

Abstract
Infiltration of immune cells in primary tumors and metastatic sites is known to influence tumor progression and metastasis. Macrophages represent the most abundant immune cells in tumor microenvironment. Accumulating evidence has shown that macrophages promote seeding, extravasation and persistent growth of tumor cells at metastatic sites. miR-155 plays an essential role in immune cell development and function, and its aberrant expression is associated with lymphomas and several types of solid tumors. However, how miR-155 expression in immune cells affects solid tumor growth and metastasis is unknown. To investigate this, bone marrow transplantation was performed by using wild type (WT) or miR-155−/− mice as bone marrow donors and WT mice as recipients, and the chimeric mice were inoculated with tumor cells. We found that bone marrow miR-155 deficiency significantly enhanced lung metastasis without a substantial effect on primary tumor growth. Next, we found that there were more macrophages accumulated in the spleen and lungs of tumor-bearing mice with miR-155-deficient bone marrow, than in those of mice with WT bone marrow. Further analysis showed that miR-155−/− macrophages in metastatic sites exhibited a tumor-promoting M2 phenotype. In vitro study suggested that compared to WT macrophages, miR-155−/− macrophages were prone to M2 polarization upon incubation with tumor cell-conditioned medium, due to elevated expression of C/EBPβ, an identified miR-155 target. Taken together, our data, for the first time, demonstrate that miR-155 in host immune cells plays a vital role in modulating solid tumor metastasis through affecting the recruitment and polarization of bone marrow-derived macrophages.
Keywords: miR-155, tumor metastasis, Lewis lung cancer (LLC), macrophage polarization, C/EBPβ
Introduction
Tumor progression and metastasis positively correlate with the infiltration of immune cells, including myeloid-derived suppressor cells (MDSCs), macrophages, neutrophils, dendritic cells, granulocytes, T cells and B cells (1–7). Among them, macrophage is the most abundant leukocyte type present in neoplastic stroma (5, 8). Accumulating clinical and experimental evidences have shown that tumor-associated macrophages (TAMs) promote tumor progression at the primary tumor sites (5, 9), and enhance tumor metastasis in distant organs (4, 10, 11). In addition, metastasis-associated macrophage is another population that is identified in experimental models of tumor metastasis (4, 5). Recent ex vivo imaging studies of metastatic lungs showed that macrophages were recruited towards extravasating tumor cells and that depletion of these macrophages dramatically reduced the seeding and extravasation efficiency and the subsequent survival of tumor cells (4).
microRNA-155 (miR-155) was among the first miRNAs that have been demonstrated to play roles in immunity and inflammation. Its expression is significantly increased in activated B cells (12), T cells (13), macrophages and dendritic cells (DCs) (14–16), and is also up-regulated in multiple immune cell lineages, including macrophages, in inflammatory responses induced by Toll-like receptor (TLR) ligands, cytokines, and specific antigens (13–15, 17). miR-155-deficient mice showed impaired immune responses (17, 18); while miR-155 overexpression in CD34+ hematopoietic progenitor cells resulted in a defective differentiation of these cells into mature myeloid and erythroid cells (19). Altogether, these observations strongly indicate miR-155 as a central regulator of the immune system.
In addition to its essential role in immunity, dysregulation of miR-155 is closely related to cancer. miR-155 transgenic mice develop B cell malignancy and elevated miR-155 expression was observed in several types of human B cell lymphomas (12, 20, 21). Altered miR-155 expression has also been found in pediatric Burkitt's lymphoma and chronic lymphocytic leukemia (21–23). Moreover, elevated miR-155 levels were also found associated with several types of solid tumor including colon cancer, breast cancer, hepatocellular carcinoma and lung cancer (24–26). Therefore, targeting miR-155 has been proposed to be a promising approach to combat cancers of hematopoietic source as well as several solid tumors (25). However, it has not been examined if miR-155 expression in host immune cells influences growth and metastasis of solid tumors. To test this, we performed a bone marrow transplantation study by using WT and miR-155−/− mice as bone marrow donors and using WT mice as recipients, and examined the effects of miR155 deficiency in bone marrow on solid tumor growth and metastasis. Our data demonstrate that miR-155 deficiency in bone marrow cells enhanced lung metastasis through increasing macrophage infiltration, which exhibited a M2 phenotype, in metastatic lungs of tumor-bearing mice, and that miR-155 deficient macrophages were prone to M2 polarization in the presence of tumor-derived factors due to elevated C/EBPβ expression.
Materials and methods
Cell culture
Lewis lung carcinoma (LLC) and B16-F10 melanoma cell lines (both syngeneic to C57BL/6 mice) were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM, Invitrogen Life Technologies, Grand Island, NY) with 10% fetal bovine serum (FBS, Invitrogen) and penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere.
Animals
C57BL/6 (WT) and bic/mir-155 knockout mice (miR-155−/−) were obtained from Jackson Laboratories (Bar Harbor, Maine). Experimental animal procedures were approved by IACUC at University of South Carolina. Mice were maintained at the University of South Carolina according to National Institutes of Health guidelines.
Bone marrow transplantation
Bone marrow transplantation (BMT) was performed as described previously (27). Briefly, recipient WT mice (6–8 weeks old, female) were lethally irradiated (900 rad). Bone marrow cells were harvested from WT and miR-155−/− donor mice by flushing the femurs and tibias with phosphate buffered saline (PBS) supplemented with 2% FBS. The flushed bone marrow cells were resuspended in PBS, and 5×106 nucleated cells were injected retro-orbitally into irradiated WT mice (8 mice/group) within 6 h after irradiation.
Tumor cell injection
For tumor growth experiment, 1×107 LLC cells (in 200 μl PBS) were subcutaneously (s.c.) implanted in the back of mice 4 weeks after bone marrow transplantation. Tumor growth was monitored by measurement of tumor size with a caliper every three days. Tumor volume was determined by the formula: length × width2/2. Primary tumors were surgically resected when they reached a length of 10 mm (14 days after subcutaneous injection of tumor cells). To promote lung metastasis, 2×105 LLC cells (in 200 μl PBS) were intravenously (i.v.) injected 10 days after subcutaneous implantation of LLC cells.
Immunohistochemistry
The lungs were perfused with PBS to eliminate circulating tumor and blood cells. Primary tumor tissues and whole lungs were fixed in 4% paraformaldehyde at 4°C for 12 h, dehydrated in 30% sucrose overnight, and embedded in OCT. Serial sections (8 μm thick) were cut throughout the entire tumor tissues and lungs. For F4/80 staining, frozen sections were fixed in acetone, incubated with 5% BSA/PBS, and incubated with FITC-conjugated rat anti-F4/80 antibody (1:100, eBioseicence, San Diego, CA) overnight at 4°C. For Ym1 staining, frozen sections were incubated with anti-mouse Ym1 rabbit polyclonal antibodies (1:200, StemCell Technologies, Vancouver, BC). The Alexa Fluor 488-conjugated secondary antibodies (Invitrogen) were used at a 1:1000 dilution and incubated for 1 h at room temperature. Slides were mounted in ProLong Gold Mounting Medium containing DAPI (Invitrogen), and the tissue sections were visualized using a Nikon ECLIPSE E600 microscope (Nikon Inc. Melville, NY). For quantitative analysis of F4/80 and Ym1 staining in lungs, we counted the number of positively stained cells in 6–12 random fields of indicated magnification per section. For F4/80 staining in tumor tissues, we determined the area occupied by immunostained cells normalized by area of DAPI-stained cells using Image-Pro Plus 6.0 analysis software (28). For vWF staining, the tumor tissue sections were fixed in 4% paraformaldehyde, rinsed with PBS and incubated with 0.3% H2O2 in methanol for 10 min. After washing with PBS, anti-mouse vWF antibody (1:2000, Abcam, Cambridge, MA) was applied. Immunocomplexes were detected with biotin-conjugated secondary antibodies and AEC chromogen/HRP substrate kit (GeneTex, Irvine, CA). The sections were counterstained with hematoxylin and mounted with a permanent mounting medium. Microvessel density was assessed with vWF staining and counted on 200× magnification fields. vWF positive endothelial cell or cell cluster clearly separate from adjacent structures was considered a single vessel. For hematoxylin and eosin (H & E) staining, sections were stained using standard procedures. H & E stained lung sections were analyzed for tumor microscopically under 4 × magnification.
Conditioned medium collection
To obtain conditioned medium, LLC and B16-F10 cells were seeded at 5×106 cells per dish of 75 cm2 and cultured till 90% confluence. The media were then replaced with serum-free DMEM. After 24 h, the supernatants were collected and filtered through a 0.22 μm filter.
In vitro macrophage treatment
Mice were injected with 3 ml of 3% thioglycollate in sterile PBS intra-peritoneally. Three days later, mice were euthanized and peritoneal macrophages were harvested by lavaging the peritoneal cavity with 2×10 ml of PBS. Cells were suspended with DMEM media containing 10% FBS; and plated in 6-well or 12-well plates. After 2 h, the non-adherent cells were removed by PBS, and the adhered macrophages were further cultured in serum-free DMEM overnight, followed by treatment with control medium, tumor cell conditioned medium or IL-4 (20 ng/ml, AASN BioAbChem Inc. Ladson, SC) for indicated period of time.
Bone marrow-derived macrophages (BMDM)
Bone marrow cells were harvested from WT or miR-155−/− mice by flushing the femurs and tibias with PBS supplemented with 2% FBS, and resuspended in bone marrow differentiation media (DMEM supplemented with 10% FBS, 20% LCCM, 100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-glutamine) at 6×105 cells/ml. Ten milliliters of cells were seeded on each 10-cm tissue culture plate (BD Biosciences, San Jose, CA). Four days after seeding, an extra 10 ml of fresh media were added to each plate and incubated for an additional 3 days. To obtain BMDMs, cells were trypsinized, centrifuged at 200× g for 5 min and then resuspended in 10 ml of BMDM cultivation media, which is composed of DMEM, 10% FBS, 5% LCCM and 2 mM L-glutamine. The cells were counted, seeded and cultivated in tissue culture plates 12 h before any further experimental procedure.
Quantitative Real-time PCR (qPCR)
Total RNA was extracted using the TRIzol reagent (Invitrogen). RNA (1 μg) was reverse-transcribed using iScript™ cDNA Synthesis Kit (Bio-Rad, Life Science, Hercules, CA). qPCR was performed on a CFX96 system (Bio-Rad) using iQ™ SYBR® Green Supermix (Bio-Rad). All primers used for qPCR analysis were synthesized by Integrated DNA Technologies (Coralville, IA). All assays were performed following the manufacturer's instructions. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of 18 S. PCR thermal cycling conditions contained 3 min at 95 °C, and 40 cycles of 15 s at 95 °C and 58 s at 60 °C. Samples were run in triplicate. The primer sequences were listed in Supplemental Table 1.
Western blot
Cells were lysed in RIPA buffer (Pierce) supplemented with protease inhibitor cocktail (Sigma). Total cellular extracts (30 μg) were separated in 10% SDS-PAGE pre-cast gels (Bio-rad) and transferred onto Nitrocellulose membranes (Millipore Corp., Bedford, MA). Membranes were first probed with C/EBPβ (1:1000, Santa Cruz Biotechnology, Inc. Santa Cruz, CA), SOCS-1 (1:1000, Abcam) or β-actin (1:1000, Sigma, St. Louis, MO) antibodies, followed by Goat anti-rabbit secondary antibody conjugated with HRP (Millipore). The Protein detection was performed using Pierce ECL Western Blotting Substrate (Pierce, Rockford, IL). Image-Pro Plus 6.0 analysis software was used to quantify signal intensities.
Flow cytometry
Bone marrow cells were harvested from mice by flushing the femurs and tibias with PBS supplemented with 2% FBS. Splenocytes were prepared by mechanical dissociation. Cells were stained with anti-CD11b PE mAb, anti-F4/80 FITC mAb, anti-Gr-1 FITC mAb, anti-ly6C FITC mAb, anti-ly6C FITC mAb, anti-CD3 PE mAb, anti-CD4 FITC mAb, anti-CD8 FITC mAb, or anti-CD19 FITC mAb (all from eBioscience) in staining buffer (PBS containing 2% FBS) for 30 min on ice in the dark. Samples were washed twice in staining buffer, analyzed by flow cytometry using a Cytomics FC 500 flow cytometer and CXP software version 2.2 (Beckman coulter, Brea CA). Data were collected for 10,000 live events per sample.
Bio-Plex cytokine/chemokine assay
Sera from WT and miR-155−/− chimeric mice were diluted by 1:4. Concentrations of 23 mouse cytokines/chemokines were measured by Bio-Plex Pro™ Mouse Cytokine 23-plex Assay (Bio-rad, M60-009RDPD) following the manufacturer's instructions.
mir155 inhibitor transfection
mirVana™ mir155 inhibitor (MH4464084) and negative control (MH4464076) were purchased from Invitrogen. LLC cells (2×105 cells) were seeded into 6-well plates. Twelve hours later, LLC cells were transfected with mir155 inhibitor or negative control using Lipofectamine® RNAiMAX Reagent (Invitrogen) according to the manufacturer's instruction. Fourty-eight hours after transfection, total RNAs were extracted and cDNAs were prepared to confirm the miR155 inhibition or the cells were used for further experiments. Gene expression of mir155 targets (HIF-1α, Bcl-6, c-Maf and SOCS1) was measrued by real-time PCR.
[3H] Thymidine incorporation assay
LLC (5×104 cells) or B16-F10 (4×104 cells) were seeded into the 24-well plates and transfected with negative control inhibitor or mir155 inhibitor. Twenty-four hours later, peritoneal macrophages (5×105 cells) from WT and miR-155−/− mice were added into LLC and B16-F10 cells. After 48 h of coculture, [3H] Thymidine (1 μCi/well) was added and further incubated for 4 h. Cultures were harvested and thymidine incorporation was measured by scintillation counting (Perkin Elmer, Waltham, MA). Data are expressed as CPM (mean ± SEM) of six cultures. Three independent experiments were performed.
Transwell migration assay
Peritoneal macrophages (1×106 cells) from WT and miR-155−/− mice were seeded into 24-well plates. Two hours after seeding, the non-adherent cells were removed by washing with PBS, and the adhered macrophages were treated with serum-free DMEM and LLC or B16-F10 derived conditioned medium. LLC or B16-F10 cells (2 × 105) were seeded onto the top chamber of transwell insert with 8 μm pore size (Corning Incorporated Life Sciences, Tewksbury, MA). Migrated tumor cells were stained with Calcein (1 μg/ml, Invitrogen) and counted under an inverted widefield fluorescence microscope at 40 × magnification (twenty fields per well, triplicate for each experimental group).
Statistical analyses
Data were presented as mean ± standard error of the mean (SEM). Statistical significance was calculated by use of Student's t test (two-group comparison) or one-way analysis of variance (ANOVA) (multi-group comparison) using the GraphPad Prism statistical program (GraphPad Prism, GraphPad Software, Inc., San Diego, CA). p < 0.05 was considered significant.
Results
miR-155 deficiency in bone marrow enhanced tumor metastasis in the lungs
To examine if miR-155 deficiency in bone marrow affects solid tumor growth and metastasis, bone marrow transplantation was performed. Wild type (WT) or miR-155−/− bone marrow cells were transplanted into lethally irradiated WT mice. Four weeks after bone marrow transplantation, WT and miR-155−/− chimeric mice (referred as WT-BMT and miR-155−/−-BMT hereafter, respectively) were inoculated with LLC cells in the back. We began to measure tumor size after the xenografts became palpable. We found that both WT-BMT and miR-155−/−-BMT mice showed a similar tumor growth rate (Fig. 1A). In addition, the tumor size showed no difference between WT-BMT and miR-155−/−-BMT mice when tumors were removed two weeks after inoculation (Fig. 1B). However, miR-155−/−-BMT mice had significantly more tumor nodules in lungs compared to WT-BMT mice (Fig. 1C). Further analysis showed that the number of micro-metastases but not macro-metastases was remarkably increased in miR-155−/−-BMT mice (Fig. 1C and E). Consistently, there was a larger total metastatic area in lungs of miR-155−/−-BMT mice than in lungs of WT-BMT counterparts (Fig. 1D). Several studies demonstrated that LLC cells metastasize to the lungs and occasionally the liver (2, 29, 30). However, in our current study, LLC tumor metastases were observed only in lung but not in liver or other organs. Quantitative real-time PCR showed that the miR-155 level in spleen of miR-155−/−-BMT mice was only 1/6 of that of WT-BMT mice (Supplementary Fig. S1), confirming the successful bone marrow reconstitution.
Figure 1.

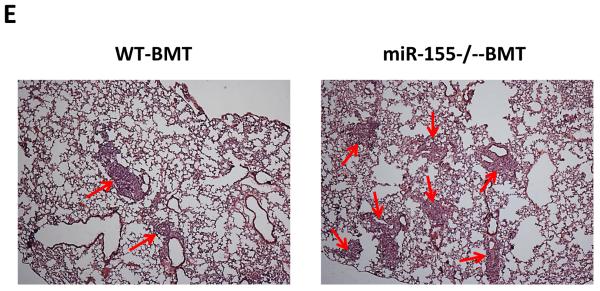
Enhanced lung metastasis in miR-155−/− chimeric mice. A, Growth rate of LLC primary tumors in WT and miR-155−/− chimeric mice. 1×107 LLC cells were subcutaneously implanted in the back of WT and miR-155−/− chimeric mice and tumor size was measured with a caliper. Tumor volume was shown as mm3. B, Average tumor weight at day 14 after LLC inoculation. C, Quantification of average number of nodules in lung of WT and miR-155−/− chimeric mice on 14 days post LLC cell implantation. Nodules smaller than 70 μm are defined as micro-metastases (micro-). Nodules larger than 70 μm are defined as macro-metastases (macro-). D, The percentage of metastatic area in lung tissues was calculated (n=8 mice per group). E, Representative H&E staining sections of the lungs from WT and miR-155−/− chimeric mice (n=8) carrying LLC tumors. The red arrows point to the metastatic nodules in the lungs. Magnification, × 4. Data are presented as the mean ± SEM of 8 mice. *p<0.05 by Student's t test.
miR-155−/− chimeric mice produced higher levels of tumor-promoting factors
Cytokines and chemokines derived from inflammatory cells as well as tumor cells can promote tumor growth and metastasis in a variety of tumor models (31, 32). In light of a higher frequency of metastases in miR-155−/−-BMT mice, we postulated that these mice may produce more tumor-promoting cytokines and chemokines. To test this, a bio-plex assay (23-plex) was performed in tumor-bearing mice. The concentrations of IL-1β, IL-6 and IL-10 in sera were dramatically increased in miR-155−/−-BMT mice than in WT-BMT mice (Fig. 2A). Moreover, a higher amount of CCL3, a chemokine for macrophage infiltration (33), was also detected in miR-155−/−-BMT mice than in WT-BM mice (Fig. 2A). IL-17 and G-CSF levels in miR-155−/−-BMT mice were greatly increased as well (Fig. 2A). Our data indicate that miR-155−/−-BMT mice produced more tumor-promoting factors, which may create a favorable microenvironment for tumor cell seeding and survival in metastatic sites. However, other cytokine (Fig. 2B–C) or chemokine (Fig. 2D) levels did not show statistical difference between these two groups.
Figure 2.

Higher production of tumor-promoting factors in miR-155−/− chimeric mice. A–D, 23 cytokines in sera of WT and miR-155−/− chimeric mice were detected by Bio-Plex Pro™ Mouse Cytokine 23-plex Assay. Data are represented as the mean ± SEM (n=8 per group). *p<0.05 by Student's t test.
miR-155 deficiency in bone marrow resulted in more macrophage mobilization in tumor-bearing mice
Bone marrow-derived cells (BMDCs) play an indispensable role in the establishment of tumor microenvironment, primarily through producing and secreting protumor factors, including cytokines, chemokines and matrix-degrading enzymes (2, 30, 34). Since miR-155 is critical for the host immune system (17, 18), we aimed to investigate if the increases in serum cytokine and chemokine levels and lung metastasis in miR-155−/−-BMT mice were due to the mobilization and accumulation of BMDCs in peripheral tissues. We thus examined bone marrow cell mobilization in tumor-bearing mice using flow cytometrical analyses. The subpopulation of cells in bone marrow, including CD11b+, Gr-1+, F4/80+, CD11b+/ly6G+, CD11b+/ly6C+, CD3+ T cells and CD19+ B cells did not show any statistical difference between WT-BMT and miR-155−/−-BMT mice (Supplementary Fig. S2); however, we found that the absolute splenocyte number in miR-155−/−-BMT mice was increased by 1.67-fold compared to that in WT-BMT counterparts although the splenic weight showed no significant difference between the two groups (Fig. 3A and B). Strikingly, not only the percentage but also the absolute number of F4/80+ macrophages in spleen of miR-155−/−-BMT mice were greatly increased compared to those in WT-BMT mice (Fig. 3C–E). Although there were increases in the percentage and the absolute number of CD11b+/ly6G+ and CD11b+/ly6C+, both have been shown to promote tumor progression, in miR-155−/−-BMT mice compared to WT-BMT counterparts, the differences were not statistically significant (Fig. 3C and D). In addition, there were more CD3+/CD4+ and CD19+ B cells in tumor-bearing miR-155−/−-BMT mice compared to WT-BMT counterparts (Supplementary Fig. S3).
Figure 3.


Increased accumulation of macrophages in the speens of miR-155−/− chimeric mice. Spleen weight (A) and total splenocytes (B) were calculated and shown. The percentage of leukocytes in spleen from WT and miR-155−/− chimeric mice was analyzed by flow cytometry using antibodies for specific surface markers (C); and the absolute number of leukocytes in spleen was calculated (D). Data were presented as the mean ± SEM of 8 mice. *p<0.05 by Student's t test. E, Representative flow cytometric dot plot of F4/80 macrophages in spleen. Percentages indicate mean values in each group.
miR-155 deficiency in bone marrow did not affect macrophage infiltration in primary tumor
Since the spleen serves as a reservoir for circulating leukocytes, we next examined if increased splenic macrophages resulted in enhanced recruitment of macrophages to the primary tumor. Tumor-associated macrophages have been known to support tumor-associated angiogenesis, promote tumor cell invasion, migration, and intravasation, as well as suppress antitumor immune responses (5). To our surprise, there appeared to be no significant difference of F4/80+ macrophage accumulation in primary tumors in WT-BMT and miR-155−/−-BMT mice (Fig. 4A and B). Moreover, the numbers of infiltrating Ym1 (a M2 macrophage marker) staining positive cells also showed no difference between WT-BMT and miR-155−/−-BMT groups (data not shown). Furthermore, we performed immunohistochemistry to detect the expression of mouse endothelial cell antigen vWF, a specific marker of angiogenesis; and found no difference in the density of intratumoral microvessels in these two groups (Fig. 4C and D). Moreover, quantitative real-time PCR was performed to detect the expression of proangiogenic and growth factors in tumor tissues. In line with the above observations, there were no significant differences in MMP9 and PIGF expression in primary tumors from WT-BMT and miR-155−/−-BMT mice (Supplementary Fig. S4), albeit MMP2 expression was decreased in the tumors of miR-155−/−-BMT mice.
Figure 4.


Lack of effects of bone marrow miR-155 deficiency on macrophage infiltration in primary tumor. A, Representative fluorescence images of tumor tissues from WT and miR-155−/− chimeric mice at the time of primary tumor resection (14 days after implantation of LLC cells). DAPI was used to stain the nuclei of cells. Magnification, 10 ×. B, F4/80 cells (shown in green) were calculated as the ratio of green fluorescence protein-surface area to DAPI-surface area. Each of the six fields was averaged per mouse. C, Immunohistochemistry for vWF expression in primary tumor tissues from WT and miR-155−/− chimeric mice at day 14 after LLC inoculation. Magnification, 20 ×. D, Quantification of vWF microvessel density (MVD). vWF positive endothelial cell or cell cluster clearly separate from adjacent structures was considered a single microvessel. Each of the eight fields was averaged per mouse. Data are presented as the mean ± SEM of 8 mice.
miR-155 deficiency in bone marrow increased macrophage infiltration in the lungs
While the recruitment of macrophages to tumors has been well documented in many tumor models (5, 8, 31, 35), the role of macrophages in promoting metastatic cell seeding, enhancing tumor cell extravasation and subsequent growth of metastatic lesions in distant sites has only recently started to be appreciated. We thus examined if macrophages were recruited to the lungs in our tumor model and contributed to the increased lung metastasis. We found that the infiltration of F4/80+ macrophages into lung tissues was increased by 3.2-fold in miR-155−/−-BMT mice compared to that in WT-BMT counterparts (Figs. 5A and 5C, left panel). In response to microenvironmental signals, macrophages undergo different activation, including `classic' activation to produce a pro-inflammatory phenotype (also called M1) and the `alternative' activation to yield an anti-inflammatory phenotype (also called M2) (5, 36). We therefore investigated whether macrophages in the lungs of miR-155−/−-BMT mice exhibited more of a `M2' phenotype than those in the lungs of WT-BMT counterparts. Immunostaining was performed in lung tissues using an antibody against Ym1, a M2 macrophage marker. Interestingly, the lung tissues of miR-155−/−-BMT mice contained significantly more Ym1+ cells than those of WT-BMT mice (Figs. 5B and 5C, right panel). These results suggest that bone marrow miR-155 deficiency promoted infiltration and M2 polarization of macrophages in metastatic lungs under tumor burden.
Figure 5.


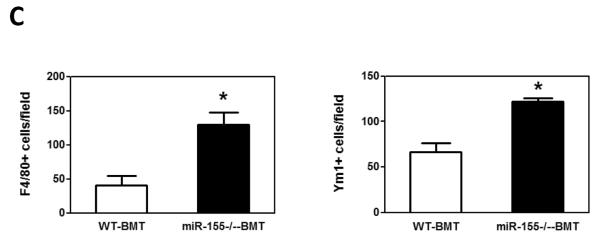
Elevated macrophage infiltration in the lungs of miR-155−/− chimeric mice. Representative fluorescence images of lung tissues from WT and miR-155−/− chimeric mice immunostained for F4/80 (A) (using FITC-conjugated rat anti-F4/80 antibody) and Ym1 (B) (using rabbit anti-mouse YM1 polyclonal antibody and Alexa Fluor 488-conjugated secondary antibody). Sections were counterstained with DAPI nuclear dye (in blue). A, magnification, 20 x; B, magnification, 10 ×; C, The numbers of F4/80 (left panel) or Ym1 (right panel) positive cells were counted in 6–12 random fields per section and the average was used for the calculation. Data are presented as the mean ± SEM of 8 mice. *p<0.05 by Student's t test.
miR-155−/− macrophages were prone to M2 polarization in vitro
To test if miR-155 deficiency promotes M2 polarization of macrophages, peritoneal macrophages from WT or miR-155−/− mice were stimulated with LLC cell conditioned medium (LCM) and the expression of Arg1, a potent M2 marker gene, was measured by quantitative real-time PCR. We found that, upon LCM treatment, mRNA level of Arg1 was significantly increased in miR-155−/− macrophages compared to WT macrophages (Fig. 6A). We also treated WT and miR-155−/− macrophages with IL-4 or B16-F10 cell conditioned medium (B16-CM). We found that both IL-4 and B16-CM treatment resulted in higher levels of Arg1 expression in miR-155−/− macrophages than in WT ones (Fig. 6B). Furthermore, we replicated the experiments using bone marrow-derived macrophages (BMDMs), and obtained similar results (Fig. 6C and D).
Figure 6.


miR-155−/− macrophages exhibit M2 phenotype. Quantitative real-time PCR analysis of Arg1 mRNA levels in WT and miR-155−/− macrophages incubated with SFM or LCM for 24 h (A) and in WT and miR-155−/− macrophages incubated with IL-4 (20 ng/ml) or B16-CM for 24 h (B). BMDM from WT and miR-155−/− mice were incubated with LCM (C) and IL-4 (D) for 24 h and Arg1 mRNA level was detected by quantitative real-time PCR. Data are presented as the mean ± SEM of three replicates. *, p<0.05 by Student's t test. E C/EBPβ is responsible for M2 polarization of miR-155−/−macrophages. WT and miR-155−/− macrophages were treated with SFM or LCM for 30 min, 8 h and 48 h; expression of C/EBPβ, SOCS1, and β–actin proteins was determined by Western blotting. β–actin was used as a loading control. The numbers under the protein bands indicate the normalized relative intensity; the band intensity relative to β–actin of the untreated WT sample at 30 min time-point was set as 1. Representative data from three independent experiments are shown.
The polarization of M2 macrophage is modulated primarily through transcription factors and their effector molecules, such as C/EBPβ, SOCS-1 and STAT6 (37–39). To identify the signaling pathways responsible for the M2 polarization of miR155−/− macrophages, we evaluated the protein levels of C/EBPβ, SOCS-1 and STAT6. C/EBPβ has been identified as a direct target of miR-155 in B cells and macrophages (40, 41). As expected, the basal level of C/EBPβ protein in miR-155−/− macrophages was higher than that in WT macrophages; LCM did not increase C/EBPβ levels in both WT and miR-155−/− macrophages at 30 min of incubation; however, C/EBPβ protein level was significantly increased 8 h after LCM treatment (Fig. 6E). Importantly, LCM-treated miR155−/− macrophages exhibited a significantly elevated level of C/EBPβ than LCM-treated WT macrophages (Fig. 6E). LCM treatment for 48 h resulted in much higher level of C/EBPβ expression in miR-155−/− macrophages than in WT ones (Fig. 6E). Increased expression of SOCS1, another confirmed target of miR-155, has been shown to re-polarize pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages (42). However, we did not observe an increase of SOCS1 expression in miR-155−/− macrophages compared to WT macrophages at any of the 30 min, 8 h and 48 h time-points (Fig. 6E). In addition, the levels of phosphorylated and total STAT6 did not show apparent difference between WT and miR-155−/− macrophages with or without LCM treatment (data not shown). Our data suggest that the enhanced M2 polarization of miR-155−/− macrophages compared to WT macrophages may be mainly due to increased C/EBPβ signaling pathway resulted from the loss of repression of C/EBPβ by miR155.
miR155 expression was not increased in LLC and B16-F10 cells and their conditioned medium reduced macrophage miR155 expression
It has been reported that miR-155 may function as an oncogene in several solid tumors. We examined the miR155 expression in cultured LLC and B16-F10 cells using quantitative real-time PCR, and we found that, compared to that in lung tissue of WT mice, miR155 expression was not increased in both of the cell types. Instead, miR155 expression was slightly but not significantly reduced in LLC cells, whereas it was significantly reduced in B16-F10 cells (sFig. 5A). To investigate if tumor cells may influence miR155 expression in macrophages, we measured the expression level of miR-155 in bone marrow-derived macrophages upon treatment with tumor cell derived conditioned medium. As shown in sFig. 5B, both LLC-CM and B16-CM resulted in a significant reduction of miR-155 expression in macrophages (99% and 77%, respectively), indicating a reciprocal regulation of tumor cells and mcarophages.
miR155 deficient MΦ promoted tumor cell migration and proliferation in vitro
Macrophages in tumor microenvironment may directly modify tumor cell behavior, such as migration and proliferation, which will facilitate tumor development (5, 31, 43). Firstly, to test if miR-155−/− macrophages promotes tumor cell migration, transwell migration assays were performed. As shown in Fig. 7A–B, migration of LLC cells towards serum-free medium treated miR-155−/− macrophages was significantly enhanced compared to that towards WT macrophages, and LLC cells migrating towards LLC-CM-treated miR-155−/− macrophages was significantly increased than that towards LLC-CM-treated WT macrophages, indicating that miR-155−/− macrophages possess much more potent chemoattracant ability for tumor cells. Similar results were obtained when B16-F10 cells were used for the assyas (Suppl. Fig. S6). The promoting effect of miR-155−/− macrophages on tumor cell migration might at least partially explain the increased lung metastasis in bone marrow miR-155 deficient mice.
Figure 7.

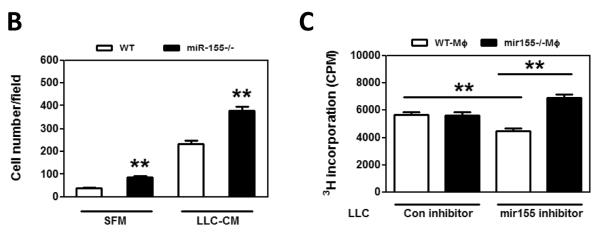
miR-155−/− macrophages promote tumor cell migration and proliferation. A, Transwell migration assay of LLC cells migrating towards WT and miR-155−/− MΦ treated with SFM and LLC-CM. Representative fluorescence images of migrated LLC cells. Calcein was used to stain the cells. Magnification, 200 ×. B Data were presented as the mean ± SEM of 20 fields/group. **, p<0.001 by Student's t test. C, [3H] Thymidine incorporation assay of LLC cell proliferation. LLC cells were transfected with Negative control (con inhibitor) and mir155 inhibitor for 24 h and were cocultured with WT MΦ and miR-155−/− MΦ for another 48 h. Data are represented as the mean ± SEM (n=6 per group). **, p<0.001 by Student's t test.
We next tested if miR-155−/− macrophages promote tumor cell proliferation. When LLC cells were co-cultured with either WT or miR155−/− macrophages, we did not found difference in the proliferation of LLC cells (data not shown). Then we sought to examine if miR155 expression level in LLC cells influences their proliferative response to macrophages. LLC cells were transfected with miR155 inhibitor or control inhibitor and the miR155 inhibition effeiciency was confimed by qPCR measurement of miR155 target gene expression, such as HIF-1α, Bcl-6, c-Maf and SOCS1 (Suppl. Fig. 7A). [3H] Thymidine incorporation assay was performed in the following groups: tumor cells transfected with control inhibitor co-cultured with WT macrophages (con inhibitor+WT MΦ), tumor cells transfected with control inhibitor co-cultured with miR155−/− macrophages (con inhibitor+miR-155−/− MΦ), tumor cells transfected with miR155 inhibitor co-cultured with WT macrophages (mir155 inhibitor+WT MΦ), and tumor cells transfected with miR155 inhibitor co-cultured with miR155−/− macrophages (mir155 inhibitor+miR-155−/− MΦ). There was no significant difference in LLC cell proliferation between con inhibitor+WT MΦ group and con inhibitor+miR-155−/− MΦ group (Fig. 7C, column 1 and 2). However, in mir155 inhibitor transfected LLC cells, addition of miR-155−/− MΦ significantly promoted LLC proliferation by 54% as compared with WT MΦ (Fig. 7C, column 3 and 4). Our data indicated that lower expression level of miR-155 in tumor cells may sensitize tumor cells to miR-155−/− MΦ. Interestingly, the LLC proliferation was significantly even though not dramatically inhibited by 19% in mir155 inhibitor transfected cells compared to con inhibitor group in the presence of WT macrophages (Fig. 7C, column 1 and 3). This phenomenon may be correlated with the oncogenic role of miR-155 in solid tumor cells. We also performed proliferation assays using B16-F10 cells, and found that even without miR155 inhibition, the proliferation of B16-F10 cells was enhanced by miR155−/− macrophages (Suppl. Fig. 7B)
Discussion
miRNAs have emerged as a class of gene expression regulators that are involved in many pathophysiology processes including cancer development. miR155 is one of miRNAs that have attracted much attention recently. Aberrant expression of miR-155 in hematopoietic cells drives development of several leukocyte-derived tumors. In addition, miR-155 expressed in certain types of solid tumor cells are increasingly implicated in the regulation of tumor growth and metastasis. Therefore, targeting miR-155 has been proposed as a promising strategy for treatment of cancers (25). However, how host hematopoietic miR-155 expression affects solid tumor development and metastasis remains unexplored. Here we showed that miR-155 deficiency in hematopoietic cells in tumor-bearing mice promoted macrophage infiltration in lungs and enhanced lung metastasis, although without obvious influence on primary tumor progression (Fig. 1 and 5). Therefore, our data uncover an important role for hematopoietic miR-155 in modulating the establishment of pro-metastatic microenvironment in the lungs, raising the concern that targeting miR-155 in host immune cells may enhance the metastasis of established cancer.
Metastasis is responsible for 90% of death in cancer patients. Metastasis requires not only the release of cells from the primary sites, but also tumor cell seeding, extravasation, and subsequent survival at distant sites as well as metastatic colonization, which are the major rate-limiting events for tumor metastasis. Hence, a truly effective anti-metastatic therapy must be capable of interfering multiple steps of the invasion-metastasis cascade. Unfortunately, many anti-metastatic agents currently in preclinical or clinical development stages can only block the dissemination of neoplastic cells from primary tumors but cannot sufficiently suppress metastasis formation in distant organs once tumor cells escape the primary tumors (44). For this reason, the development of new therapeutic agents that alter the microenvironment where the neoplastic cells proliferate and survive in the distant tissues represents an intriguing strategy to suppress tumor metastasis.
It is well recognized that metastasis of solid tumors requires collaborative interactions between malignant cells and a variety of “activated” stromal cells at both primary and metastatic sites (2, 3, 6, 30). Among them, macrophages are the most abundant leukocytes that present in neoplastic stroma (5, 8, 45). Emerging evidence has shown that macrophages potentiate the seeding and establishment of metastatic cells in distant tissues in addition to its well-defined functions in primary tumors (4, 11, 45, 46). Said et al. showed that tumor endothelin-1 (ET-1) expression in bladder cancer cells was necessary for metastatic lung colonization which was dependent on macrophage infiltration in the lungs (46). Gil-Bernabe AM et al. found that tissue factor (TF) expression by tumor cells enhanced tumor cells survival in the lungs by recruiting macrophages (45). Although there are only few studies focused on metastasis-associated macrophages, existing data indicate that infiltration of macrophages in metastatic sites is essential for effective metastasis. Our current study suggests that miR-155 expression in metastasis-associated macrophages may serve as a defense against cancer metastasis through halting macrophage polarization toward a pro-tumor M2 phenotype.
Macrophages in tumor microenvironment may directly modify tumor cell behavior, such as migration and proliferation, which will facilitate tumor development (5, 31, 43). Since tumor cell migration is a critical step of the metastatic cascade, we investigated the ability of WT and miR-155−/− macrophages to attract tumor cells by a transwell migration assay. We verified that miR-155−/− macrophages exerted a stronger chemotactic effect than WT macrophages on both LLC (Figure 7A–B) and B16-F10 cells (Suppl. Fig. 6), which may partially explain the increased lung metastasis in bone marrow miR-155 deficient mice. We also found that miR155−/− macrophages affected proliferation of LLC cells and B16-F10 cells. Interestingly, inhibition of miR155 expression in LLC cells reduced proliferation by 19% under the influence of WT macrophages (Fig 7C column 3 vs column 1), indicating inhibition of LLC miR155 expression could be beneficial; however, when these cells were incubated with miR155−/− macrophages, the proliferation was increased by 54% (Fig 7C column 4 vs column 3). Given the fact that LLC-CM suppressed macrophage miR155 expression by 99% (Suppl. Fig. 5B), it is conceivable that universal inhibition of miR155 expression in both tumor cells and macrophages will more likely favor metastasis of LLC. In the case of B16-F10 cells, even without miR155 inhibition, miR155−/− macrophages promoted their proliferation (Suppl. Fig. 7B). The difference between LLC cells and B16-F10 cells may be due to their expression levels of endogenous miR155; B16-F10 cells express much lower level of miR155 than LLC cells (Suppl. Fig. 5A).
By flow cytometry analysis, we found that macrophage was the only cell type that the percentage was increased in miR-155−/−-BMT mice, which indicated that macrophages might be recruited into primary tumor sites and metastatic lungs. However, in our study, macrophages specifically appeared in the lungs but not the primary tumors. We postulate that the expression level of chemokines that are capable of recruiting macrophages are specifically increased in lungs but not in tumor sites. During tumor development, tumor cell-secreted factors educate distant metastatic organs through activating stromal cells, such as resident macrophages, fibroblasts and endothelial cells, which creates a favorable microenvironment for the recruitment of bone marrow derived cells (BMDCs) (2, 29, 30, 32, 47). A variety of inflammatory factors, such as fibronectin, SDF1, MMP9, S100A8 and S100A9, upregulated in lung stromal cells, are responsible for the recruitment of macrophages and BMDCs from circulation into metastatic sites in tumor-bearing mice. Hiratsula et al. demonstrated that S100A8 and S100A9 were secreted by endothelial cells or Mac1+-myeloid cells in metastatic lungs whereas its expression was undetectable in primary tumors (30). Similarly, in our study, we found that S100A8 and S100A9, which are strong chemotactic agonists for monocytes and macrophages, were strongly induced in lung tissues after tumor cell inoculation but not in primary tumors. In addition, we also found that macrophages expressed TLR4, the receptor for S100A8 and S100A9, which indicated that TLR4/ S100A9 interaction may be responsible for the recruitment of macrophages into lung tissues. In addition, Kaplan et al. showed that fibronectin expression in resident fibroblasts was significantly increased in lungs of tumor-bearing mice compared to tumor-free mice (2). Increased fibronectin expression attracts VLA-4-expressing BMDCs migrating into lung tissues. In our model, we also observed an increase of fibronectin expression in lung of tumor-bearing mice.
Several genes that are involved in the regulation of macrophage phenotype have been identified or predicted as targets of miR-155, such as C/EBPβ, SOCS1 and c-Maf (18, 42, 48). C/EBPβ plays a critical role in regulation of Arg1 expression and M2 macrophage polarization (39). It has been demonstrated that miR-155 reduces the endogenous expression of C/EBPβ in macrophages through interacting with the 3'-untranslated region of the C/EBPβ mRNA (40, 48). Downregulation of miR-155 in Akt2−/− macrophages caused an increase of C/EBPβ expression which resulted in M2 polarization (49). In addition, SOCS1 is up-regulated in M2 macrophages and its rapid increase is critical for sustaining the anti-inflammatory phenotype and function of M2 macrophages (42). c-Maf, an identified target of miR-155 in Th2 cells (18), strongly activates IL-10 and IL-4 expression in macrophages (50, 51), potentially contributing to the state of anti-inflammatory function of macrophages. All these evidences indicate that miR-155 may modulate macrophage polarization. In our current study, we not only confirmed the high responsiveness of miR-155−/− macrophages to IL-4 stimulation (a potent M2 macrophage inducer) (Fig. 6), but further showed that miR-155-deficient macrophages were prone to M2 polarization under the treatment with tumor cell conditioned medium and that C/EBPβ played a crucial role in the increased M2 polarization of miR-155−/− macrophage (Fig. 6E). However, although the basal level of SOCS1 was slightly higher in miR-155−/− macrophages than in WT ones, tumor cell-conditioned medium failed to induce a further increase of SOCS1 level (Fig. 6E), indicating that SOCS1 may not be involved in the miR-155 modulation of macrophage polarization induced by tumor-derived factors. Our data are in agreement with two recent in vitro studies. Cao X et al. reported that overexpression of miR-155 could re-program anti-inflammatory, pro-tumorial M2 TAMs to pro-inflammatory, antitumor M1 macrophages (52). He et al. showed that induction of miR-155 suppressed C/EBPβ protein expression as well as cytokine production in tumor-activated monocytes (40). Importantly, our current work provides the first evidence that the modulation of macrophage polarization by miR-155 may operate in vivo in cancer metastasis.
Recent studies highlighted the oncogenic function of tumor cell-expressed miR-155, since miR-155 was found upregulated in multiple types of human cancers and ectopic expression of miR-155 significantly promoted tumor development in many experimental mouse models (24–26). These findings suggested that therapeutic targeting of miR-155 in malignant cells may provide a novel strategy for delaying tumor development. However, our current study suggests that universal inhibition of miR-155 in cancer patients may backfire. On the one hand, suppression of miR-155 in tumor cells may inhibit tumor cell growth; on the other hand, impaired expression of miR155 in host macrophages may promote tumor metastasis.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by grants NIH R21HL106325 (DF), AHA SDG4110005 (DF) and NIH K01DK069489 (WA).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.DeNardo DG, Barreto JB, Andreu P, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–7. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. 2006;13:118–37. doi: 10.1159/000092969. [DOI] [PubMed] [Google Scholar]
- 4.Qian B, Deng Y, Im JH, et al. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One. 2009;4:e6562. doi: 10.1371/journal.pone.0006562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20:300–14. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acharyya S, Oskarsson T, Vanharanta S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–78. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doedens AL, Stockmann C, Rubinstein MP, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–75. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–52. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest. 2012;122:1503–18. doi: 10.1172/JCI61392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eis PS, Tam W, Sun L, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–32. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haasch D, Chen YW, Reilly RM, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol. 2002;217:78–86. doi: 10.1016/s0008-8749(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 14.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu C, Huang X, Zhang X, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood. 2011;117:4293–303. doi: 10.1182/blood-2010-12-322503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–8. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–11. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Georgantas RW, 3rd, Hildreth R, Morisot S, et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A. 2007;104:2750–5. doi: 10.1073/pnas.0610983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costinean S, Zanesi N, Pekarsky Y, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–9. doi: 10.1073/pnas.0602266103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Berg A, Kroesen BJ, Kooistra K, et al. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer. 2003;37:20–8. doi: 10.1002/gcc.10186. [DOI] [PubMed] [Google Scholar]
- 22.Kluiver J, Haralambieva E, de Jong D, et al. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer. 2006;45:147–53. doi: 10.1002/gcc.20273. [DOI] [PubMed] [Google Scholar]
- 23.Kluiver J, Poppema S, de Jong D, et al. BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005;207:243–9. doi: 10.1002/path.1825. [DOI] [PubMed] [Google Scholar]
- 24.Shibuya H, Iinuma H, Shimada R, Horiuchi A, Watanabe T. Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology. 2011;79:313–20. doi: 10.1159/000323283. [DOI] [PubMed] [Google Scholar]
- 25.Jiang S, Zhang HW, Lu MH, et al. MicroRNA-155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119–27. doi: 10.1158/0008-5472.CAN-09-4250. [DOI] [PubMed] [Google Scholar]
- 26.Chang S, Wang RH, Akagi K, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17:1275–82. doi: 10.1038/nm.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yancey PG, Ding Y, Fan D, et al. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory Ly-6Chigh monocytosis: evidence that the effects are not apolipoprotein E dependent. Circulation. 124:454–64. doi: 10.1161/CIRCULATIONAHA.111.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dawson MR, Duda DG, Chae SS, Fukumura D, Jain RK. VEGFR1 activity modulates myeloid cell infiltration in growing lung metastases but is not required for spontaneous metastasis formation. PLoS One. 2009;4:e6525. doi: 10.1371/journal.pone.0006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hiratsuka S, Nakamura K, Iwai S, et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289–300. doi: 10.1016/s1535-6108(02)00153-8. [DOI] [PubMed] [Google Scholar]
- 30.Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–75. doi: 10.1038/ncb1507. [DOI] [PubMed] [Google Scholar]
- 31.Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol. 2011;21:139–46. doi: 10.1016/j.semcancer.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Wu Y, Li YY, Matsushima K, Baba T, Mukaida N. CCL3-CCR5 axis regulates intratumoral accumulation of leukocytes and fibroblasts and promotes angiogenesis in murine lung metastasis process. J Immunol. 2008;181:6384–93. doi: 10.4049/jimmunol.181.9.6384. [DOI] [PubMed] [Google Scholar]
- 34.Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13:193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 36.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–12. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 37.Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM., Jr. Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene. 2005;353:98–106. doi: 10.1016/j.gene.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Ruffell D, Mourkioti F, Gambardella A, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A. 2009;106:17475–80. doi: 10.1073/pnas.0908641106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–61. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 40.He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Immunol. 2009;6:343–52. doi: 10.1038/cmi.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costinean S, Sandhu SK, Pedersen IM, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood. 2009;114:1374–82. doi: 10.1182/blood-2009-05-220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whyte CS, Bishop ET, Ruckerl D, et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J Leukoc Biol. 2011;90:845–54. doi: 10.1189/jlb.1110644. [DOI] [PubMed] [Google Scholar]
- 43.Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20:538–49. doi: 10.1016/j.ccr.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith SC, Theodorescu D. Learning therapeutic lessons from metastasis suppressor proteins. Nat Rev Cancer. 2009;9:253–64. doi: 10.1038/nrc2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gil-Bernabe AM, Ferjancic S, Tlalka M, et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood. 2012;119:3164–75. doi: 10.1182/blood-2011-08-376426. [DOI] [PubMed] [Google Scholar]
- 46.Said N, Smith S, Sanchez-Carbayo M, Theodorescu D. Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. J Clin Invest. 2010;121:132–47. doi: 10.1172/JCI42912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–93. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Connell RM, Rao DS, Chaudhuri AA, et al. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med. 2008;205:585–94. doi: 10.1084/jem.20072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arranz A, Doxaki C, Vergadi E, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A. 2012;109:9517–22. doi: 10.1073/pnas.1119038109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao S, Liu J, Song L, Ma X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol. 2005;174:3484–92. doi: 10.4049/jimmunol.174.6.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho IC, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85:973–83. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 52.Cai X, Yin Y, Li N, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012;4:341–3. doi: 10.1093/jmcb/mjs044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.