFMS selection, we immobilized biotinylated IL-6 onto nonporous superparamagnetic beads (epoxy M-270) through neutravidin, which displays particularly low background library binding (Figure S2). We performed the first round of selection with a library containing greater than 10 copies of 1 × 1012 unique sequences. Round one was performed with 5 × 106 beads in 5 mL binding buffer, and all subsequent rounds used 1 × 106 beads in 500 μL binding buffer. After binding the library against IL-6-labeled beads for one hour at room temperature, we applied the sample to the device and washed for 5 minutes at 30 mLhr−1. We then transferred the minimal segment of tubing containing the trapped, library-bound beads into a microcentrifuge tube containing PCR buffer and eluted the beads for direct amplification.
We performed CFMS selection under two slightly different conditions. After round three under the initial selection conditions (selection A), we noted that the number of PCR cycles required to amplify affinity-enriched fusions had decreased by three, indicating that pool 2A was becoming enriched for binding over background. We sequenced a random sampling of ten clones from pool 3A and found that approximately 70% of these represented a single sequence, eFn-3A02 (Figure 2A). However, this sequence, as well as two nondominant sequences from this pool, contained cysteines, and we were concerned that disulfide bond formation may mediate binding to IL-6, which contains two solvent-exposed disulfide bonds, one of which is important for function.[21] While eFn-3A02 has clear target specificity, we felt that a noncysteine-containing ligand would eliminate the possibility of nonspecific disulfide bond formation and preserve an orthogonal chemical handle, a useful feature of this scaffold.[8]
Figure 2.
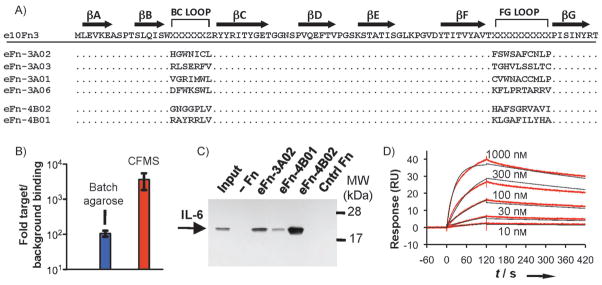
Characterization of selected IL-6-binding sequences. A) The sequence of the e10Fn3 library (x =random, z =L, I, or V). After three rounds, pool 3A converged to clone eFn-3A02, which represented 70% of selected clones. Selection B was performed to avoid cysteine-containing clones, and converged after four rounds to eFn-4B02 (16 out of 20 clones) and eFn-4B01 (3 out of 20 clones). B) Quantitative PCR-based binding assay to demonstrate increased selectivity of CFMS for pool 4B, which bound to IL-6 target beads more than 3000-fold more efficiently than a non-enriched pool (background binding); the selectivity of pool 4B using cross-linked agarose beads was approximately 100-fold. C) Pull-down results of IL-6 using immobilized e10Fn3 samples, as visualized by western blot (input =5% of total). D) SPR measurements to determine affinity of eFn-4B02 for IL-6 (koff = 8.8 × 10−4 s−1; kon = 4.2 × 104 M−1 s−1; KD = 21 nM).
We thus repeated the selection against pool 1 while blocking cysteines in our library with iodoacetamide (selection B) to convert free thiols into thioethers (Figure S3). By round 4B, the number of PCR cycles required to amplify enriched binders had decreased by four, thus indicating pool convergence. eFn-4B02 was the dominant sequence, representing approximately 80% of the 20 clones we sequenced. Neither this clone nor Fn-4B01 (composing ca. 15% of pool 4B) contained cysteines (Figure 2A).
Our results show that CFMS can generate highly convergent pools in considerably fewer rounds (3–4) compared to previous mRNA display experiments (6–10 rounds).[5,6] With CFMS, we observed much lower background binding, based on semiquantitative PCR during initial rounds of selection (ca. 27 PCR cycles for detection after CFMS compared to ca. 20 in previous studies). As long as target binding is not significantly less efficient, this improved signal-to-noise ratio will result in higher selection efficiency. To quantify this, we utilized quantitative PCR to directly compare the binding of pool 4B relative to a naïve pool in binding to beads with or without IL-6 using either CFMS or a conventional agarose bead format (Figure 2B). Pool 4B showed approximately a 100-fold enhanced binding to the target over background in the agarose bead format, which is in agreement with previous measurements.[5,6] However, the background binding in the CFMS format is significantly lower, whereas for pool 4B target binding remains nearly as efficient (ca. 30% lower), thus leading to a greater than 3000-fold target-to-background binding ratio. This difference in selectivity explains the improved rate of enrichment observed in CFMS and associated reduction in the number of selection rounds needed.
To determine if our clones can efficiently and specifically bind IL-6, we used bacterially-expressed e10Fn3 variants immobilized onto nickel affinity resin to pull down IL-6 from solution (Figure 2C) in the presence of a 500-fold excess of bovine serum albumin (BSA). It is unlikely that the cysteine-containing eFn-3A02 binds as a result of nonspecific disulfide bonding in this assay since BSA contains 35 cysteines (17 disulfides and one free cysteine), and should therefore prevent such association. eFn-4B02 was the most efficient binder and was chosen for subsequent analysis by surface plasmon resonance (SPR; Figure 2D). eFn-4B02 exhibited an excellent off-rate, koff = 8.8 × 10−4 s−1, which gives a half-life of approximately 13 minutes; this illustrates the advantages of continuous flow washing. The on-rate (kon) was 4.2 × 104 M−1 s−1, thus giving a calculated equilibrium binding constant of KD = 21 nM.
Finally, we explored whether our selected 10Fn3 variants could inhibit IL-6 signaling in human hepatocytes (HuH 7.5.1), which express both the IL-6-specific co-receptor IL-6Ra and the signaling receptor gp130. We performed a western blot to analyze signaling inhibition by measuring phosphorylation of STAT3, a downstream transducer of gp130 activation (Figure 3). As expected, eFn-4B02 was a more effective inhibitor than eFn-3A02, and a nonselected control e10Fn3 variant had little effect (Figure 3A). eFn-4B02 inhibits IL-6 signaling in a concentration-dependent manner (based on three independent experiments) with an IC50 = 419 nM (Figures 3B and C).
Figure 3.

Inhibition of IL-6 function. IL-6 signaling inhibition was assayed by western blot for STAT3 phosphorylation at Y705. A) IL-6 (50 ngmL−1) was added to HuH-7 cells pre-incubated with 500 nM of various e10Fn3s for 12 min. B) Quantitative western blot to determine concentration-dependent inhibition; imaged using an infrared dyelabeled secondary antibody. Shown is one of three independent experiments. eFn-4B02 (10 nM−10 μM) and IL-6 (10 ngmL−1) were pre-incubated for 30 min before applying to HuH-7 cells for 12 min. C) Four-parameter nonlinear regression curve fit of quantitative western blot analysis (IC50 = 422 nM).
In summary, we report a simple and cost-effective micro-fluidic method for the rapid generation of functional non-immunoglobulin affinity reagents. Increased partitioning efficiencies in CFMS result in a greater than 30-fold increase in enrichment over conventional, agarose-based selections, thus enabling faster selection. Notably, CFMS requires no special materials or devices, involves minimal cost, and the miniaturized format decreases the amount of target material needed for the selection. Continuous washing in the CFMS format should allow selection of ligands with better binding kinetics, as demonstrated by the excellent off-rate of the IL-6-binding eFn-4B02 ligand. Importantly, CFMS allows many affinity enrichment experiments to be carried out in parallel and therefore represents a significant step toward the goal of massively parallel discovery of affinity reagents for a wide range of molecular targets.
Experimental Section
For detailed methods see the Supporting Information.
Supplementary Material
Footnotes
We thank Prof. Vaithilingaraja Arumugaswami and Prof. Christopher T. Denny for helpful comments and discussions as well as Jenny L. Anderson for technical assistance. We are grateful for the financial support from the National Institutes of Health, ARO Institute for Collaborative Biotechnologies (ICB), Armed Forces Institute of Regenerative Medicine (AFIRM), and California Institute for Regenerative Medicine.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201101149.
Contributor Information
Dr. C. Anders Olson, Email: rsun@mednet.ucla.edu, Department of Molecular and Medical Pharmacology, University of California, Los Angeles (USA)
Dr. Jonathan D. Adams, Department of Physics, University of California, Santa Barbara (USA)
Prof. Terry T. Takahashi, Email: richrob@usc.edu, Department of Chemistry, University of Southern California (USA)
Hangfei Qi, Email: rsun@mednet.ucla.edu, Department of Molecular and Medical Pharmacology, University of California, Los Angeles (USA).
Shannon M. Howell, Email: richrob@usc.edu, Department of Chemistry, University of Southern California (USA)
Prof. Ting-Ting Wu, Email: rsun@mednet.ucla.edu, Department of Molecular and Medical Pharmacology, University of California, Los Angeles (USA)
Prof. Richard W. Roberts, Email: richrob@usc.edu, Department of Chemistry, University of Southern California (USA)
Prof. Ren Sun, Email: rsun@mednet.ucla.edu, Department of Molecular and Medical Pharmacology, University of California, Los Angeles (USA)
Prof. H. Tom Soh, Email: tsoh@engr.ucsb.edu, Materials Department, Department of Mechanical Engineering, University of California, Santa Barbara, CA 93106 (USA)
References
- 1.Margulies DH. J Immunol. 2005;174:2451. doi: 10.4049/jimmunol.174.5.2451. [DOI] [PubMed] [Google Scholar]
- 2.Stoevesandt O, Taussig MJ. Proteomics. 2007;7:2738. doi: 10.1002/pmic.200700155. [DOI] [PubMed] [Google Scholar]
- 3.Binz HK, Amstutz P, Pluckthun A. Nat Biotechnol. 2005;23:1257. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 4.Koide A, Bailey CW, Huang X, Koide S. J Mol Biol. 1998;284:1141. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 5.Liao HI, Olson CA, Hwang S, Deng H, Wong E, Baric RS, Roberts RW, Sun R. J Biol Chem. 2009;284:17512. doi: 10.1074/jbc.M901547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson CA, Liao HI, Sun R, Roberts RW. ACS Chem Biol. 2008;3:480. doi: 10.1021/cb800069c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bloom L, Calabro V. Drug Discovery Today. 2009;14:949. doi: 10.1016/j.drudis.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Ishikawa FN, Chang HK, Curreli M, Liao HI, Olson CA, Chen PC, Zhang R, Roberts RW, Sun R, Cote RJ, Thompson ME, Zhou C. ACS Nano. 2009;3:1219. doi: 10.1021/nn900086c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levin AM, Weiss GA. Mol Biosyst. 2006;2:49. doi: 10.1039/b511782h. [DOI] [PubMed] [Google Scholar]
- 10.Roberts RW, Szostak JW. Proc Natl Acad Sci USA. 1997;94:12297. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi TT, Roberts RW. Methods Mol Biol. 2009;535:293. doi: 10.1007/978-1-59745-557-2_17. [DOI] [PubMed] [Google Scholar]
- 12.Wilson DS, Keefe AD, Szostak JW. Proc Natl Acad Sci USA. 2001;98:3750. doi: 10.1073/pnas.061028198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seelig B, Szostak JW. Nature. 2007;448:828. doi: 10.1038/nature06032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Adams JD, Turner K, Cochran FV, Gambhir SS, Soh HT. Lab Chip. 2009;9:1033. doi: 10.1039/b820985e. [DOI] [PubMed] [Google Scholar]
- 15.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Nat Biotechnol. 1999;17:691. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 16.Minor DL, Jr, Kim PS. Nature. 1994;367:660. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 17.Smith CK, Withka JM, Regan L. Biochemistry. 1994;33:5510. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 18.Cota E, Steward A, Fowler SB, Clarke J. J Mol Biol. 2001;305:1185. doi: 10.1006/jmbi.2000.4378. [DOI] [PubMed] [Google Scholar]
- 19.Taga T, Kishimoto T. Annu Rev Immunol. 1997;15:797. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 20.Rose-John S, Scheller J, Elson G, Jones SA. J Leukocyte Biol. 2006;80:227. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 21.Rock FL, Li X, Chong P, Ida N, Klein M. Biochemistry. 1994;33:5146. doi: 10.1021/bi00183a018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.