

Abstract
Autophagy has emerged as a critical lysosomal pathway that maintains cell function and survival through the degradation of cellular components such as organelles and proteins. Investigations specifically employing the liver or hepatocytes as experimental models have contributed significantly to our current knowledge of autophagic regulation and function. The diverse cellular functions of autophagy, along with unique features of the liver and its principal cell type the hepatocyte, suggest that the liver is highly dependent on autophagy for both normal function and to prevent the development of disease states. However, instances have also been identified in which autophagy promotes pathological changes such as the development of hepatic fibrosis. Considerable evidence has accumulated that alterations in autophagy are an underlying mechanism of a number of common hepatic diseases including toxin-, drug- and ischemia/reperfusion-induced liver injury, fatty liver, viral hepatitis and hepatocellular carcinoma. This review summarizes recent advances in understanding the roles that autophagy plays in normal hepatic physiology and pathophysiology with the intent of furthering the development of autophagy-based therapies for human liver diseases.
Keywords: autophagy, liver, hepatocyte, hepatitis, ischemia/reperfusion, liver injury, hepatotoxin, hepatocellular carcinoma, drug toxicity
Introduction
Autophagy is an intracellular degradative pathway that targets cytosolic components to lysosomes to be degraded for the purposes of maintaining cellular homeostasis and supplying substrates for energy generation. Our understanding of the functions and regulation of this lysosomal degradative pathway has grown tremendously over the past few years in large part through investigations conducted in yeast and Drosophila, but increasingly from studies in mammalian cells and tissues as well. Of the three known primary types of autophagy, macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy, most of our current knowledge is concentrated on macroautophagy (hereafter referred to as autophagy), which is the focus of this review. In autophagy, cytosolic constituents are encircled by a double-membrane structure termed an autophagosome that fuses with a lysosome. In the resulting autolysosome, enzymes degrade the cargo of the autophagosome for release back into the cytosol. The factors controlling this pathway are complex and include more than 30 autophagy-related (ATG) genes. The components of the autophagic pathway and their regulation have been well described in other recent reviews.1,2
Features and functions peculiar to the liver identify it as an organ in which autophagy potentially plays an important role. For these reasons the liver has served as a frequent model for very basic investigations of autophagy. As a result, several selective forms of autophagy, mitophagy3,4 and lipophagy,5 were first described in studies of cultured hepatocytes and whole liver. A number of features of hepatocytes and the liver as a whole make this organ particularly dependent on autophagy. First, the liver is unique in its regenerative properties in that hepatocytes are normally in a quiescent state but retain the ability to quickly enter the cell cycle when there is a loss of liver mass from injury or surgical resection. The lack of cell turnover makes hepatocytes particularly vulnerable to the effects of impaired autophagy, as long-lived cells have time to accumulate high levels of products that are normally disposed of by cell division and/or autophagy. When excessive levels of damaged organelles and oxidized or aggregated proteins are allowed to accumulate, cellular injury or transformation can occur. In addition, autophagy may have other metabolic or proliferative functions during the rapid regeneration that occurs in the liver, but this possibility has not yet been examined. Second, the liver is an important metabolic organ, not only in terms of the requirements of its own cells which have a large number of mitochondria, but also for the whole body as the liver produces glucose and stores fat. The recently described ability of autophagy to regulate these metabolic pathways implicates autophagy as an important modulator of hepatic metabolism. The anatomy of the liver also makes it unique in that it directly receives all of the portal blood supply from the intestines. As a result, the liver is the largest immune organ in the body and faces the continual challenge of exposure to orally-ingested antigens as well as products released by intestinal bacteria such as lipopolysaccharide (LPS). The potent innate immune response of the liver, which is normally protective against exogenous antigens, can become overactivated during liver injury, and promote cellular damage. Finally, the hepatocyte is the primary site of infection for a number of various liver trophic viruses that cause some of the most common infectious diseases worldwide. The emerging importance of autophagy in regulation of the immune response6 is another important area of involvement for autophagy in the liver.
This review discusses some of the critical functions of autophagy in normal hepatic physiology as well as the evidence for mechanistic roles of autophagy in diseases of the liver. Considerable data already link autophagic function with the pathophysiology underlying the most common diseases of the liver. A critical question in these investigations is whether autophagic function is impaired in these disease states, which has important implications for whether therapeutic strategies designed to alter hepatic levels of autophagy may be effective treatments for these diseases. The careful demonstration of a defect in autophagy in the genetic liver disease of SERPINA1/α1-antitrypsin deficiency (ATD) has already prompted a human clinical trial of the effects of an autophagy-inducing drug in this disease. However, basal levels of autophagy are critical, particularly in the liver, as evidenced by the spontaneous development of liver disease in mice with a hepatocyte knockout of autophagy.7 Thus, maintaining or augmenting constitutive levels of autophagy may be an important therapeutic strategy as well.
Basic Functions of Autophagy in the Liver
Autophagy in hepatic protein degradation
The lysosomal pathway was first implicated in the turnover of cellular proteins in the 1940s, and it was recognized subsequently that proteins along with other intracellular constituents were degraded in lysosomes by the process of autophagy.8 Initially autophagy was considered a pathway in which long-lived cytosolic proteins and organelles were degraded nonselectively. More recently it has been recognized that autophagy is also able to target specific cytosolic components such as aggregated proteins, damaged/excess organelles and lipids for selective degradation.
The liver has a basal level of autophagic function that is significantly increased in response to starvation. In rodents housed in environmentally controlled rooms, the liver is undergoing basal levels of autophagy during the day when the animals are not feeding. Using perfused liver systems it has been estimated that basal autophagic function degrades 1.5% of total hepatic protein per hour.9 In response to starvation, the rate of protein degradation increases to 4.5% of total liver protein per hour.9 When rodents are starved for 48 h, increased levels of autophagy can account for the degradation of 40% of total liver protein.10 Thus, changes in hepatic autophagic function can have dramatic effects on hepatic physiology. Hepatocytes are thought to have higher levels of autophagy than other cell types because of their increased abundance of lysosomes and lysosomal enzymes such as CTSL/cathepsin L.11 This increased autophagy is another reason why the liver is a convenient organ in which to examine autophagic regulation and function.
Early studies of the regulation of hepatic autophagy demonstrated that the nutritional control of autophagy could be explained in part due to its suppression by amino acids and insulin, and its stimulation by glucagon.12 Studies in cultured primary hepatocytes have shown that autophagy is inhibited by 3-methyladenine, implicating the class III phosphatidylinositol 3-kinase (PtdIns3K) as a regulator of hepatic autophagy.12,13 The suppressive effect of amino acids becomes apparent when their levels reach 2–4 times normal,9 and leucine and glutamine are the most potent of the amino acids having this effect.14,15 The mechanism by which glucagon exerts its activating effect on hepatic autophagy is not completely understood, but the inhibition of autophagy by insulin signaling is mediated by effects on the kinase mechanistic target of rapamycin (MTOR). Binding to the insulin receptor leads to phosphoinositide 3-kinase (PI3K)-AKT activation and inactivation of the tuberous sclerosis TSC1-TSC2 complex which, in turn, releases inhibition of RHEB that then activates MTOR.16 Studies in perfused liver have suggested that the G protein GNAI3/Giα3 is required for suppression of autophagy by insulin, as Gαi3 knockout mice fail to downregulate autophagy in response to insulin.17
A more definitive understanding of the role of autophagy in hepatic protein degradation has come from studies of a liver-specific autophagy-knockout mouse model generated by conditional deletion of the autophagy gene Atg7.7 The livers of these mice are markedly enlarged, taking up to 30% of the body weight of the mouse, and the cells are characterized by marked structural alterations of mitochondria and peroxisomes, and the intracellular accumulation of polyubiquitinated proteins. These findings therefore provide sophisticated evidence for the essential role of autophagy in the disposal of damaged and aggregated proteins and turnover of organelles. Interestingly, the aggregates of polyubiquitinated proteins disappeared when the liver-specific ATG7-knockout mouse was bred to a mouse null for SQSTM1/p62, a scaffolding protein degraded by autophagy. These findings demonstrate that SQSTM1 is essential to direct damaged or aggregated cytosolic proteins into the autophagic pathway.18
The increase in autophagy that occurs in response to starvation plays an essential role in supplying amino acids and substrates for energy production that hepatocytes need to survive nutrient deprivation. There is also evidence that starvation-induced autophagy contributes to hepatic glucose production through the action of glucogenic amino acids. In the liver-specific Atg7-null mouse, blood glucose levels decline during starvation to a significantly greater extent than in control mice.19 Taken together, these studies establish the essential role of basal autophagy in the turnover of intracellular organelles, degradation of long-lived cytosolic proteins and damaged proteins, and the contribution of induced autophagy to cell survival by supplying amino acids, glucose and energy needed for cellular integrity during stress.
Selective mitochondrial autophagy (mitophagy)
Mitophagy during nutrient deprivation and mitochondrial turnover
Mitochondria are a major source of the substrates supplied by hepatocytes from the increase in autophagy during starvation. Mitochondria are particularly rich in protein and lipids, and approximately 85% of autophagic events during nutrient deprivation of cultured hepatocytes involve selective autophagy of mitochondria, a process termed mitophagy.20,21 In healthy liver, despite minimal cell proliferation, individual mitochondria turn over with a half-life of 10 to 25 d, as basal levels of mitophagy remove worn out mitochondria in balance with the biogenesis of new mitochondria.22,23 Elimination of aged and damaged mitochondria protects cells against mitochondrial release of pro-apoptotic proteins, generation of toxic reactive oxygen species (ROS) and futile hydrolysis of ATP after mitochondrial depolarization.3,21,24,25 Mitophagy also eliminates mitochondria during cytoplasmic remodeling under nutrient-replete conditions and degrades mitochondrial DNA, including DNA that has been damaged or mutated.20,26,27 Both inadequate and excessive mitophagy promote cell injury and death.24,28,29 Thus, a balanced regulation of mitophagy is vital for cellular homeostasis.
Time course of mitophagy
Microtubule-associated protein 1 light chain 3 (LC3) associates with forming and newly formed autophagosomes, and the fusion protein, green fluorescent protein-LC3 (GFP-LC3), is a fluorescent marker of autophagosome formation.30 Mostly diffuse in the cytosol under nutrient-replete conditions, with an induction of autophagy GFP-LC3 incorporates into small (0.2–0.3 μm) pre-autophagic structures in proximity to mitochondria.20 After nutrient deprivation, these pre-autophagic structures grow into cup-shaped phagophores that envelop and then sequester individual mitochondria within autophagosomal vesicles termed mitophagosomes (Fig. 1A). Sequestration frequently occurs coordinately with mitochondrial fission and once initiated is complete within 6–7 min. Mitochondria maintain their membrane potential during sequestration and depolarize only after sequestration is complete, as indicated by loss of the red fluorescence of the membrane potential-indicating fluorophore tetramethylrhodamine methylester. After sequestration, mitophagosomes fuse with lysosomes and acidify. Mitochondrial contents are then digested within approximately 10 min.4,20 The pharmacological PI3K and PtdIns3K inhibitors wortmannin and 3-methyladenine block mitophagic sequestration almost completely, signifying that nutrient deprivation-induced mitophagy involves the classical BECN1-PIK3C3/VPS34 autophagic pathway.4,20,31
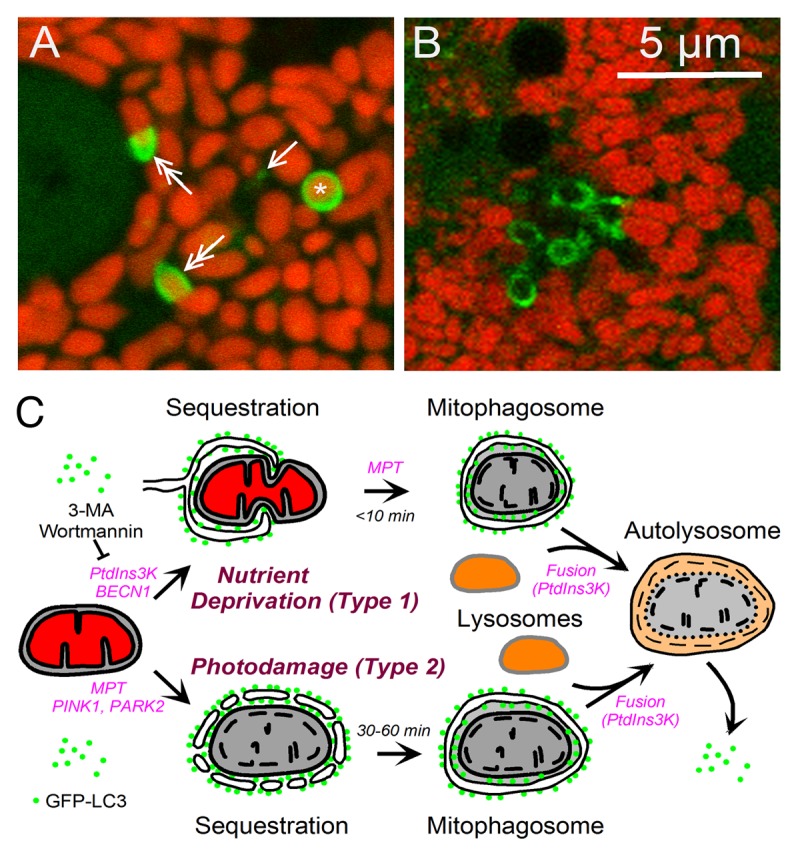
Figure 1. Type 1 and type 2 mitophagy. In (A and B), GFP-LC3 transgenic hepatocytes were loaded with red-fluorescing tetramethylrhodamine methylester, an indicator of mitochondrial polarization. (A) Nutrient deprivation-induced (type 1) mitophagy in a GFP-LC3 transgenic hepatocyte. Note the presence of a pre-autophagic structure (arrow), phagophores forming around mitochondria (double arrows) and a mitophagosome containing a red-fluorescing polarized mitochondria (asterisk). (B) Photodamage-induced (type 2) mitophagy in a wortmannin-treated GFP-LC3 transgenic hepatocyte. In this form of mitophagy, mitophagosomes indicated by green rings contain depolarized mitochondria, which therefore lack fluorescence. (C) Scheme of type 1 and 2 mitophagy. In type 1 mitophagy induced by nutrient deprivation, PtdIns3K-BECN1 activation leads to formation of a GFP-LC3-labeled phagophore, which sequesters a polarized mitochondrion into a mitophagosome, often in coordination with mitochondrial fission. Mitochondrial depolarization follows sequestration, which can be blocked by inhibitors of the MPT. The mitophagosome then undergoes PtdIns3K-dependent fusion with lysosomes, and hydrolytic digestion of the entrapped mitochondrion occurs. In Type 2 mitophagy induced by photodamage, photoirradiation causes MPT onset and sustained mitochondrial depolarization. GFP-LC3 attaches to the depolarized mitochondrion and by coalescence forms a mitophagosome in a PtdIns3K-independent fashion. Further mitophagosome processing occurs identically to the type 1 pathway.
Damage-induced mitophagy
Global mitochondrial injury from mitochondrial uncoupling (depolarization) and oxidative stress induces a robust autophagic response.32,33 Evidence that depolarization of single mitochondria induces mitophagy comes from photodamage experiments in cultured hepatocytes where small groups of mitochondria are exposed to 488-nm laser light which damages mitochondrial flavoproteins and promotes ROS production.3,34-36 Light exposure in this way depolarizes mitochondria transiently at lower illumination, but with a stronger light exposure sustained irreversible depolarization occurs, which is accompanied by inner membrane permeabilization akin to the mitochondrial permeability transition (MPT).
In nutrient-replete hepatocytes, GFP-LC3 fluorescence begins to decorate the edges of depolarized mitochondria approximately 30 min after laser-induced photodamage when depolarization is sustained and not transient. Subsequently, the GFP-LC3 fluorescence coalesces, and individual mitophagosomes form that then acidify. However, no stimulation of autophagy occurs outside the region of photoirradiation. Surprisingly, PI3K and PtdIns3K inhibitors do not block photodamage-induced mitophagy (Fig. 1B).
Variants of mitophagy: Type 1 and type 2
These observations suggest that mitophagy has two variants (Fig. 1C). Nutrient deprivation-induced mitophagy typifies type 1 mitophagy in which pre-autophagic structures grow to envelop and sequester mitochondria into mitophagosomes, often in coordination with mitochondrial fission. In type 2 mitophagy, as exemplified by photodamage-induced mitophagy, aggregates of GFP-LC3 decorate the periphery of damaged depolarized mitochondria and coalesce into mitophagosomes. In type 2 mitophagy, cup-shaped phagophores do not appear to form, and mitochondrial fission is absent. Nonetheless, once formed, mitophagosomes acidify and degrade their contents in both variants of mitophagy. Importantly, PI3K and PtdIns3K inhibition with 3-methyladenine or wortmannin blocks type 1 mitophagy completely, but type 2 mitophagy not at all.
In type 1 mitophagy, mitochondrial depolarization does not occur until after a mitochondrion is captured inside a mitophagosome, whereas depolarization is required to initiate sequestration in type 2 mitophagy. In mammalian cells, PTEN-induced putative kinase 1 (PINK1) and PARK2 (an E3 ubiquitin ligase) are implicated in mitophagy induced by uncoupling.33,37-40 PINK1 and PARK2 are proteins in which mutations cause familial forms of Parkinson disease. PINK1 recruits PARK2 to the outer membranes of depolarized mitochondria, which in turn ubiquitinates outer membrane proteins to target mitochondria for mitophagy. Future work will be needed to determine whether involvement of PINK1 and PARK2 is unique to type 2 mitophagy or whether PINK1 and PARK2 also play a role in type 1 mitophagy where mitochondrial depolarization follows rather than precedes autophagic sequestration. Nonetheless, an important distinction between type 1 and type 2 mitophagy is that type 1 mitophagy sequesters mitochondria that are polarized and apparently normal, as during nutrient deprivation and cytoplasmic remodeling, whereas type 2 mitophagy specifically targets depolarized mitochondria as a mechanism to clear cells of the dysfunctional organelles. Another open question is the role of the MPT in type 1 and 2 mitophagy. Although mitochondrial depolarization after MPT onset seems sufficient to induce type 2 mitophagy, MPT inhibitors such as cyclosporin A and nonimmunosuppressive N-methyl-4-isoleucine cyclosporine (NIM811) also block type 1 mitophagy, apparently by preventing mitochondrial depolarization after sequestration.4,26,41-43 Future studies will be needed to address these and other questions about mitophagy.
Autophagy mediates hepatocellular lipid metabolism
Another one of the organ-specific functions of the liver that make it highly dependent on autophagy is that the liver serves as the second largest repository of stored lipids in the body after adipose tissue. Hepatocytes are a major cellular storehouse for neutral lipids in the form of triglycerides (TGs) and cholesterol esters contained in specialized organelles termed lipid droplets (LDs).44,45 Until recently the breakdown of these lipid stores had been thought to occur exclusively from the actions of cytosolic lipases. A curious difference between liver and adipose tissue, despite their common function in lipid storage, is the relative paucity of cytosolic lipases in hepatocytes as compared with adipocytes. For this reason it had been previously unclear as to how hepatocytes could rapidly mobilize their lipid stores in times of metabolic need.46 Lipids can be degraded in lysosomes which contain acidic lipases and break down exogenous lipoproteins. This fact, together with the realization that autophagy and lipolysis have similar functions and hormonal control, suggested that autophagy may degrade endogenous lipids as well. Studies have now clearly demonstrated that autophagy mediates the breakdown of intracellular LD stores through the process of lipophagy.5
Lipophagy was first identified in the liver by studies of a pharmacological or genetic inhibition of autophagy in hepatocytes in culture or in vivo. A block in autophagy increases cultured hepatocyte TG content and LD number and size in response to a lipid challenge.5 The trafficking of intracellular lipids and LD proteins through autophagosomes and lysosomes was demonstrated by fluorescence and electron microscopy and the movement of lipid through the autophagic pathway increases with lipid supplementation.5 These findings were confirmed in mice with a hepatocyte-specific knockout of Atg7. The loss of hepatocyte autophagy leads to a marked increase in hepatic TG and cholesterol content, demonstrating that lipophagy limits hepatocyte lipid accumulation in vivo.5 In addition to serving as a mechanism to regulate intracellular lipid stores, lipophagy controls cellular energy homeostasis by providing free fatty acids (FFAs) from the breakdown of TGs. FFAs drive rates of mitochondrial β-oxidation and cellular ATP generation.5 Thus, lipophagy not only regulates amounts of passively stored lipid, but also controls active rates of cellular metabolism and energy generation.
Increased movement of lipid droplets into autophagic compartments occurs in cultured hepatocytes with lipid supplementation, and in mouse livers during nutrient deprivation. Biochemical and electron microscopy findings of an increased association of the autophagosomal protein LC3 with LDs were seen in mouse livers in response to starvation. With starvation a remarkable switch of cargo selection occurs in autophagosomes as the number with lipid cargo increases markedly with the lengthening time of starvation.5 These findings identified lipophagy as another selective form of autophagy that could mobilize cellular lipid stores at variable rates that depend on the exogenous supply of lipids and other nutrients.
How LDs are selectively targeted for autophagy in response to nutritional signals is not yet known, but likely involves protein-protein interactions between membrane proteins on the phagophore and the LD. Numerous proteins have been identified that are part of the phospholipid coating of LDs.44,45 Recent studies have demonstrated that the autophagosomal protein LC3, which is critical for autophagosome membrane formation,2 associates with LDs. The association of LC3 with LDs in the apparent absence of a phagophore membrane5,47 suggests an additional possible function for this protein in the recognition of LDs by the autophagic pathway. Other possibilities include the soluble NSF attachment protein receptors (SNAREs) which have been co-implicated in LD fusion48 and autophagosome biogenesis.49,50 The eventual identification of the structural components that trigger the selective process of lipophagy may suggest new therapeutic targets to prevent hepatocyte steatosis.
By supplying FFAs that can be incorporated into lipoproteins for export, hepatic lipophagy may serve as a mechanism to regulate whole body metabolism. This possibility implies that extrahepatic controls must exist to integrate levels of hepatic lipophagy with global nutritional status. Studies have begun to identify such pathways of crosstalk between hepatic autophagy and external metabolic signals. One example is hormonal control of hepatic lipophagy by thyroid hormone.51 This association was suggested by the known function of the active form of thyroid hormone, 3,3′5-triiodo-thyronine (T3), as a critical regulator of tissue metabolism including the induction of mitochondrial β-oxidation in the liver.52 T3 induces autophagy in cultured hepatocellular carcinoma cells and mouse liver.51 The ability of T3 to increase hepatic β-oxidation is dependent on autophagy, as an ATG5 knockdown prevents this effect. T3 induces lipophagy that increases the delivery of FFAs to mitochondria to elevate rates of β-oxidation. The mechanism of induction of autophagy by T3 remains to be determined, but AMP-activated protein kinase is likely involved, as this kinase modulates mitophagy in response to changes in cellular energy levels.53 The ability of T3 to cause lipid breakdown by inducing lipophagy may explain the known but unexplained inverse relationship in humans between thyroid hormone levels and the development of steatosis in nonalcoholic fatty liver disease.54
Another example of the responsiveness of hepatic lipophagy to external signals is that which occurs from circadian rhythms. Hepatic lipophagy is regulated by changes in ATG14 mediated by the forkhead box O (FOXO) family of transcription factors and circadian rhythms,55 which will be discussed in more detail subsequently. Additional regulatory pathways of hepatic lipophagy will likely be delineated, including ones mediated by the central nervous system, as part of a complex regulation of hepatic lipid metabolism.
The finding that autophagy mediates endogenous lipid metabolism provides a new mechanism by which cellular levels of autophagy may regulate liver physiology and pathophysiology. The most obvious implication is that levels of autophagy may modulate the excessive cellular lipid accumulation that underlies the steatotic liver diseases of alcoholic and nonalcoholic fatty liver.56,57 However, the critical involvement of lipophagy in the maintenance of mitochondrial β-oxidation suggests a much broader function for lipophagy in any instance in which lipid metabolism is supporting cellular energy homeostasis. Lipophagy may therefore be an important survival mechanism against cellular injury, and a reduction in autophagy sensitizes hepatocytes to cell death from oxidant stress as the result of ATP depletion from impaired β-oxidation.58 Lipophagy is also essential to sustain levels of β-oxidation for the energy-dependent process of hepatic stellate cell activation that occurs in response to fibrogenic stimuli.59 This finding emphasizes the fact that lipophagy functions in all cells, not just the fat-storing hepatocyte, and studies are needed in other liver cell types to fully define the role of lipophagy in the liver. The function of this selective form of autophagy in specific examples of hepatic pathophysiology will be discussed in more detail in subsequent sections.
Autophagy modulates cell death
Autophagy has long been recognized as a critical pathway in the regulation of cell death and survival.60-63 The role of autophagy in cell death can be confusing. Frequently, the same stimulus triggers autophagy and cell death simultaneously. In fact, a category of cell death termed autophagic cell death was proposed early on based on the coexistence of these two phenomena in cells.64 However, the true role of autophagy, which can be prodeath or prosurvival, was often not clearly defined in these early studies due to a reliance on nonspecific pharmacological inhibitors of autophagy. The currently recommended approach to differentiate the role of autophagy in cell death is to inhibit key autophagy genes through genetic deletion or RNAi-mediated knockdown. In this way it can be determined whether cell death or long-term cell survival is suppressed, enhanced or not changed at all by autophagy.65
Recent studies with careful genetic inhibition of autophagy have established that autophagy functions mainly as a prosurvival pathway. Obvious functions of autophagy that may be mechanisms of cell survival are the removal of damaged or harmful intracellular components or factors, or the supply of nutrients to maintain cellular energy homeostasis under adverse conditions. However, autophagy may promote cell death when the process is dysfunctional, resulting in excessive catabolism, cargo misrecognition and/or activation of the apoptotic machinery.
Autophagy promotes cell survival
Autophagy can play a prosurvival role under normal physiological conditions or pathological stress. In neonatal mice, autophagy is required for the endogenous generation of nutrients in energy-dependent organs like heart and diaphragm as the newborn adapts to taking in nutrients from an exogenous source, the mother’s milk.66 A global inhibition of autophagy therefore leads to the rapid postnatal death of newborn mice.66 At the cellular level, the importance of autophagy for survival during nutrient or growth factor deprivation has been well defined in mammalian and yeast cells.67
Autophagy is also important for cellular survival under stressful conditions. In mammalian cells, autophagy is activated in response to metabolic stress, ischemia or hypoxia.68 Suppression of autophagy in these instances can result in increased cell death. In the context of liver injury, autophagy is protective against liver injury caused by alcohol, acetaminophen (APAP) and ischemia/reperfusion injury as will be discussed in subsequent sections. Thus, it may be beneficial to promote autophagic function in these conditions. In other instances the involvement of autophagy in cell death may make it advantageous to inhibit autophagic function. Cytotoxic compounds, including many chemotherapeutic agents, can activate autophagy, likely secondary to their induction of cellular damage. For example, the multikinase inhibitor sorafenib is a beneficial treatment for advanced hepatocellular carcinoma (HCC). However, the response of tumor cells to ER stress or MTOR suppression during treatment can induce protective autophagy, which reduces efficacy of the drug.69,70 Simultaneous suppression of autophagy can be used to enhance sorafenib-induced tumor cell death and tumor regression.
Autophagy promotes cell survival through its basic function of degrading intracellular components. In nutrient/growth factor depletion, autophagic degradation recycles the cellular proteins and glycogen to provide amino acids and glucose for ATP generation.71 Under pathological conditions, autophagy may promote cell survival by the clearance of misfolded proteins, accumulated lipids and/or damaged mitochondria. Removal of misfolded proteins resulting from ER stress, proteasome inhibition or genetic mutation is an important mechanism by which autophagy maintains cell viability,72 and will be discussed later in the context of liver injury and cellular toxicity in SERPINA1/α1-antitrypsin deficiency. In alcoholic liver injury, autophagy may remove lipid droplets and damaged mitochondria to reduce oxidative stress and lipid peroxidation to protect hepatocytes.73 Thus, considerable interest exists in the modulation of autophagy as both a mechanism of hepatic cell death, and as a potential pathway to exploit in order to prevent liver injury.
Autophagy can mediate cell death
Although the function of autophagy is mainly prosurvival, the possibility exists that autophagy promotes cell death in some situations. The clearest example in which autophagy mediates cell death is in the development of the salivary glands in Drosophila.74 In mammalian cells, cell death related to autophagy has been reported in stressful conditions in response to certain chemotherapeutic drugs, radiation, hypoxia and ischemia.62 In these cases, deletion or RNAi-mediated knockdown of key autophagy genes can significantly reduce cell death, while overexpressing these genes promotes death. The mere ability of a knockdown of autophagy to protect against cell death is not sufficient proof that autophagy is promoting death, as cells lacking macroautophagy may be resistant to cell death because of crosstalk among autophagic pathways that leads to the protective upregulation of CMA.75
How autophagy promotes cell death is not entirely clear. Although it is tempting to assume that excessive self-digestion could lead to the depletion of key molecules or organelles essential to cell survival, the mechanisms of killing may be as diverse as the stress signals that induce autophagy in the first place. The autophagic machinery may directly interface with apoptotic factors or necrotic pathways to promote cell death. For example, ATG5 has been reported in nonhepatic cells to bind to FADD [Fas (TNFRSF6)-associated via death domain] and activate CASP8 and downstream caspases after death receptor engagement.76 Another example is that ATG5 overexpression leads to its cleavage by calpains into a 24-kDa ATG5 N-terminal fragment that translocates to mitochondria. There, this cleavage product binds to BCL2L1 and inactivates it, resulting in cytochrome c release and cell death.77 Finally, the autophagy factor BECN1, which possesses the conserved BH3 domain of the BCL2 family proteins, can interact with multiple antideath BCL2 family members, such as BCL2 and BCL2L1,78 which leads to mutual suppression. In these cases, autophagy is linked to the classical apoptosis pathway and cell death is actually mediated by the apoptotic machinery. Whether such mechanisms mediate hepatic cell death remains uninvestigated.
Factors that determine whether autophagy is prosurvival or prodeath
The role of autophagy in cell death could switch between promotion and inhibition depending on the context. One determinant could be the level of autophagy. In C. elegans, physiological levels of autophagy during starvation are prosurvival, whereas excessive autophagy can be prodeath.79 The presence of a compensatory mechanism, such as CMA, may also determine whether inhibition of macroautophagy renders cells sensitive or resistant to certain stressful signals. Murine fibroblasts prepared from ATG5-knockout embryos have increased levels of death receptor-initiated death, but are more resistant to menadione- and UV radiation-induced death due to a compensatory increase in CMA.75 Cellular transformation could also affect how autophagy functions. Autophagy induced by ER stress,80 or proteasome inhibitors,81 is protective in tumor cells, but indifferent or detrimental in nontransformed cells. The context-dependent function of autophagy in cell death needs to be better understood so that this role of autophagy may be manipulated for the control of liver injury and treatment of liver cancer.
Regulation of the immune response by autophagy
Autophagy plays multiple roles in immunity, both in the sensing of infection and as an effector of the immune response.82,83 As a mechanism of delivering cytoplasmic content to endosomes/lysosomal compartments that are enriched in immune sensors, autophagy functions in the detection of microbial infection. Interferon production in response to vesicular stomatitis virus infection requires the autophagic delivery of cytosolic replication intermediates to the endosome and subsequent activation of toll-like receptor (TLR) 7.84 Similarly, autophagy is required for TLR3 stimulation in coxsackievirus B3 infection.85 Additionally, activation of innate immune signaling molecules, such as TLRs or EIF2AK2/protein kinase R stimulates autophagy.86-89 Thus, autophagy is part of a feedforward mechanism in innate immune sensing, wherein autophagy stimulates TLR signaling, which in turn increases the induction of autophagy. The LPS-TLR pathway is important to many pathophysiological conditions in the liver such as hepatocellular injury and fibrosis,90 suggesting that TLR-mediated effects on autophagy may affect many of these processes.
In addition to sensing infection, autophagy performs two related antimicrobial effector functions: microbial destruction, and the processing of antigen for MHC presentation. The specific degradation of microbes is by a process of selective autophagy termed xenophagy. Although the mechanism is likely to vary somewhat depending on the infectious agent, the process is typified by the post-translational modification of microbial protein(s), the binding of an autophagy adaptor protein such as SQSTM1, and the association of an autophagosome with the adaptor-microbe complex.91 The degraded microbial peptides can then be delivered for MHC presentation to stimulate the adaptive immune response. Roles for autophagy have been proposed in the cross-presentation of peptides to MHC-I and the presentation of endogenous peptides to MHC-II.92-96 Autophagy or components of the autophagic machinery can also degrade membrane compartments that protect microbes, including phagosomes, vacuoles and microbe-induced membrane compartments.91,97 Xenophagy has been described in epithelial cells, macrophages and neurons, whereas roles for autophagy in antigen presentation have been observed in epithelial cells, macrophages, lymphocytes and dendritic cells.
Studies of autophagic function in liver immunity have been limited and generally restricted to hepatitis viruses. Many successful pathogens have evolved ways to inhibit autophagy and blunt the immune response; or alternatively, redirect autophagy for promicrobial purposes.83 Interestingly, viruses that target the liver, including hepatitis B virus (HBV), hepatitis C virus (HCV) and dengue virus (DENV), all usurp autophagy for proviral functions. Multiple roles for autophagy have been proposed for these viruses. One of the potential roles for HCV-induced autophagy is the suppression of innate immunity by an unknown mechanism.98-100 It is currently unclear whether this is a unique feature of HCV-induced autophagy. It may relate to the observation that HCV requires the autophagy immune effector IRGM (immunity-related GTPase family, M) for autophagy induction and replication.101 Additionally, studies of mice transgenic for hepatocyte-specific HCV NS3/4A expression indicate differential effects of type I interferon on autophagy. IFN/interferon, α 1 induces amphisomes, which may stimulate TLR recognition of viral antigen, whereas IFNB1 stimulates autolysosome formation and viral protein degradation.102 Thus, the regulation of immune responses in the liver by autophagy has several layers of complexity.
Circadian regulation of autophagy
Many biological processes in mammals exhibit robust diurnal rhythms, particularly pathways involved in nutrient and energy metabolism.103-105 The restriction of metabolic functions to a certain time window during the day may provide advantages for organisms as they anticipate and synchronize their body metabolism to feeding and activity cycles. At the molecular level, the biological clock is comprised of transcriptional activators and repressors that are assembled into positive and negative feedback loops that act in concert to drive rhythmic gene transcription.106 The temporal synchronization of tissue metabolism is achieved by reciprocal signaling between the clock and metabolic regulatory networks in response to light and nutrient cues. For example, the transcriptional coactivator PPARGC1A (peroxisome proliferator-activated receptor gamma. coactivator 1 α) integrates clock and metabolic gene programs and is modulated by CSNK1D/casein kinase 1, delta, an integral clock component.107,108
In the 1970s, a series of electron microscopy studies by Pfeifer and colleagues demonstrated that the abundance of autophagic vacuoles varies throughout the day in several tissues, including hepatocytes, retinal rod cells, cardiomyocytes, pancreatic acinar cells and the renal proximal tubules in rats.109,110 In addition, certain lysosomal hydrolases exhibit rhythmic activities in the liver.111 Using more specific molecular markers for autophagy, recent work has demonstrated that autophagic activity is temporally restricted in several mouse tissues, including the liver, heart and skeletal muscle.112 Autophagic flux, as measured by the rate of LC3-II degradation, peaks at noon and decreases to lower levels in the dark phase. A cell-autonomous role of clock in autophagy regulation is supported by the observations that mice lacking liver clock have aberrant autophagy gene expression and activity. These findings add a temporal dimension to the regulation of autophagy in normal physiology.
The cyclic activation of autophagic flux in the liver is associated with rhythmic mRNA and protein expression of genes involved in different aspects of autophagy, including Ulk1, Bnip3, Gabarapl1, Ctsl and Atp6v1d.112 Transcriptional control is emerging as an important aspect of autophagy regulation. To date, several transcription factors have been identified that regulate autophagy gene expression in cultured cells and in vivo, including FOXO3,113,114 TFEB (transcription factor EB),115 CEBPB [CCAAT/enhancer-binding protein (C/EBP), β]112 and SREBF2 (sterol regulatory element binding transcription factor 2).116 In the context of circadian autophagy, CEBPB appears to play a critical role. Adenoviral expression of CEBPB is sufficient to stimulate the autophagy gene program and autophagic protein degradation in cultured primary hepatocytes, whereas RNAi knockdown of this factor in the liver impairs autophagy gene expression and leads to significant accumulation of SQSTM1.112 CEBPB is required for autophagy induction in response to starvation as well as during light/dark cycles, suggesting that this factor links nutritional and circadian signals to autophagy.
Close coupling of autophagic degradation to the biological clock may provide distinct advantages for multicellular organisms to maintain nutrient homeostasis.117 In fact, the concentrations of plasma amino acids and metabolites exhibit robust circadian oscillations that are partially mediated through autophagy.19,118 The expression of genes involved in de novo lipogenesis, cholesterol biosynthesis and fatty acid β-oxidation is highly rhythmic in the liver, suggesting that the circadian regulation of these metabolic cycles is synchronized with autophagy to optimize nutrient storage or fuel oxidation. Cyclic activation of autophagy by the mechanisms discussed (Fig. 2) may also play an important role in temporal remodeling of hepatic cellular proteomes and organelles as well as reconfiguration of their bioenergetic properties during light/dark cycles.
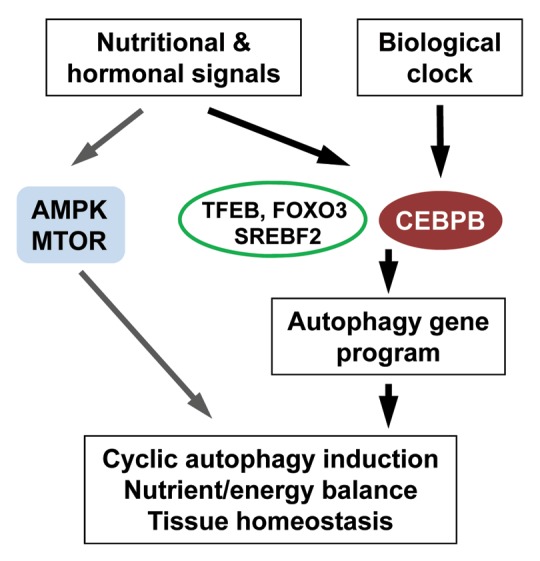
Figure 2. Circadian regulation of autophagy. The expression of autophagy genes is regulated by the biological clock through a CEBPB-mediated transcriptional pathway. In parallel, autophagy activity is modulated by nutrient- and energy-sensing pathways to drive rhythmic autophagy induction that maintains homeostasis. These pathways include the AMPK and MTOR pathways as well as the transcription factors TFEB, FOXO3 and SREBF2.
Role of Autophagy in Hepatic Diseases
SERPINA1/α1-anti-trypsin deficiency
The classical form of ATD caused by homozygosity for the SERPINA1 (α1-antitrypsin) Z allele SERPINA1-Z is the most common genetic liver disease in children.119 This childhood disease then leads to cirrhosis and HCC in many more adults than previously recognized.120 The deficiency is caused by a point mutation that renders the hepatic secretory glycoprotein SERPINA1 prone to misfolding, polymerization and aggregation. The mutant SERPINA1-Z molecule accumulates in hepatocytes, and levels of SERPINA1 in the blood and body fluids are reduced to 10–15% of normal. Accumulation of mutant SERPINA1-Z in the endoplasmic reticulum (ER) of hepatocytes leads to liver damage by a gain-of-function, proteotoxic mechanism as demonstrated by the presence of liver damage in the PiZ transgenic mouse model of ATD. The PiZ mouse was generated with a transgene that consists of a genomic fragment encompassing coding regions, introns and extensive upstream and downstream flanking regions of the human SERPINA1-Z gene.121 The marked accumulation of polymerized and aggregated SERPINA1-Z in mouse hepatocytes leads to liver damage closely resembling what is seen in the human disease with steatosis, hepatocyte hyperproliferation and carcinoma.122,123 These mice express the endogenous murine ortholog of SERPINA1 so there is no loss of function, and the liver damage must be the result of a gain-of-function effect.
Although liver disease in ATD is caused by a toxic mechanism, studies of a human ATD cohort have shown that only a subpopulation of those with the genetic defect develop liver disease.124,125 This fact implies that additional genetic and/or environmental modifiers determine whether an affected individual is susceptible to, or protected from, liver disease. It had been theorized that these modifiers influence the fate of mutant SERPINA1-Z once it accumulates in the ER. These modifiers could be working by either altering intracellular degradative mechanisms or activating cellular response pathways that protect the cell from the consequences of SERPINA1-Z accumulation in the ER. Investigations have led to the recognition that intracellular degradation of SERPINA1-Z involves both the proteasomal and autophagic pathways.126 SERPINA1-Z accumulation leads to a distinct set of cellular responses including induction of autophagy,127 activation of CASP3, 7, 8 and 9 and the ER-localized CASP12,128 and increased NFKB signaling without eliciting the unfolded protein response (UPR).129
Autophagy was first implicated in ATD with the observation of a marked increase in autophagosomes in fibroblast cell lines engineered to express mutant SERPINA1-Z.130 Increased numbers of autophagosomes were also seen in the livers of both PiZ mice and patients with ATD.130 The mechanistic involvement of autophagy in SERPINA1-Z degradation was demonstrated by the finding that there was a marked delay in degradation of SERPINA1-Z expressed in an ATG5-null fibroblast cell line that was reversed by reconstitution of wild-type ATG5.127 Furthermore, massive accumulation of SERPINA1-Z with very large cytoplasmic inclusions was observed in ATG5-null cells. In addition to providing definitive evidence that autophagy contributes to the disposal of SERPINA1-Z, these results suggest that autophagy plays a proteostatic role in the SERPINA1-deficient state, preventing the toxic cytoplasmic accumulation of SERPINA1-Z through piecemeal digestion of insoluble aggregates.
The importance of autophagy in the disposal of SERPINA1-Z has been confirmed in yeast using a completely different experimental approach.131 A library of yeast mutants was engineered to express human SERPINA1-Z and then screened for its impaired degradation. One strain with defective SERPINA1-Z degradation had a mutation of a yeast gene that is orthologous with mammalian BECN1/VPS30/ATG6. In the absence of this VPS30 ortholog or the ortholog of ATG16, there was a marked delay in SERPINA1-Z disposal. This study was particularly revealing because delayed SERPINA1-Z degradation was most apparent when SERPINA1-Z was expressed at high levels. At lower levels of expression, SERPINA1-Z degradation was not significantly different from that in wild-type yeast. These results indicate that at lower levels of expression SERPINA1-Z in the ER is predominantly soluble and degraded by the proteasome. At higher levels of expression SERPINA1-Z accumulates as insoluble polymers/aggregates that require autophagy for disposal. Studies in yeast by Kruse et al.,132 also discovered that autophagy degrades a mutant subunit of fibrinogen that forms insoluble aggregates in the ER of hepatocytes in an inherited form of fibrinogen deficiency. Degradation of the mutant fibrinogen was slowed in yeast strains lacking Vps30/Atg6 and Atg16. The fibrinogen deficiency that was modeled in these studies has been associated with chronic liver disease characterized by distinct fibrillar aggregates in the ER of hepatocytes, similar to what is seen in ATD. The results of these yeast studies substantiate the concept that autophagy is specialized for the disposal of aggregation-prone proteins that cause liver disease by the proteotoxic consequences of their accumulation in the ER of hepatocytes.
Accumulation of SERPINA1-Z in the ER also activates hepatic autophagy in a mouse model of ATD. A novel mouse model with hepatocyte-specific inducible expression of SERPINA1-Z, the Z mouse, was bred with the GFP-LC3 mouse to generate a Z mouse with green fluorescent autophagosomes.131 Green fluorescent autophagosomes appear in the livers of GFP-LC3 mice only after 24 h of starvation. In the Z × GFP-LC3 mouse, fluorescent autophagosomes appear spontaneously after induction of hepatocyte expression of the SERPINA1-Z gene.127 GFP+ autophagosomes are not seen in the liver of the Saar × GFP-LC3 mouse, which has hepatocyte-specific inducible expression of the SERPINA1 Saar variant that accumulates in the ER but does not polymerize. Thus, autophagy is activated when SERPINA1-Z polymerizes and aggregates, and plays a critical role in disposing of SERPINA1-Z to prevent massive intracellular aggregates.
The ability of autophagy to degrade ER SERPINA1-Z aggregates suggested that this pathway would be an ideal therapeutic target in ATD. Recently a drug that enhances autophagy, carbamazepine (CBZ), was found to be effective in cell line and mouse models of ATD.123 CBZ increases autophagic degradation of SERPINA1-Z in cultured cells, and when administered by oral gavage to the PiZ mouse model of ATD reduces the hepatic load of SERPINA1-Z. Importantly, CBZ treatment reduces hepatic fibrosis in vivo as demonstrated by immunohistochemical staining for fibrous tissue and by quantification of hepatic hydroxyproline. The mechanism by which CBZ enhances autophagic degradation has not been described, but the lack of effectiveness of rapamycin suggests that an MTOR-independent mechanism may be involved.123 CBZ is currently being investigated in a phase II/III trial for severe liver disease due to ATD.
Recently a novel C. elegans model of ATD was adapted to a high content screening platform for identification of potential therapeutic agents.133 An initial screen of the LOPAC drug library provided additional evidence for the potential strategy of employing autophagy enhancer drugs because four of the five most impressive hit compounds appear to act by increasing autophagy.133 Administration of each of these drugs induced the formation of autophagosomes in a C. elegans line engineered for expression of red fluorescent-LGG-1, a worm autophagosomal membrane-specific protein. One of these drugs, pimozide, was also identified in a mammalian cell-based assay for enhancing autophagic degradation of HTT (huntingtin).134,135 One of the newly identified drugs, fluphenazine, appears to reduce the cellular load of SERPINA1-Z in a cell line model and reduces hepatic fibrosis in the PiZ mouse model of ATD (Perlmutter, D., personal communication).
Taken together, these studies show that autophagy plays a key role in the proteostatic response in ATD. Genetic and/or environmental modifiers that alter autophagic function may be at least partially responsible for the wide variation in the incidence and severity of liver disease among patients with ATD. Drugs that enhance autophagy are therefore attractive candidates for ameliorating the liver disease that develops in some patients with ATD.
Nonalcoholic fatty liver disease
Nonalcoholic fatty liver disease (NAFLD) is an important component of the metabolic syndrome together with obesity and diabetes. NAFLD encompasses a spectrum of hepatic abnormalities ranging from simple fatty liver or steatosis, to fatty liver with hepatocellular injury and inflammation, termed nonalcoholic steatohepatitis (NASH).57 NAFLD is now the most prevalent liver disease in the United States,136,137 accounting for 75% of all chronic liver disease.138 The previously described functions of autophagy in the liver suggest a number of mechanisms by which autophagy may affect the development or progression of NAFLD.139 Autophagy may modulate the excessive storage of lipid in this disease, development of inflammation, the progression to hepatocyte injury and cell death and the chronic complications of NASH such as fibrosis and HCC.
With the description of lipophagy, the most important role of autophagy in fatty liver disease could be to regulate the process of excessive lipid accumulation. High fat diet (HFD)-fed mice with a hepatocyte-specific knockout of Atg7 develop markedly increased liver TGs and cholesterol content, clearly indicating that defects in autophagy can promote hepatic steatosis.5,140 Insulin resistance is thought to be critical to the development of NAFLD,141,142 and a complex interrelationship exists between autophagy and both insulin resistance and lipid accumulation. Insulin downregulates autophagy in response to nutrient supplies, but autophagy modulates insulin sensitivity as well. Hyperinsulinemic, HFD-fed mice have decreased levels of autophagy,143 which is not surprising given the ability of insulin to inhibit autophagy. However, the direct effect of insulin occurs through MTOR signaling, and in these studies levels of ATG5 and ATG7 were decreased, suggesting a different mechanism for the effects of insulin on liver autophagy in obesity. In addition, reduced levels of ATG7 and autophagic function have been demonstrated in the livers of genetically obese Ob/Ob mice. ATG7 levels were not restored to normal by the reversal of the hyperinsulinemia,140 suggesting that the defect in autophagy is not secondary to insulin. In both diet-induced and genetically obese mice, impaired autophagy has been associated with insulin resistance with decreased hepatic insulin signaling occurring in concert with increased ER stress.140 Adenoviral-mediated ATG7 overexpression decreases ER stress and improves insulin sensitivity in these animals. Defective autophagy may lead to insulin resistance from increased ER stress, a known mechanism of insulin resistance, but this remains to be directly proven. Adding further complexity to the relationship between autophagy and steatosis is that not only does autophagy regulate cellular lipid stores, but also levels of lipid content in turn affect autophagic function. HFD feeding leads to a defect in the movement of lipids into the autophagic pathway.5 The mechanism of this effect remains unclear. Decreases in autophagic pathway proteins have been reported;140,143 however, other studies of diet-induced obesity have failed to reveal any decrease in levels of these proteins at times when significant amounts of hepatic lipid accumulation had occurred (Czaja. M.J., personal communication). Alternative reported mechanisms include defects in the process of autophagosome-lysosome fusion,144 and reduced levels of lysosomal enzymes.145 Likely, the mechanism is multifactorial, but the important implication is that a harmful cycle may exist in which independent factors promote both impaired autophagy and hepatic steatosis, but then the decrease in autophagy exacerbates steatosis, which further impairs autophagy. This cycle thus creates a perpetual worsening of both cellular autophagic function and lipid accumulation. The ability of lipid accumulation to depress autophagic function extends to other forms of autophagy such as CMA.146 Interestingly, aging is a risk factor for the development of the metabolic syndrome including NAFLD,147 and a cause of decreased autophagy.148 Aging may therefore promote NAFLD development in part through decreased autophagy, although a study of HFD-fed aged mice indicates that aging promotes liver injury, but does not affect the degree of steatosis.149
Simple steatosis is a benign condition, but progression to inflammation and hepatocellular injury marks the development of NASH that can then progress to chronic liver disease and liver failure. Inflammation in adipose tissue as well as in liver is not just a passive marker of NASH but considered critical to its pathogenesis.150 Proinflammatory signaling mediated by LPS through the TLR4 pathway has been implicated in NASH development,151 suggesting a mechanism by which immune cell autophagy may mediate the inflammatory reaction in NASH. Further studies are needed to address the possibility that the previously discussed effects of autophagy on TLR4 signaling may affect NASH development. Whether obesity and insulin resistance affect levels of autophagy in macrophages similar to hepatocytes needs to be determined as well.
The mechanisms of hepatocellular injury and cell death in NASH are unknown, but FFA-induced lipotoxicity, oxidative stress and cytotoxic cytokines, particularly TNF, have all been implicated.152 As previously discussed, autophagy is involved in all of these forms of death. Specifically in hepatocytes there is evidence that hepatocyte autophagy mediates resistance to injury from FFAs and oxidant stress. Studies in HepG2 hepatocellular carcinoma cells demonstrated that the saturated FFA palmitate inhibits autophagy, which contributes to its ability to induce apoptosis.153 In contrast, the nontoxic unsaturated FFA oleate induces autophagy. Cell death from oxidant stress is increased in a rat hepatocyte cell line in the absence of either macroautophagy or CMA.58 Further studies in primary hepatocytes and in vivo rodent models are needed to confirm these findings and to specifically examine whether autophagy mediates these forms of death in the setting of steatosis.
The numerous potential mechanisms of involvement of autophagy in NAFLD suggest that autophagy may be a potent therapeutic target in NASH treatment or prevention. Therapeutic efforts to increase hepatic autophagy may not only reverse the hepatic manifestations of NAFLD such as hepatocellular steatosis and injury, but also some of the underlying metabolic abnormalities of the disease through effects on insulin resistance. In addition, altering autophagy may prevent common end-stage complications of NAFLD including HCC, as will be discussed in subsequent sections.
Alcoholic liver disease
Alcoholic liver disease (ALD) is a major cause of chronic liver disease in the United States and globally. Similar to NAFLD, ALD has a wide spectrum of pathogenic features ranging from steatosis to more severe acute alcoholic hepatitis, fibrosis, cirrhosis and even HCC.154 Although more than 90% of alcohol drinkers develop steatosis, only 30% develop fibrosis and cirrhosis.56,155 This fact has led to attempts to identify genetic factors that may affect human susceptibility to the development of advanced ALD. For example, genetic variants of PNPLA3 (patatin-like phospholipase domain-containing 3), a protein that regulates hepatic lipid metabolism,156 have been linked to ALD development.157,158 Individual variability in protective pathways that mitigate against the harmful hepatic effects of ethanol may be particularly important in modifying disease susceptibility. Recent findings have implicated autophagy induction by ethanol as a cellular protective mechanism against acute ethanol-induced steatosis and liver injury (Fig. 3).73,155 Ethanol is oxidized mainly by alcohol dehydrogenase (ADH) and partially by CYP2E1 (cytochrome P450, family 2, subfamily E, polypeptide 1). A chemical inhibitor of both ADH and CYP2E1, 4-methylpyrazole, significantly blocks ethanol-induced autophagosome formation. This suggests that the induction of autophagy requires ethanol metabolism and is mediated by ethanol’s reactive metabolites.73,159 This concept is further supported by the finding that ethanol induces autophagy only in HepG2 cells that stably express ADH and CYP2E1 and not in control HepG2 cells expressing vector alone.73 Moreover, ethanol oxidation generates ROS that are required for autophagy induction because ethanol-induced autophagy is blocked by antioxidants. Finally, both acute and chronic ethanol exposure suppresses AKT in vitro and in mouse liver.160,161 AKT is a positive regulator of MTOR, suggesting that decreased MTOR signaling from reduced AKT activity contributes to the increase in autophagy from ethanol in mouse liver.

Figure 3. Proposed model for the role of autophagy in alcohol- and APAP-induced liver injury. Both ethanol and APAP are first metabolized in the liver by the enzymes CYP2E1 (ethanol and APAP) and ADH (ethanol). The metabolism of APAP generates reactive metabolites which deplete hepatic GSH and bind to cellular and mitochondrial proteins to initiate mitochondrial damage. Consequently, the metabolism of both ethanol and APAP lead to increased ROS production and damaged mitochondria. Damaged mitochondria can lead to necrotic/apoptotic cell death and further ROS production. ROS may inactivate MTOR to trigger autophagy, which helps to remove ethanol-induced excessive lipid droplets and damaged mitochondria and in turn attenuate alcohol-induced liver injury. Pharmacological induction of autophagy by rapamycin and Torin 1 significantly protects against ethanol- and APAP-induced liver injury in mice. In hepatocytes exposed to APAP, damaged mitochondria can be removed by canonical mitophagy resulting in reduced necrosis. A portion of damaged mitochondria can also form mitochondrial spheroids which may also attenuate APAP-induced liver injury.
Intriguingly, ethanol-induced autophagy does not target proteins for degradation, but selectively removes damaged mitochondria and lipid droplets that accumulate in liver cells with ethanol treatment.73,155 Pharmacological induction of autophagy by rapamycin significantly suppresses acute alcohol-induced steatosis. Torin 1, a more potent, selective and ATP-competitive MTOR inhibitor, almost completely blocks acute ethanol-induced steatosis and liver injury in mice (Ding W-X, personal communication). However, although acute ethanol can induce autophagy in cultured primary hepatocytes and mouse liver, the effect of chronic ethanol exposure on autophagy is not yet clear. Mice chronically fed an ethanol-containing liquid diet have increased liver weight and hepatic protein content, suggesting impaired hepatic catabolism.162 Chronic ethanol consumption impairs proteasome function resulting in the retention of proteins that may contribute to increased liver mass. However, increased protein retention is also consistent with an ethanol-induced decrease rather than increase in autophagy.163 Also suggestive of a decrease in autophagy with chronic ethanol is that one of the typical features of chronic alcohol abuse is the formation of the hepatic protein aggregates Mallory-Denk bodies which are cytosolic inclusion bodies enriched with KRT8/keratin 8 and KRT18, as well as other proteins including ubiquitin and SQSTM1. Treatment with rapamycin significantly decreases the number of Mallory-Denk bodies in proteasome inhibitor-treated KRT8 transgenic mice.164 Therefore, regardless of the effects of acute or chronic ethanol exposure on hepatocellular autophagy, pharmacologically enhancing hepatic autophagy seems to be beneficial in alcohol-induced liver disease. However, rapamycin has multiple effects other than to increase autophagy,165,166 and off-target effects of rapamycin may have accounted for the beneficial effects in these studies. More investigations are needed to determine the effects of chronic ethanol use on autophagic function, as well as the ability of more specific enhancers of autophagy to prevent or reverse ALD.
Drug-induced liver injury
Most drugs are metabolized and detoxified in the liver, making this organ the principal target for drug damage. Drug-induced liver injury is a major problem in drug development, and a common cause for the withdrawal of approved drugs from the market. In the United States, drug-induced hepatotoxicity is the etiology of more than 50% of the cases of acute liver failure. APAP, a widely used antipyretic and analgesic drug, is the most common source of severe drug hepatotoxicity. While APAP is safe at therapeutic levels, an overdose can cause severe liver injury in animals and in humans.167 It has been well documented that APAP-induced hepatotoxicity is mediated mainly by its reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI) which is generated from APAP metabolism by cytochrome P450, mainly by the CYP2E1 isoform. NAPQI can deplete hepatic stores of glutathione (GSH), an intracellular antioxidant that regulates cellular redox homeostasis. Once GSH is depleted, NAPQI reacts with many cellular proteins, including mitochondrial proteins, to form protein adducts.168 APAP-induced mitochondrial protein adducts may lead to mitochondrial damage and subsequent necrosis. APAP induces autophagy to remove damaged mitochondria which prevents APAP-induced necrosis (Fig. 3).169 When autophagy is further enhanced by treatment with rapamycin, APAP-induced necrosis is significantly inhibited in cultured primary hepatocytes and in mouse liver. Rapamycin does not affect APAP-induced GSH depletion, indicating that the effect of rapamycin is downstream of APAP metabolism. Treatment with rapamycin 2 h after APAP administration significantly ameliorates APAP-induced liver injury, despite the fact that APAP metabolism and hepatic GSH depletion have already occurred.169 This finding is particularly important because most patients at risk for serious hepatotoxicity from an acute APAP overdose do not receive medical care until they are past the metabolic phase. Therefore, pharmacological induction of autophagy may have a potential therapeutic application in humans with APAP hepatotoxicity because of this advantage.
Currently, the mechanism by which mitophagy protects against APAP cell death is unknown. APAP-induced ROS production is suppressed by rapamycin but exacerbated by chloroquine,169 indicating that mitophagy may attenuate mitochondrial ROS formation and release of prodeath factors. Mitochondria are central regulatory components in cell death and common targets for drug injury, suggesting that increased mitophagy could be a central mechanism of cell survival after drug-induced liver injury. Efavirenz, a non-nucleoside reverse transcriptase inhibitor used to treat HIV infection, induces mitochondrial damage and subsequent hepatocyte death. Similar to APAP, efavirenz triggers mitophagy and pharmacological suppression of autophagy enhances efavirenz-induced cell death.170,171 Although these studies must be extended to other forms of drug hepatotoxicity, levels of mitophagy may prove to be a critical determinant of the development of drug-induced hepatocyte injury.
Although mitophagy seems to be important in many pathophysiological contexts including drug hepatotoxicity, the molecular mechanisms that mediate mitophagy in mammalian cells in general, or specifically in hepatocytes, are not completely understood. Multiple molecules and signaling pathways have been implicated in mitophagy in mammalian cells, including the BH3-only BCL2 family proteins BNIP3 and BNIP3L, FUNDC1 (a mitochondrial outer membrane protein that directly interacts with LC3), and the MPT and PINK1-PARK2 signaling pathway as previously discussed.172-175 Among these factors, PINK1-PARK2 signaling has been most extensively studied. Cytosolic PARK2 is recruited to damaged mitochondria by PINK1, a mitochondrial serine/threonine protein kinase.33,176-178 Once on mitochondria, PARK2 promotes the ubiquitination of a subset of mitochondrial outer membrane proteins including VDAC1, TOMM20, MFN1 (mitofusin 1) and MFN2.179-181 PARK2 also recruits SQSTM1 to mitochondria likely through the direct binding of SQSTM1 with ubiquitinated mitochondrial proteins. Although SQSTM1 is required for mitochondrial perinuclear clustering, this protein is not essential for mitophagy in mammalian cells.178,182 It should be noted that in all these studies, immunostaining for TOMM20, a mitochondrial outer membrane protein, was used as a mitophagy marker to assess the role of SQSTM1 in mitophagy. It has recently been demonstrated that PARK2-dependent degradation of mitochondrial outer membrane proteins such as TOMM20, MFN1 and MFN2 is mediated by the proteasome and not autophagy.180,181 Therefore, a current challenge in investigations of mitophagy is to develop more reliable, quantitative assays to assess mitophagy, in particular for in vivo studies of the liver
Mitochondria are dynamic organelles that constantly undergo fission and fusion, and it has been speculated that mitochondrial fission may promote mitophagy because smaller mitochondria are more easily enveloped by autophagosomes than larger mitochondria. Mitochondrial fusion is regulated by MFN1 and MFN2, and their levels are controlled by PARK2-mediated ubiquitination and proteasomal degradation. In various mammalian cells, exogenous overexpression of PARK2 promotes MFN1 and MFN2 degradation resulting in mitochondrial fragmentation and mitophagy.180,181 However, in APAP-treated primary mouse hepatocytes and in mouse liver, there is no significant degradation of MFN1 and MFN2 even though APAP treatment increases mitochondrial translocation of PARK2 (Ding, W.-X., et al., unpublished observations). The E3 ligase function of PARK2 is regulated by post-translational modifications such as phosphorylation, ubiquitination and S-nitrosylation,183 and it is possible that APAP may induce some of these modifications resulting in inactivation of PARK2. Further work is needed to identify the post-translational modifications of PARK2 that are induced by APAP in mouse liver. It is also likely that the lack of degradation of MFN1 and MFN2 after APAP treatment could be due to the uneven lobular distribution of CYP2E1 and NAPQI in mouse liver. Indeed, autophagy and mitophagy are only induced in areas adjacent to the APAP-induced necrotic areas, which are mainly located near the central vein. Moreover, APAP has been demonstrated to induce a unique dynamic change in mouse hepatic mitochondria, which has been termed the mitochondrial spheroid.184,185 Under conventional electron microscopy, a mitochondrial spheroid is a ring-like or cup-like spherical structure which forms a lumen surrounded by mitochondrial membranes, similar to a phagosome-like structure. The lumen contains cytosolic proteins, ER membranes, lipid droplets or other mitochondria. Mitochondrial spheroids are formed in response to APAP-induced oxidative mitochondrial damage independent of mitophagy, require MFN1 or MFN2, and are negatively regulated by PARK2. The mitochondrial spheroids acquire lysosomal markers and limited degradation capacity for some mitochondrial intermembrane space and inner membrane proteins. Although the physiological significance of the mitochondrial spheroid is not clear, it may represent a different mechanism of maintaining mitochondrial homeostasis in response to APAP-induced mitochondrial damage. A better understanding of these structures may offer a promising approach for drug-induced hepatotoxicity in which damaged mitochondria play an essential role.
Ischemia/reperfusion injury
The liver is dependent on oxygen to maintain function and cell survival, and is highly susceptible to hypoxic and ischemic stress, particularly in the pericentral zone. Although prolonged ischemia induces severe tissue acidosis and by itself eventually causes liver cell death, a recovery of blood flow and return to normal pH paradoxically worsens short-term ischemic damage, an event termed reperfusion injury. Ischemia/reperfusion (I/R) injury is a causal factor contributing to the morbidity and mortality in hepatic sinusoidal obstruction syndrome, hemorrhagic shock, trauma and cardiac arrest.186 In addition, the vulnerability of the liver to I/R injury is a major obstacle to liver resection and transplantation surgery where reperfusion after sustained ischemia is unavoidable during hepatectomy and vascular reconstruction.
Hepatocellular death after reperfusion has been attributed to numerous mechanisms that include reactive oxygen and nitrogen species generation, disruption of Ca2+ homeostasis, loss of cellular antioxidants, stimulation of catabolic enzymes, ATP depletion and mitochondrial dysfunction.186-188 In isolated rat hepatocytes, I/R sequentially induces an increase in mitochondrial Ca2+ and ROS, onset of the MPT and hepatocyte death.189 Thus, mitochondrial dysfunction is a critical downstream event leading to I/R-mediated cell death.188,190,191
The mitochondrial inner membrane is virtually impermeable to all solutes except for those with specific transporters. Some pathological conditions including I/R trigger the opening of high conductance permeability transition pores in the mitochondrial inner membrane leading to the MPT.190,192-196 Loss of the permeability barrier in the inner membrane following the onset of MPT causes an abrupt nonselective diffusion of various solutes.197 Barrier breakdown results in an uncoupling of electron transport chains from ATP production, and ultimately both the pH gradient and mitochondrial membrane potential collapse, which causes ATP depletion and necrosis.192 The MPT can also trigger apoptosis by releasing pro-apoptotic mitochondrial proteins such as cytochrome c that are normally sequestered in the intermembranous space.190,191,198,199 Apoptosis is an active ATP-dependent process, and therefore the levels of cellular ATP serve as a molecular switch that determines the mode of cell death after I/R.190
Autophagy and I/R injury in young livers
Autophagy clears abnormal or dysfunctional mitochondria to ensure optimal cellular function and survival. In addition to supplying cellular energy, mitochondria are the major source of ROS generation,200 and mitochondrial DNA is prone to ROS-mediated damage.201 With impaired or insufficient mitophagy, the cell accumulates damaged mitochondria, leading to uncontrolled ROS formation, mitochondrial DNA mutation, energetic failure and ultimately cell death. Furthermore, the release of cell death-signaling molecules from one mitochondrion to neighboring mitochondria rapidly propagates this injurious signal cascade throughout the cell.202,203 The failure of mitophagy to remove even a small subset of damaged mitochondria during I/R can therefore have a significant impact on hepatocellular function and viability. The function of mitophagy is therefore essential for hepatic function and cell survival after I/R.
Ischemia exposes hepatocytes to nutrient deprivation, acidosis and ATP depletion. Although starvation rapidly stimulates autophagy in normal livers, the lack of ATP during ischemia suppresses the induction of autophagy, which is a highly energy-dependent process.204,205 Prolonged ischemia also substantially reduces the levels of key autophagic proteins, particularly ATG7 and BECN1, which in turn further decreases autophagic function.204 Pharmacological studies revealed that the decreased expression of ATG7 and BECN1 during ischemia is caused at least in part by the Ca2+-dependent proteases, CAPN/calpains. The causality of Ca2+ overloading and subsequent activation of calpains in I/R injury has been well documented in a variety of organs.189,206,207 With the combination of ATP depletion and ATG loss, the formation of autophagic vesicles is impeded and autophagic flux becomes minimal during prolonged ischemia.
During reperfusion the hepatocellular pH recovers and the supply of oxygen resumes. In the early phase of reperfusion the mitochondria temporarily repolarize and begin generating ATP, which induces autophagy.204,205 At the same time, reperfusion of ischemic hepatocytes triggers Ca2+ and ROS accumulation in a subset of mitochondria.189 Prominent hepatocyte injury occurs when the increase in autophagy is insufficient to neutralize the reperfusion stress. When the capacity of autophagic clearance counterbalances or surpasses reperfusion-induced mitochondrial changes, altered mitochondria are eliminated in a timely fashion by autophagy, and hepatocyte viability is maintained. Determinations of autophagic flux with chloroquine or bafilomycin A1 and fluorescence imaging of GFP-LC3 show that reperfused hepatocytes have increased autophagic flow and mitophagy during the early phase of reperfusion.205 However, when intramitochondrial loading of Ca2+ and ROS exceed autophagic clearance during the later phase, autophagy fails to remove all dysfunctional mitochondria and widespread onset of the MPT ensues. The MPT leads to irreversible uncoupling of oxidative phosphorylation, ATP depletion, energetic failure and ultimately hepatocyte death. Confirming this sequence of events, strategies that enhance autophagy, including pre-ischemia nutrient depletion, and ATG7 or BECN1 overexpression, all suppress the MPT and increase hepatocyte survival after reperfusion.205 Thus, impaired or insufficient autophagy is a crucial mechanism underlying I/R injury to the liver.
Autophagy and I/R injury in aged livers
Life expectancy has increased 1.6-fold over the past century,208 and aging is strongly associated with an increased incidence and severity of disease. Clinically, the steady rise in life span has increased the number of elderly patients who require surgical treatment for hepatic malignancies. However, the aged liver has significantly less reparative capacity following I/R injury associated with hepatectomy and liver transplantation.209-211
As in young livers, autophagy plays a paramount role in I/R injury in aged livers. Among the three types of autophagy, CMA declines with aging in the liver.212,213 In contrast, studies with livers from 3- and 26-mo old mice demonstrated that levels of some autophagy-related proteins and basal autophagic flux are increased in aged hepatocytes, suggesting that old hepatocytes acquire an enhanced basal autophagy as a protective or adaptive response to aging.205 Moreover, hepatocytes from both ages show a comparable autophagic response to a mild stress such as normoxia or starvation.205 Therefore, constitutive hepatocellular autophagy is less likely to be compromised by aging alone, as is widely thought, and other studies have similar findings.214 A striking reduction in autophagy is evident when aged cells succumb to moderate I/R from short-term ischemia. Whereas young livers initiate a strong autophagic response to moderate ischemia and reperfusion and tolerate this stress, aged livers fail to increase autophagy due to I/R-mediated depletion of ATG4B, a key autophagic protein necessary for the formation of autophagosomes and the recycling of LC3.205,215 As a consequence, aged hepatocytes and livers are highly prone to I/R injury. Similar to young livers after prolonged ischemia, aged livers after short-term ischemia accumulate dysfunctional mitochondria, undergo MPT and lose viability soon after reperfusion.
Future perspectives
A growing body of evidence has accumulated indicating that autophagy is protective against hepatic I/R injury.204,205,215-218 Enhancing autophagy has emerged as a new potential strategy to improve liver function after I/R. Despite its therapeutic potential, many unanswered questions remain, such as how autophagy in nonparenchymal cells is affected by I/R and how different liver cell types respond to cold vs. warm ischemia. In addition, current approaches to increase hepatocellular autophagy, including pretreatment with inducers of autophagy before ischemia and the viral delivery of specific autophagy genes, are limited in their clinical applications. Future studies are warranted to better understand the relationship between autophagic dysfunction and hepatic I/R injury in order to better target therapeutics to address this problem.
Hepatitis B virus
HBV is a hepatotropic virus which can cause severe liver disease including hepatitis, liver cirrhosis and HCC. HBV is a small DNA virus with a circular, partially double-stranded 3.2-kb genome. Upon infection of hepatocytes, this DNA is converted into a covalently closed circular DNA in the nucleus where it directs the transcription of viral RNAs. The HBV genome contains only four genes. The S gene codes for three co-C-terminal envelope proteins named large, middle and small HBV surface antigens (HBsAgs). The C gene codes for the viral core protein and a related protein termed the precore protein. The core protein packages its own mRNA, which is also known as the pregenomic RNA, to form the viral core particle. In contrast, the precore protein is a nonstructural protein with immunomodulatory functions. The P gene codes for the viral DNA polymerase which is also a reverse transcriptase. The X gene codes for a multifunctional protein (HBx) that enhances viral replication. After its packaging by the core protein, the pregenomic RNA, which contains the entire genomic sequence, is converted into the viral DNA in the core particle by the viral DNA polymerase that is also packaged. The core particle subsequently interacts with the surface antigens in intracellular membranes for the formation of the mature virus which is then released from infected cells via a pathway that likely involves multivesicular bodies.219
HBV has been consistently demonstrated to activate autophagy in cell culture, in the livers of transgenic mice that carry the HBV genome and during natural infection.220-222 However, differences in the mechanism by which HBV activates the autophagic pathway have been reported. HBx has been shown to activate the promoter of the BECN1 gene to stimulate its expression and increase autophagy in response to starvation.223 However, it has also been demonstrated that HBx can bind to and activate PtdIns3K.220,224 PtdIns3K activation increases cellular levels of phosphatidylinositol-3-phosphate and the number of autophagic vacuoles including autophagosomes and autolysosomes. It is conceivable that HBx may induce autophagy via several pathways due to its multiple activities. In contrast, Li et al.,221 demonstrated that HBx plays a lesser role in the induction of autophagy than the small HBsAg protein. Small HBsAg increases autophagy via the induction of ER stress and activation of the UPR, and an HBV genome incapable of expressing small HBsAg does not activate the UPR or autophagy. This finding is surprising, as previous studies have implicated HBx,225,226 large HBsAg protein,227 and an HBV mutant with a deletion in the preS2 region,228,229 in the induction of ER stress by HBV. Small HBsAg has not been reported in the past to induce ER stress, and also a reduction in levels of this protein has been shown to lead to large HBsAg accumulation.230,231 Increased large HBsAg levels can cause ER stress and hepatocellular injury.232,233 Since small HBsAg mutations can promote the retention of this protein in the ER and Golgi,234 a possible explanation is that the specific HBV strains used by Li et al.221 in their studies harbor mutations that induce ER stress secondary to their ER retention. However, this possibility would not explain why they did not observe an induction of ER stress when they abolished the expression of the small HBsAg by a missense mutation of its initiation codon, which should lead to accumulation of the large HBsAg in the ER.221 Further research is needed to resolve these disparate results from different laboratories regarding the mechanism by which HBV induces autophagy. Interestingly, although HBV could induce autophagosomes and autolysosomes,220,221 it could not enhance autophagic protein degradation, possibly due to the inability of HBV to enhance the sequestration of protein cargos destined for autophagic degradation, which is a selective process.235
An additional factor that influences the induction of autophagy by HBV is the specific viral genotype. HBV genotype C, which is associated with an increased risk of HCC,236 is more potent than genotype B in the induction of autophagy.222 It is unclear whether the greater ability of genotype C HBV to induce autophagy is related to its increased virulence.
Autophagy functions to promote HBV replication, as inhibition of autophagy reduces HBV replication in cells.220,221,224 Autophagy marginally affects HBV RNA transcription and does not regulate the encapsidation of viral pregenomic RNA. In one study, the inhibition of autophagy abolished HBV DNA replication.220 In another study, however, the inhibition of autophagy only marginally affected viral DNA replication, but prevented the envelopment of HBV.221 The reason for this discrepancy is unclear, but the recent observation that the ablation of autophagy by a liver-specific knockout of Atg5 abolishes HBV DNA replication in mouse liver strongly supports a role for autophagy in HBV DNA replication.237 How autophagy enhances HBV DNA replication or envelopment is unclear, but the observation that HBsAg and the core protein colocalize with autophagic vacuoles suggests that these membrane vesicles participate in HBV DNA replication and/or morphogenesis.220 Although HBx is required to enhance HBV replication in vivo and in HepG2 hepatocellular carcinoma cells, HBx is not required for HBV replication in Huh7 hepatocellular carcinoma lines. In addition, inhibition of autophagy has no effect on the replication of an HBx-negative HBV mutant in Huh7 cells, indicating that HBV may employ autophagy-independent pathways for its replication under certain conditions.220 Thus far, HBV is the only DNA virus known to capitalize on the autophagic pathway for the benefit of its own replication.238 It remains to be determined, however, whether the activation of the autophagic pathway by HBV plays any role in the pathogenesis of HBV-induced liver disease.
Hepatitis C virus
Similar to HBV, HCV is hepatocyte-trophic virus that is a major worldwide health problem due to the high prevalence of HCV chronic infection that can progress to cirrhosis and HCC.239 HCV is a member of the Flaviviridae family, and its genome is a positive-strand RNA ~9.6-kb long. The HCV genome encodes a polyprotein precursor of ~3,000 amino acids which is cleaved by both viral and host proteases into structural (core, E1, E2 and p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins.240-242 HCV-infected cells accumulate lipid droplets that play an important role in the assembly of virus particles.
The ability of HCV to induce autophagy was first reported in HCV-infected cell cultures. Autophagic vacuoles are increased in HCV-infected human hepatocytes, and HCV genotype 1a (clone H77)- or genotype 2a (clone JFH1)-infected hepatocytes display LC3-positive puncta on the autophagic vacuoles.243 A similar increase in autophagic vacuoles has been reported in hepatocytes harboring subgenomic or full-length replicons, and with infection with cell culture-grown HCV.244-248 In HCV infection, autolysosome formation is observed, although contradictory findings have been reported for autophagy maturation in hepatocytes bearing HCV subgenomic replicons or individual proteins.247,249-252 Furthermore, elimination of HCV RNA by treatment with IFN abrogates the lipidation of LC3 and puncta formation in cells harboring a subgenomic HCV replicon.248,253 Increased numbers of autophagic vacuoles have been observed by electron microscopy in the livers of chronically infected HCV patients, but the number of vesicles does not correlate with the infecting HCV genotype or viral load.248,250,253
Confocal microscopy of HCV-infected cells has not revealed a significant colocalization of HCV core or nonstructural proteins with the autophagic marker proteins LC3 or ATG5.243,244 In contrast, an association between HCV RNA and autophagosomes, or between nonstructural proteins and autophagic proteins, has been demonstrated.101,246,254,255 These differences could be due to distinct experimental conditions used in these studies, including the cell types employed for transfection of HCV RNA or infection with cell culture-grown HCV, and the incubation times examined for the association of viral and autophagic proteins. Future studies need to clarify the association of specific viral proteins and RNA with the autophagosome in HCV infection. Several HCV proteins, including NS3, NS4B and NS5A, induce autophagy,101,251,252 implicating the involvement of multiple HCV proteins in autophagosome formation.
Autophagic machinery promotes HCV replication
HCV-mediated induction of autophagy is well established, however the precise role of autophagy in HCV biology is poorly understood. It is clear that an inhibition of infectious HCV particle production occurs in hepatocytes with a knockdown of autophagy.100,244,256 The IFNG-inducible IRGM protein directly associates with several autophagy proteins, and favors infectious viral particle production.101 Silencing of the autophagy genes encoding LC3 or ATG7 inhibits HCV genomic RNA replication in Huh7.5 cells.247,257 However, Tanida et al.256 observed that the intracellular expression of HCV mRNA and proteins remains unchanged in ATG7 and BECN1 knockdown HCV-infected cells. In contrast, a requirement for complete autophagosome maturation for promotion of HCV RNA replication has also been reported.249 Thus, it is clear that autophagy is necessary for virus production, because all of the studies are in agreement with the fact that an impairment of autophagy inhibits infectious HCV particle release. One additional mechanism by which autophagy may have this effect is that a knockdown of autophagy proteins in HCV-infected cells induces apoptosis, suggesting that increased autophagy promotes the survival of infected cells.100,248 The ability of autophagy to promote HCV infection is clear, but further studies need to clarify the mechanism(s) of this effect and specifically whether autophagy mediates HCV replication and assembly.
Mechanism of HCV-mediated autophagy induction
The signaling pathways implicated in the induction of autophagy by HCV vary with the infected cell type. HCV infection induces autophagy independent of BECN1-BCL2 dissociation. BECN1 expression is increased at the transcriptional level in HCV-infected cells.251 On the other hand, knockdowns of RAB5 or PIK3C3/VPS34 do not completely block autophagosome formation.252,255 HCV-mediated enhancement of the UPR has also been implicated as a mechanism for autophagy induction,247,249 and the UPR is increased in liver biopsies of HCV-infected patients.247,249,258 The MTOR signaling pathway is a negative regulator of autophagy;1,259 however, HCV infection in hepatocytes activates MTOR.1,251,259,260 Paradoxically, an inhibition of MTOR and EIF4EBP1 phosphorylation has been reported in autophagy knockdown HCV-infected cells, suggesting that alternatively HCV-mediated autophagy acts upstream of MTOR signaling.251 Only transient PtdIns3K-AKT activation early in HCV infection is seen, raising the possibility that MTOR is not an important regulator of autophagy in HCV infection.261 In total, these findings suggest that multiple signaling pathways are involved in HCV induction of autophagy.
Autophagy regulates the innate immune signaling pathway
Autophagy regulates the innate immune response as previously discussed, and can either activate or inactivate antiviral molecules in virus-infected cells.84,262,263 Altered autophagy therefore may affect the immune response to HCV infection. The ability of BECN1 or ATG7 knockdowns in HCV-infected hepatocytes to increase interferon signaling,100 may be secondary to mitochondrial ROS generation.264 Mitochondrial ROS production is increased in BECN1-knockdown cells infected with HCV or transduced with HCV NS5A as compared with control cells.251 HCV pathogen-associated molecular pattern induced RIG-I signaling is also enhanced in ATG5 and LC3 knockdown virus-infected cells.249 In contrast, silencing of ATG7 in hepatocytes harboring the HCV replicon does not alter the induction of OAS1 (2′,5′-oligoadenylate synthetase 1, 40/46Da), an interferon-stimulated gene.255 The differences in these findings may again represent the nature of the experimental conditions, particularly the use of different cell lines and infectious HCV vs. a replicon.
Future perspectives
The underlying mechanism of HCV-mediated autophagy induction is still emerging. HCV-mediated autophagy promotes infectious virus production and evades the innate immune response. Several viruses, such as influenza virus and HIV-1, have evolved strategies to prevent lysosomal degradation of viral particles by blocking the fusion of autophagosomes with lysosomes.265,266 However, HCV-mediated autophagosome maturation is poorly understood. Furthermore, infection of HCV in autophagy-impaired cells inhibits virus growth, suggesting a close connection between autophagy induction and HCV replication. With the emerging knowledge of the intersection of autophagic signaling pathways and HCV infection, a better understanding of the role of autophagy in the HCV life cycle will hopefully be achieved.
Dengue virus
Like HCV, DENV is a Flaviviridae family member. It is transmitted to humans via the mosquito vectors Aedes aegypti and Aedes albopictus. Although hepatic DENV infection does not lead to chronic hepatitis, DENV is a major global health problem, producing 50–100 million infections annually, resulting in the clinical syndromes of dengue fever, dengue hemorrhagic fever and dengue shock syndrome.267 DENV infects many cell types including hepatocytes; however, cells of the myeloid lineage are thought to be the primary target in vivo.268 Multiple findings have demonstrated that autophagy plays a proviral role in DENV replication in hepatocytes.269-273 DENV infection induces autophagy, and an inhibition of autophagy reduces viral replication and titers.
Both direct and indirect roles for autophagy in DENV replication have been proposed.274 DENV, like all positive-stranded RNA viruses, remodels cellular membranes to form sites of replication. It was initially hypothesized that DENV may replicate on the membranes of amphisomes, which are autophagosomes that have fused with endosomes.271 However, electron microscopy tomography of DENV replication complexes revealed that the virally-induced membrane structures are ER invaginations.275 Subsequently, it was demonstrated that DENV-induced autophagy stimulates lipid metabolism.5,269 The form of autophagy induced by DENV has the hallmarks of lipophagy.5 DENV infection not only increases the number of autophagosomes per cell, but also enhances the percentage of autophagosomes associated with lipid droplets. This association results in decreased lipid droplet size and number. DENV-induced lipophagy degrades TGs, producing FFAs that then undergo β-oxidation. The generation of FFAs by lipophagy promotes viral replication, since the requirement of autophagy for DENV replication could be supplanted by the addition of FFAs. This complementation of inhibited autophagy by FFAs requires β-oxidation, suggesting that autophagy functions to increase energy production for DENV replication.269 It is possible that autophagy performs other functions as well, and DENV-induced autophagy has also been proposed to play an indirect role in DENV replication by inhibiting apoptosis.276
The mechanism by which DENV induces lipophagy is unclear, as is its function. Typically cells respond to energy needs by inhibiting anabolic processes and stimulating catabolic ones to conserve energy and promote cell survival. In the case of DENV infection, both lipid anabolism and catabolism are stimulated together. DENV increases lipid catabolism by lipophagy while also enhancing lipid anabolism through viral stimulation of fatty acid synthesis.270 This raises the possibility that DENV infection deregulates lipid metabolism, thereby depleting cellular energy stores, which may contribute to the cytopathic effect of DENV on its host cell. The findings also suggest unique aspects of lipid metabolic regulation in DENV infection. The overexpression of at least one viral protein, NS4A, induces autophagy and protects cells from apoptosis.276 It has not been examined whether NS4A-induced autophagy stimulates lipid metabolism.
Hepatic fibrosis
Hepatic fibrosis represents the accumulation of extracellular matrix (ECM) that accompanies chronic tissue injury in a range of organs including liver, lung, kidney, skin, pancreas, bone marrow and heart. Although this scarring response is thought to have evolved for the beneficial effect of encapsulating and thereby limiting tissue injury, the detrimental consequences of fibrosis can be profound and ultimately lead to organ failure. It has been estimated that up to 45% of all deaths in the industrialized world are attributable to chronic fibrotic diseases including those in the liver.277
Efforts to prevent fibrotic diseases hinge on an understanding of the cell types and regulatory mechanisms that generate excessive ECM. Recent progress has been made in identifying the cellular sources of ECM in experimental models of tissue injury. All fibrotic tissues contain a specialized mesenchymal cell type, termed a myofibroblast, that is derived from resident cell types, primarily pericytes,278 and to a lesser extent from circulating bone marrow-derived cells. While the relative contribution from these sources varies among tissues, the generalized features of these cells—a contractile phenotype, well-developed rough endoplasmic reticulum and ECM secretion—indicate that many core features of fibrosis are conserved across all tissues.279 The mesenchymal cell source of myofibroblasts in liver injury is the hepatic stellate cell. These resident perisinusoidal cells are the best studied among tissue myofibroblast precursors because well-established methods exist to isolate them to purity from rodent and human liver.280-282 The most characteristic feature of these cells in normal liver is the presence of perinuclear membrane-bound droplets filled with retinyl esters, primarily retinyl acetate and retinyl palmitate.283
The isolation of hepatic stellate cells has led to a clearer understanding of their response to injury through a process termed “activation.” Hepatic stellate cell activation occurs during liver injury in vivo, as well as following their primary culture on plastic or other stiff substrata. The most unique and reproducible feature of stellate cell activation is the progressive loss of their intracellular retinoid-containing droplets and adoption of a more contractile and fibroblast-like appearance. Other features of stellate cell activation include increased proliferation, contractility, fibrogenesis and proinflammatory signaling.284
How retinyl esters are lost during stellate cell activation has been poorly understood, and it has been especially difficult to clarify whether this event is required for cellular activation. The loss of retinoids during cellular activation in culture is dependent on serum and platelet-derived growth factor,285 and leads to extracellular accumulation of free retinol, indicating that hydrolysis of retinyl esters probably occurs prior to cellular export. Interestingly, animals lacking the enzyme LRAT (lecithin retinol acyltransferase [phosphatidylcholine–retinol O-acyltransferase)], whose stellate cells lack lipid droplets due to their inability to esterify all-trans-retinol into retinyl esters, still mount a normal fibrogenic response to liver injury.286 These findings have suggested that hepatic stellate cell lipid storage is irrelevant to the process of cellular activation.
The recent discovery of lipophagy as a cellular mechanism of lipid breakdown,5 suggested that the loss of lipid droplets during stellate cell activation might result from increased autophagic activity, and this possibility has now been experimentally supported.59 Hepatic stellate cell activation in vitro and in rodent models of liver injury is associated with features of autophagy induction including a marked increase in autophagic vacuoles, LC3-II levels and autophagic flux. Similar findings of increased autophagy are also present in activated human stellate cells. Blocking autophagy in cultured cells either with 3-methyladenine,59,287 or specific siRNAs to Atg5 or Atg7, all lead to attenuated stellate cell activation and fibrogenesis. Importantly, mice with a genetic deletion of Atg7 specifically in stellate cells have attenuated fibrosis following sustained liver injury due to either carbon tetrachloride or thioacetamide. The link between lipophagy and stellate cell activation raised the possibility that activation of autophagy results in increased energy production by liberating FFAs from retinyl esters to serve as an energy source. Blockage of fatty acid oxidation prevents stellate cell activation, whereas provision of the exogenous FFA oleate to cultured autophagy-defective cells overcomes the block in activation conferred by loss of autophagic signaling through Atg7 depletion.
These findings were extended to other mesenchymal cell types from kidney and lung, demonstrating that autophagy is a generalized response to tissue injury in other organs besides liver. Renal mesangial cells from mice treated with siRNA to Atg7 also have reduced fibrogenic gene expression, whereas human lung fibroblasts from patients with pulmonary fibrosis have enhanced autophagic activity compared with cells derived from normal patients.59
Together, these data support a model wherein activation of fibrogenic cells in injured tissue leads to an energy requirement that is met by liberating FFAs through autophagy. Whereas this process is most obvious in hepatic stellate cells because of their abundant lipid content, the same concept may apply to other mesenchymal cells. The capacity of lecithin-retinol acetyl transferase-deficient cells to adopt and maintain a fibrogenic phenotype indicates that esterification of retinol into retinyl esters is not required for activation, yet it is possible that these cells simply contain more FFAs or other sources of cellular energy to fuel activation.
This newly uncovered link between autophagy and mesenchymal cell activation opens new opportunities for therapeutic antagonism of autophagy as an antifibrotic strategy. However, whereas the blockage of autophagy in mesenchymal cells may attenuate fibrosis, autophagy is essential to preserve energy homeostasis in other cell types, especially hepatocytes. Thus, therapeutic efforts to inhibit autophagy would need to be highly selective. From a mechanistic perspective, the findings also raise interesting questions about what are the upstream drivers of the profibrotic autophagic response in mesenchymal cells. One candidate is ER stress, which has been linked to the induction of autophagy in other situations.288
Hepatocellular cancer
The function of autophagy in carcinogenesis has been controversial, with experimental evidence suggesting that autophagy both prevents and promotes tumor development.289,290 These discrepancies are explained by current evidence of two different cellular functions of autophagy depending on the stage of carcinogenesis. In normal cells, autophagy functions to prevent neoplastic transformation through the removal of damaged organelles and specific proteins such as SQSTM1. In contrast, in established tumors autophagy supports tumor growth by satisfying the increased metabolic demands of the proliferating transformed cells by providing nutrient support in the form of amino acids, FFAs and glucose. Thus, autophagy functions as a double-edged sword in cancer, initially acting as a tumor suppressor, but once the tumor is established promoting its survival by meeting its metabolic demands (Fig. 4). Considerable findings have begun to define the mechanisms that underlie the two functions of autophagy in carcinogenesis in general, and specifically in the primary liver tumor HCC.

Figure 4. Functions of autophagy in tumorigenesis. Autophagy has a bifunctional role in the development of liver cancer. Prior to cellular transformation, autophagy functions to suppress tumor formation by preventing genomic instability, promoting cell senescence and limiting inflammation. After transformation has occurred, autophagy promotes tumorigenesis by satisfying the high metabolic demands of the tumor cells and aiding their survival.
Inhibition of autophagy promotes liver tumor development
Autophagy is suppressed by mutation or deletion of oncogenes and tumor suppressor genes that regulate the insulin signaling pathway because persistent activation of MTOR complex 1 (MTORC1) by insulin is an important inhibitory check on autophagy. Reduced autophagy from constant MTORC1 activation may promote neoplastic transformation. A hepatocyte-specific mouse knockout of PTEN or TSC1 leads to constitutively activated MTOR, a resultant decrease in autophagic activity and spontaneous HCC.291-293 However, MTOR controls multiple physiological processes in addition to autophagy, including gene transcription and protein synthesis. Therefore, these studies provide only indirect evidence that increased MTOR signaling promotes tumorigenesis through an autophagy-dependent mechanism.
The finding that the autophagy gene Becn1 is a tumor suppressor gene, particularly for HCC, provides more definitive evidence for a tumor suppressive role for autophagy.294 Autophagy is dependent on PtdIns3K generation of phosphatidylinositol-3-phosphate, and the PtdIns3K complex consisting of PIK3R4/p150, PIK3C3/VPS34, BECN1 and ATG14 (complex I) mediates early autophagosome formation.295-297 Another PtdIns3K complex composed of PIK3R4, PIK3C3, BECN1 and UVRAG (UV radiation resistance associated) protein (complex II) facilitates autophagosome and endosome maturation.298,299 In contrast, when KIAA0226/rubicon associates with a subpopulation of UVRAG-containing PtdIns3K complexes, this complex (complex III) negatively regulates a later step of autophagy and the endocytic pathway.300,301 Recruitment of BECN1 to the PtdIns3K complex is sensitive to nutrient conditions. BECN1 forms a complex with ER-associated BCL2 under nutrient-rich conditions and is released after BCL2 is phosphorylated by MAPK8/JNK1 during starvation.302 Becn1+/– mice therefore have significantly reduced autophagic activity and increased risk for cancer. These mice develop three types of spontaneous tumors, one of which is HCC, and more frequently develop preneoplastic lesions when crossed with mice containing a hepatic HBV transgene.303,304 The mouse knockout of SH3GLB1/BIF-1, a protein that activates PtdIns3K complex II through an interaction with UVRAG, causes lymphoma and HCC in mice.305 This series of reports suggests that inactivation of PtdIns3K is a strong promoter of HCC. However, it has been unclear whether findings in Becn1 and Sh3glb1-knockout mice really reflect a phenotype of defective autophagy, because PtdIns3K is involved in other cellular pathways.
The most conclusive findings of a protective role for autophagy in hepatic carcinogenesis are the long-term studies of mice with a deletion of ATG5 or ATG7, which develop multiple liver tumors. Both ATG5 and ATG7 are part of two ubiquitin-like conjugation systems essential for autophagosome formation.2 The ATG5 and ATG7 proteins basically have a specialized function for autophagy, although recent studies have revealed novel roles of ATG12-ATG5-ATG16L1 and ATG7 distinct from autophagosome formation in specialized cells such as macrophages and neuroendocrine cells.97,306,307 Small tumors form in the livers of globally mosaic Atg5 null mice and hepatocyte-specific Atg7 knockout mice at the ages of 7–9 mo.308,309 Importantly, Atg5 mosaic knockouts develop tumors only in the liver,308 suggestive of a specific reliance of hepatocytes on the tumor suppressive function of autophagy. As the mice age, tumor number and size increase until the livers are almost covered by tumors at 16–19 mo of age. Atg5- and Atg7-knockout hepatocytes/tumor cells have enlarged mitochondria and a large number of peroxisomes, probably due to impaired organelle quality control, and increased protein oxidation and oxidative stress.308,309 As a result, cells in autophagy-deficient livers are thought to have genomic instability. The resulting DNA damage in autophagy-deficient cells activates the DNA damage sensor ATM, and CHEK2/CHK2 then phosphorylates Ser20 of TRP53/p53 which induces a series of pro-apoptotic genes and cell death.310 The expression of pro-apoptotic genes observed in Atg7-knockout neonate livers is completely blocked by the additional knockout of Chek2.310 The phosphorylation of TRP53 and enhanced cell death observed in atg7−/− murine embryonic fibroblasts are restored by simultaneous depletion of Chk2.310 A reason for the greater propensity of hepatocytes to form tumors in the absence of autophagy as compared with other tissue cell types may be that the proliferative capacity of hepatocytes leads to a marked imbalance between cell proliferation and death. Further studies need to better examine the role of the ATM-CHEK2-TRP53 pathway in tumorigenesis in autophagy-deficient livers.
Regulation of autophagy in tumors
To develop successful autophagy-modulating strategies against cancer, how the function of autophagy differs based on the tumor stage, cell type and genetic factors must be better understood. To this end, the specific autophagy pathways regulated in tumor development and progression and by cancer therapy need to be more clearly established. The ability of both oncogenes and tumor suppressor genes to regulate autophagy and have an impact on tumor development has already been discussed. Whereas oncogene products usually inhibit autophagy, tumor suppressor proteins positively regulate autophagy, highlighting how autophagy can be involved in both tumor suppression and promotion. Demonstrating the complexity of autophagy regulation in tumors, the tumor suppressor TRP53/TP53 can function either as an inhibitor or simulator of autophagy, depending on its cellular location.289,290 In addition to these factors, microRNAs (miRNAs), endogenous 22-nucleotide RNAs that suppress gene expression by mRNA cleavage and/or translational repression, have also been linked to tumorigenesis by modulating cell proliferation, differentiation and invasion.311 Several miRNAs modulate autophagy in tumor cells through the targeting of autophagy-related genes,312-314 and tumor cells often have abnormal miRNA expression. In hepatic carcinogenesis, the regulation of autophagy by MIR375 in HCC under hypoxic conditions has been described.315 Hypoxia is not only one of the most pervasive microenvironmental stresses in solid tumors, but also a canonical activator of autophagy. MIR375 is primarily an inhibitor of autophagy in HCC cells under hypoxic stress and is downregulated in HCC. Decreased MIR375 impairs HCC cell viability by attenuating the protective role of autophagy through inhibition of ATG7 expression. Since the dual role of autophagy in tumor suppression and promotion presents a challenge in designing autophagy-based cancer therapy, understanding the signaling pathways that regulate autophagy in specific contexts, such as hypoxia-induced autophagy for HCC, may be the best way to utilize positive and negative regulators of autophagy as therapeutic targets in cancer.
Mechanisms of HCC development in the absence of autophagy
As discussed, mice haploinsufficient for Becn1, globally mosaic for Atg5 deficiency, or lacking Atg7 specifically in the liver, develop liver tumors probably secondary to the dysregulation of signal transduction pathways as well as to impaired organelle quality control. Critical to this process in hepatic tumors is the hepatocyte accumulation of SQSTM1 that occurs in the absence of autophagy. Liver adenoma growth in mice with a liver-specific knockout of Atg7 is markedly suppressed by a concomitant knockout of Sqstm1 because SQSTM1 accumulation from the loss of autophagy leads to dysregulation of NFKB signaling and NFE2L2/NRF2 activation.308 Activation of NFKB, an important cell survival factor, due to excess SQSTM1 accumulation, may mediate tumor development. In agreement with this hypothesis, suppression of NFKB signaling by a SQSTM1 knockout prevents growth and development of RAS-induced lung adenocarcinoma,316 and inducing Sqstm1 expression via constitutive activation of KRAS contributes to the development of pancreatic adenocarcinoma.317 The dysregulation of NFKB signaling in autophagy-incompetent cells is due at least in part to increased SQSTM1 levels.318
NFE2L2 activation also contributes to tumorigenesis in autophagy-deficient mice. NFE2L2 is a transcription factor responsible for the expression of a battery of genes encoding antioxidant proteins and detoxification enzymes. Somatic mutations in either Nfe2l2, or its inhibitory binding partner Keap1, have been identified in patients with lung, head and neck or gallbladder cancers.319,320 These mutations make tumor cells resistant to oxidative damage and anticancer agents because of sustained NFE2L2 activation. NFE2L2 can also be activated in certain types of cancer by mechanisms other than genetic mutation,321,322 and oncogene-driven Nfe2l2 transcription serves as an early event in tumorigenesis.323 NFE2L2 also redirects glucose and glutamine into anabolic pathways, especially under the sustained activation of PI3K-AKT signaling.324 An active PI3K-AKT pathway augments the nuclear accumulation of NFE2L2, enabling it to promote metabolic activities that support cell proliferation and enhance cytoprotection.324 Aggregates positive for SQSTM1 are often detected in HCC, and increased expression of NFE2L2 target genes has been observed in these tumors,309 suggesting that persistent activation of NFE2L2 in response to increased levels of SQSTM1 may contribute to HCC development. Tumors in Atg5- or Atg7-knockout livers are monoclonal with regular arrangements and patterns, and metastasis is not observed, indicating that these tumors are benign adenomas.308,309 These results are consistent with the concept of autophagy serving as a double-edged sword in hepatic carcinogenesis. In the absence of autophagy, tumor initiation is promoted, but full malignant transformation is not achieved, perhaps because the metabolic functions of autophagy are required for the survival of malignant cells. Tumor cells in the knockout livers have decreased mitochondrial function compatible with metabolic compromise.309 The findings are in contrast to BECN1-haplodeficient mice which develop frank HCC.303,304 As discussed, loss of BECN1 may have effects other than to decrease autophagy, such as blocking apoptosis. BECN1-haplodeficient fibroblasts are equally sensitive to several cell death stimuli,304 but limited death inducers were examined and studies were not performed in hepatocytes. Nonetheless, all of the studies of tumor formation in the absence of autophagy clearly point to a particular sensitivity of the liver to develop tumors in the absence of autophagic capacity.
Although autophagy functions as a tumor suppressor in nontumor cells or in the early stages of tumor cell development, autophagy becomes important for cancer cell survival once tumors are established. Cancer cells have increased metabolic demands (for both energy substrates and building blocks) for proliferation, and often grow under hypoxic conditions until angiogenesis is sufficiently established. Therefore, cancer cells, particularly those with RAS mutations such as pancreatic cancer, rely heavily on autophagy.325,326 Whether nutritional support is the molecular mechanism remains unclear, but a blockage of autophagy is sufficient to inhibit proliferation in pancreatic and other cancer cell types.327,328 Furthermore, loss of autophagy in these cells is accompanied by impaired oxidative phosphorylation likely secondary to a decreased supply of intermediates for the tricarboxylic acid cycle.325,326 Although the failure of Atg5 and Atg7 knockout mice to progress from adenomas to HCC is suggestive of the requirement for autophagy to complete hepatocyte malignant transformation, the dependency of HCC for autophagy to meet this tumor’s metabolic needs has not been examined.
Future Directions
The accidental yet fortuitous discovery of lysosomes by Christian de Duve’s laboratory nearly 60 years ago set the stage for the elucidation of the autophagic pathways.329 It is significant that these first investigations were conducted in liver, as have many of the studies that have subsequently delineated the functions of autophagy. Autophagy is the major cellular process by which distinct portions of liver cytoplasm are enveloped in autophagosomes, and the enclosed cargo is broken down inside lysosomes. Autophagy generally promotes cell survival in quiescent liver cells, which continually turn over nucleic acids, proteins, complex carbohydrates and lipids to generate nucleotides, amino acids, simple sugars and fatty acids, respectively. These smaller molecules are not only used to generate energy, but also certain nucleotides (AMP), amino acids (most prominently leucine) and fatty acids (oleate and palmitate) that are produced regulate the rate of autophagy by controlling autophagosome formation. Such regulation is dependent not only on nutrient intake, but also on the liver’s internal circadian rhythms. Autophagy also selectively targets substrates in the liver. This specificity is clearly illustrated in type 2 mitophagy as with drug (APAP)- and I/R-induced mitophagy, and the rather selective degradation of lipid droplets in response to long-term nutrient deprivation, a lipid challenge or acute alcohol administration. Autophagy’s vital function in innate immunity against bacterial pathogens is well documented. Curiously, however, in the liver the hepatitis B and C viruses and dengue virus have each evolved unique mechanisms to enhance autophagic function in order to commandeer this pathway for their propagation in the liver.
In the diseased liver, autophagy is clearly a therapeutic target. In ATD, the mutant form of SERPINA1 is retained inside hepatocytes where it aggregates, eventually causing cell injury and death, fibrosis and liver cancer. The drug carbamazepine, which is normally used to treat neurological disorders, shows promise as an autophagy enhancer, thereby accelerating the intracellular clearance of aggregated SERPINA1-Z. The therapeutic potential of CBZ is now being tested in clinical trials while other potential drugs to augment autophagy are undergoing screening by high-throughput autophagy assays. Similarly, the discovery of autophagy’s function in hepatic lipid catabolism implies that the most prevalent form of human liver disease, NAFLD, and its more severe form, NASH, both represent deficits in lipophagy. It is therefore reasonable that drugs now undergoing testing for their ability to stimulate autophagy may have therapeutic value for patients with NAFLD and/or NASH. Similarly, some of these compounds might be effective in ablating steatosis and accelerating the clearance of Mallory-Denk bodies in patients with alcoholic liver disease. In this review, studies of autophagy in liver parenchymal cells or hepatocytes have been emphasized. However, the perisinusoidal stellate cells that secrete extracellular matrix components and which initiate liver fibrosis also have an active autophagic pathway. Recent findings indicate that stellate cell activation depends on the oxidation of fatty acids derived from lipophagy of retinyl esters. Thus, enhancement of autophagy in this instance initiates stellate cell activation, bringing about a fibrotic response. These findings imply that, in contrast to the pathologies just mentioned, therapeutic measures to limit or block autophagy specifically in stellate cells, would attenuate fibrosis after liver injury. It will be important for future investigations to better address the functions of autophagy in non-parenchymal cells as findings of conflicting roles of autophagy in different cell types has important implications for the use of therapies that increase autophagic function. Finally, rather strong evidence indicates that autophagy has a tumor suppressor function in liver, based on findings that mice lacking the autophagy gene Atg5 develop liver tumors that increase in size and frequency as the animals age. These findings imply that the deficiency in autophagosome formation promotes genomic instability that leads to cellular transformation. This finding might be somewhat specific to HCC and not apply universally to all forms of cancer, since in pancreatic ductal adenocarcinoma cells, for example, autophagy is higher than in normal pancreas.326 Thus, the mechanisms that trigger and sustain neoplasia in a specific tissue vary with the tissue’s genomic activity and specific physiology.
In summary, tremendous strides in our understanding of the importance of autophagy in the liver have been made over the past few years in large part due to the availability of genetically altered models of autophagy. Although major questions remain, the critical importance of autophagy in many hepatic physiological and pathological conditions has been clearly established. Future investigations are likely to further elucidate the importance of the autophagic pathways in the liver, which will hopefully provide new therapeutic approaches to the most common liver diseases.
Acknowledgments
This work was supported in part by the National Institutes of Health (DK044234 and DK061498 to M.J.C., AA020518 and RR021940 to W.-X.D., AA020709 and DK056621 to S.L.F., DK079879, DK090115 and AG028740 to J.-S.K., DK037034, DK073336, CA119079, CA138313, GM103542 and RR015455 to J.J.L., DK077086 and HL097738 to J.D.L., DK094652, AI083025 and CA123328 to J.H.J.O., DK096990, DK084512 and DK076918 to D.H.P., AI080703 and AI057153 to G.R., DK081817 to R.B.R., GM095566 to A.T. and CA083817 to X.-M.Y.) and a Howard Hughes Medical Institute Physician-Scientist Award (A.T.).
Glossary
Abbreviations:
- ADH
alcohol dehydrogenase
- ALD
alcoholic liver disease
- APAP
acetaminophen
- ATD
SERPINA1/α1-antitrypsin deficiency
- ATG
autophagy-related
- CBZ
carbamazepine
- CEBPB
CCAAT/enhancer-binding protein (C/EBP), β
- CMA
chaperone-mediated autophagy
- CYP2E1
cytochrome P450, family 2, subfamily E, polypeptide 1
- DENV
dengue virus
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- FFAs
free fatty acids
- FOXO
forkhead box O
- GFP
green fluorescent protein
- HBsAg
hepatitis B virus surface antigen
- HBV
hepatitis B virus
- HBx
hepatitis B virus protein X
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HFD
high-fat diet
- I/R
ischemia/reperfusion
- LC3
microtubule-associated protein 1 light chain 3
- LD
lipid droplet
- LPS
lipopolysaccharide
- MFN
mitofusin
- miRNA
microRNA
- MPT
mitochondrial permeability transition
- MTOR
mechanistic target of rapamycin
- NAFLD
nonalcoholic fatty liver disease
- NAPQI
N-acetyl-p-benzoquinone imine
- NASH
nonalcoholic steatohepatitis
- PI3K
class I phosphoinositide 3-kinase
- PINK1
PTEN-induced putative kinase 1
- PPARGC1A
peroxisome proliferator-activated receptor gamma, coactivator 1 α
- PtdIns3K
class III phosphatidylinositol 3-kinase
- ROS
reactive oxygen species
- SERPINA1
serpin peptidase inhibitor, clade A (α-1 antiproteinase, antitrypsin), member 1
- SERPINA1-Z
SERPINA1 Z allele
- SQSTM1
sequestosome 1 (p62)
- T3
3,3′5-triiodo-thyronine
- TGs
triglycerides
- TLR
toll like receptor
- UPR
unfolded protein response
- UVRAG
UV radiation resistance associated
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/25063
References
- 1.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 2.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 3.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ueno T, Ezaki J, Kominami E. Metabolic contribution of hepatic autophagic proteolysis: old wine in new bottles. Biochim Biophys Acta. 2012;1824:51–8. doi: 10.1016/j.bbapap.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 9.Schworer CM, Shiffer KA, Mortimore GE. Quantitative relationship between autophagy and proteolysis during graded amino acid deprivation in perfused rat liver. J Biol Chem. 1981;256:7652–8. [PubMed] [Google Scholar]
- 10.Mortimore GE, Hutson NJ, Surmacz CA. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc Natl Acad Sci U S A. 1983;80:2179–83. doi: 10.1073/pnas.80.8.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirschke H, Langner J, Wiederanders B, Ansorge S, Bohley P. Cathepsin L. A new proteinase from rat-liver lysosomes. Eur J Biochem. 1977;74:293–301. doi: 10.1111/j.1432-1033.1977.tb11393.x. [DOI] [PubMed] [Google Scholar]
- 12.Seglen PO, Gordon PB, Poli A. Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim Biophys Acta. 1980;630:103–18. doi: 10.1016/0304-4165(80)90141-5. [DOI] [PubMed] [Google Scholar]
- 13.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pösö AR, Wert JJ, Jr., Mortimore GE. Multifunctional control of amino acids of deprivation-induced proteolysis in liver. Role of leucine. J Biol Chem. 1982;257:12114–20. [PubMed] [Google Scholar]
- 15.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gohla A, Klement K, Piekorz RP, Pexa K, vom Dahl S, Spicher K, et al. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci U S A. 2007;104:3003–8. doi: 10.1073/pnas.0611434104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 19.Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7:727–36. doi: 10.4161/auto.7.7.15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim I, Lemasters JJ. Mitochondrial degradation by autophagy (mitophagy) in GFP-LC3 transgenic hepatocytes during nutrient deprivation. Am J Physiol Cell Physiol. 2011;300:C308–17. doi: 10.1152/ajpcell.00056.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 22.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–9. [PubMed] [Google Scholar]
- 23.Pfeifer U. Inhibition by insulin of the formation of autophagic vacuoles in rat liver. A morphometric approach to the kinetics of intracellular degradation by autophagy. J Cell Biol. 1978;78:152–67. doi: 10.1083/jcb.78.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Enriquez S, Kai Y, Maldonado E, Currin RT, Lemasters JJ. Roles of mitophagy and the mitochondrial permeability transition in remodeling of cultured rat hepatocytes. Autophagy. 2009;5:1099–106. doi: 10.4161/auto.5.8.9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107:11835–40. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemasters JJ. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol. 1999;276:G1–6. doi: 10.1152/ajpgi.1999.276.1.G1. [DOI] [PubMed] [Google Scholar]
- 29.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 30.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201–14. doi: 10.1089/ars.2010.3482. [DOI] [PubMed] [Google Scholar]
- 32.Kim EH, Choi KS. A critical role of superoxide anion in selenite-induced mitophagic cell death. Autophagy. 2008;4:76–8. doi: 10.4161/auto.5119. [DOI] [PubMed] [Google Scholar]
- 33.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aggarwal BB, Quintanilha AT, Cammack R, Packer L. Damage to mitochondrial electron transport and energy coupling by visible light. Biochim Biophys Acta. 1978;502:367–82. doi: 10.1016/0005-2728(78)90057-9. [DOI] [PubMed] [Google Scholar]
- 35.Alexandratou E, Yova D, Handris P, Kletsas D, Loukas S. Human fibroblast alterations induced by low power laser irradiation at the single cell level using confocal microscopy. Photochem Photobiol Sci. 2002;1:547–52. doi: 10.1039/b110213n. [DOI] [PubMed] [Google Scholar]
- 36.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14:1919–28. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–55. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–31. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–42. doi: 10.1128/MCB.02246-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–46. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6:462–72. doi: 10.4161/auto.6.4.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cui T, Fan C, Gu L, Gao H, Liu Q, Zhang T, et al. Silencing of PINK1 induces mitophagy via mitochondrial permeability transition in dopaminergic MN9D cells. Brain Res. 2011;1394:1–13. doi: 10.1016/j.brainres.2011.01.035. [DOI] [PubMed] [Google Scholar]
- 43.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–7. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 44.Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006;7:373–8. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- 45.Thiele C, Spandl J. Cell biology of lipid droplets. Curr Opin Cell Biol. 2008;20:378–85. doi: 10.1016/j.ceb.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 46.Zechner R, Madeo F. Cell biology: Another way to get rid of fat. Nature. 2009;458:1118–9. doi: 10.1038/4581118a. [DOI] [PubMed] [Google Scholar]
- 47.Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun. 2009;382:419–23. doi: 10.1016/j.bbrc.2009.03.039. [DOI] [PubMed] [Google Scholar]
- 48.Boström P, Andersson L, Rutberg M, Perman J, Lidberg U, Johansson BR, et al. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat Cell Biol. 2007;9:1286–93. doi: 10.1038/ncb1648. [DOI] [PubMed] [Google Scholar]
- 49.Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen W-L, et al. SNARE proteins are required for macroautophagy. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell. 2011;146:303–17. doi: 10.1016/j.cell.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428–38. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jackson-Hayes L, Song S, Lavrentyev EN, Jansen MS, Hillgartner FB, Tian L, et al. A thyroid hormone response unit formed between the promoter and first intron of the carnitine palmitoyltransferase-Ialpha gene mediates the liver-specific induction by thyroid hormone. J Biol Chem. 2003;278:7964–72. doi: 10.1074/jbc.M211062200. [DOI] [PubMed] [Google Scholar]
- 53.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ittermann T, Haring R, Wallaschofski H, Baumeister SE, Nauck M, Dörr M, et al. Inverse association between serum free thyroxine levels and hepatic steatosis: results from the Study of Health in Pomerania. Thyroid. 2012;22:568–74. doi: 10.1089/thy.2011.0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–14. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–85. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–71. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Singh R, Xiang Y, Czaja MJ. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology. 2010;52:266–77. doi: 10.1002/hep.23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–46. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 61.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–10. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao W, Kang J-H, Liao Y, Li M, Yin X-M. Autophagy and cell death. In: Yin X-M, Dong Z, eds. Essentials of apoptosis: a guide for basic and clinical research. New York: Springer, 2009:671-90 [Google Scholar]
- 63.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–81. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 65.Thorburn A. Studying autophagy’s relationship to cell death. Autophagy. 2008;4:391–4. doi: 10.4161/auto.5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 67.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–7. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 68.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011;7:1159–72. doi: 10.4161/auto.7.10.16818. [DOI] [PubMed] [Google Scholar]
- 70.Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131:548–57. doi: 10.1002/ijc.26374. [DOI] [PubMed] [Google Scholar]
- 71.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 72.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–50. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 73.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–52. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Singh R, Massey AC, Kane SS, Kaushik S, Grant T, et al. Loss of macroautophagy promotes or prevents fibroblast apoptosis depending on the death stimulus. J Biol Chem. 2008;283:4766–77. doi: 10.1074/jbc.M706666200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–9. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 77.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 78.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–6. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kang C, Avery L. To be or not to be, the level of autophagy is the question: dual roles of autophagy in the survival response to starvation. Autophagy. 2008;4:82–4. doi: 10.4161/auto.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–10. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 81.Ding WX, Ni HM, Gao W, Chen X, Kang JH, Stolz DB, et al. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther. 2009;8:2036–45. doi: 10.1158/1535-7163.MCT-08-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol. 2012;189:15–20. doi: 10.4049/jimmunol.1102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jordan TX, Randall G. Manipulation or capitulation: virus interactions with autophagy. Microbes Infect. 2012;14:126–39. doi: 10.1016/j.micinf.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 85.Gorbea C, Makar KA, Pauschinger M, Pratt G, Bersola JL, Varela J, et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J Biol Chem. 2010;285:23208–23. doi: 10.1074/jbc.M109.047464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tallóczy Z, Virgin HW, 4th, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–9. doi: 10.4161/auto.2176. [DOI] [PubMed] [Google Scholar]
- 87.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–21. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283:33175–82. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–35. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 91.Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13:1319–27. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol. 2009;10:480–7. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–39. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, et al. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33:1250–9. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- 95.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–6. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 96.Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012;11:397–409. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Estrabaud E, De Muynck S, Asselah T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J Hepatol. 2011;55:1150–3. doi: 10.1016/j.jhep.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 99.Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology. 2011;53:406–14. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grégoire IP, Richetta C, Meyniel-Schicklin L, Borel S, Pradezynski F, Diaz O, et al. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011;7:e1002422. doi: 10.1371/journal.ppat.1002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Desai MM, Gong B, Chan T, Davey RA, Soong L, Kolokoltsov AA, et al. Differential, type I interferon-mediated autophagic trafficking of hepatitis C virus proteins in mouse liver. Gastroenterology. 2011;141:674–85, 685.e1-6. doi: 10.1053/j.gastro.2011.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–37. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 104.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–54. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rutter J, Reick M, McKnight SL. Metabolism and the control of circadian rhythms. Annu Rev Biochem. 2002;71:307–31. doi: 10.1146/annurev.biochem.71.090501.142857. [DOI] [PubMed] [Google Scholar]
- 106.Ukai H, Ueda HR. Systems biology of mammalian circadian clocks. Annu Rev Physiol. 2010;72:579–603. doi: 10.1146/annurev-physiol-073109-130051. [DOI] [PubMed] [Google Scholar]
- 107.Li S, Chen XW, Yu L, Saltiel AR, Lin JD. Circadian metabolic regulation through crosstalk between casein kinase 1δ and transcriptional coactivator PGC-1α. Mol Endocrinol. 2011;25:2084–93. doi: 10.1210/me.2011-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu C, Li S, Liu T, Borjigin J, Lin JD. Transcriptional coactivator PGC-1α integrates the mammalian clock and energy metabolism. Nature. 2007;447:477–81. doi: 10.1038/nature05767. [DOI] [PubMed] [Google Scholar]
- 109.Pfeifer U, Scheller H. A morphometric study of cellular autophagy including diurnal variations in kidney tubules of normal rats. J Cell Biol. 1975;64:608–21. doi: 10.1083/jcb.64.3.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pfeifer U, Strauss P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. J Mol Cell Cardiol. 1981;13:37–49. doi: 10.1016/0022-2828(81)90227-3. [DOI] [PubMed] [Google Scholar]
- 111.Bhattacharya R, von Mayersbach H. Histochemistry of circadian changes of some lysosomal enzymes in rat liver. Acta Histochem Suppl. 1976;16:109–15. [PubMed] [Google Scholar]
- 112.Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 2011;30:4642–51. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 114.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–83. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 115.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seo YK, Jeon TI, Chong HK, Biesinger J, Xie X, Osborne TF. Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 2011;13:367–75. doi: 10.1016/j.cmet.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ma D, Li S, Molusky MM, Lin JD. Circadian autophagy rhythm: a link between clock and metabolism? Trends Endocrinol Metab. 2012;23:319–25. doi: 10.1016/j.tem.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Minami Y, Kasukawa T, Kakazu Y, Iigo M, Sugimoto M, Ikeda S, et al. Measurement of internal body time by blood metabolomics. Proc Natl Acad Sci U S A. 2009;106:9890–5. doi: 10.1073/pnas.0900617106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360:2749–57. doi: 10.1056/NEJMcp0900449. [DOI] [PubMed] [Google Scholar]
- 120.Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in α-1-antitrypsin deficiency. Cell Death Differ. 2009;16:39–45. doi: 10.1038/cdd.2008.103. [DOI] [PubMed] [Google Scholar]
- 121.Carlson JA, Rogers BB, Sifers RN, Hawkins HK, Finegold MJ, Woo SL. Multiple tissues express alpha 1-antitrypsin in transgenic mice and man. J Clin Invest. 1988;82:26–36. doi: 10.1172/JCI113580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Carlson JA, Rogers BB, Sifers RN, Finegold MJ, Clift SM, DeMayo FJ, et al. Accumulation of PiZ α 1-antitrypsin causes liver damage in transgenic mice. J Clin Invest. 1989;83:1183–90. doi: 10.1172/JCI113999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–32. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 124.Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976;294:1316–21. doi: 10.1056/NEJM197606102942404. [DOI] [PubMed] [Google Scholar]
- 125.Piitulainen E, Carlson J, Ohlsson K, Sveger T. α1-antitrypsin deficiency in 26-year-old subjects: lung, liver, and protease/protease inhibitor studies. Chest. 2005;128:2076–81. doi: 10.1378/chest.128.4.2076. [DOI] [PubMed] [Google Scholar]
- 126.Perlmutter DH. Alpha-1-antitrypsin deficiency: importance of proteasomal and autophagic degradative pathways in disposal of liver disease-associated protein aggregates. Annu Rev Med. 2011;62:333–45. doi: 10.1146/annurev-med-042409-151920. [DOI] [PubMed] [Google Scholar]
- 127.Kamimoto T, Shoji S, Hidvegi T, Mizushima N, Umebayashi K, Perlmutter DH, et al. Intracellular inclusions containing mutant α1-antitrypsin Z are propagated in the absence of autophagic activity. J Biol Chem. 2006;281:4467–76. doi: 10.1074/jbc.M509409200. [DOI] [PubMed] [Google Scholar]
- 128.Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G Signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant α1-antitrypsin Z in the classical form of α1-antitrypsin deficiency. J Biol Chem. 2007;282:27769–80. doi: 10.1074/jbc.M704330200. [DOI] [PubMed] [Google Scholar]
- 129.Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant α1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280:39002–15. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- 130.Teckman JH, Perlmutter DH. Retention of mutant α(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279:G961–74. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- 131.Kruse KB, Brodsky JL, McCracken AA. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: one for soluble Z variant of human α-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol Biol Cell. 2006;17:203–12. doi: 10.1091/mbc.E04-09-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kruse KB, Dear A, Kaltenbrun ER, Crum BE, George PM, Brennan SO, et al. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am J Pathol. 2006;168:1299–308, quiz 1404-5. doi: 10.2353/ajpath.2006.051097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gosai SJ, Kwak JH, Luke CJ, Long OS, King DE, Kovatch KJ, et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One. 2010;5:e15460. doi: 10.1371/journal.pone.0015460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A. 2007;104:19023–8. doi: 10.1073/pnas.0709695104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tsvetkov AS, Miller J, Arrasate M, Wong JS, Pleiss MA, Finkbeiner S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–7. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960–7. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 137.Ruhl CE, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology. 2003;124:71–9. doi: 10.1053/gast.2003.50004. [DOI] [PubMed] [Google Scholar]
- 138.Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9:524–30.e1; quiz e60. doi: 10.1016/j.cgh.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 139.Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:159–66. doi: 10.1586/egh.11.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–50. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 142.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 143.Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284:31484–92. doi: 10.1074/jbc.M109.033936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–65. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Inami Y, Yamashina S, Izumi K, Ueno T, Tanida I, Ikejima K, et al. Hepatic steatosis inhibits autophagic proteolysis via impairment of autophagosomal acidification and cathepsin expression. Biochem Biophys Res Commun. 2011;412:618–25. doi: 10.1016/j.bbrc.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 146.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705–14. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–9. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 148.Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–40. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 149.Fontana L, Zhao E, Amir M, Dong H, Tanaka K, Czaja MJ. Aging promotes the development of diet-induced murine steatohepatitis but not steatosis. Hepatology. 2013;57:995–1004. doi: 10.1002/hep.26099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maher JJ, Leon P, Ryan JC. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology. 2008;48:670–8. doi: 10.1002/hep.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–9. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339:487–98. doi: 10.1124/jpet.111.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–9. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- 155.Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011;236:546–56. doi: 10.1258/ebm.2011.010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130–44. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42:21–3. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]
- 158.Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53:86–95. doi: 10.1002/hep.24017. [DOI] [PubMed] [Google Scholar]
- 159.Thomes PG, Trambly CS, Thiele GM, Duryee MJ, Fox HS, Haorah J, et al. Proteasome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem Biophys Res Commun. 2012;417:262–7. doi: 10.1016/j.bbrc.2011.11.097. [DOI] [PubMed] [Google Scholar]
- 160.Yeon JE, Califano S, Xu J, Wands JR, De La Monte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38:703–14. doi: 10.1053/jhep.2003.50368. [DOI] [PubMed] [Google Scholar]
- 161.Shulga N, Hoek JB, Pastorino JG. Elevated PTEN levels account for the increased sensitivity of ethanol-exposed cells to tumor necrosis factor-induced cytotoxicity. J Biol Chem. 2005;280:9416–24. doi: 10.1074/jbc.M409505200. [DOI] [PubMed] [Google Scholar]
- 162.Baraona E, Leo MA, Borowsky SA, Lieber CS. Alcoholic hepatomegaly: accumulation of protein in the liver. Science. 1975;190:794–5. doi: 10.1126/science.1198096. [DOI] [PubMed] [Google Scholar]
- 163.Donohue TM, Jr., Zetterman RK, Zhang-Gouillon ZQ, French SW. Peptidase activities of the multicatalytic protease in rat liver after voluntary and intragastric ethanol administration. Hepatology. 1998;28:486–91. doi: 10.1002/hep.510280228. [DOI] [PubMed] [Google Scholar]
- 164.Harada M, Hanada S, Toivola DM, Ghori N, Omary MB. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology. 2008;47:2026–35. doi: 10.1002/hep.22294. [DOI] [PubMed] [Google Scholar]
- 165.Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, Fujimura S, et al. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem. 2004;279:8452–9. doi: 10.1074/jbc.M306337200. [DOI] [PubMed] [Google Scholar]
- 166.Schliess F, Reissmann R, Reinehr R, vom Dahl S, Häussinger D. Involvement of integrins and Src in insulin signaling toward autophagic proteolysis in rat liver. J Biol Chem. 2004;279:21294–301. doi: 10.1074/jbc.M313901200. [DOI] [PubMed] [Google Scholar]
- 167.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, et al. Acute Liver Failure Study Group Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 2005;42:1364–72. doi: 10.1002/hep.20948. [DOI] [PubMed] [Google Scholar]
- 168.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 169.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology. 2012;55:222–32. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Apostolova N, Gomez-Sucerquia LJ, Gortat A, Blas-Garcia A, Esplugues JV. Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival-promoting autophagy. Hepatology. 2011;54:1009–19. doi: 10.1002/hep.24459. [DOI] [PubMed] [Google Scholar]
- 171.Apostolova N, Gomez-Sucerquia LJ, Gortat A, Blas-Garcia A, Esplugues JV. Autophagy as a rescue mechanism in efavirenz-induced mitochondrial dysfunction: a lesson from hepatic cells. Autophagy. 2011;7:1402–4. doi: 10.4161/auto.7.11.17653. [DOI] [PubMed] [Google Scholar]
- 172.Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–81. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 173.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–5. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–85. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 176.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–42. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–33. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285:27879–90. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–70. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286:19630–40. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–37. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Walden H, Martinez-Torres RJ. Regulation of Parkin E3 ubiquitin ligase activity. Cell Mol Life Sci. 2012;69:3053–67. doi: 10.1007/s00018-012-0978-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ding WX, Guo F, Ni HM, Bockus A, Manley S, Stolz DB, et al. Parkin and mitofusins reciprocally regulate mitophagy and mitochondrial spheroid formation. J Biol Chem. 2012;287:42379–88. doi: 10.1074/jbc.M112.413682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ding WX, Li M, Biazik JM, Morgan DG, Guo F, Ni HM, et al. Electron microscopic analysis of a spherical mitochondrial structure. J Biol Chem. 2012;287:42373–8. doi: 10.1074/jbc.M112.413674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jaeschke H. Mechanisms of reperfusion injury after warm ischemia of the liver. J Hepatobiliary Pancreat Surg. 1998;5:402–8. doi: 10.1007/s005340050064. [DOI] [PubMed] [Google Scholar]
- 187.Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 188.Kim JS, He L, Qian T, Lemasters JJ. Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Curr Mol Med. 2003;3:527–35. doi: 10.2174/1566524033479564. [DOI] [PubMed] [Google Scholar]
- 189.Kim JS, Wang JH, Lemasters JJ. Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: role of Ca2+-dependent mitochondrial formation of reactive oxygen species. Am J Physiol Gastrointest Liver Physiol. 2012;302:G723–31. doi: 10.1152/ajpgi.00082.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 191.Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304:463–70. doi: 10.1016/S0006-291X(03)00618-1. [DOI] [PubMed] [Google Scholar]
- 192.Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol. 1997;273:C1783–92. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- 193.Nieminen AL, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem J. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ. Nitric oxide: a signaling molecule against mitochondrial permeability transition- and pH-dependent cell death after reperfusion. Free Radic Biol Med. 2004;37:1943–50. doi: 10.1016/j.freeradbiomed.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 195.Kon K, Kim JS, Jaeschke H, Lemasters JJ. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 2004;40:1170–9. doi: 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- 196.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–55. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 198.Candé C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, et al. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–22. doi: 10.1016/S0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- 199.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57. doi: 10.1016/S0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 200.Richter C, Gogvadze V, Laffranchi R, Schlapbach R, Schweizer M, Suter M, et al. Oxidants in mitochondria: from physiology to diseases. Biochim Biophys Acta. 1995;1271:67–74. doi: 10.1016/0925-4439(95)00012-S. [DOI] [PubMed] [Google Scholar]
- 201.Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic Biol Med. 2002;32:804–12. doi: 10.1016/S0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- 202.Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2:156–62. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- 203.Pacher P, Hajnóczky G. Propagation of the apoptotic signal by mitochondrial waves. EMBO J. 2001;20:4107–21. doi: 10.1093/emboj/20.15.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kim JS, Nitta T, Mohuczy D, O’Malley KA, Moldawer LL, Dunn WA, Jr., et al. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47:1725–36. doi: 10.1002/hep.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wang JH, Ahn IS, Fischer TD, Byeon JI, Dunn WA, Jr., Behrns KE, et al. Autophagy suppresses age-dependent ischemia and reperfusion injury in livers of mice. Gastroenterology. 2011;141:2188–99.e6. doi: 10.1053/j.gastro.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–6. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 207.Shi Y, Melnikov VY, Schrier RW, Edelstein CL. Downregulation of the calpain inhibitor protein calpastatin by caspases during renal ischemia-reperfusion. Am J Physiol Renal Physiol. 2000;279:F509–17. doi: 10.1152/ajprenal.2000.279.3.F509. [DOI] [PubMed] [Google Scholar]
- 208.Selzner M, Selzner N, Jochum W, Graf R, Clavien PA. Increased ischemic injury in old mouse liver: an ATP-dependent mechanism. Liver Transpl. 2007;13:382–90. doi: 10.1002/lt.21100. [DOI] [PubMed] [Google Scholar]
- 209.Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138:513–21, 521.e1-6. doi: 10.1053/j.gastro.2009.09.067. [DOI] [PubMed] [Google Scholar]
- 210.Okaya T, Blanchard J, Schuster R, Kuboki S, Husted T, Caldwell CC, et al. Age-dependent responses to hepatic ischemia/reperfusion injury. Shock. 2005;24:421–7. doi: 10.1097/01.shk.0000181282.14050.11. [DOI] [PubMed] [Google Scholar]
- 211.Huber N, Sakai N, Eismann T, Shin T, Kuboki S, Blanchard J, et al. Age-related decrease in proteasome expression contributes to defective nuclear factor-kappaB activation during hepatic ischemia/reperfusion. Hepatology. 2009;49:1718–28. doi: 10.1002/hep.22840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Vittorini S, Paradiso C, Donati A, Cavallini G, Masini M, Gori Z, et al. The age-related accumulation of protein carbonyl in rat liver correlates with the age-related decline in liver proteolytic activities. J Gerontol A Biol Sci Med Sci. 1999;54:B318–23. doi: 10.1093/gerona/54.8.B318. [DOI] [PubMed] [Google Scholar]
- 213.Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008;14:959–65. doi: 10.1038/nm.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–92. doi: 10.1089/rej.2006.0535. [DOI] [PubMed] [Google Scholar]
- 215.Wang JH, Behrns KE, Leeuwenburgh C, Kim JS. Critical role of autophagy in ischemia/reperfusion injury to aged livers. Autophagy. 2012;8:140–1. doi: 10.4161/auto.8.1.18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cardinal J, Pan P, Dhupar R, Ross M, Nakao A, Lotze M, et al. Cisplatin prevents high mobility group box 1 release and is protective in a murine model of hepatic ischemia/reperfusion injury. Hepatology. 2009;50:565–74. doi: 10.1002/hep.23021. [DOI] [PubMed] [Google Scholar]
- 217.Degli Esposti D, Sebagh M, Pham P, Reffas M, Poüs C, Brenner C, et al. Ischemic preconditioning induces autophagy and limits necrosis in human recipients of fatty liver grafts, decreasing the incidence of rejection episodes. Cell Death Dis. 2011;2:e111. doi: 10.1038/cddis.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Minor T, Stegemann J, Hirner A, Koetting M. Impaired autophagic clearance after cold preservation of fatty livers correlates with tissue necrosis upon reperfusion and is reversed by hypothermic reconditioning. Liver Transpl. 2009;15:798–805. doi: 10.1002/lt.21751. [DOI] [PubMed] [Google Scholar]
- 219.Patient R, Hourioux C, Roingeard P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell Microbiol. 2009;11:1561–70. doi: 10.1111/j.1462-5822.2009.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A. 2010;107:4383–8. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, et al. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol. 2011;85:6319–33. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang J, Shi Y, Yang H. [Infection with hepatitis B virus enhances basal autophagy] Wei Sheng Wu Xue Bao. 2010;50:1651–6. [PubMed] [Google Scholar]
- 223.Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology. 2009;49:60–71. doi: 10.1002/hep.22581. [DOI] [PubMed] [Google Scholar]
- 224.Sir D, Ann DK, Ou JH. Autophagy by hepatitis B virus and for hepatitis B virus. Autophagy. 2010;6:548–9. doi: 10.4161/auto.6.4.11669. [DOI] [PubMed] [Google Scholar]
- 225.Cho HK, Cheong KJ, Kim HY, Cheong J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem J. 2011;435:431–9. doi: 10.1042/BJ20102071. [DOI] [PubMed] [Google Scholar]
- 226.Li B, Gao B, Ye L, Han X, Wang W, Kong L, et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007;124:44–9. doi: 10.1016/j.virusres.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 227.Xu Z, Jensen G, Yen TS. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. J Virol. 1997;71:7387–92. doi: 10.1128/jvi.71.10.7387-7392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, Lai MD, et al. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis. 2004;25:2023–32. doi: 10.1093/carcin/bgh207. [DOI] [PubMed] [Google Scholar]
- 229.Na B, Huang Z, Wang Q, Qi Z, Tian Y, Lu CC, et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS One. 2011;6:e26240. doi: 10.1371/journal.pone.0026240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ou JH, Rutter WJ. Regulation of secretion of the hepatitis B virus major surface antigen by the preS-1 protein. J Virol. 1987;61:782–6. doi: 10.1128/jvi.61.3.782-786.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Persing DH, Varmus HE, Ganem D. Inhibition of secretion of hepatitis B surface antigen by a related presurface polypeptide. Science. 1986;234:1388–91. doi: 10.1126/science.3787251. [DOI] [PubMed] [Google Scholar]
- 232.Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, et al. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell. 1989;59:1145–56. doi: 10.1016/0092-8674(89)90770-8. [DOI] [PubMed] [Google Scholar]
- 233.Xu Z, Yen TS. Intracellular retention of surface protein by a hepatitis B virus mutant that releases virion particles. J Virol. 1996;70:133–40. doi: 10.1128/jvi.70.1.133-140.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chua PK, Wang RY, Lin MH, Masuda T, Suk FM, Shih C. Reduced secretion of virions and hepatitis B virus (HBV) surface antigen of a naturally occurring HBV variant correlates with the accumulation of the small S envelope protein in the endoplasmic reticulum and Golgi apparatus. J Virol. 2005;79:13483–96. doi: 10.1128/JVI.79.21.13483-13496.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44:279–89. doi: 10.1016/j.molcel.2011.07.039. [DOI] [PubMed] [Google Scholar]
- 236.Lin CL, Kao JH. Hepatitis B viral factors and clinical outcomes of chronic hepatitis B. J Biomed Sci. 2008;15:137–45. doi: 10.1007/s11373-007-9225-8. [DOI] [PubMed] [Google Scholar]
- 237.Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol. 2011;85:13453–6. doi: 10.1128/JVI.06064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sir D, Ou JH. Autophagy in viral replication and pathogenesis. Mol Cells. 2010;29:1–7. doi: 10.1007/s10059-010-0014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 240.Bartenschlager R, Frese M, Pietschmann T. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res. 2004;63:71–180. doi: 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 241.Bartenschlager R, Sparacio S. Hepatitis C virus molecular clones and their replication capacity in vivo and in cell culture. Virus Res. 2007;127:195–207. doi: 10.1016/j.virusres.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 242.Brass V, Moradpour D, Blum HE. Molecular virology of hepatitis C virus (HCV): 2006 update. Int J Med Sci. 2006;3:29–34. doi: 10.7150/ijms.3.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol. 2008;82:2241–9. doi: 10.1128/JVI.02093-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A. 2009;106:14046–51. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chu VC, Bhattacharya S, Nomoto A, Lin J, Zaidi SK, Oberley TD, et al. Persistent expression of hepatitis C virus non-structural proteins leads to increased autophagy and mitochondrial injury in human hepatoma cells. PLoS One. 2011;6:e28551. doi: 10.1371/journal.pone.0028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ferraris P, Blanchard E, Roingeard P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J Gen Virol. 2010;91:2230–7. doi: 10.1099/vir.0.022186-0. [DOI] [PubMed] [Google Scholar]
- 247.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–61. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Taguwa S, Kambara H, Fujita N, Noda T, Yoshimori T, Koike K, et al. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J Virol. 2011;85:13185–94. doi: 10.1128/JVI.06099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest. 2011;121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Rautou PE, Cazals-Hatem D, Feldmann G, Mansouri A, Grodet A, Barge S, et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am J Pathol. 2011;178:2708–15. doi: 10.1016/j.ajpath.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol. 2012;86:8705–12. doi: 10.1128/JVI.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011;85:10561–71. doi: 10.1128/JVI.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Vescovo T, Romagnoli A, Perdomo AB, Corazzari M, Ciccosanti F, Alonzi T, et al. Autophagy protects cells from HCV-induced defects in lipid metabolism. Gastroenterology. 2012;142:644–53.e3. doi: 10.1053/j.gastro.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 254.Guévin C, Manna D, Bélanger C, Konan KV, Mak P, Labonté P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology. 2010;405:1–7. doi: 10.1016/j.virol.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, et al. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem. 2012;287:18036–43. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009;5:937–45. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]
- 257.Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J Gastroenterol. 2010;45:195–203. doi: 10.1007/s00535-009-0132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Asselah T, Bièche I, Mansouri A, Laurendeau I, Cazals-Hatem D, Feldmann G, et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J Pathol. 2010;221:264–74. doi: 10.1002/path.2703. [DOI] [PubMed] [Google Scholar]
- 259.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J Virol. 2012;86:6315–22. doi: 10.1128/JVI.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Liu Z, Tian Y, Machida K, Lai MM, Luo G, Foung SK, et al. Transient activation of the PI3K-AKT pathway by hepatitis C virus to enhance viral entry. J Biol Chem. 2012;287:41922–30. doi: 10.1074/jbc.M112.414789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104:14050–5. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol. 2010;189:925–35. doi: 10.1083/jcb.201002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A. 2009;106:2770–5. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6:367–80. doi: 10.1016/j.chom.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–68. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Gubler DJ. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 2002;10:100–3. doi: 10.1016/S0966-842X(01)02288-0. [DOI] [PubMed] [Google Scholar]
- 268.Harris E, Holden KL, Edgil D, Polacek C, Clyde K. Molecular biology of flaviviruses. Novartis Found Symp. 2006;277:23–39, discussion 40, 71-3, 251-3. doi: 10.1002/0470058005.ch3. [DOI] [PubMed] [Google Scholar]
- 269.Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8:422–32. doi: 10.1016/j.chom.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A. 2010;107:17345–50. doi: 10.1073/pnas.1010811107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J Gen Virol. 2009;90:448–56. doi: 10.1099/vir.0.005355-0. [DOI] [PubMed] [Google Scholar]
- 272.Khakpoor A, Panyasrivanit M, Wikan N, Smith DR. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J Gen Virol. 2009;90:1093–103. doi: 10.1099/vir.0.007914-0. [DOI] [PubMed] [Google Scholar]
- 273.Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008;374:240–8. doi: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Heaton NS, Randall G. Dengue virus and autophagy. Viruses. 2011;3:1332–41. doi: 10.3390/v3081332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–75. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.McLean JE, Wudzinska A, Datan E, Quaglino D, Zakeri Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J Biol Chem. 2011;286:22147–59. doi: 10.1074/jbc.M110.192500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens. 2011;20:297–305. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- 279.Mehal WZ, Iredale J, Friedman SL. Scraping fibrosis: expressway to the core of fibrosis. Nat Med. 2011;17:552–3. doi: 10.1038/nm0511-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, Yamasaki G. Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology. 1992;15:234–43. doi: 10.1002/hep.1840150211. [DOI] [PubMed] [Google Scholar]
- 282.Friedman SL, Roll FJ. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with Stractan. Anal Biochem. 1987;161:207–18. doi: 10.1016/0003-2697(87)90673-7. [DOI] [PubMed] [Google Scholar]
- 283.Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Goodman DS. Biochemical characteristics of isolated rat liver stellate cells. Hepatology. 1987;7:1224–9. doi: 10.1002/hep.1840070609. [DOI] [PubMed] [Google Scholar]
- 284.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Friedman SL, Wei S, Blaner WS. Retinol release by activated rat hepatic lipocytes: regulation by Kupffer cell-conditioned medium and PDGF. Am J Physiol. 1993;264:G947–52. doi: 10.1152/ajpgi.1993.264.5.G947. [DOI] [PubMed] [Google Scholar]
- 286.Kluwe J, Wongsiriroj N, Troeger JS, Gwak GY, Dapito DH, Pradere JP, et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut. 2011;60:1260–8. doi: 10.1136/gut.2010.209551. [DOI] [PubMed] [Google Scholar]
- 287.Thoen LF, Guimarães EL, Dollé L, Mannaerts I, Najimi M, Sokal E, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–60. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 288.Shimada Y, Kobayashi H, Kawagoe S, Aoki K, Kaneshiro E, Shimizu H, et al. Endoplasmic reticulum stress induces autophagy through activation of p38 MAPK in fibroblasts from Pompe disease patients carrying c.546G>T mutation. Mol Genet Metab. 2011;104:566–73. doi: 10.1016/j.ymgme.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 289.Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3:366–75. doi: 10.1016/j.molonc.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.White EJ, Martin V, Liu JL, Klein SR, Piya S, Gomez-Manzano C, et al. Autophagy regulation in cancer development and therapy. Am J Cancer Res. 2011;1:362–72. [PMC free article] [PubMed] [Google Scholar]
- 291.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113:1774–83. doi: 10.1172/JCI20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–6. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 293.Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E, et al. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal. 2012;5:ra24. doi: 10.1126/scisignal.2002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 295.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–21. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–67. doi: 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–99. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 299.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10:776–87. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–96. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 301.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–76. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ishibashi K, Uemura T, Waguri S, Fukuda M. Atg16L1, an essential factor for canonical autophagy, participates in hormone secretion from PC12 cells independently of autophagic activity. Mol Biol Cell. 2012;23:3193–202. doi: 10.1091/mbc.E12-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127:1267–76.e6. doi: 10.1016/j.jaci.2010.12.1078. [DOI] [PubMed] [Google Scholar]
- 308.Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–84. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–8. doi: 10.1126/science.1218395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–14. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–41. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Huang Y, Chuang AY, Ratovitski EA. Phospho-ΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle. 2011;10:3938–47. doi: 10.4161/cc.10.22.18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–23. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Chang Y, Yan W, He X, Zhang L, Li C, Huang H, et al. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143:177–87.e8. doi: 10.1053/j.gastro.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 316.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 317.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–20. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011;16:123–40. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 320.Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–88. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 321.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–37. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kinch L, Grishin NV, Brugarolas J. Succination of Keap1 and activation of Nrf2-dependent antioxidant pathways in FH-deficient papillary renal cell carcinoma type 2. Cancer Cell. 2011;20:418–20. doi: 10.1016/j.ccr.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 325.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest. 2008;118:79–88. doi: 10.1172/JCI33700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–27. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60:604–17. doi: 10.1042/bj0600604. [DOI] [PMC free article] [PubMed] [Google Scholar]